

Abstract
Interactions between TGFβ1 and ras signaling pathways play an important role in cancer development. Here we show that in primary mouse keratinocytes, v-rasHa does not block the early biochemical events of TGFβ1 signal transduction but does alter global TGFβ1 mediated gene expression in a gene specific manner. Expression of Smad3 dependent TGFβ1 early response genes and the TGFβ1 cytostatic gene expression response were not altered by v-rasHa consistent with an intact TGFβ1 growth arrest. However, TGFβ1 and v-rasHa cause significant alteration in genes regulating matrix remodeling as the TGFβ1 induction of extracellular matrix genes was blocked by v-rasHa but specific matrix proteases associated with cancer progression were elevated. Smad3 deletion in keratinocytes repressed normal differentiation maker expression and caused expression of Keratin 8 a simple epithelial keratin and marker of malignant conversion. Smad3 was required for the TGFβ1 cytostatic response in v-rasHa keratinocytes, but also for protease induction, keratinocyte attachment and migration. These results show that pro-oncogenic activities of TGFβ1 can occur early in carcinogenesis before loss of its tumor suppressive function and that selective regulation rather than complete inactivation of Smad3 function may be crucial for tumor progression.
Keywords: TGFβ1, ras oncogene, skin carcinogenesis, Smad3, keratinocytes, transcriptional profiling
Introduction
Transforming growth factor β1 (TGFβ1) is a cytokine with context dependent tumor suppressor and tumor promoting activity in cancer development. For most normal epithelial cells, and at early stages of tumor development TGFβ1 is believed to regulate gene expression in ways that are consistent with its function as a tumor suppressor [1]. This largely occurs through activation of the surface serine-threonine receptor complex and phosphorylation intracellular Smads, although other signaling pathways are also engaged [2]. In contrast elevated expression of TGFβ1 may enhance malignant properties of tumor cells through effects on invasion, metastasis, epithelial to mesenchymal transition or anti-tumor immunity [3]. This alteration in function of TGFβ1 during tumor progression may in part depend on the interaction with other signaling pathways such as ras that become activated during tumor progression [3]. Thus overexpression of an oncogenic ras or activation of the ERK pathway has been reported to block TGFβ1 or TGFβ superfamily signaling through several distinct mechanisms [4–9]. However, oncogenic ras and TGFβ1 can cooperate to enhance epithelial to mesenchymal transition and invasive behavior of neoplastic breast cancer cells [10,11]. However it is not clear if these studies done in immortal and cancer cell lines are applicable to normal epithelial cells.
Two-stage chemical carcinogenesis of the mouse epidermis is a widely used model to study the interaction of TGFβ1 and oncogenic ras. Mutations in the c-Ha-ras gene occur in most benign and malignant squamous tumors derived from 7,12-di methylbenz[α]anthracene (DMBA) initiation and 12-O-tetradecanoylphorbol-13-acetate (TPA) promotion [12], and this can be modeled in vitro using a retrovirus expressing v-rasHa to infect primary mouse keratinocytes (MEK). v-rasHa causes MEK hyperproliferation, altered expression of differentiation-related genes [13], and formation of papillomas in a skin graft [14]. TGFβ signaling can suppress squamous tumor formation and progression initiated by an activated cellular or viral ras oncogene [15–18] but overexpression of TGFβ1 in normal skin causes formation of highly malignant spindle cell carcinomas [19] and enhances a metastatic phenotype of benign squamous tumor cells [20]. The different roles of TGFβ1 in skin carcinogenesis may depend on the interaction of Smad2 and Smad3 with ras signaling. Both Smad2 and Smad3 expression are lost during progression of chemically induced tumors [21], and expression of v-rasHa in Smad3 null MEK causes rapid progression to squamous cell carcinoma in a skin graft system [22]. However other studies have shown that Smad3 is required for tumor formation in the 2-stage carcinogenesis model [23], suggesting context dependent interactions between ras and Smads that are critical for tumor suppression or progression in this model.
In this report we have used normal and Smad3 null MEK to examine the effect of oncogenic ras on TGFβ1 signaling in a primary epithelial cell and identify how Smad3 impacts cancer progression. Here we show that oncogenic ras alters TGFβ1 regulated gene expression in a gene specific manner producing a phenotype with characteristics of tumor suppression and tumor progression. Loss of Smad3 both enhances and inhibits TGFβ1 responses associated with progression, suggesting that selective rather than global inactivation of key TGFβ1 responses is an essential component of tumor progression.
Materials and Methods
Cell Culture
Primary keratinocytes were isolated from newborn BALB/c mice and cultured as described previously [24]. On day 3 of culture, the keratinocytes were transduced with a replication-defective retrovirus containing the v-rasHa gene [14] at a moi of 1–2 and are referred to as v-rasHa keratinocytes. The high titer of this retrovirus ensures that all keratinocytes in the culture are infected without selection. Smad3 wildtype and null keratinocytes (C57Bl/6 × SVJ) were isolated and cultured as described [22]. For induction of differentiation, confluent cultures were switched to medium containing 0.12 mM CaCl2 for 24 hours which induces a normal pattern of squamous differentiation in vitro [25]. In all experiments keratinocytes were treated with 1 ng/ml TGFβ1 (R&D) after 4 days of v-rasHa infection and RNA or protein was harvested at the indicated times. Luciferase assays to measure TGFβ1 induced gene expression were done by transfecting MEK in quadruplicate with the luciferase reporter pGL-SBE4-luc containing a TGFβ1 responsive Smad binding element [26] and a renilla luciferase internal control. Cells were treated with or without 1 ng/ml TGFβ1 for 24 hrs before analysis of luciferase activity using a dual luciferase system (Promega).
Gelatin Zymography
Conditioned media was made by incubating keratinocytes in 0.2% chelex-treated serum and 0.05mM Ca2+ with or without TGFβ1 (1ng/ml) for 24 hours. Equal volumes of supernatant was added to non-reducing sample buffer (2% SDS, 50mM Tris-HCl, 20% glycerol, and 0.01% bromophenol blue) and protease activity was detected using 10% SDS-PAGE gels copolymerized with gelatin (1mg/ml) as described [27].
3H-Thymidine Incorporation
DNA synthesis in MEK or v-rasHa MEK of indicated genotypes treated with 1 ng/ml TGFβ1 for 24 hrs was measured in quadruplicate as previously described [22].
Migration Assay
To measure keratinocyte migration, confluent monolayers of either normal or v-rasHa transduced MEK were treated for 3 hrs with 10ug/ml mitomycin C (Sigma) in PBS to prevent cell proliferation and then washed and allowed to recover for 1 hr in media. A scratch was made in the monolayer using a plastic pipette tip, and a photographic record made of the scratch distance using a 10X objective and a Spot digital camera (Nikon). Cells were treated with 1 ng/ml TGFβ1 and photographs were taken at the same location as the zero time measurement to record distance migrated into the scratch. Gap closure was measured in photographs using Spot Advanced Software, and the distance migrated was determined by subtracting the gap distance measured at 10 or 24 hrs from the initial measurement divided by 2. Replicates were measured at two different regions along the gap with at least 8 measurements made at each region. Each treatment group was done in triplicate and the experiment was repeated twice.
Attachment Assay
4 days after virus infection keratinocytes and controls were treated with 1 ng/ml TGFβ1 for 24 hrs, and then trypsinized, and plated at 5x104 cells/well into 96 well trays that had been precoated overnight with purified type I laminin, type IV collagen or fibronectin (BD Biosciences) at 10 ug/well and blocked with BSA. After incubation at 37 degrees for 3 hrs in a CO2 incubator, the wells were washed and attached cells were measured using an MTT assay (Promega). Each treatment group was done in quadruplicate.
Immunostaining
Cells were fixed with ice-cold methanol for 10 min, blocked with 5% donkey serum in 1% BSA-TBS, and stained with Smad2/3 (N-19, Santa Cruz) or H-Ras (C-20, Santa Cruz) antibodies overnight at 4 degrees. After incubation with labeled secondary antibodies cells were mounted with VECTASHIELD with DAPI (Vector Labs.) and analyzed by Zeiss LSM 510 microscope (Confocal Core Facility, NCI).
Immunoblotting
Nuclear and cytoplasmic extracts were isolated and immunoblotting was carried out as described [22]. Total cell extracts to measure keratins were isolated as described [25]. The primary antibodies used were as follows: Smad2, Smad3, and Smad4 (Zymed); H-Ras (C-20), Lamin A/C (Santa Cruz); pERK1/2, ERK1/2, pSmad2 (Cell Signaling Technology); β-actin (Chemicon); Keratin 8 (University of Iowa Hybridoma bank); and Keratin 1, 10, Loricrin (Covance).
Microarray Analysis
Total RNA was isolated from 5 independent cultures using TRIzol (Gibco BRL) and purified using the QIAGEN RNeasy Midi Kit (Qiagen), with on column DNase digestion. RNA from primary and ras MEK treated for 1, 6, 24, and 48 hrs with TGFβ1 at 1ng/ml was reverse transcribed and compared in dual fluor hybridizations to RNA from the corresponding untreated control using NCI 9000 feature cDNA microarrays. For each time point sample from primary and ras cells, 5–8 arrays including dye swaps were analyzed. To examine the role of Smad3 in the effects of oncogenic ras on TGFβ1 mediated transcriptional response, RNA was isolated from primary keratinocytes derived from Smad3 wildtype and null mice and mock infected or infected with the v-rasHa retrovirus. Microarray analyses were performed according to a published protocol [28]. Signal intensity was detected using the Axon GenePix 4000 scanner and analyzed with GenePix Pro 5.0 software (Axon). The image and data were uploaded into the NCI microarray database system supported by the Center for Information Technology of NIH for data interpretation and visualization. Gene expression ratios for each time point for a given gene were averaged after preliminary filtering, and then TGFβ1 regulated genes were identified as those with ratios greater than 1.8 or less than 0.56-fold in at least 1/8 averaged arrays across the time course. For Smad3 wild type and null data set, genes with expression ratio greater than 1.8 or less than 0.56 in at least 1/4 averaged arrays were identified as the Smad3 data set. Two independent RNA samples were analyzed for this data set. The data were further analyzed using K-means clustering to identify similar and/or distinct expression patterns in primary and ras keratinocytes following TGFβ1 treatment. All array data are deposited for public viewing in the Gene Expression Omnibus (GEO) at www.ncbi.nlm.nih.gov/geo under GEO accession GSE 3228. Expression of selected genes was measured using the LightCycler (Roche) or MyIQ system (BioRad) to validate the results of microarray data as described previously [28]. The primers for quantitative PCR were designed by PrimerSelect (DNASTAR Inc.) and sequences used are available in Supplemental Table I. Expression of Gapdh or β-actin was used as a normalization control for quantitative PCR.
Results
Oncogenic ras does not block initial TGFβ1 induced biochemical responses in primary mouse keratinocytes
To examine the effects of oncogenic ras on the TGFβ1 signaling pathway in a normal epithelial cell, we infected primary mouse epidermal keratinocytes (MEK) with a replication defective retrovirus expressing v-rasHa [14] and analyzed the initial Smad dependent steps in the TGFβ1 signal transduction pathway. High levels of Ras protein and hyperactivation of ERK1/2 were confirmed by immunoblotting (Fig.1A) and elevated Ras protein expression in all cells was demonstrated by indirect immunofluorescence (Fig.1D). Figure 1B shows that as expected v-rasHa expression elevated mRNA levels of several cell growth associated genes such as proliferin (4-fold), TGFα(17-fold), and cyclin D1 (6-fold). When control and v-rasHa transduced MEK were treated with TGFβ1 there was an increase Smad2, 3 and 4, increased phosphorylation of p-Smad2 that followed similar patterns of induction and decay in both cell types (Fig.1C), and similar nuclear translocation of Smad2/3 in both normal and v-rasHa transformed keratinocytes (Fig.1D). Western analysis (Fig.1C) showed that the total levels of Smad3 and 4 were slightly elevated in the v-rasHa MEK, but no change in levels of p-ERK1/2 were observed after treatment with TGFβ1. Thus at this level of analysis oncogenic ras does not alter TGFβ1 induced Smad phosphorylation or translocation, and TGFβ1 does not alter v-rasHa induced hyperactivation of ERK1/2.
Figure 1. Oncogenic v-rasHa does not block Smad phosphorylation or translocation.
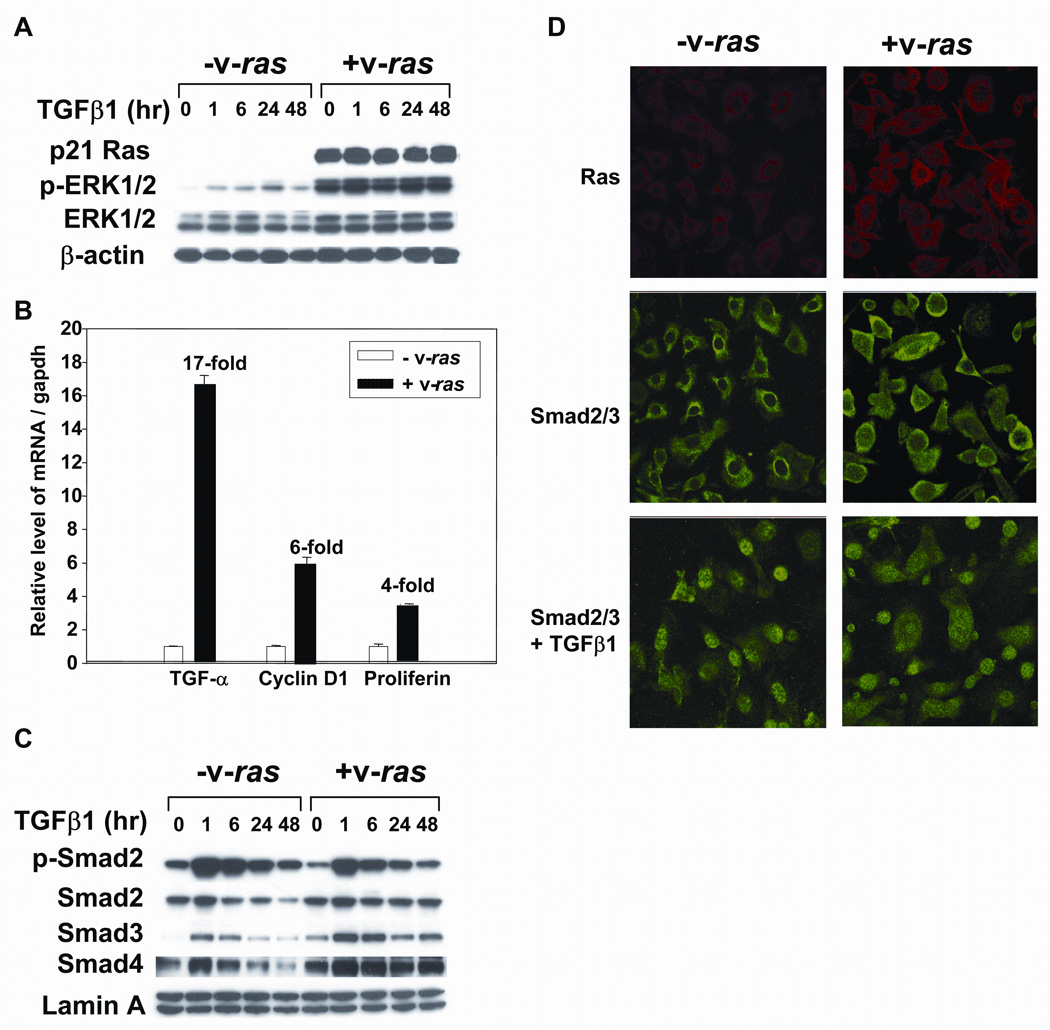
Balb/c primary keratinocytes were transduced with the v-rasHa retrovirus and then control and v-rasHa keratinocytes treated with TGFβ1 at 1ng/ml and samples taken for immunoblotting, quantitative PCR or immunofluorescence. A. Immunblotting of cellular extracts showing high levels of ras protein expression in transduced keratinocytes and elevated phosphorylation of ERK1/2 that is not altered by TGFβ1 treatment. B. Increased expression of proliferation associated genes in MEK expressing v-rasHa. RNA was isolated from control and v-rasHa transduced cells and expression of the indicated genes determined by quantitative PCR as described in Materials and Methods. The level of each transcript was normalized to that of Gapdh, and expressed as relative to the expression level in control keratinocytes. C. TGFβ1 induces similar alterations in Smad levels and phosphorylation in control and v-rasHa transduced keratinocytes. Nuclear extracts were isolated from keratinocytes at the indicated times after treatment with TGFβ1 and immunoblotted for the indicated Smads. D. v-rasHa does not prevent nuclear translocation of Smad2/3. Indirect immunofluorescence for Smad2/3 (FITC) or ras (Texas Red) in MEK or v-rasHa transduced MEK treated with TGFβ1 (1 ng/ml) for 1 hour. Nuclei were detected using DAPI staining.
Gene specific effects of oncogenic ras on TGFβ1 signaling
To assess the effects of v-rasHa on generic TGFβ1-induced gene expression we transfected MEK and v-rasHa transduced MEK with a SBE (Smad binding element)-luciferase reporter that is dependent on TGFβ1 and Smad signaling for activation. TGFβ1 stimulated SBE driven luciferase activity 6 fold in the MEK but there was only a 2 fold increase in the v-rasHa transduced MEK (Fig. 2), suggesting that despite similar immediate biochemical responses as observed in Figure 1, v-rasHa was able to inhibit TGFβ1 mediated transcriptional activation. To determine if oncogenic ras blocked all or a subset of TGFβ1 transcriptional responses we analyzed global patterns of TGFβ1 mediated gene expression in the presence or absence of v-rasHa. Gene expression patterns were compared in control and v-rasHa expressing keratinocytes after 1, 6, 24 and 48 hrs of treatment with TGFβ1. 274 TGFβ1-responsive genes were identified (Supplemental Table IA) using the average expression ratio from at least 5 arrays for each treatment and filtering for genes whose expression ratio was greater than 1.8 or less than 0.56 for at least 1 time point. A hierarchical clustering method utilizing the K-means algorithm was used to group genes based on similar expression patterns throughout the time course (Fig. 3A). For each cluster the average expression ratio of all the genes was calculated and plotted against time, as shown in Figure 3B. Two major clusters were observed: the first cluster containing 94 genes whose induction (cluster 1, n=42 genes) or repression (cluster 3, n=52 genes) by TGFβ1 was not altered by v-rasHa, and there was no statistically significant difference in the average expression ratio of genes in these clusters associated with the presence or absence or v-rasHa and a second cluster of 140 genes whose induction (cluster 2, n=66 genes) or suppression (cluster 4, n=74 genes) by TGFβ1 was reduced or blocked by v-rasHa. For these clusters by 48 hrs post TGFβ1 treatment where maximal induction or repression of gene expression by TGFβ1 occurred, the presence of v-rasHa significantly altered the expression ratio (p<.001). Thus these results show that rather than simply suppressing all TGFβ1 signaling, ras oncogene signaling in keratinocytes alters TGFβ1 mediated gene expression in a gene specific manner.
Figure 2. Oncogenic ras suppresses SBE-luciferase reporter transactivation by TGFβ1.
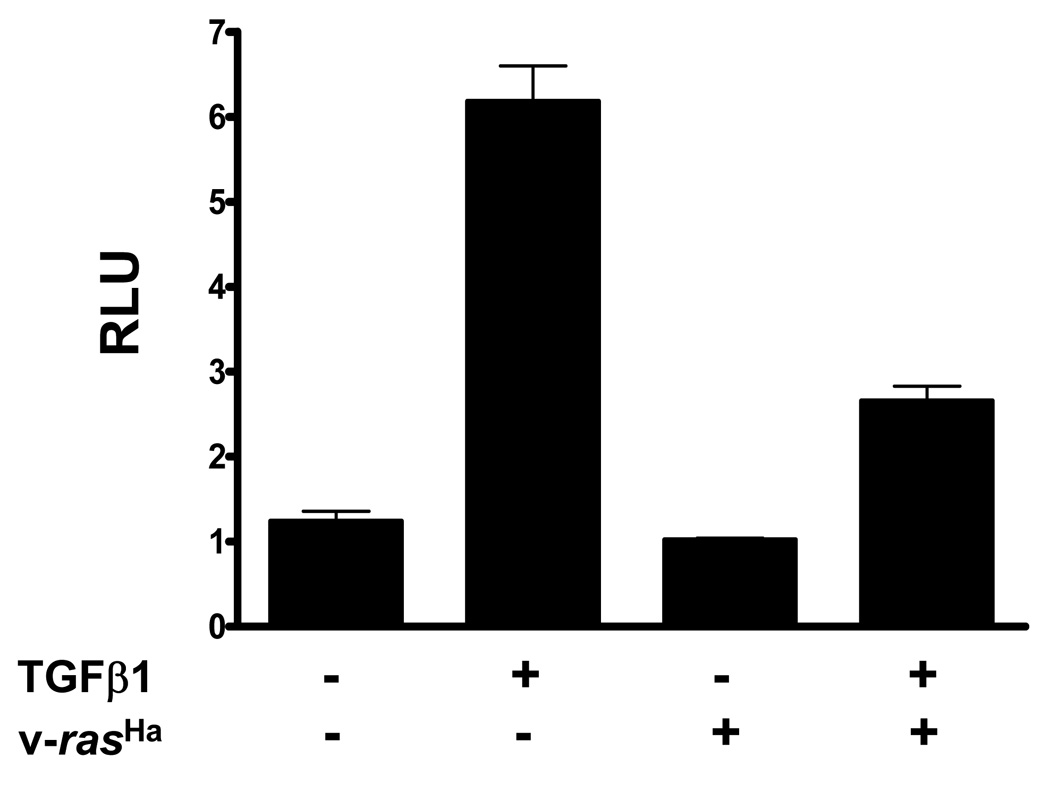
MEK and v-rasHa transduced MEK were transfected with pGL-SBE4-luc and pRLTK and luciferase activity measured in a dual luciferase assay after 24 hrs in the presence or absence of 1ng/ml TGFβ1. RLU represent SBE-luciferase values normalized to renilla luciferase for each well.
Figure 3. Gene specific alteration of TGFβ1 transcriptome by v-rasHa.
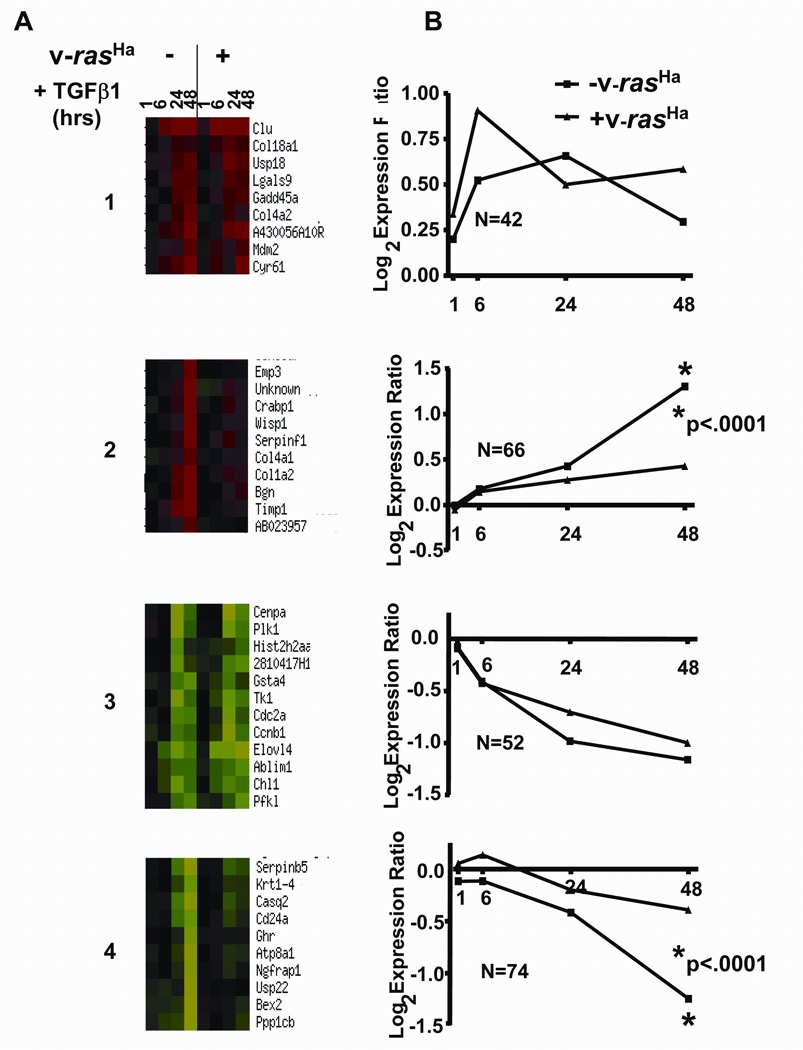
Primary and v-rasHa transduced keratinocytes were treated with TGFβ1 and microarray analyses were performed on RNA isolated at the indicated times as described in Materials and Methods. A Representative portions of major clusters of expression patterns obtained using the K-means cluster program from the NCI mAdbweb site. Cluster1: Similar induction by TGFβ1 in control and v-rasHa expressing MEK; Cluster 2: Induction by TGFβ1 suppressed in v-rasHa expressing MEK; Cluster 3: Similar repression by TGFβ1 in control and v-rasHa expressing MEK; Cluster 4: Suppression by TGFβ1 blocked in v-rasHa expressing MEK. Each time point in the heat map represents the averaged (5–8 arrays) log2 transformed expression ratio calculated from TGFβ1 treated cells over untreated control cells. Red indicates overexpression in TGFβ1 treated cells while green indicates underexpression in TGFβ1 treated cells as compared to controls. B. Plots of the average log2 expression ratio at each time point for all genes within the corresponding cluster in A. N is the number of genes in each cluster used for the analysis, and p values were computed using a nonparametric T test from the values of the 48 hr time points.
Early transcriptional responses to TGFβ1 are not blocked by oncogenic Ras
Several studies have identified a set of genes that are rapidly activated or repressed following activation of TGFβ1 signaling [29]. Figure 4 shows results from the microarray analysis (A) and quantitative PCR (B) expressed as log2 ratios for each treated time point relative to untreated control for several TGFβ early response genes. The initial pattern of TGFβ1 induction or repression of Skil, Smad7, Tieg1, Idb1, Idb2, Ier3 and c-Myc was not significantly altered by v-rasHa, although Smad7 expression was sustained through 6 hrs and there was a more rapid return of c-Myc levels to baseline in the v-rasHa keratinocytes. Since previous studies have shown that these TGFβ1 primary response genes are largely dependent on Smad3 we tested how loss of Smad3 altered gene expression of Smad7 in the v-rasHa keratinocytes. TGFβ1 induced a 4 fold increase in smad7 expression within 1 hr in the Smad3 wildtype v-rasHa MEK that was significantly reduced in the Smad3 null v-rasHa MEK as well as control MEK (Fig. 4C). Although non-Smad3 pathways may contribute to the slight induction of Smad7 by TGFβ1 in the Smad3 null keratinocytes, these results show that the TGFβ1 induction of Smad7 is not affected by overexpression of oncogenic ras.
Figure 4. Oncogenic ras does not block TGFβ1 early gene response.
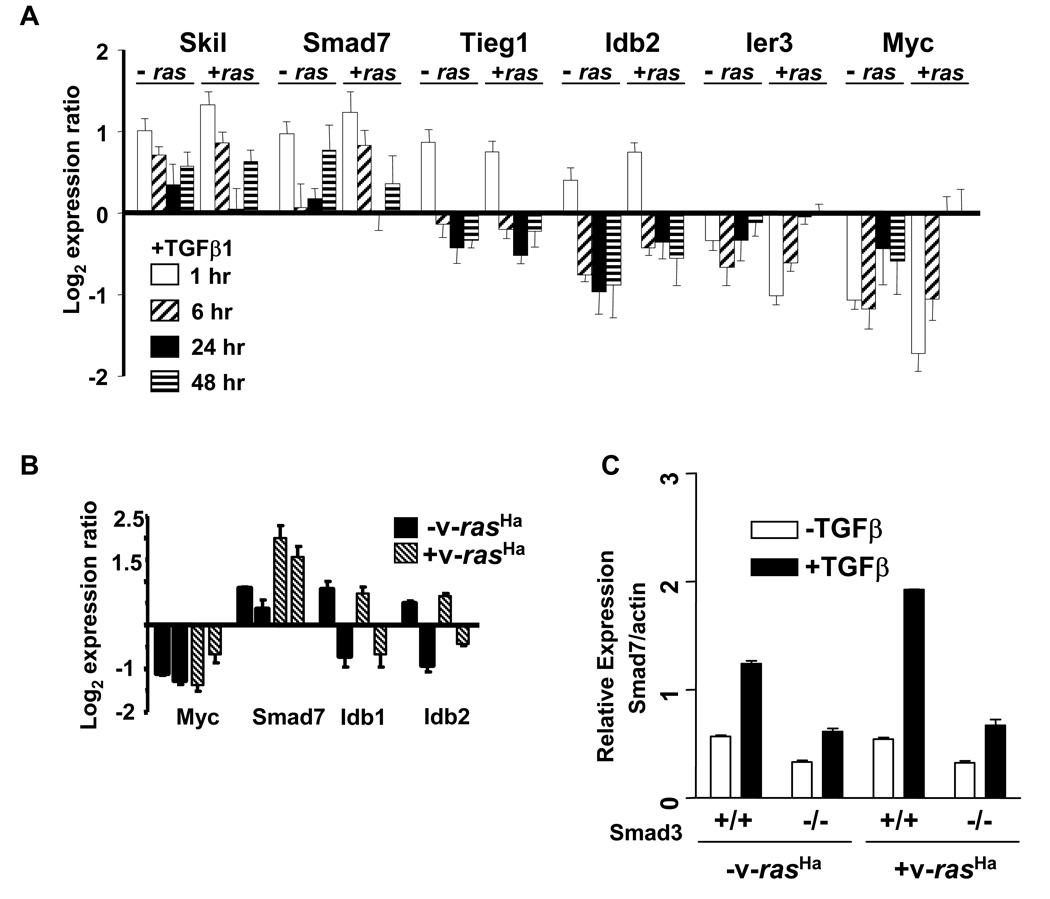
A Log2 expression ratio (TGFβ1 treated/control) from microarrays of specific early response genes at 1, 6, 24 and 48 hrs (left to right) after TGFβ1 treatment. Each histogram represents the average expression ratio expressed as treated over control from at least 5 arrays per time point analyzed. B. Quantitative PCR validation of microarray response at 1 and 6 hrs after TGFβ1 treatment for specific early response genes. Normalized quantitative PCR data is expressed as a ratio of TGFβ1 treated/control for comparison to microarray.  MEK,
MEK,  v-rasHa transduced MEK. C. Smad7 induction by TGFβ1 is Smad3 dependent in keratinocytes. Quantitative PCR analysis of Smad7 transcript levels in Smad3 wt and Smad3 null control and v-rasHa transduced MEK treated with TGFβ1 (1 ng/ml) for 1 hour.
v-rasHa transduced MEK. C. Smad7 induction by TGFβ1 is Smad3 dependent in keratinocytes. Quantitative PCR analysis of Smad7 transcript levels in Smad3 wt and Smad3 null control and v-rasHa transduced MEK treated with TGFβ1 (1 ng/ml) for 1 hour.
Expression of matrix remodeling but not cell cycle genes are blocked by ras
TGFβ1 is a potent inhibitor of normal epithelial cell proliferation and this is mediated in part through downregulation of c-myc and induction of several cyclin dependent kinase inhibitors [30]. Cell cycle associated genes were highly represented in the cluster of genes downregulated in both primary MEK and v-rasHa MEK, including Ccnb1, Ccnd2, Cdc2a, Cenpa, Gmnn, Mad2l1, Mcm5, Mki67, Odc, Plk1, Tk1, Top2a, Ung, and Tpx2 (Fig. 5A,B), and suggest that v-rasHa cannot abrogate the TGFβ1 cell cycle arrest program. To test this we treated keratinocytes with different doses of TGFβ1 for 24 hrs and measured DNA synthesis using 3H-thymidine incorporation. Despite somewhat altered kinetics of Smad7 and c-Myc response to TGFβ1 in the v-rasHa MEK (Fig. 4A,B), Figure 5C shows that TGFβ1 inhibited DNA synthesis in the primary keratinocytes and v-rasHa MEK with the same dose response, indicating that the cytostatic response program was largely intact despite high levels of v-rasHa expression.
Figure 5. Ras does not block TGFβ1 cytostatic response.
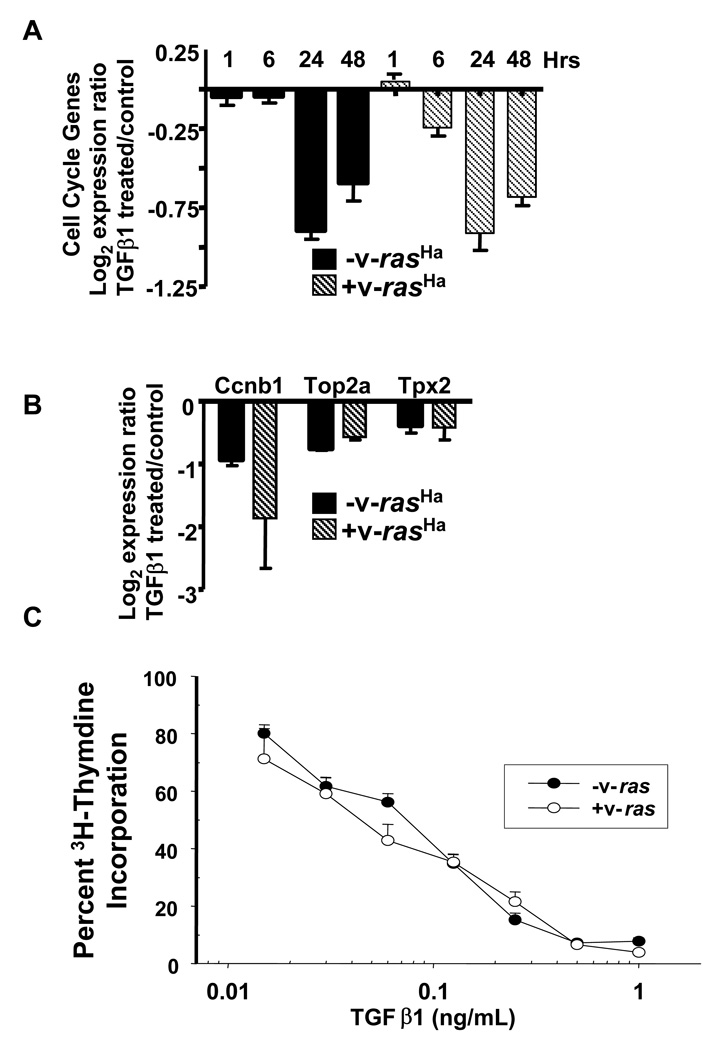
A. Histogram representing the average log2 microarray expression ratio (TGFβ1 treated/untreated) of cell cycle genes in MEK and v-rasHa transduced MEK at the indicated times after treatment. Genes in this set include: Ccnb1, Ccnd2, Cdc2a, Cenpa, Gmnn, Mad2l1, Mcm5, Mki67, Odc, Plk1, Tk1, Top2a, Ung. B. Quantitative PCR validation of Ccnb1, Top2a and Tpx2 downregulation by TGFβ1 in MEK and v-rasHa transduced MEK at 24 hrs. Histograms represent normalized expression levels expressed relative to untreated cells. C. Oncogenic ras does not block TGFβ1 cytostatic response. Normal and v-rasHa transduced MEK were treated in triplicate with the indicated concentrations of TGFβ1 and incorporation of 3H-thymidine measured after 24 hrs. Percent incorporation was measured relative to untreated control for each cell type.
While cell cycle genes were unaffected, v-rasHa blocked the TGFβ1-mediated induction of most genes encoding extracellular matrix proteins including Bgn, Col1A1, Col5A1, Pcdh7, Itbp1 and Sparc. Figure 6A shows the averaged log2 expression ratio of multiple ECM genes relative to untreated cells, at 1, 6 24 and 48 hrs after treatment with TGFβ1 from microarray analysis. Figure 6B shows validation for expression of 7 individual genes at 24 hrs post TGFβ1 treatment by quantitative PCR. TGFβ1 induced an approximately 3 fold increase in expression for these genes by 48 hrs and this induction was blocked by v-rasHa (Fig. 6A). In contrast, gene expression of two proteases critical in matrix remodeling and tumor cell invasion, Urokinase (uPA) and Mmp9 were significantly upregulated in the v-rasHa keratinocytes following treatment with TGFβ1, while induction of tissue inhibitor of matrix metalloproteinases (Timp1) was blocked by v-rasHa (Fig. 6C). We collected conditioned media from MEK and v-rasHa MEK treated with TGFβ1 for 24 hrs and analyzed protease activity using gelatin zymography. Figure 6D shows that TGFβ1 treated v-rasHa MEK expressed much higher levels compared to v-rasHa MEK and control or TGFβ1 treated MEK of a gelatinase that is likely to be pro-MMP9 based on co-migration with a recombinant standard. Higher molecular weight bands detected on the zymogram likely represent aggregates of MMP9. In addition we detected a band of gelatinase activity at 35Kd specifically in the TGFβ1 treated v-rasHa MEK that could represent a distinct MMP, although the exact identity remains to be determined. Since changes in matrix and protease expression are often linked to cell migration, we used a monolayer scratch assay to measure effects of TGFβ1 on MEK migration. v-rasHa transduced MEK migrated faster into the scratch compared to normal MEK (Fig. 6E). TGFβ1 treatment inhibited migration of MEK (p=.001) but the migration of v-rasHa MEK was reproducibly stimulated 1.5 fold by TGFβ1. Despite this we did not observe any TGFβ1 enhancement of keratinocyte invasion through a matrigel coated membrane irrespective of the presence or absence of v-rasHa (data not shown).
Figure 6. Ras alters TGFβ1 regulation of extracellular matrix and keratinocyte migration.
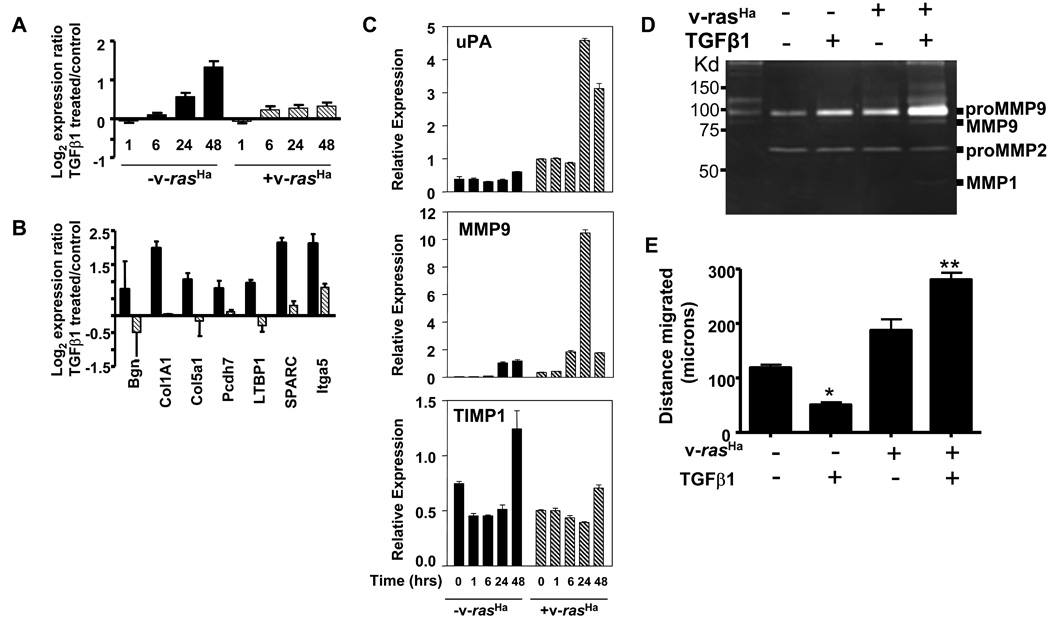
A. TGFβ1 mediated induction of extracellular matrix genes is blocked by oncogenic ras. Each histogram represents the average log2 microarray expression ratio (TGFβ1 treated/untreated) for a set of ECM genes induced by TGFβ1 at the indicated times after treatment. Genes in this set include: Bgn, Col1a1, Col1a2, Col4a1, Col4a2, Col5a1, Col6a2, Efemp2, Fbln5, Itga5, Ltbp1, Nid1, Sparc, Tnn. B. Quantitative PCR validation of the inhibition of ECM gene induction by v-rasHa for indicated genes at 24 hrs after TGFβ1 treatment. Histograms represent normalized expression levels expressed relative to untreated cells,  -v-rasHa,
-v-rasHa,  + v-rasHa C. Quantitative PCR analysis of MMP9, urokinase (uPA) and TIMP1 transcript levels in MEK and v-rasHa MEK treated with TGFβ1. Cells were treated with TGFβ1 (1ng/ml) and RNAs were isolated at the indicated times. Expression levels for each gene were normalized to β-actin levels. D. Gelatin zymography of conditioned media from MEK and v-rasHa MEK treated with TGFβ1 (1 ng/ml) for 24 hrs. Equal volumes of conditioned media were loaded on a gelatin zymogram and position of proteases determined as described in Materials and Methods. Identity of MMP9 was based on co-migration with a recombinant MMP9, and relative size based on molecular wt standards. Other arrows indicate additional bands of gelatinase activity. E. TGFβ1 inhibits migration of MEK but stimulates migration of v-rasHa MEK. Data show migration of the indicated cell type cultured in the presence or absence of TGFβ1 for 18 hrs. Confluent monolayers were treated with mitomycin C, scratched and treated with TGFβ1 for 18 hrs, and distance migrated calculated as described in Materials and Methods. Each histogram represents the average distance migrated in two defined fields for each of 4 wells per treatment group. Data are representative of three independent experiments. * significantly different from untreated MEK at p=.001, ** significantly different from untreated v-rasHa MEK, as determined by unpaired Students T Test.
+ v-rasHa C. Quantitative PCR analysis of MMP9, urokinase (uPA) and TIMP1 transcript levels in MEK and v-rasHa MEK treated with TGFβ1. Cells were treated with TGFβ1 (1ng/ml) and RNAs were isolated at the indicated times. Expression levels for each gene were normalized to β-actin levels. D. Gelatin zymography of conditioned media from MEK and v-rasHa MEK treated with TGFβ1 (1 ng/ml) for 24 hrs. Equal volumes of conditioned media were loaded on a gelatin zymogram and position of proteases determined as described in Materials and Methods. Identity of MMP9 was based on co-migration with a recombinant MMP9, and relative size based on molecular wt standards. Other arrows indicate additional bands of gelatinase activity. E. TGFβ1 inhibits migration of MEK but stimulates migration of v-rasHa MEK. Data show migration of the indicated cell type cultured in the presence or absence of TGFβ1 for 18 hrs. Confluent monolayers were treated with mitomycin C, scratched and treated with TGFβ1 for 18 hrs, and distance migrated calculated as described in Materials and Methods. Each histogram represents the average distance migrated in two defined fields for each of 4 wells per treatment group. Data are representative of three independent experiments. * significantly different from untreated MEK at p=.001, ** significantly different from untreated v-rasHa MEK, as determined by unpaired Students T Test.
Tumor suppressive and prooncogenic functions of Smad3
We previously showed that Smad3 null keratinocytes transduced with the v-rasHa retrovirus undergo rapid malignant conversion in a skin grafting bioassay [22], but others have demonstrated that Smad3 null mice are resistant to 2-stage chemical carcinogenesis [23]. To test how Smad3 functions to promote or suppress oncogenic functions of ras we examined responses to TGFβ1 in Smad3 wildtype and null keratinocytes expressing v-rasHa. Microarray analysis of Smad3 wildtype and null keratinocytes treated with TGFβ1 for 48 hours showed that most cell cycle associated genes analyzed in the earlier data set (Fig. 5) were downregulated by TGFβ1 in both Smad3 wildtype, Smad3 null and Smad3 wildtype/v-rasHa keratinocytes but this downregulation was abolished in Smad3 null/v-rasHa keratinocytes (Fig. 7A), and Supplemental Table II. In agreement with these gene expression data, TGFβ1 treatment caused 95% inhibition of DNA synthesis in Smad3 wildtype and wildtype/v-rasHa MEK and 80% inhibition of DNA synthesis in Smad3 null MEK. In contrast, while the Smad3 null/v-rasHa had only a slight increase in baseline levels of DNA synthesis compared to the Smad3 wildtype/v-rasHa cells, growth inhibition by TGFβ1 was only 40% (Fig. 7B). The microarray studies also suggested that keratinocyte differentiation was altered in the absence of Smad3 (Supplemental Table II). Expression of normal differentiation markers such as Keratins 1 and 10 are lost during progression of mouse squamous tumors and Keratin 8, a marker for simple epithelia that is reexpressed upon malignant conversion of keratinocytes [31,32]. To test if Smad3 regulates keratinocytes differentiation in vitro, we increased media calcium from 0.05mM to 0.12mM and measured expression of differentiation markers by immunoblotting. As expected Smad3 wildtype MEK switched to 0.12 mM calcium expressed elevated levels of the normal differentiation markers Keratins 1, 10 and Loricirin, but not Keratin 8. In contrast, Smad3 null MEK expressed detectable levels of Keratin 8 under basal conditions and this was induced following a calcium switch, while the induction of Keratins 1 and 10 and Loricrin was blocked (Fig. 7C). v-rasHa MEK expressed high levels of Keratin 8 under all conditions and did not show induction of these differentiation markers (data not shown).
Figure 7. Loss of Smad3 suppresses TGFβ1 cytostatic response and promotes aberrant differentiation.
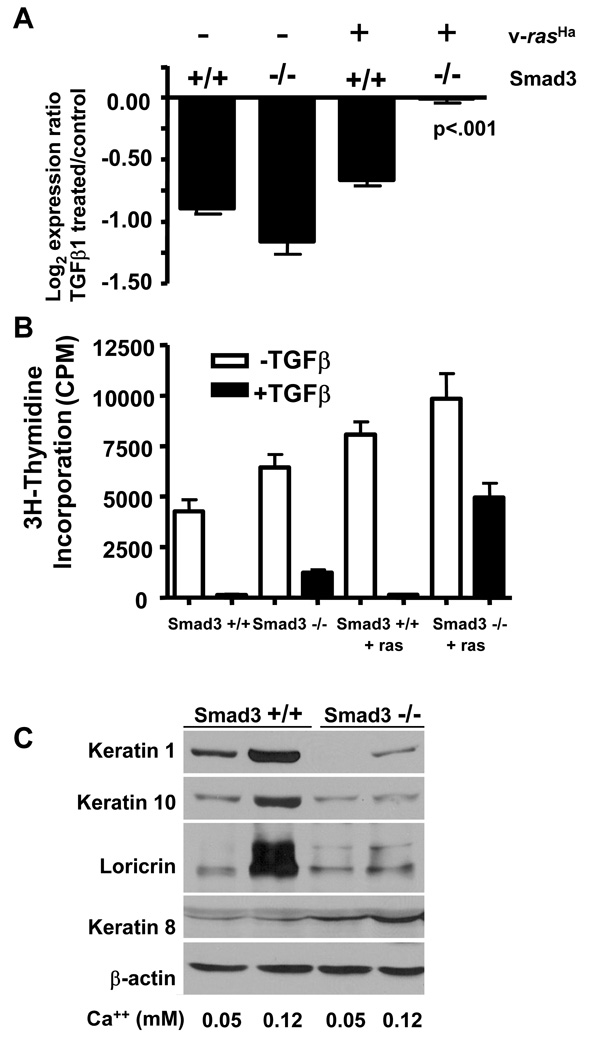
A. Downregulation of cell cycle genes by TGFβ1 is lost only in Smad3 null/v-ras keratinocytes. Averaged log2 expression ratio (TGFβ1 treated/control) of cell cycle genes identified on microarrays of the indicated genotypes. Genes in this set include: Mad2l, Pmscl1, Birc5, Tk1, Mcm5, Ung, Top2a, Cdc2a, Plk, Nusap1, Ccnb1, Stk6, Cenpa. B. Cytostatic response to TGFβ1 in Smad3 wildtype and null keratinocytes. DNA synthesis was measured using a 3H-thymidine incorporation assay in control or TGFβ1 treated cells of the indicated genotypes as described in Materials and Methods. C. Loss of Smad3 blocks normal differentiation and causes abnormal induction of Keratin 8. Smad3 wildtype or null MEK were cultured in 0.05 mM calcium media until confluent then switched to 0.12 mM calcium medium to induce differentiation. Total cellular lysates were harvested after 24 hrs and keratin expression determined by immunoblotting with antibodies specific for mouse Loricrin or Keratin 1, 10 or 8.
Unlike the cell cycle response, the loss of Smad3 did not significantly alter the reduction in ECM gene expression levels caused by expression of v-rasHa (Supplemental Table II). Thus for both biglycan (Bgn) and Ltbp1, the levels of expression in both Smad3 wildtype/v-rasHa and Smad3null/v-rasHa keratinocytes were 5 fold lower than corresponding control cells and there was no induction by TGFβ1 (data not shown). In contrast, Figure 8A shows that the ability of TGFβ1 to induce either Mmp9 or uPA gene expression in v-rasHa MEK was blocked by the loss of Smad3. It is likely that slight differences between kinetics and level of induction of these genes compared to Figure 6C reflects strain specific differences between Balb/c and Smad3 wildtype keratinocytes. In addition, TGFβ1 stimulated attachment of v-rasHa MEK was dependent on Smad3. Figure 8B shows that TGFβ1 caused a 1.5 to 2-fold increase in attachment of Smad3 wildtype/v-rasHa keratinocytes to untreated, laminin I, type IV collagen, and fibronectin coated tissue culture plastic. For all substrates attachment of TGFβ1 treated v-rasHa MEK ranged from 1.4 to 1.8 fold higher than TGFβ1 treated MEK (data not shown). Thus v-rasHa by itself enhances TGFβ1 induced attachment. In the absence of Smad3, attachment to fibronectin increased slightly, nevertheless the TGFβ1 stimulated increase was abolished or significantly reduced for each matrix substrate tested (Fig. 8B). Paralleling the accelerated wound healing in the Smad3 null mouse [33] Smad3 null MEK migrated faster than the wildtype cells in the scratch assay (data not shown) but TGFβ1 suppressed the migration of the Smad3 null/v-rasHa MEK but not Smad3 wildtype v-rasHa MEK (Fig. 8C). Taken together, these data show that Smad3 functions to both suppress and promote an oncogenic ras driven cancer phenotype.
Figure 8. Smad3 loss blocks TGFβ1 induction of proteases and migration in v-rasHa transduced keratinocytes.
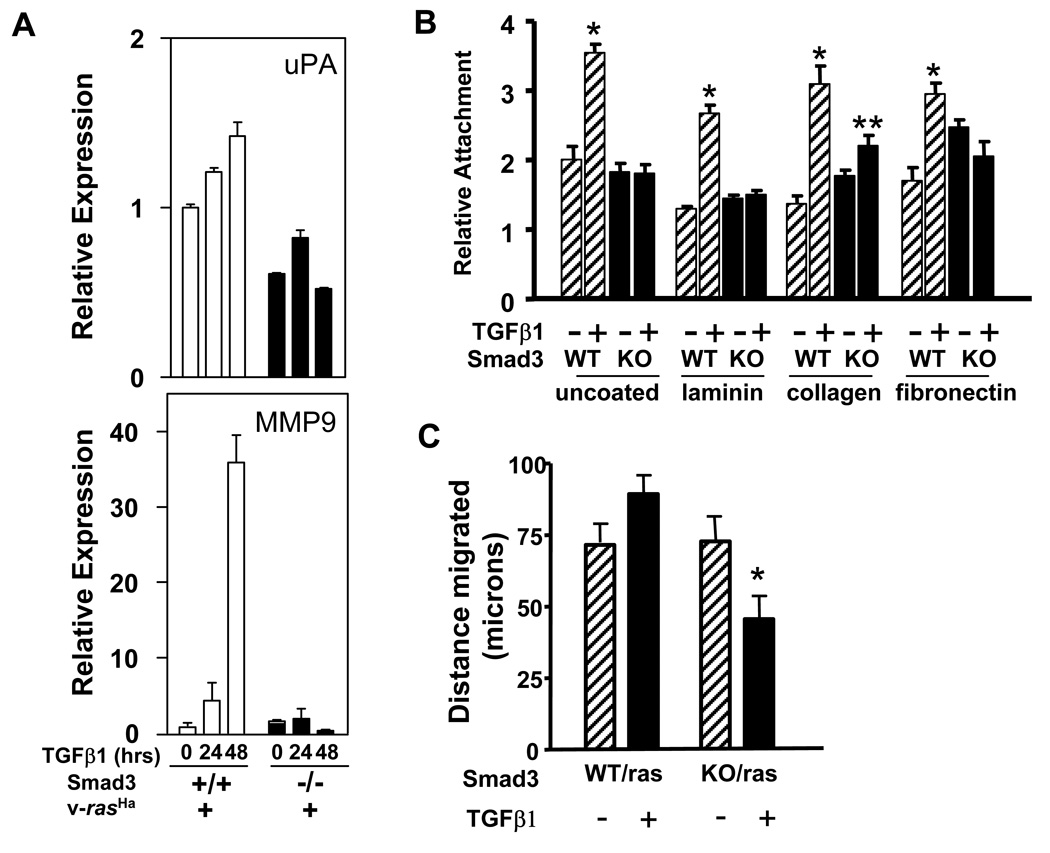
A. Measurement of the relative transcript levels of urokinase (uPA) and MMP9 in Smad3 wt and Smad3 null MEK expressing v-rasHa treated with TGFβ1 (1 ng/ml) for the indicated times by Quantitative PCR. Histograms show the relative transcript level normalized to β-actin. B. Attachment of Smad3 wildtype and null v-rasHa MEK to the indicated substrates in the presence or absence of TGFβ1 (1 ng/ml). Histograms represent the average attached cells in quadruplicate wells measured as indicated in Materials and Methods, and normalized to the attachment of uninfected wildtype keratinocytes. Experiment was repeated twice with similar results. * : significantly different from untreated on same matrix using a two-tailed T-test at p<.001, ** significantly different from untreated on same matrix at p=.006 C. Distance migrated into a denuded monolayer of Smad3 wildtype and null v-rasHa MEK in the presence and absence of TGFβ1 (1 ng/ml). Migration was measured in triplicate wells as indicated in Materials and Methods 10 hours after scratching the cell monolayer and treating with TGFβ1. * : significantly different from Smad3 wildtpe/v-rasHa using a two-tailed T-test at P<.05.
Discussion
Activation of ras in normal and neoplastic epithelial cell lines can antagonize and synergize with TGFβ1 signaling [4,6,7,10,34] providing a mechanism for subversion of TGFβ1 tumor suppressor function and pro-oncogenic switching of TGFβ1. Despite these studies, TGFβ1 clearly suppresses tumor development in vivo where ras is the activating oncogene, suggesting that immortalized cell lines may not illuminate the complex interactions between these two pathways. In this report we have utilized a primary culture model in which mouse epidermal keratinocytes are transduced with a high titer retrovirus expressing v-rasHa, to more clearly define interactions between ras and TGFβ1 signaling at the earliest stages of cancer development. Our data show that oncogenic ras and hyperactivation of the ERK1/2 pathway did not block Smad2 phosphorylation, nuclear translocation or regulation of Smad3 dependent early response genes. However, over a 48 hr time course ras significantly altered the pattern of TGFβ1 mediated gene expression, with 50% of TGFβ1 regulated genes (both induced and suppressed) blocked while 34% were unaffected by oncogenic ras. This is similar to repression of 32% of TGFβ1 induced genes in 3T3 fibroblasts by activated ras oncogene [35]. In agreement with previous studies [36,29], our data show that in primary keratinocytes the majority of TGFβ1 regulated gene expression is Smad3 dependent, since TGFβ1 induction or repression of these genes is blocked, or significantly reduced in Smad3 null keratinocytes. Surprisingly, Smad3/4 dependent early response genes [29] are unaffected by oncogenic ras, while the induction of many extracellular matrix genes, that are well defined primary targets of TGFβ1 and Smad3 is blocked by v-rasHa. These data suggest that additional factors may be important in targeting specific Smad3 dependent genes for suppression, or that secondary events occur in a TGFβ1 gene expression cascade that contribute to blocked expression of specific Smad3 dependent genes. In contrast to the blocked ECM gene expression observed in this study under conditions of hyperactivated ras and ERK1/2, TGFβ1 treatment of ras transformed 3T3 fibroblasts reversed the inhibition of matrix gene expression and the transformed phenotype caused by ras [35]. Furthermore, an array study of the HaCaT human keratinocyte cell line showed that many of these same matrix genes were dependent on ERK1/2 for responsiveness to TGFβ1 [37]. Taken together, these studies suggest that the context of ERK1/2 activation distinctly alters the readout of the TGFβ1 transcriptome.
Within the complex changes in the TGFβ1 transcriptome caused by ras we find compelling evidence that components of both the tumor suppressor and pro-oncogenic functions of TGFβ1 occur at the same time in this early stage cancer model. Thus while ras does alter the kinetics of c-myc downregulation and smad7 induction by TGFβ1, suggesting a degree of alteration in the growth arrest program, nearly all identifiable cell cycle associated genes were similarly downregulated by TGFβ1 in the presence or absence of v-rasHa, and TGFβ1 caused a nearly identical dose dependent growth arrest in both cell types. This is consistent with reduced tumor formation in mouse models of TGFβ1 overexpression [17,38], and suggests that at the earliest stage of cancer development modeled by expression of v-rasHa in primary epidermal keratinocytes, the central cytostatic effect of TGFβ1 that correlates with homeostatic growth control and tumor suppression is intact.
Regulation of extracellular matrix remodeling is one of the most significant functions of TGFβ1 that has important functions in wound healing, inflammation, fibrosis and cancer development [39]. Our data show that v-rasHa caused significant suppression of both basal and TGFβ1 induced extracellular matrix gene expression in primary keratinocytes. This decrease in matrix gene expression was accompanied by induction of at least two protease genes urokinase and Mmp9, increased gelatinolytic activity in the conditioned media of TGFβ1 treated v-rasHa MEK, and inhibition of TGFβ1 induced TIMP1. Furthermore, we found that in an in vitro monolayer migration assay, TGFβ1 stimulated v-rasHa MEK migration but blocked migration of normal MEK, the latter consistent with effects of TGFβ1 signaling in studies of keratinocyte migration and in vivo wound healing [40–42]. Similar induction of urokinase and Mmp9 by TGFβ1 has been linked to enhanced migration in transformed keratinocytes cell lines [43,44] but this is the first report to link this induction to the earliest stage of cancer development in keratinocytes. How ras signaling switches the migration response to TGFβ1 from inhibition to stimulation is not yet clear but is likely related both to extracellular matrix remodeling and to changes in expression of integrins such as α6β4 which are crucial for enhanced migration and invasion of skin cancer cells [45]. Nevertheless, these results show that in the context of a ras oncogene, TGFβ1 causes significant changes expression of genes involved in matrix protein expression and matrix remodeling enzymes that are associated with the phenotype of many neoplastic cells in vivo [46], Thus at the earliest stage of cancer development components of TGFβ1 tumor suppressive and oncogenic activities are present.
While Smad3 is a critical mediator of TGFβ1 transcriptional responses [29], it is not frequently mutated in human cancers [1]. Here we find that deletion of Smad3 both enhances and inhibits expression of a tumor progression phenotype. This is similar to xenograft and transgenic models of breast cancer, where inhibition of Smad2/3 or inactivation of the TGFβ receptor enhanced tumorigenesis of premalignant cells but blocked metastasis of malignant cell lines [50,51]. Thus while loss of Smad3 by itself modestly reduces responsiveness of keratinocytes to TGFβ1 mediated growth inhibition it significantly alters keratinocyte differentiation, blocking calcium mediated induction of suprabasal markers Keratin 1 and 10 and the granular layer marker Loricrin. Most importantly, loss of Smad3 causes induction of Keratin 8 a marker of simple epithelia and malignant conversion of squamous tumors [48]. Similar effects of Smad7, cripto-1 and the Alk5 inhibitor SB431542 on keratinocyte differentiation have been reported by us [49], and suggest that TGFβ1 through Smad3 is important for maintaining squamous differentiation and suppressing simple epithelial differentiation. Our results also show that in ras expressing keratinocytes Smad3 is essential for the TGFβ1 cytostatic response, but is also essential for the induction of protease genes, cell attachment and migration by TGFβ1. The observation that deletion of Smad3 does not completely block TGFβ1 mediated growth inhibition suggests that Smad2 or other non-Smad signaling pathways are also essential for the TGFβ1 cytostatic response and that Smad3 may be primarily responsible for regulating squamous differentiation. Indeed it is possible that ras activation acts on these other pathways but this is only evident in the absence of Smad3. In a previous study we showed that v-rasHa transduced Smad3 null keratinocytes rapidly progress to squamous cell carcinoma in an in vivo graft model due in part to abrogation of a TGFβ1 induced senescence response [22]. In contrast, Smad3 null mice are resistant to tumor promotion and skin tumor induction [47], suggesting that the effects of Smad3 on cancer progression in the skin model are complex. It is possible that loss of TGFβ1 responsiveness, altered oncogene induced senescence and suppressed squamous differentiation in grafted Smad3 null/v-rasHa keratinocytes may be sufficient to accelerate malignant conversion this may not be sufficient within the context of an intact epidermis. Taken together these data suggest that squamous tumor progression requires selective rather than complete inactivation of Smad3 function such that the cytostatic and differentiation arms of TGFβ1 signaling are lost but matrix remodeling and migratory phenotype are retained.
Supplementary Material
Acknowledgements
The authors would like to acknowledge the support and encouragement of Dr. Stuart Yuspa and members of LCBG, NCI as well as Brad Martin for initial help with migration and attachment assays.
Grant Support: NIH grant CA122109 (A.B.Glick) and the Intramural Research Program of the Center for Cancer Research, National Cancer Institute, NIH
Reference List
- 1.de Caestecker MP, Piek E, Roberts AB. Role of transforming growth factor-beta signaling in cancer. J Natl Cancer Inst. 2000;92:1388–1402. doi: 10.1093/jnci/92.17.1388. [DOI] [PubMed] [Google Scholar]
- 2.Massague J. TGF-beta signal transduction. Annu Rev Biochem. 1998;67:753–791. doi: 10.1146/annurev.biochem.67.1.753. [DOI] [PubMed] [Google Scholar]
- 3.Glick AB. TGFbeta1, Back to the Future: Revisiting its Role as a Transforming Growth Factor. Cancer Biol Ther. 2004;3:276–283. doi: 10.4161/cbt.3.3.849. [DOI] [PubMed] [Google Scholar]
- 4.Kretzschmar M, Doody J, Timokhina I, Massague J. A mechanism of repression of TGFbeta/Smad signaling by oncogenic Ras. Genes Dev. 1999;13:804–816. doi: 10.1101/gad.13.7.804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kretzschmar M, Doody J, Massague J. Opposing BMP and EGF signalling pathways converge on t he TGF-β family mediator Smad1. Nature. 1997;389:618–622. doi: 10.1038/39348. [DOI] [PubMed] [Google Scholar]
- 6.Lo RS, Wotton D, Massague J. Epidermal growth factor signaling via Ras controls the Smad transcriptional co-repressor TGIF. EMBO J. 2001;20:128–136. doi: 10.1093/emboj/20.1.128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Saha D, Datta PK, Beauchamp RD. Oncogenic ras represses transforming growth factor-beta/Smad signaling by degrading tumor suppressor Smad4. J Biol Chem. 2001;276:29531–29537. doi: 10.1074/jbc.M100069200. [DOI] [PubMed] [Google Scholar]
- 8.Liu X, Sun Y, Weinberg RA, Lodish HF. Ski/Sno and TGF-beta signaling. Cytokine Growth Factor Rev. 2001;12:1–8. doi: 10.1016/s1359-6101(00)00031-9. [DOI] [PubMed] [Google Scholar]
- 9.Dai C, Liu Y. Hepatocyte growth factor antagonizes the profibrotic action of TGF-beta1 in mesangial cells by stabilizing Smad transcriptional corepressor TGIF. J Am Soc Nephrol. 2004;15:1402–1412. doi: 10.1097/01.asn.0000130568.53923.fd. [DOI] [PubMed] [Google Scholar]
- 10.Oft M, Akhurst RJ, Balmain A. Metastasis is driven by sequential elevation of H-ras and Smad2 levels. Nat Cell Biol. 2002;4:487–494. doi: 10.1038/ncb807. [DOI] [PubMed] [Google Scholar]
- 11.Shi Y, Massague J. Mechanisms of TGF-beta signaling from cell membrane to the nucleus. Cell. 2003;113:685–700. doi: 10.1016/s0092-8674(03)00432-x. [DOI] [PubMed] [Google Scholar]
- 12.Balmain A, Brown K. Oncogene activation in chemical carcinogenesis. Adv Cancer Res. 1988;51:147–182. doi: 10.1016/s0065-230x(08)60222-5. [DOI] [PubMed] [Google Scholar]
- 13.Dlugosz AA, Cheng C, Williams EK, Dharia AG, Denning MF, Yuspa SH. Alterations in murine keratinocyte differentiation induced by activated rasHa genes are mediated by protein kinase C-alpha. Cancer Res. 1994;54:6413–6420. [PubMed] [Google Scholar]
- 14.Roop DR, Lowy DR, Tambourin PE, et al. An activated Harvey ras oncogene produces benign tumours on mouse epidermal tissue. Nature. 1986;323:822–824. doi: 10.1038/323822a0. [DOI] [PubMed] [Google Scholar]
- 15.Glick AB, Lee MM, Darwiche N, Kulkarni AB, Karlsson S, Yuspa SH. Targeted deletion of the TGF-beta 1 gene causes rapid progression to squamous cell carcinoma. Genes Dev. 1994;8:2429–2440. doi: 10.1101/gad.8.20.2429. [DOI] [PubMed] [Google Scholar]
- 16.Amendt C, Schirmacher P, Weber H, Blessing M. Expression of a dominant negative type II TGF-β receptor in mouse skin results in an increase in carcinoma incidence and an acceleration of carcinoma development. Oncogene. 1998;17:25–34. doi: 10.1038/sj.onc.1202161. [DOI] [PubMed] [Google Scholar]
- 17.Blessing M, Nanney LB, King LE, Hogan BL. Chemical skin carcinogenesis is prevented in mice by the induced expression of a TGF-β related transgene. Teratog Carcinog Mutagen. 1995;15:11–21. doi: 10.1002/tcm.1770150103. [DOI] [PubMed] [Google Scholar]
- 18.Wang XJ, Greenhalgh DA, Bickenbach JR, et al. Expression of a dominant-negative type II transforming growth factor beta (TGF-β) receptor in the epidermis of transgenic mice blocks TGF-β-mediated growth inhibition. Proc Natl Acad Sci U S A. 1997;94:2386–2391. doi: 10.1073/pnas.94.6.2386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cui W, Fowlis DJ, Bryson S, et al. TGFβ1 inhibits the formation of benign skin tumors, but enhances progression to invasive spindle carcinomas in transgenic mice. Cell. 1996;86:531–542. doi: 10.1016/s0092-8674(00)80127-0. [DOI] [PubMed] [Google Scholar]
- 20.Weeks BH, He W, Olson KL, Wang XJ. Inducible expression of transforming growth factor beta1 in papillomas causes rapid metastasis. Cancer Res. 2001;61:7435–7443. [PubMed] [Google Scholar]
- 21.He W, Cao T, Smith DA, Myers TE, Wang XJ. Smads mediate signaling of the TGFbeta superfamily in normal keratinocytes but are lost during skin chemical carcinogenesis. Oncogene. 2001;20:471–483. doi: 10.1038/sj.onc.1204117. [DOI] [PubMed] [Google Scholar]
- 22.Vijayachandra K, Lee J, Glick AB. Smad3 regulates senescence and malignant conversion in a mouse multistage skin carcinogenesis model. Cancer Res. 2003;63:3447–3452. [PubMed] [Google Scholar]
- 23.Li AG, Lu SL, Zhang MX, Deng C, Wang XJ. Smad3 knockout mice exhibit a resistance to skin chemical carcinogenesis. Cancer Res. 2004;64:7836–7845. doi: 10.1158/0008-5472.CAN-04-1331. [DOI] [PubMed] [Google Scholar]
- 24.Dlugosz AA, Glick AB, Tennenbaum T, Weinberg WC, Yuspa SH. Isolation and utilization of epidermal keratinocytyes for oncogene research. 1995:3–20. doi: 10.1016/0076-6879(95)54003-2. [DOI] [PubMed] [Google Scholar]
- 25.Yuspa SH, Kilkenny AE, Steinert PM, Roop DR. Expression of murine epidermal differentiation markers is tightly regulated by restricted extracellular calcium concentrations in vitro. J Cell Biol. 1989;109:1207–1217. doi: 10.1083/jcb.109.3.1207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zawel L, Dai JL, Buckhaults P, et al. Human smad3 and smad4 are sequence-specific transcription activators. Mol Cell. 1998;1:611–617. doi: 10.1016/s1097-2765(00)80061-1. [DOI] [PubMed] [Google Scholar]
- 27.Rhee JS, Diaz R, Korets L, Hodgson JG, Coussens LM. TIMP-1 alters susceptibility to carcinogenesis. Cancer Res. 2004;64:952–961. doi: 10.1158/0008-5472.can-03-2445. [DOI] [PubMed] [Google Scholar]
- 28.Darwiche N, Ryscavage A, Perez-Lorenzo R, et al. Expression profile of skin papillomas with high cancer risk displays a unique genetic signature that clusters with squamous cell carcinomas and predicts risk for malignant conversion. Oncogene. 2007;26:6885–6895. doi: 10.1038/sj.onc.1210491. [DOI] [PubMed] [Google Scholar]
- 29.Yang YC, Piek E, Zavadil J, et al. Hierarchical model of gene regulation by transforming growth factor beta. Proc Natl Acad Sci U S A. 2003;100:10269–10274. doi: 10.1073/pnas.1834070100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Seoane J, Pouponnot C, Staller P, Schader M, Eilers M, Massague J. TGFbeta influences Myc, Miz-1 and Smad to control the CDK inhibitor p15INK4b. Nat Cell Biol. 2001;3:400–408. doi: 10.1038/35070086. [DOI] [PubMed] [Google Scholar]
- 31.Larcher F, Bauluz C, Diaz-Guerra M, et al. Aberrant expression of the simple epithelial type II keratin 8 by mouse skin carcinomas but not papillomas. Mol Carcinog. 1992;6:112–121. doi: 10.1002/mc.2940060206. [DOI] [PubMed] [Google Scholar]
- 32.Markey AC, Lane EB, Churchill LJ, MacDonald DM, Leigh IM. Expression of simple epithelial keratins 8 and 18 in epidermal neoplasia. J Invest Dermatol. 1991;97:763–770. doi: 10.1111/1523-1747.ep12486607. [DOI] [PubMed] [Google Scholar]
- 33.Ashcroft GS, Yang X, Glick AB, et al. Mice lacking Smad3 show accelerated wound healing and an impaired local inflammatory response. Nat Cell Biol. 1999;1:260–266. doi: 10.1038/12971. [DOI] [PubMed] [Google Scholar]
- 34.Oft M, Peli J, Rudaz C, Schwarz H, Beug H, Reichmann E. TGF-beta1 and Ha-Ras collaborate in modulating the phenotypic plasticity and invasiveness of epithelial tumor cells. Genes Dev. 1996;10:2462–2477. doi: 10.1101/gad.10.19.2462. [DOI] [PubMed] [Google Scholar]
- 35.Wisdom R, Huynh L, Hsia D, Kim S. RAS and TGF-beta exert antagonistic effects on extracellular matrix gene expression and fibroblast transformation. Oncogene. 2005;24:7043–7054. doi: 10.1038/sj.onc.1208870. [DOI] [PubMed] [Google Scholar]
- 36.Piek E, Ju WJ, Heyer J, et al. Functional characterization of transforming growth factor beta signaling in Smad2- and Smad3-deficient fibroblasts. J Biol Chem. 2001;276:19945–19953. doi: 10.1074/jbc.M102382200. [DOI] [PubMed] [Google Scholar]
- 37.Zavadil J, Bitzer M, Liang D, et al. Genetic programs of epithelial cell plasticity directed by transforming growth factor-beta. Proc Natl Acad Sci U S A. 2001;98:6686–6691. doi: 10.1073/pnas.111614398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pierce DF, Gorska AE, Chytil A, et al. Mammary tumor suppression by transforming growth factor β1 transgene expression. Proc Natl Acad Sci U S A. 1995;92:4254–4258. doi: 10.1073/pnas.92.10.4254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Roberts AB, McCune BK, Sporn MB. TGF-beta: regulation of extracellular matrix. Kidney Int. 1992;41:557–559. doi: 10.1038/ki.1992.81. [DOI] [PubMed] [Google Scholar]
- 40.Tsuboi R, Sato C, Shi CM, Ogawa H. Stimulation of keratinocyte migration by growth factors. J Dermatol. 1992;19:652–653. doi: 10.1111/j.1346-8138.1992.tb03751.x. [DOI] [PubMed] [Google Scholar]
- 41.Chan T, Ghahary A, Demare J, et al. Development, characterization, and wound healing of the keratin 14 promoted transforming growth factor-beta1 transgenic mouse. Wound Repair Regen. 2002;10:177–187. doi: 10.1046/j.1524-475x.2002.11101.x. [DOI] [PubMed] [Google Scholar]
- 42.Hosokawa R, Urata MM, Ito Y, Bringas P, Jr, Chai Y. Functional significance of Smad2 in regulating basal keratinocyte migration during wound healing. J Invest Dermatol. 2005;125:1302–1309. doi: 10.1111/j.0022-202X.2005.23963.x. [DOI] [PubMed] [Google Scholar]
- 43.Santibanez JF, Frontelo P, Iglesias M, Martinez J, Quintanilla M. Urokinase expression and binding activity associated with the transforming growth factor beta1-induced migratory and invasive phenotype of mouse epidermal keratinocytes. J Cell Biochem. 1999;74:61–73. [PubMed] [Google Scholar]
- 44.Santibanez JF, Guerrero J, Quintanilla M, Fabra A, Martinez J. Transforming growth factor-beta1 modulates matrix metalloproteinase-9 production through the Ras/MAPK signaling pathway in transformed keratinocytes. Biochem Biophys Res Commun. 2002;296:267–273. doi: 10.1016/s0006-291x(02)00864-1. [DOI] [PubMed] [Google Scholar]
- 45.Tennenbaum T, Yuspa SH, Grover A, et al. Extracellular matrix receptors and mouse skin carcinogenesis: altered expression linked to appearance of early markers of tumor progression. Cancer Res. 1992;52:2966–2976. [PubMed] [Google Scholar]
- 46.Larsen M, Artym VV, Green JA, Yamada KM. The matrix reorganized: extracellular matrix remodeling and integrin signaling. Curr Opin Cell Biol. 2006;18:463–471. doi: 10.1016/j.ceb.2006.08.009. [DOI] [PubMed] [Google Scholar]
- 47.Li AG, Lu SL, Zhang MX, Deng C, Wang XJ. Smad3 knockout mice exhibit a resistance to skin chemical carcinogenesis. Cancer Res. 2004;64:7836–7845. doi: 10.1158/0008-5472.CAN-04-1331. [DOI] [PubMed] [Google Scholar]
- 48.Cheng C, Kilkenny AE, Roop D, Yuspa SH. The v-ras oncogene inhibits the expression of differentiation markers and facilitates expression of cytokeratins 8 and 18 in mouse keratinocytes. Mol Carcinog. 1990;3:363–373. doi: 10.1002/mc.2940030608. [DOI] [PubMed] [Google Scholar]
- 49.Shukla A, Ho Y, Liu X, Ryscavage A, Glick AB. Cripto-1 alters keratinocyte differentiation via blockade of transforming growth factor-beta1 signaling: role in skin carcinogenesis. Mol Cancer Res. 2008;6:509–516. doi: 10.1158/1541-7786.MCR-07-0396. [DOI] [PubMed] [Google Scholar]
- 50.Tian F, DaCosta BS, Parks WT, et al. Reduction in Smad2/3 signaling enhances tumorigenesis but suppresses metastasis of breast cancer cell lines. Cancer Res. 2003;63:8284–8292. [PubMed] [Google Scholar]
- 51.Siegel PM, Shu W, Cardiff RD, Muller WJ, Massague J. Transforming growth factor beta signaling impairs Neu-induced mammary tumorigenesis while promoting pulmonary metastasis. Proc Natl Acad Sci U S A. 2003;100:8430–8435. doi: 10.1073/pnas.0932636100. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.