

Abstract
Botulinum neurotoxins (BoNTs), proteins secreted by the bacteria genus Clostridium, represent a group of extremely lethal toxins and a potential bioterrorism threat. As the current therapeutic options are of a predominantly prophylactic nature and cannot be used en masse, new strategies and ultimately potential treatments are desperately needed to combat any widespread release of these neurotoxins. In these regards, our laboratory has been working on developing new alternatives to treat botulinum intoxication through the development of inhibitors of the light chain proteases, the etiological agent which causes BoNT intoxication. Such a strategy has required the construction of two high throughput screens and small molecule non-peptidic libraries; excitingly, inhibitors of the BoNT/A protease have been uncovered and are being optimized via structure activity relationship studies.
Keywords: botulism, botulinum neurotoxin A, botulinum neurotoxin B, light chain metalloprotease, protease inhibitors
Introduction
Botulinum neurotoxin (BoNT), a protein secreted by the bacteria Clostridium botulinum, C. botyricum and C. barati is a highly potent substance with a lethal dose of only 1 ng/kg of body weight for BoNT/A (Bossi et al., 2006). It is classified as a Category A agent by the Centers for Disease Control, representing agents which are easily disseminated, cause high mortality and require special action for public health preparedness. Hence, the intentional use of this toxic substance for lethal purposes is a concern among public health officials. As the world enters a new era of bioterrorism, the use of biological weapons, such as the widespread release of BoNT would have a devastating effect on any population exposed to the toxin.
Clostridium botulinum is a Gram-positive anaerobic rod-shaped bacteria, which forms spores and is found in the soil (Shukla and Sharma, 2005; Arnon et al., 2001). There are seven serotypes of Clostridium botulinum (A–G) (Table 1) classified by the immunological differences of the neurotoxins each strain produces (BoNT/A–G) (Shukla and Sharma, 2005). Of the seven serotypes, BoNT/A is the most poisonous to humans followed by BoNT/B and BoNT/E. These three serotypes of BoNTs are also the most common cause of human botulism (Franciosa et al., 2003). Exposure to the neurotoxins typically occurs by the consumption of spoiled home canned food. The bacteria can also be cultured in the laboratory for large scale production of toxin for clinical purposes (Schantz and Johnson, 1992). Yet, it is the ease of production and transport that causes major concerns of the malicious use of BoNT.
Table 1.
List of the 7 serotypes of the botulinum neurotoxin, including the cleavage site of the protein cleaved by each light chain of the serotype and which type of host they affect. VAMP (vesicle associated membrane protein) also known as synaptobrevin; SNAP-25 (synaptosomal associated protein).
| Serotype | Host | Protein substrate | Cleavage site |
|---|---|---|---|
| A | Human | SNAP-25 | Gln197-Arg198 |
| B | Human | VAMP | Gln76-Phe77 |
| C | Animal | SNAP-25, Syntaxin | Arg198-Ala199 Lys253-Ala254 |
| D | Animals | VAMP | Lys59-Leu60 |
| E | Human, Fish | SNAP-25 | Arg180-Ile181 |
| F | Human | VAMP | Gln58-Lys59 |
| G | Human | VAMP | Ala81-Ala82 |
BoNTs are lethal due to the high specificity and efficiency with which they cleave proteins important for neurotransmitter release. The mechanism of BoNT intoxication is a four step process that results in muscular and respiratory paralysis which, if not treated in a timely manner will ultimately lead to death (Finkelstein, 1990; Hambleton, 1994; Montecucco and Schiavo, 1994; Montecucco et al., 1996; Rossetto et al., 2001). The BoNTs are produced by Clostridium botulinum as a single 150 kDa polypeptide chain with three functional domains (binding, translocation and catalytic). (Figure 1) Cleavage of the polypeptide chain results in the formation of two polypeptide chains: a light (LC) and heavy (HC) chain linked by a disulfide bond and noncovalent interactions (Schiavo et al., 1992 (a)). (Fig. 1) The LC (50 kDa) is a zinc metalloprotease that cleaves soluble N-ethylmaleimide-sensitive fusion proteins (SNARE) located at the nerve endings (Baldwin et al., 2007). The SNARE proteins including synaptosomal associated protein (SNAP-25), syntaxin and synaptobrevin also known as vesicle associated membrane protein (VAMP) are required for synaptic vesicle membrane fusion (Sutton et al, 1998). The fusion of the synaptic vesicle is necessary for release of acetylcholine into the synaptic cleft for normal muscle function. The BoNT LC cleaves these important proteins resulting in flaccid paralysis. Interestingly, each BoNT LC serotype cleaves an unique peptide bond located on the SNARE proteins. BoNT/A, C, and E cleave SNAP-25 (Binz et al., 1994), BoNT/B, D, G and E cleave VAMP, (Barr et al., 2005; Schiavo et al., 1992 (b)), whereas BoNT/C exclusively cleaves syntaxin (Table 1; Figure 2).
Fig. 1.

BoNT/A holotoxin (reprinted with permission from Trends Biochem. Sci. 2002, 57, 552.) The light chain endopeptidase (catalytic domain) is depicted in red color.
Fig. 2.

Overview of the SNARE proteins targeted by the 7 different serotypes of botulinum neurotoxin.
Structurally, BoNT LCs are globular proteins sharing 36.5 % sequence identity between the serotypes (Lacy and Stevens, 1999). The active sites of the 7 serotypes contain the HEXXH (X is any amino acid) Zn-binding motif. The catalytic machinery of the LCs are very similar to each other, therefore the specificity for the substrate is due to the residues forming the cavity that provides access to the active site.
Once a person is suspected of suffering from botulism, treatment must be administered immediately. Depending on the severity of botulism, a person may need medical assistance for weeks or even months. A severe case of BoNT poisoning requires the use of mechanical ventilation, due to paralysis of the respiratory system. Along with physical treatment, antitoxins are available for humans suffering from botulism. There are antitoxins for each type of BoNT, which can sequester the neurotoxin and the complex is then cleared from the body. The antitoxin, that is the only pharmacological treatment available, is an equine product, which is commonly administered to patients as a trivalent form with antibodies specific to BoNT/A, B and E. There is a risk of an allergic reaction with the use of the antitoxin and patients must be monitored closely for any reaction to the antitoxin (Black and Gunn, 1980). Along with the risk of an allergic reaction, the long term effects of continued use of antitoxins are unknown (Villar et al., 2006). Furthermore, antitoxin supply is limited and a large scale biological attack with BoNT on a population would be devastating (Arnon et al., 2001; Villar et al., 2006). For these reasons, other treatments for botulism, especially those effective after BoNT enters neuronal cells, are highly desirable. In this light, small molecule inhibitors of BoNT targeting the toxin after cellular entry would represent a very attractive approach (Dickerson and Janda, 2006; Burnett et al., 2007).
Botulinum Neurotoxin A Inhibitors
With the BoNT/A light chain being a zinc protease, we have focused on designing small molecules containing a zinc coordinating moiety. Among the variety of groups that coordinate zinc via mono or bidentate modes (Jacobsen et al., 2007), the moiety that is of particular interest to our laboratory is the hydroxamate. The hydroxamate functionality is a monoanionic bidentate chelator of zinc, a widely used inhibitor motif of matrix metalloproteinases (Skiles et al., 2004). Unfortunately, this moiety has also been tagged as a “non-druggable” functional group because of potential indiscriminate metal chelation (O’Brien et al., 2000) and potential high clearance due to hydroxamate hydrolysis and proteolysis (Summers et al., 1987). However, even with these issues, suberoylanilide hydroxamic acid (Vorinostat) was approved by the FDA to treat patients with cutaneous T-cell lymphoma (Grant et al., 2007). Therefore, we sought to develop inhibitors containing the hydroxamate “warhead” on different scaffolds that would bind tightly to the active site of the enzyme, thus minimizing their promiscuity for other proteases and/or metals.
To achieve our goals, we established and optimized a high-throughput FRET-based assay (Boldt et al., 2006 (a)) using the commercially available SNAPtide™ (List biological Laboratories, Inc., Campbell, CA), a truncated sequence of the native BoNT/A LC substrate, SNAP-25 (Shine, 2003). Using this assay, we have analyzed a library of hydroxamates, generated in situ from 150 randomly chosen carboxylic acids (Boldt et al., 2006(b)). From the initial screen, five compounds were found to give 50% or more inhibition at 50 μM concentration, and out of these five lead structures, para-chlorocinnamic hydroxamate (1) displayed an IC50 value of 15 μM, (Figure 3) and was thus selected for further structure-activity relationship (SAR) studies. Thus, a series of compounds were synthesized, to reveal that while replacement of the chloro substituent was not tolerated, interestingly introduction of another chloro substituent in the ortho-position (Figure 4, compound 6) resulted in the most potent nonpeptidic BoNT/A inhibitor to date, with IC50 value <1 μM (Boldt et al., 2006 (b)).
Fig. 3.

Structures of ‘hit’ compounds from the initial in situ screen. With an IC50 of 15 μM, 4-chloro-cinnamic hydroxamate (1) was the most potent one.
Fig. 4.
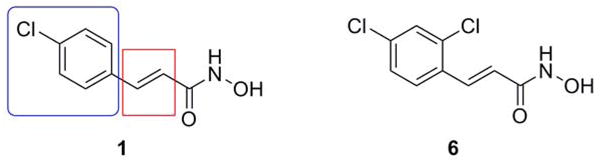
Structure-activity relationship (SAR) study sectors on the original ‘hit’ (1) and the structure of the new lead structure with improved potency (6).
Subsequently, the X-ray crystallographic structures of BoNT/A light chain with both 4-chlorocinnamic hydroxamate (1) and 2,4-dichlorocinnamic hydroxamate (6) were reported (Silvaggi et al., 2007). Apart from the expected coordination of the hydroxyl oxygen of the hydroxamate moiety to the Zn(II) atom (Figure 5), the phenyl ring of the inhibitors were observed to bind into a pocket formed by the hydrophobic residues Ile161, Phe194 and Phe369. According to the crystal structure, the increased potency of 6 compared to 1 results from the favorable interaction of the additional chlorine atom with the Arg 363 residue, making it an almost “perfect fit” with the active site of the enzyme (Silvaggi et al., 2007).
Fig. 5.
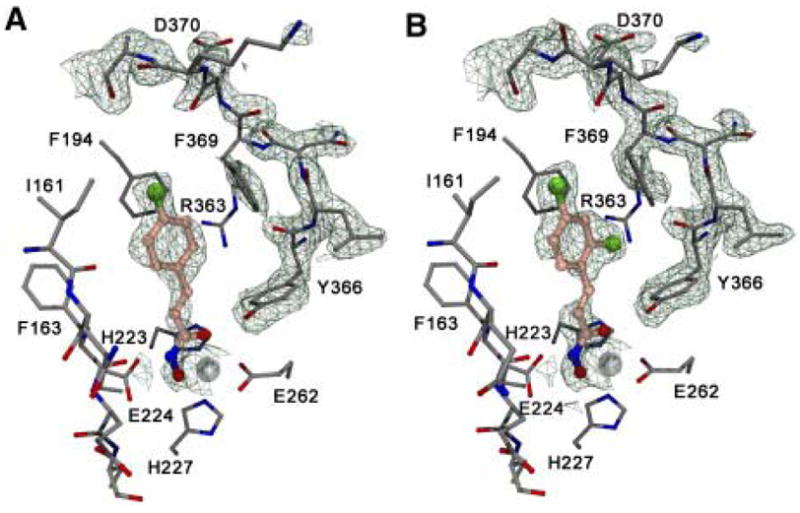
Crystal structures of 1 (A) and 6 (B) in the active site of BoNT/A LC protease (adapted with permission from Chem. Biol. 2007, 14, 533).
To further explore the flexibility of the active site and the nature of the interaction of the substituent in the ortho-position, and at the same time trying to improve the potency of the lead structure 6 (Capkova et al., 2007), a new series of SAR’s were conducted. For this purpose, we chose two structural modifications: (a) fusing an additional aromatic ring to the benzene moiety of the parent lead structure and (b) replacing the ortho-Cl atom with substituents of varying electronegativity and steric dimensions (Figure 6). In the case of the ring fused derivatives, we rationalized that ‘freezing out’ the conformational dynamics could contribute to increased stability of the enzyme-inhibitor complex (Choi and Silverman, 2002). In order to enhance the potential for hydrogen bonding interactions within the catalytic cleft, we introduced a series of fused ring derivatives with varying hydrogen bonding capability (Figure 6). In the case of the derivatives modified in the ortho-position, we hypothesized that a strong dipole/electronegative character between Arg363 and the substituent at the ortho position would result in a tighter binding thereby increasing the inhibition of the derivative (Silvaggi et al., 2007). To verify this hypothesis, we designed a series of compounds bearing ortho-substituents with varying electronegativities (Figure 6). While the increased conformational constraint imported by the inclusion of another aromatic ring resulted in dramatic loss of inhibitory activity, thus refuting our first hypothesis, some of the more electronegative ortho-substituted derivatives displayed significant inhibitory activity in the low micromolar or submicromolar range. The best substituents proved to be the electron withdrawing groups, namely Br, CF3 and NO2. However, similar inhibitory activity was observed in case of the methyl-substituted derivative, which questions the nature of the interaction between the substituent in the ortho-position and the Arg363 residue. Unfortunately, no clear trend was observed between the inhibitory activity and the electron donating/withdrawing character of the substituent, but this study yielded three new small molecule inhibitors with comparable potency to the parent molecule (Capkova et al., 2007).
Fig. 6.

Second-generation SAR of BoNT/A protease hydroxamate inhibitors.
Additionally, the flexibility of the active site of BoNT/A LC and its ability to undergo dramatic rearrangements in order to accommodate different inhibitors, uncovered by the X-ray studies (Silvaggi et al., 2007), was once more confirmed when we performed another high-throughput screen of a second library of hydroxamates, which yielded the adamantane acetic acid hydroxamate (10) (Figure 7) as a new lead scaffold for the development of BoNT/A inhibitors (Capkova et al., 2008). A rigorous kinetic analysis of inhibitor 10 showed a KI of 460 ± 80 nM. Our results suggested a competitive mechanism of inhibition, however, due to the nature of the adamantane scaffold we were concerned about the possibility of aggregation of the inhibitor. This effect has been recently thoroughly investigated by Shoichet (Feng and Shoichet, 2006) and detergents were found to disrupt these nonspecific interactions. Gratifyingly, there was no difference in inhibitory activity observed either in the presence or in the absence of detergent, indicating that no aggregation takes place (Capkova et al., 2008).
Fig. 7.
Structure of adamantane acetic acid hydroxamate (10), the most potent inhibitor of BoNT/A LC protease to date (KI = 460 nM) and a new lead structure for inhibitor development.
11: 60LSELDRADALQAGAS(K(Dnp))76FE(Dpa(Mca))79SAAKLKRKYWWKNLK94
12: 60LSELDDRADALQAG(Pya)74SQ(Nop)77ESSAAKLKRKYWWKNLK94
Native: 60LSELDRADALQAGASQFETSAAKLKRKYWWKNLK94
Closely connected to any search for protease inhibitors is the development of reliable assay for evaluating their biological activity. In the case of high-throughput assays, the method of choice is one employing a fluorescence resonance energy transfer (FRET) substrate (Ullmann, 2006). For BoNT/A, the most widely used FRET substrate is the SNAPtide™, but it suffers from some major drawbacks, including: (a) SNAPtide™ has a KM of more than 1 mM, resulting in any assay with this substrate being conducted under substrate limiting conditions; (b) the low catalytic efficiency of BoNT/A with SNAPtide™ (kcat/KM approx. 4300 M−1s−1) generally requires enzyme concentrations approaching 100 nM to provide adequate reproducibility (Boldt et al. 2006(a)). Thus, while the SNAPtide™ has proved suitable for the screening of large libraries and for crude IC50 determinations, it is inadequate for precise kinetic analyses such as the evaluation of the mechanism of inhibition. Other, HPLC based assays, make use of a substrate developed by Schmidt (Schmidt and Bostian, 1995; Schmidt and Bostian, 1997) which employs either the BoNT/A LC or the nicked and reduced holotoxin (Sukonpan et al., 2004). However, again these assays suffer from similar drawbacks as the SNAPtide™ assay due to the nature of the substrate, a 17-amino acid truncated sequence of SNAP-25 with a KM of 0.8 mM (Sukonpan et al., 2004).
We sought to develop an assay wherein the substrate could be varied from below its KM to at least 5-fold above KM as well as operating with enzyme concentrations in the low to subnanomolar range. To meet these requirements, a truncated SNAP-25 (141–206) was engaged. This 66-amino acid sequence thought to contain the majority of the recognition elements of the native substrate (Breidenbach and Brunger, 2004) was chemically synthesized. BoNT/A LC cleaves SNAP-25 between residues Gln197 and Arg198; using HPLC, the 9-amino acid peptide product of BoNT/A catalysis may be separated from other components within the reaction mixture. Furthermore, peptide product detection may be accomplished using either UV absorbance or mass spectral analysis. The limit of quantitation (LOQ) for the UV analysis is approximately 100 nM, thus requiring either high concentrations of enzyme or excessive incubation times. On the other hand, mass spectral detection coupled with the use of a stable isotopically labeled (SIL) internal standard achieved an LOQ for the peptide product of less than 10 nM, allowing a 10-fold reduction in the working enzyme concentration. By these means, we have developed an accurate, sensitive and reliable assay for the kinetic analysis of BoNT/A small molecule inhibitors (Capkova et al., 2008).
Working Towards a High Throughput Botulinum Neurotoxin B Assay
As stated (vide supra), our laboratory has spent considerable efforts on developing inhibitors targeting the proteolytic light chain of BoNT/A (Boldt et al., 2006 (b); Capkova et al., 2007). Following this success we have focused our attention to the next most common serotype causing botulism, BoNT/B. The lethal dose of BoNT/B is about three times less potent than A, at 3 ng/kg of body weight (Kozaki et al., 1974). Hence, this BoNT serotype has also fueled fear among the global community that this neurotoxin could be also developed into a weapon with deadly results. Yet, while considerable attention has been centered on the development of assays to identify inhibitors of the BoNT/A LC protease, scant research has been devoted to the discovery of BoNT/B LC protease inhibitors. We view this paucity mainly from the standpoint of a lack of high throughput assays for this serotype. Indeed, inhibitors that have been reported, are either small molecules with modest potency (Adler et al., 1998;Hanson et al., 2000) or peptides (Anne et al., 2003(a); Anne et al., 2003(b); Blommaert et al., 2004), which have typically poor pharmacokinetic/dynamic properties (Smith et al., 1994).
To address these deficiencies our group has begun to validate a high throughput assay which would allow us to identify inhibitors of the BoNT/B LC protease. Our strategy has focused upon a cognate peptide substrate to synaptobrevin, which has a FRET pair attached between the cleavage site of a chemically prepared peptide. In this context we note that FRET pairs which bracket the cleavage sites of targeted peptides have been employed frequently as a mechanism to monitor enzymatic activity of numerous proteins (Hemmila and Hurskainen, 2002). Careful analysis of the BoNT/B LC substrate synaptobrevin has allowed us to design and synthesize a proprietary peptide substrate based on the core sequence of VAMP. Accordingly, the length of the substrate, along with the location of the FRET pair was devised based on published results for the BoNT/B light chain substrate requirements (Shone et al., 1993; Shone et al., 1994). Based on these hypotheses cleavage of the chemically synthesized substrate by the BoNT/B protease should cause an increase of fluorescence, which can be monitored in real time. Finally, the combination of a FRET-peptide substrate combined with a fluorescence plate reader will engender a high throughput assay.
To date our laboratory has developed a peptide (11) significantly different from the FRET-peptide substrate developed by the Roques laboratory (12) (Anne, et al., 2001) in an attempt to improve on the kinetic parameters of their substrate (Figure 8). A 35 amino acid substrate was synthesized that is virtually identical to the central region of synaptobrevin (Figure 8), which is important for binding and cleavage by the BoNT/B protease (Shone and Roberts, 1994). Thus, we initiated this effort by embedding a FRET pair using conserved amino acid substitutions within this sequence. Hence, glutamine at position 76 was replaced with a lysine so that 2,4-dinitrophenyl (DNP) could be coupled to the free amine of lysine to form half of the FRET pair. Lysine was also chosen as the amino acid to couple DNP, this was undertaken so that the side chain of lysine would have the ability to orientate the DNP away from the catalytic activity of the BoNT/B LC and thus catalytic efficiency would not be diminished by the addition of DNP to the peptide. The other half of the FRET pair was to contain 7-methoxycoumarin acetic acid (MCA), which would be appended to the unnatural amino acid diaminopropionic acid (Dpa). Our choice of Dpa stems from the fact that this unnatural amino acid, Dpa, is structurally similar to serine, the native residue found in synaptobrevin at position 79. In comparison, 12, incorporates pyrenylalanine (pyr) and p-nitrophenylalanine (Nop) as the FRET pair at positions 74 and 77, respectively. We note our preference for MCA, over pyr due to the smaller chemical structure of MCA. Our hypothesis was that the smaller fluorophore, would enhance the catalytic efficiency of the BoNT/B LC for 11. We also hoped that the location where we inserted the FRET pair would not disrupt the catalytic activity of the enzyme.
Fig. 8.

Peptides synthesized and incorporated into high throughput assays for the detection of inhibitors of BoNT/B LC. (11) Developed in our laboratory: K(DNP) = 2,4-dinitrophenyl (DNP) attached to lysine, Dpa(MCA)= diaminopropionic acid (Dpa) attached to 7-methoxycoumarin acetic acid (MCA); (12) Developed by the Roques laboratory (Anne, et al., 2004): Pya = pyrenylalanine, Nop = p-nitrophenylalanine; Native: A fragment of the native substrate containing the core central region (syb 60–94) of synaptobrevin (the substrate of the BoNT/B LC).
Unfortunately, the kinetic analysis of our FRET-peptide substrate (11) gave rather poor kinetic parameters when compared to the Roques’ synthetic FRET-substrate and the native substrate (Table 2). The KM of 11 (63 μM) was similar to the value reported for 12 (47 μM), however, there is a large discrepancy between the two substrates in terms of kcat. The kcat of 12 was 45 sec−1, while our substrate had a value of 0.17 sec−1. Thus, 11 binds as well as the 12 and better than the native substrate, however, the catalytic turnover of 11 was only modest. This is evident by comparing the catalytic efficiency of the three peptides, 12 has the highest catalytic efficiency (9.6 × 105 s−1M−1) which is one order of magnitude greater than the native substrate (7.2 × 104 s−1M−1) and two orders of magnitude greater than 11 (2.7 × 103 s−1M−1). An explanation for the difference in catalytic efficiency between our substrate and the native substrate is the addition of the FRET pair which is what typically occurs when molecules, such as chromophores are added to a peptide that is cleaved by an enzyme (Hayes and Mellor, 2002). A more perplexing question is the increase in catalytic efficiency of 12 compared to the native substrate. An explanation for this difference in catalytic efficiency is a single amino acid substitution made within 12. The Roques’ substrate contains a single amino acid substitution at position 79 where a threonine is replaced with a serine. This small substitution, even though located well away from the cleavage site of the peptide has been reported to double the kcat (Shone and Roberts, 1994). Hence, taking advantage of this substitution resulted in the Roques laboratory producing a FRET-substrate with a catalytic efficiency greater than the native substrate.
Table 2.
Kinetic parameters of the FRET peptide substrate of the central region of synaptobrevin: the substrate of BoNT/B LC produced by our laboratory (11), the Roques laboratory (12) (Anne, et al., 2001) and the native substrate (syb 60–94) (Shone and Roberts, 1994).
| Substrate | KM(μM) | kcat(s−1) | kcat/KM (s−1M−1) |
|---|---|---|---|
| 11a | 63 | 0.17 | 2.7 × 103 |
| 12b | 47 | 45 | 9.6 × 105 |
| Nativeb | 330 | 24 | 7.3 × 104 |
kinetic experiments of the substrate were performed at 24°C
kinetic experiments of the substrate were performed at 37°C.
Our laboratory will continue to search for ways to improve the kinetic parameters of our synthetic FRET substrate, which ultimately will feed into a high throughput assay. We believe that our FRET pair (DNP and MCA) is still an excellent choice for incorporating into a peptide substrate cleaved by the BoNT/B protease due to the relative small chemical structure of the pair. However, to improve on the catalytic proficiency we must re-position the FRET pair within the sequence, most notably the position of MCA at residue 79. As observed by the Roques’ substrate, residue 79 is important for catalytic efficiency, as a minor substitution such as serine for threonine has a large effect. Thus, the approach our laboratory is taking in developing a high throughput assay that will incorporate a synthetic substrate that is structurally similar to the native substrate should be achievable, and a central tenet in the development of future inhibitors for BoNT/B protease.
Conclusion
In an ever growing world of global terrorism countermeasures must be developed to combat attacks via biological weapons such as BoNT. An avenue of treatment that has been lacking is the development of therapeutics to treat BoNT intoxication. Our laboratory has sought to explore opportunities for treatment using small molecule inhibitors of the BoNT proteases. We believe that our current research provides a strong foundation for the development of future therapeutic avenues for the treatment of BoNT intoxication.
Acknowledgments
This work is supported by National Institutes of Health (AI30500) and the Skaggs Institute for Chemical Biology.
Footnotes
Ethical Statement
This statement is to verify that the paper has not been published previously, that it is not under consideration for publication elsewhere, and that if accepted, it will not be published elsewhere in the same form, in English or in any other language, without the written consent of the publisher. The authors declare that there are no conflicts of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Adler M, Nicholson JD, Hackley BE. Efficacy of a novel metalloprotease inhibitor on botulinum neurotoxin B activity. FEBS Lett. 1998;429 (3):234–238. doi: 10.1016/s0014-5793(98)00492-x. [DOI] [PubMed] [Google Scholar]
- Anne C, Cornille F, Lenoir C, Roques BP. High-throughput fluorogenic assay for determination of botulinum type B neurotoxin protease activity. Anal Biochem. 2001;291 (2):253–261. doi: 10.1006/abio.2001.5028. [DOI] [PubMed] [Google Scholar]
- Anne C, Turcaud S, Quancard J, Teffo F, Meudal H, Fournie-Zaluski MC, Roques BP. Development of potent inhibitors of botulinum neurotoxin type B. J Med Chem. 2003a;46(22):4648–4656. doi: 10.1021/jm0300680. [DOI] [PubMed] [Google Scholar]
- Anne C, Blommaert A, Turcaud S, Martin A-S, Meudal H, Roques BP. Thio-derived disulfides as potent inhibitors of botulinum neurotoxin type B: implications for zinc interaction. Bioorg Med Chem. 2003b;11(21):4655–4660. doi: 10.1016/s0968-0896(03)00450-4. [DOI] [PubMed] [Google Scholar]
- Arnon SS, Schechter R, Inglesby TH, Henderson DA, Bartlett JG, Ascher MS, Eitzen E, Fine AD, Hauer J. Botulinum toxin as a biological weapon: medical and public health management. JAMA. 2001;285(8):1059–1079. doi: 10.1001/jama.285.8.1059. [DOI] [PubMed] [Google Scholar]
- Baldwin MR, Kim JJP, Barbieri JT. Botulinum neurotoxin B-host receptor recognition: it takes two receptors to tango. Nat Struct Mol Biol. 2007;14(1):9–10. doi: 10.1038/nsmb0107-9. [DOI] [PubMed] [Google Scholar]
- Barr JR, Moura H, Boyer AE, Woolfitt AR, Kalb SR, Pavlopoulos A, McWilliams LG, Schmidt JG, Martinez RA, Ashley DL. Botulinum neurotoxin detection and differentiation by mass spectrometry. Emerg Infect Dis. 2005;11(10):1578–1583. doi: 10.3201/eid1110.041279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Binz T, Blasi J, Yamasaki S, Baumeister A, Link F, Sudhof TC, Jahn R, Niemann H. Proteolysis of SNAP-25 by types E and A botulinal neurotoxins. J Biol Chem. 1994;269(23):1617–1620. [PubMed] [Google Scholar]
- Black RE, Gunn RA. Hypersensitivity reactions associated with botulinal antitoxin. Am J Med. 1980;69(4):567–570. doi: 10.1016/0002-9343(80)90469-6. [DOI] [PubMed] [Google Scholar]
- Blommaert A, Turcaud S, Anne C, Roques BP. Small tripeptide surrogates with low nanomolar affinity as potent inhibitors of the botulinum neurotoxin B metallo- proteolytic activity. Bioorg Med Chem. 2004;12 (11):3055–3062. doi: 10.1016/j.bmc.2004.03.006. [DOI] [PubMed] [Google Scholar]
- Boldt GE, Kennedy JP, Hixon MS, McAllister LA, Barbieri JT, Tzipori S, Janda KD. Synthesis, characterization and development of a high-throughput methodology for the discovery of botulinum neurotoxin A inhibitors. J Comb Chem. 2006a;8(4):513–521. doi: 10.1021/cc060010h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boldt GE, Kennedy JP, Janda KD. Identification of a potent botulinum neurotoxin A protease inhibitor using in situ lead identification chemistry. Org Lett. 2006b;8(8):1729–1732. doi: 10.1021/ol0603211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bossi P, Garin D, Guihot A, Gay F, Crance JM, Debord T, Autran B, Bricaire F. Bioterrorism:management of major biological agents. Cell Mol Life Sci. 2006;63 (19):2196–2212. doi: 10.1007/s00018-006-6308-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breidenbach MA, Brunger AT. Substrate recognition strategy for botulinum neurotoxin serotype A. Nature. 2004;432(7019):925–929. doi: 10.1038/nature03123. [DOI] [PubMed] [Google Scholar]
- Burnett JC, Opsenica D, Sriraghavan K, Panchal RG, Ruthel G, Hermone AR, Nguyen TL, Kenny TA, Lane DJ, McGrath CF, Schmidt JJ, Vennerstrom JL, Gussio R, Solaja BA, Bavari S. A refined pharmacophore identifies potent 4-amino-7-chloroquinoline-based inhibitors of the botulinum neurotoxin serotype A metalloprotease. J Med Chem. 2007;50(9):2127–2136. doi: 10.1021/jm061446e. and references cited therein. [DOI] [PubMed] [Google Scholar]
- Capkova K, Yoneda Y, Dickerson TJ, Janda KD. Synthesis and structure-activity relationships of second-generation hydroxamate botulinum neurotoxin A protease inhibitors. Bioorg Med Chem Lett. 2007;17(23):6463–6466. doi: 10.1016/j.bmcl.2007.09.103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capkova K, Hixon MS, McAllister LA, Janda KD. Toward the discovery of potent inhibitors of botulinum neurotoxin A: development of a robust LC-MS based assay operational from low to subnanomolar enzyme concentrations. Chem Commun. 2008:3525–3527. doi: 10.1039/b808305c. [DOI] [PubMed] [Google Scholar]
- Choi S, Silverman RB. Inactivation and inhibition of γ-aminobutyric acid aminotransferase by conformationally restricted vigabatrin analogues. J Med Chem. 2002;45(20):4531–4539. doi: 10.1021/jm020134i. [DOI] [PubMed] [Google Scholar]
- Dickerson TJ, Janda KD. The use of small molecules to investigate molecular mechanisms and therapeutic targets for treatment of botulinum neurotoxin A intoxication. ACS Chem Biol. 2006;1(6):359–369. doi: 10.1021/cb600179d. and references cited therein. [DOI] [PubMed] [Google Scholar]
- Feng BY, Shoichet BK. A detergent-based assay for the detection of promiscuous inhibitors. Nat Protoc. 2006;1(2):925–929. doi: 10.1038/nprot.2006.77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finkelstein A. Channels formed in phospholipids bilayer membranes by diphtheria, tetanus, botulinum and anthrax toxin. J Physiol. 1990;84(2):188–190. [PubMed] [Google Scholar]
- Franciosa G, Aureli P, Schechter R. Clostridium botulinum. Food Sci Technol. 2003;125:61–90. [Google Scholar]
- Grant S, Easley C, Kirkpatrick P. Vorinostat. Nat Rev Drug Disc. 2007;6 (1):21–22. doi: 10.1038/nrd2227. [DOI] [PubMed] [Google Scholar]
- Hallis B, James BA, Shone CC. Development of novel assays for botulinum type A and B neurotoxins based on their endopeptidase activities. J Clin Microbiol. 1996;34 (8):1934–1938. doi: 10.1128/jcm.34.8.1934-1938.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hambleton P. Botulinum toxin: structure and pharmacology. Eur Arch Otorhinolaryngol. 1994;(Suppl):S200–S202. doi: 10.1007/978-3-642-85090-5_71. [DOI] [PubMed] [Google Scholar]
- Hemmilä IA, Hurskainen P. Novel detection strategies for drug discovery. Drug Disc Today. 2002;7 (18):S150–S156. doi: 10.1016/s1359-6446(02)02390-5. [DOI] [PubMed] [Google Scholar]
- Hayes D, Mellor G. High throughput screening- considerations for enzyme assays. In: Eisenthal R, Danson MJ, editors. Enzyme Assays. 2. University Press; Oxford: pp. 235–247. [Google Scholar]
- Jacobsen FE, Lewis JA, Cohen SM. The design of inhibitors for medicinally relevant metalloproteins. ChemMedChem. 2007;2(2):152–171. doi: 10.1002/cmdc.200600204. [DOI] [PubMed] [Google Scholar]
- Kozaki S, Sakaguchi S, Sakaguchi G. Purification and some properties of progenitor toxins of Clostridium botulinum type B. Infect Immun. 1974;10(4):750–756. doi: 10.1128/iai.10.4.750-756.1974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lacy DB, Stevens RC. Sequence homology and structural analysis of the clostridial neurotoxins. J Mol Biol. 1999;291(5):1091–1104. doi: 10.1006/jmbi.1999.2945. [DOI] [PubMed] [Google Scholar]
- Montecucco C, Schiavo G. Mechanism of action of tetanus and botulinum neurotoxins. Mol Microbiol. 1994;13(1):1–8. doi: 10.1111/j.1365-2958.1994.tb00396.x. [DOI] [PubMed] [Google Scholar]
- Montecucco C, Schiavo G, Rossetto O. The mechanism of action of tetanus and botulinum neurotoxins. Arch Toxicol Suppl. 1996;18:342–354. doi: 10.1007/978-3-642-61105-6_32. [DOI] [PubMed] [Google Scholar]
- O’Brien EC, Farkas E, Gil MJ, Fitzgerald D, Castineras A, Nolan KB. Metal complexes of salicylhydroxamic acid (H2Sha), anthranilic hydroxamic acid and benzohydroxamic acid. Crystal and molecular structure of [Cu(phen)2(Cl)]Cl·H2Sha, a model for a peroxidase-inhibitor complex. J Inorg Biochem. 2000;79 (1–4):47–51. doi: 10.1016/s0162-0134(99)00245-7. [DOI] [PubMed] [Google Scholar]
- Rossetto O, Seveso M, Caccin M, Schiavo G, Montecucco C. Tetanus and botulinum neurotoxins: turning bad guys into good by research. Toxicon. 2001;39(1):27–41. doi: 10.1016/s0041-0101(00)00163-x. [DOI] [PubMed] [Google Scholar]
- Schantz EJ, Johnson EA. Properties and use of botulinum toxin and other microbial neurotoxins in medicine. Microbiol Rev. 1992;56(1):80–99. doi: 10.1128/mr.56.1.80-99.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schiavo G, Rossetto O, Santucci A, DasGupta BR, Montecucco C. Botulinum neurotoxin are zinc proteins. J Biol Chem. 1992a;267(33):23479–23483. [PubMed] [Google Scholar]
- Schiavo GG, Benfenati F, Poulain B, Rossetto O, de Laureto PP, DasGupta BR, Montecucco C. Tetanus and botulinum-B neurotoxins block neurotransmitter release by proteolytic cleavage of synaptobrevin. Nature. 1992b;359(6398):832–835. doi: 10.1038/359832a0. [DOI] [PubMed] [Google Scholar]
- Schmidt JJ, Bostian KA. Proteolysis of synthetic peptides by type A botulinum neurotoxin. J Protein Chem. 1995;14(8):703–708. doi: 10.1007/BF01886909. [DOI] [PubMed] [Google Scholar]
- Schmidt JJ, Bostian KA. Endoproteinase activity of type A botulinum neurotoxin: substrate requirements and activation by serum albumin. J Protein Chem. 1997;16(1):19–26. doi: 10.1023/a:1026386710428. [DOI] [PubMed] [Google Scholar]
- Silvaggi NR, Boldt GE, Hixon MS, Kennedy JP, Tzipori S, Janda KD, Allen KA. Structures of Clostridium botulinum neurotoxin serotype A light chain complexed with small-molecule inhibitors highlight active-site flexibility. Chem Biol. 2007;14(5):533–542. doi: 10.1016/j.chembiol.2007.03.014. [DOI] [PubMed] [Google Scholar]
- Shine NR. Patent application. 2003. U.S. 6,504,006 B1. [Google Scholar]
- Shone CC, Quinn CP, Wait R, Hallis B, Fooks SG, Hambleton P. Proteolytic cleavage of synthetic fragments of vesicle-associated membrane protein, isoform-2 by botulinum type B neurotoxin. Eur J Biochem. 1993;217(3):965–971. doi: 10.1111/j.1432-1033.1993.tb18327.x. [DOI] [PubMed] [Google Scholar]
- Shone CC, Roberts AK. Peptide Substrate Specificity and Properties of the zinc-endopeptidase Activity of Botulinum Type B Neurotoxin. Eur J Biochem. 1994;225(1):263–270. doi: 10.1111/j.1432-1033.1994.00263.x. [DOI] [PubMed] [Google Scholar]
- Shukla H, Sharma S. Clostridium botulinum: a bug with beauty and weapon. Crit Rev Microbiol. 2005;31(1):11–18. doi: 10.1080/10408410590912952. [DOI] [PubMed] [Google Scholar]
- Skiles JW, Gonnella NC, Jeng AY. The Design, Structure, and Clinical Update of Small Molecular Weight Matrix Metalloproteinase Inhibitors Curr. Med Chem. 2004;11(22):2911–2977. doi: 10.2174/0929867043364018. [DOI] [PubMed] [Google Scholar]
- Smith PL, Yeulet SE, Citerone DR, Drake F, Cook M, Wall DA, Marcello J. SK&F 110679: comparison of absorption following oral or respiratory administration. J Control Release. 1994;28 (1–3):67–77. [Google Scholar]
- Sukonpan C, Oost T, Goodnough M, Tepp W, Johnson EA, Rich DH. Synthesis of substrates and inhibitors of botulinum neurotoxin type A metalloprotease. J Peptide Res. 2004;63 (2):181–183. doi: 10.1111/j.1399-3011.2004.00124.x. [DOI] [PubMed] [Google Scholar]
- Summers JB, Gunn BP, Mazdiyasni H, Goetze Young PR, Bouska JB, Dyer RD, Brooks DW, Carter GW. In vivo characterization of hydroxamic acid inhibitors of 5-lipoxygenase. J Med Chem. 1987;30 (11):2121–2126. doi: 10.1021/jm00394a032. [DOI] [PubMed] [Google Scholar]
- Sutton RB, Fasshauer D, Jahn R, Brunger AT. Crystal structure of a SNARE complex involved in synaptic exocytosis at 2.4 Å resolution. Nature. 1998;395:347–353. doi: 10.1038/26412. [DOI] [PubMed] [Google Scholar]
- Ullmann D. Fluorescence screening assays. In: Taylor JB, Triggle DJ, editors. Comprehensive Medicinal Chemistry. Vol. 3. Elsevier; Oxford: 2006. pp. 599–616. [Google Scholar]
- Villar Elliott, Davenport Botulism: The Many Faces of Botulinum Toxin and its Potential for Bioterrorism. Infectious Disease Clinics of North America. 2006;20 (2):313–327. doi: 10.1016/j.idc.2006.02.003. [DOI] [PubMed] [Google Scholar]