

Abstract
Kaposi's sarcoma-associated herpesvirus (KSHV) has been linked to the development of Kaposi's sarcoma, a major AIDS-associated malignancy, and to hematologic malignancies including primary effusion lymphoma and multicentric Castleman's disease. Like other herpesviruses, KSHV is capable of both latent and lytic replication. Understanding the molecular details associated with this transition from latency to lytic replication is key to controlling virus spread and can impact the development of intervention strategies. Here, we report that KAP-1/TIF1β, a cellular transcriptional repressor that controls chromosomal remodeling, participates in the process of switching viral latency to lytic replication. Knockdown of KAP-1 by siRNA leads to KSHV reactivation mediated by K-Rta, a key transcriptional regulator. In cells harboring latent KSHV, KAP-1 was associated with the majority of viral lytic-gene promoters. K-Rta overexpression induced the viral lytic cycle with concomitant reduction of KAP-1 binding to viral promoters. Association of KAP-1 with heterochromatin was modulated by both sumoylation and phoshorylation. During lytic replication of KSHV, KAP-1 was phosphorylated at Ser824. Several lines of evidence directly linked the viral protein kinase (vPK) to this post-translational modification. Additional studies demonstrated that this phosphorylation of KAP-1 produced a decrease in its sumoylation, consequently decreasing the ability of KAP-1 to condense chromatin on viral promoters. In summary, the cellular transcriptional repressor KAP-1 plays a role in regulating KSHV latency, and vPK modulates the chromatin remodeling function of this repressor.
Keywords: KAP-1, heterochromatin, sumoylation, phosphorylation, herpesvirus latency, viral protein kinase
Introduction
The KRAB domain-associated protein-1/transcriptional intermediary factor 1β (KAP-1/TIF1β) was initially identified as a universal transcriptional co-repressor for Kruppel-associated box-domain-containing zinc finger protein (KRAB-ZFP), the largest family of transcriptional silencers in the human genome (1). KAP-1 recruits and coordinates the assembly of several chromatin-remodeling proteins, such as histone deacetylase multiprotein complexes (e.g., nucleosome remodeling and deacetylase (NuRD) and nuclear receptor corepressor (N-CoR1)), histone methyltransferases (e.g., SET domain, bifurcated 1 (SETDB1)), and heterochromatin protein 1 (HP1) through its plant homeo domain (PHD), bromo domain, and PXVXL motif (2-5). These proteins, together with trimethylated histone 3 lysine 9 (H3K9m3) and histone 3 lysine 27 (H3K27m3), are hallmarks of heterochromatin. As a corepressor, KAP-1 interacts with murine double minute 2 (Mdm2), melanoma antigen (MAGE) and signal transducer and activator of transcription 3 (STAT3), thereby modulating transcriptional activity of protein 53 (p53) and STAT3 (6-8). Increasing evidence suggests that post-translational modifications, such as phosphorylation and sumoylation, are important for regulating the repression function of KAP-1 (9). Phosphorylation of KAP-1 at serine 824 (Ser824) by phosphatidylinositol-3 kinase-like (PIKK) protein kinases, such as ataxia telangiectasia mutated (ATM), is critical to chromatin relaxation in response to DNA damage (10, 11). Sumoylation of KAP-1 at lysine 554, 779 and 804 generates binding platforms for SETDB1 and histone deacetylase 1 (HDAC1), thus enhancing the co-repression function of KAP-1 (12-14). Importantly, phosphorylation of Ser824 is antagonistic to sumoylation at these three sites. Thus, these post-translational modifications affect the ability of KAP-1 to condense or relax chromatin (9).
A common property of herpesviruses is their capacity to establish latency, whereby the majority of viral genes are silenced and the genome persists in cells as an episome which is matintained in a condensed chromatin state. Upon induction by certain viral gene products or chemicals, the viral episosme gradually relaxes its compact chromatin structure, leading to expression of all viral genes and lytic replication. KSHV, also known as human herpesvirus 8, is an oncogenic gamma-herpesvirus involved in the pathogenesis of Kaposi's sarcoma (KS), primary-effusion lymphoma (PEL), and multicentric Castleman's disease (15, 16). Like other herpesviruses, KSHV undergoes both lytic and latent infections, with strong tendency toward the latter (17). Latent to lytic transition can be triggered by a single viral gene product, K-Rta (18), or by chemicals that activate cell signaling pathways and influence chromatin structure such as phorbol esters and butyrate (19). These findings indicate that this transition involves global remodeling of viral chromatin from the heterochromatin to the euchromatin state (20). Although K-Rta functions to switch from latency to lytic replication, additional early viral gene products are also important for optimal transcription of the viral episome during the lytic phase of replication. The exact sequence of events associated with chromosomal remodeling of KSHV is not well understood. Given the prominent role of KAP-1 in regulation of heterochromatin formation, we explored its role in the life cycle of KSHV. Using two different cell culture systems as models for KSHV reactivation, we showed that modulation of KAP-1 levels exhibited a profound effect on maintaining viral latency. Our studies also demonstrated that viral protein kinase (vPK/ORF-36) phosphorylates KAP-1 and thereby modulates its chromatin remodeling activity.
Materials and Methods
Cell culture and plasmid DNA
293, 293T, TREx-F3H3-K-Rta and TREx-F3H3-vPK BCBL-1 cells were cultured as previously described (21). KAP-1 knockdown cell lines were generated by transduction of TREx-F3H3-K-Rta BCBL-1 cells with lentiviral particles expressing shRNA targeting KAP-1 (CCGGGAGGACTACAACCTTATTGTTCTCGAGAACAATAAGGTTGTAGTCCTCTTTTT), then selected with puromycin (1μg/ml) (Invitrogen). To overexpress wild-type KAP-1 and its mutant in KAP-1 knockdown cells, shRNA resistant KAP-1 construct was developed by mutating the shKAP-1 target site to GAGGACTACAATTTGATTGTT. Expression vectors containing shRNA-resistant KAP-1* and KAP-1*-Ser824D were transiently transfected into cells using FugeneHD and selected with G418 (150μg/ml) (Cellgro). The Vero-rKSHV.219 cell line was kindly provided by Dr. Jeffrey Vieira (University of Washington, Seattle, WA) (22). Expression vectors pCMV-Tag2A-Flag-KAP-1 (9), pcDNA3-T7-vPK-wt and pcDNA3-T7-vPK-K108Q were described previously (23). pGL3-Basic (Promega) and pGEX-2T (Amersham Pharmacia Biotech) were used to construct reporter and GST-fusion proteins. The pLK0.1 and pLK0.1-shKAP-1 vectors (Open BioSystems) were used to generate lentivirus expressing KAP-1 shRNA.
Immunoprecipitation and western blot analysis
Nuclear extracts of TREx-F3H3-vPK BCBL-1 cells were prepared as previously described (2). Transfected 293 cells were collected in modified RIPA buffer (24). Cell lysates were incubated with anti-Flag agarose (Sigma) overnight at 4°C. Agarose were washed, and proteins were analyzed by immunoblotting. The following antibodies were used: anti-Flag and anti-β-actin (Sigma), anti-KAP-1 (Cell Signaling Technologies), anti-phospho-KAP-1-S824 (Bethyl Laboratories), and anti-vPK antibody (24).
Mass spectrometry analysis of vPK interacting proteins
TREx-F3H3-vPK BCBL-1 cells were treated with doxycycline (0.1μg/ml) to induce vPK expression. Nuclear extracts were prepared from vPK-inducible BCBL-1 cells at 48hrs after doxycycline treatment. vPK-interacting protein complexes were co-immunoprecipitated using anti-Flag agarose, and eluted by flag peptide. Eluted complexes were separated by 6-14% SDS-PAGE, detected by Coomassie blue staining, and identified using LC-MS/MS (Protein and Nucleic Acid Facility, Stanford University).
Reporter assay
293 cells were cultured in 24-well plates. Plasmids were transfected using TransFectin (Bio-Rad). Forty-eight hours after transfection, cell lysate was prepared and luciferase activity was determined by the Luciferase Assay System (Promega).
In vitro protein kinase assay
vPK kinase activity was measured as described previously (24); purified wild-type vPK (vPK-wt) or kinase-dead vPK-K108Q(0.1μg) were incubated with GST-KAP-1 or GST-KAP-1-S824A substrates.
ChIP-on-vChip assay
ChIP assay was performed according to the protocol provided at http://genomics.ucdavis.edu/farnham. Antibodies used were anti-KAP-1 (Abcam), anti-HP1α (Upstate), anti-H3K9m3 (Abcam), and rabbit IgG (Alpha Diagnostic International). ChIP DNA and 10% input were amplified using a whole genome amplification kit (Sigma). ChIP sample was labeled with Cy3, and input sample was labeled with Cy5 using the 3DNA array 900DNA kit (Genisphere). After co-hybridization of labeled DNA samples to the viral chip, the slides were scanned with the Agilent DNA microarray scanner at a resolution of 10 μm. Images were captured and quantified using Scanalyze software (http://rana.lbl.gov/EisenSoftware.htm). The ChIP signal of the experimental sample was normalized and compared with control input.
Assays of KSHV growth and gene expression in KAP-1 knockdown BCBL-1 cells
To assess viral growth, supernatant from 7.5×105 of control and doxycycline-induced (0.2μg/ml) KAP-1 knockdown, siRNA-resistant KAP-1 and KAP-1-Ser824A overexpressed TREx-F3H3-K-Rta and TREx-F3H3-vPK BCBL-1 cells were collected at 0 and 48hrs. DNA from virions was prepared (25) and quantified by real-time PCR (TaqMan) as previously described (17). For measuring viral protein expression, total cell lysates (TCLs) were prepared at 0 and 48hrs after K-Rta induction and immunoblotted.
Results
KAP-1 knockdown enhances KSHV replication
To explore the role of KAP-1 in KSHV reactivation, we stably expressed KAP-1 shRNA in a BCBL-1 cell line carrying the Tet-inducible K-Rta viral transactivator (TREx-F3H3-K-Rta BCBL-1) (21). Mixed populations of puromycin-resistant cells were isolated, and knockdown of KAP-1 was assessed by immunoblotting (Fig.1A-upper-panel). To determine the effect of KAP-1 on production of virus, supernatants were collected after doxycycline induction of K-Rta and analyzed for levels of virion-associated DNA by quantitative PCR (qPCR) amplification. KAP-1 knockdown significantly enhanced viral reactivation and increased virus production by ∼5-fold over vector-control cells at 48hrs after induction (Fig.1A-lower-panel). In agreement with increased virus release, levels of immediate-early/early genes, K-Rta and K-bZIP, and late gene, K8.1, were elevated in KAP-1 knockdown cells (Fig.1B). The effect of KAP-1 knockdown was also examined in the Vero-rKSHV.219 cell line, which harbors recombinant KSHV genome expressing green fluorescent protein (GFP) from the cellular EF-1α promoter and red fluorescent protein (RFP) from the viral lytic PAN promoter. All cells expressed GFP with very few producing RFP, indicating tight control of latency. However, in KAP-1 knockdown cells, RFP positive cells increased 25-50 fold (Fig.1D). These data suggest that KAP-1 acts to suppress lytic replication presumably by compacting and silencing viral chromatin.
Figure 1. KAP-1 knockdown increases KSHV replication and lytic gene expression.
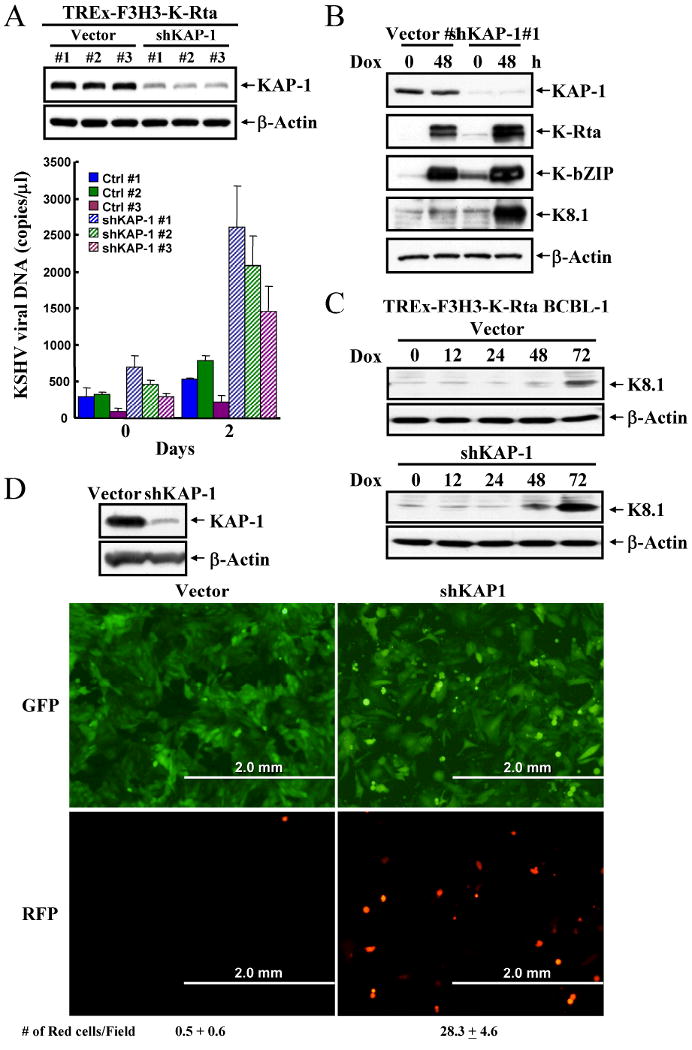
A, TCLs from vector-control and KAP-1 knockdown TREx-F3H3-K-Rta BCBL-1 cells were analyzed by immunoblotting. Supernatants were collected at day 0 and 2 after doxycycline (0.1μg/ml) induction and levels of virion-associated DNA were determined by real-time qPCR. Values are reported as mean±SD. B, TCLs from the cells described above were immunoblotted. C, TCLs were collected from vector-control and KAP-1 knockdown BCBL-1 cells at 0, 12, 24, 48, 72hrs after doxycycline (0.1μg/ml) induction and immunoblotted with anti-K8.1 antibody. D, TCLs from Vero-rKSHV.219 cells, transduced by control and KAP-1 shRNA lentiviruses, were analyzed by immunoblotting. Photomicrographs of cells described in left-panel: vector-control cells; right-panel: shKAP-1 knockdown cells; top-panel: GFP-fluorescence (latent-infected cells); lower-panel: RFP-fluorescence (lytic-infected cells).
KAP-1 is associated with latent KSHV genomes and dissociates after reactivation
To determine whether KAP-1 binds to KSHV chromatin, we performed a ChIP-on-vChip assay by hybridizing DNA from KAP-1 associated viral chromatin with a viral promoter array chip (vChip) designed by our laboratory (26). This array contains upstream sequence regions (USR) of 83 KSHV genes; each USR, about 500bp, contains sites for assembly of transcription factors (26). Cross-linked viral chromatin was prepared by sonication of uninduced and doxycycline-induced TREx-F3H3-K-Rta BCBL-1 cells. Expression of K-Rta was verified by immunoblot (Fig.2A-upper-panel). Chromatin was immunoprecipitated from cell lysates with anti-KAP-1 antibody. To verify this approach, we first demonstrated that KAP-1 binds the ZNF433 promoter, a known KAP-1 target (27) (data not shown). DNA from KAP-1 immunoprecipitates and input were hybridized on the vChip. The enrichment value of KAP-1 binding to promoters was obtained by dividing normalized levels of immunoprecipitated DNA intensity by the input DNA for each array spot. Taking 2-fold difference as significant, KAP-1 was associated with more than two-thirds of viral USRs in uninduced cells (Fig.2A-middle-panel), whereas only two USRs scored significantly after K-Rta-induction for 24hrs (Fig.2A-lower-panel). This finding suggests that KAP-1 associates with latent viral chromatin and dissociates during reactivation. The ChIP-on-vChip data were confirmed by gene-specific real-time qPCR, using three USRs representing early (K-bZIP/K8, vPK/ORF36) and latent genes (LANA/ORF73). Consistent with our vChip data, KAP-1 bound to transcription complexes assembled at all three promoters before K-Rta induction and dissociated after reactivation (Fig.2B).
Figure 2. Chromatin immunoprecipitation assay to determine KAP-1, HP1 and H3K9m3 binding to KSHV promoters during viral reactivation.
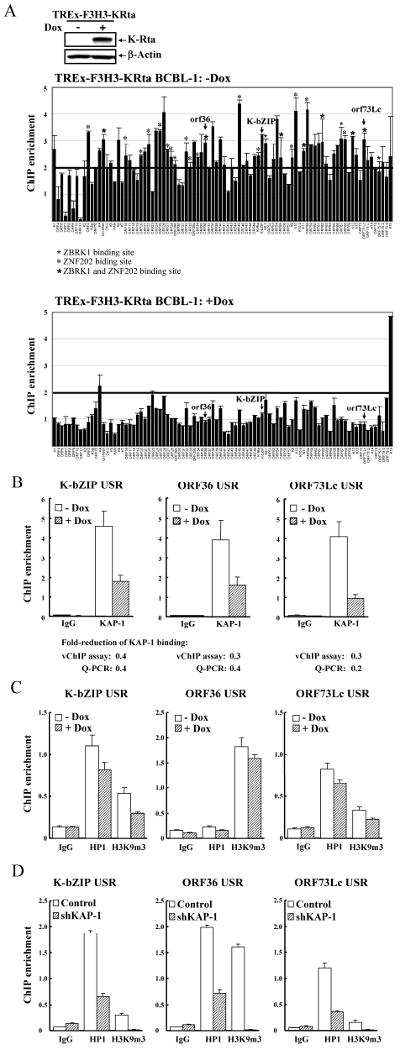
A, Immunoblotting of K-Rta expression in TREx-F3H3-K-Rta BCBL-1 cells treated with doxycycline (0.1μg/ml) for 24hrs. β-actin was used as control. ChIP assay was performed on non-induced and K-Rta-induced BCBL-1 cells using anti-KAP-1 antibody or IgG. The KAP-1 binding pattern along KSHV promoters before (middle-panel) and after (lower-panel) viral reactivation is shown. Mean±SD is the result of replicate spots. B, ChIP-on-vChip data was verified by real-time qPCR using primers specific for USR of KSHV K-bZIP, vPK/ORF36 and LANA/ORF73Lc. Input DNA was normalized to 1. Values reported as mean±SD. C, ChIP assay was performed on non-induced and K-Rta-induced BCBL-1 cells using IgG, anti-HP1, or anti-H3K9m3 antibody. ChIP DNA was analyzed as described in B. D, Analysis of ChIP DNA of HP1 and H3K9m3 bound to KSHV promoters in control and KAP-1 knockdown BCBL-1 cells was done as described in B.
Effect of KAP-1 dissociation on KSHV heterochromatin formation
Heterochromatin is characterized by high levels of H3K9m3 and co-recruitment of HP1. KAP-1 interacts with HP1 and SETDB1, which is a H3K9m3 methylase. Accordingly, KAP-1 can facilitate formation of heterochromatin (28). We determined whether HP1 association or H3K9m3 changed during viral reactivation as predicted by this model. TREx-F3H3-K-Rta BCBL-1 cells were used for analysis of transcription complexes assembled on the USRs of K-bZIP, vPK, and LANA. A general trend of decreasing HP1 and H3K9m3 association was observed 24hrs after K-Rta induction. Although the decrease in the vPK USR was marginal, the reduction of H3K9m3 binding to these USRs was accompanied by an increase in H3K9Ac as we recently reported. (26) (Fig.2C). To test whether KAP-1 downmodulation is, in part, responsible for decreasing HP1 and H3K9m3 association, we examined HP1 and H3K9m3 binding levels on these three USRs using KAP-1 shRNA-tranduced TREx-F3H3-K-Rta BCBL-1 cells. KAP-1 knockdown significantly lowered HP1 and K3K9m3 levels in all three regions (Fig.2D).
Role of KAP-1 serine 824 phosphorylation in viral reactivation
To explore the mechanism by which KAP-1 modulates viral reactivation, levels of KAP-1 and KAP1(Ser824) phosphorylation were monitored. Ser824 phosphorylation by ATM is of particular interest because this post-translational modification is responsible for relaxing chromatin that surrounds sites of DNA damage (10, 11). Levels of KAP-1(Ser824) phosphorylation increased after 24hrs of induction, while KAP-1 levels remained unchanged (Fig.3A). To test the hypothesis that KAP-1(Ser824) phosphorylation plays a role in maintaining open viral chromatin, we developed a shRNA-resistant KAP-1*-S824D, which mimics KAP-1 phosphorylation. This mutant, as well as shRNA-resistant KAP-1*, was introduced into the KAP-1 knockdown TREx-F3H3-K-Rta BCBL-1 cell line. Expression of each of these constructs was confirmed in transfected cells (Fig.3B). As before, depleting KAP-1 by shRNA increased lytic replication at 48hrs after K-Rta induction (Fig.3C). Introduction of shRNA-resistant wild-type KAP-1* restored lytic replication to original levels. By contrast, shRNA-resistant KAP-1-S824D lacked the ability to repress lytic replication and instead, further enhanced release of virus. These data taken together suggest that phosphorylation of KAP-1 at S824 affects its ability to repress chromatin.
Figure 3. Phosphorylation of KAP-1 Ser824 site plays an essential role in KSHV reactivation.
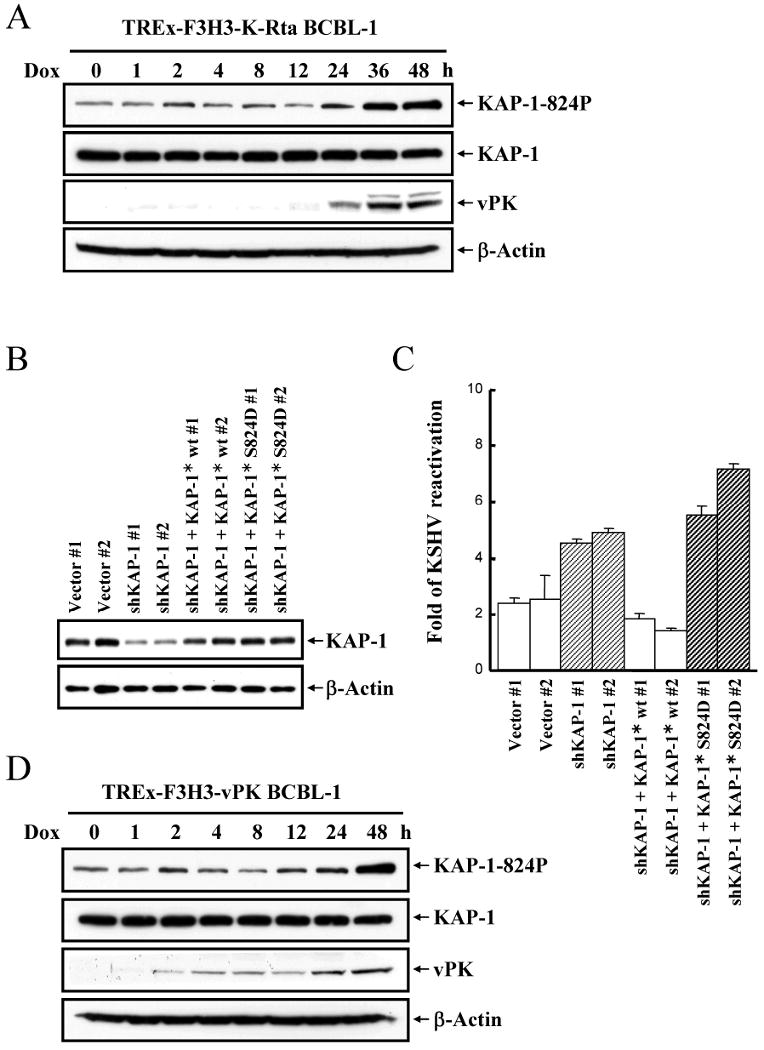
A, Phosphorylation of KAP-1 Ser824 site during KSHV reactivation. TREx-F3H3-K-Rta. BCBL-1 cells were treated with doxycycline (0.1μg/ml) and harvested at the indicated time points shown for Phospho-Ser824 KAP-1, KAP-1, vPK and β-actin immunoblots. B Immunoblotting of KAP-1 expression in vector-control, KAP-1 knockdown and shRNA-resistant KAP-1 and KAP-1-S824D mutant transfected TREx-F3H3-K-Rta BCBL-1 cells. C, Virion-associated DNA from supernatants of cells described in B were collected at day 0 and 2 after doxycycline (0.1μg/ml) induction and determined as described in Fig.1A. KSHV reactivation at day 2 was normalized against day 0. D, Phosphorylation of KAP-1(Ser824) during vPK overexpression. TREx-F3H3-vPK BCBL-1 cells were treated and analyzed as in Fig.3A.
vPK interacts with and phosphorylates KAP-1 at Ser824
The correlation of KAP-1 phsophorylation with vPK expression prompted us to investigate whether vPK may modulate KAP-1 activity. A Flag-HA-tagged, vPK-inducible BCBL-1 cell line was generated (TREx-F3H3-vPK BCBL-1). During vPK induction, phosphorylation of KAP-1(Ser824) increased, whereas steady-state levels of KAP-1 remained unchanged (Fig.3D). This finding suggests that vPK induces phosphorylation of KAP-1, either directly or indirectly. Taking advantage of the doubly-tagged vPK in conjunction with tandem mass spectroscopy, we identified proteins associated with affinity purified vPK. Interestingly, KAP-1 was amongst the major proteins associated with vPK (Fig.4A). Other cellular proteins identified by this methods included HSP chaperones, which are known to interact with protein kinases. To confirm this interaction, 293T cells were co-transfected with T7-vPK and Flag-KAP-1 or Flag-vPK alone. Transfected proteins were immunoprecipitated using anti-Flag agarose. Immunoblotting of KAP-1 and vPK complexes confirmed the interaction of these two proteins (Fig.4B). This interaction was also demonstrated in naturally infected BCBL-1 cells after K-Rta induction (Fig.4C). Together, these data showed that KAP-1 and vPK form a protein complex.
Figure 4. KAP-1 forms a complex with vPK.
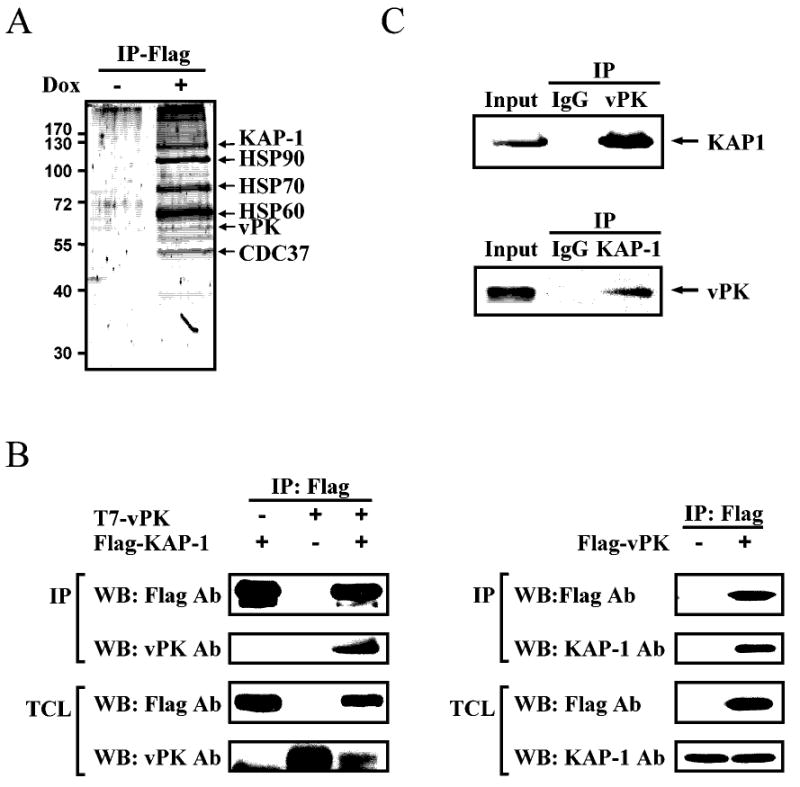
A, Coomassie blue staining of vPK-interacting proteins. B, 293T cells were transiently transfected with expression vectors encoding Flag-tagged-KAP-1, T7-tagged-vPK or both (left-panel) or Flag-tagged-vPK (right-panel). Flag-KAP-1 or Flag-vPK was immunoprecipitated with anti-Flag agarose followed by immunoblotting with the indicated antibodies. TCLs were probed for protein expression. C, TCLs from non-induced and K-Rta-induced BCBL-1 cells were subjected to immunoprecipitation with anti-KAP-1, anti-vPK antibody or IgG followed by immunoblotting with the indicated antibodies.
To determine whether vPK directly phosphorylated KAP-1, we performed an in vitro kinase reaction using recombinant purified wild-type vPK and kinase-dead vPK-K108Q, with GST-KAP-1 substrate. Wild-type vPK readily phosphorylated KAP-1, whereas kinase-dead mutant did not (Fig.5A-left-panel). The data described previously imply that vPK phosphorylates KAP-1(Ser824). In vitro kinase assay was performed using either wild-type KAP-1 or KAP-1-S824A. The results show that KAP-1 phosphorylation was reduced in the S824A mutant as compared to wild-type (Fig.5A-right-panel), indicating that Ser824 is a major phosphorylation site for vPK. Residual phosphorylation of the KAP-1 mutant indicates additional sites of vPK phosphorylation. To further demonstrate that this phosphorylation occurs in vivo, Flag-KAP-1 or Flag-KAP-1-S824A were co-transfected with vPK wild-type or K108Q into 293 cells. vPK phosphorylation of KAP-1 was monitored by immunoblot using anti-phospho-KAP-1-S824 antibody. Consistent with the in vitro results, vPK phosphorylated wild-type KAP-1 at Ser824 but not its S824A mutant in vivo (Fig.5B).
Figure 5. vPK phosphorylates KAP-1, reduces KAP-1 sumoylation, and alleviates KAP-1 transcriptional repression function.
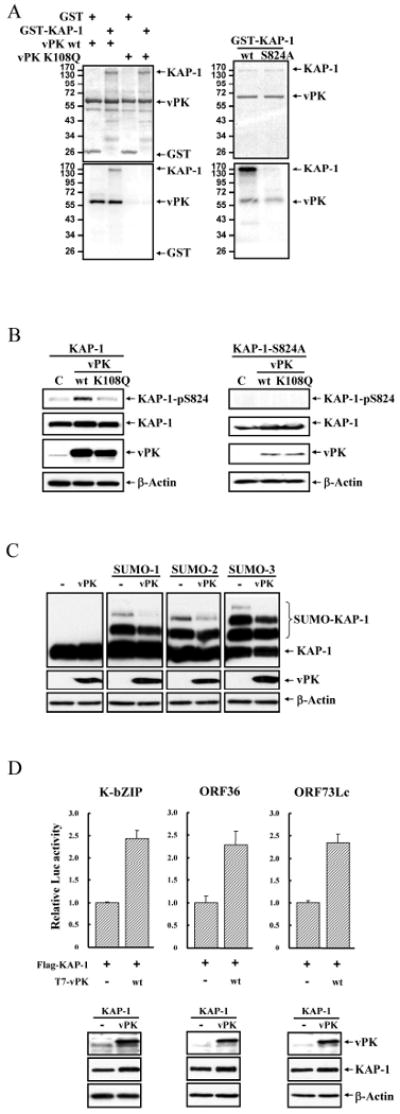
A, Left-panel: Purified GST or GST-KAP-1 was incubated with vPK-wt or vPK-vPK-K108Q in the in vitro kinase reaction. Phosphorylated substrates were resolved on SDS-PAGE and detected by autoradiography. Equal amounts of input proteins were confirmed by Coomassie blue staining. Right-panel: in vitro kinase assay was performed as described using GST-KAP-1 and GST-KAP-1-S824A. B, Control or vector expressing T7-vPK or T7-vPK-K108Q were co-transfected with Flag-KAP-1 or Flag-KAP-1-S824A, and TCL were immunoblotted with the indicated antibodies. C, 293 cells were co-transfected with T7-vPK-wt, Flag-KAP-1 and T7-SUMO-1, -2 or -3. 48 hrs post-transfection, TCLs were separated by SDS-PAGE and immunoblotted using specific antibodies. D, 293 cells were co-transfected with KSHV K-bZIP, vPK/ORF36, or LANA/ORF73Lc promoter luciferase reporter and T7-vPK-wt together with Flag-KAP-1. After forty-eight hours, luciferase activities were determined. Values are reported as mean±SD. Protein expression was confirmed by immunoblotting.
Sumoylation of KAP-1 is affected by vPK
KAP-1 trans-repression and recruitment of SETDB1 histone methylase depends on its sumoylation status, while Ser824 phosphorylation reduces the degree of KAP-1 sumoylation (9, 13). Accordingly, we asked whether vPK also modulated KAP-1 sumoylation as well as its transcriptional functions. Flag-KAP-1 and T7-vPK were co-transfected into 293 cells together with plasmids expressing three SUMO species. vPK reduced the extent of KAP-1 sumoylation in all these settings (Fig.5C). In parallel, we examined whether vPK affects KAP-1-mediated transcriptional repression by reporter assay using three KSHV promoters. The repressive effect of KAP-1 on K-bZIP, ORF36 and ORF73Lc promoters was relieved by wild-type vPK (Fig.5D).
vPK enhances KSHV viral replication
Since KAP-1 regulates KSHV latency and vPK modulates KAP-1 activity via post-translational modification, the affect of vPK on regulation of KSHV latency was investigated. Using the vPK-inducible BCBL-1 cell lines, we tested whether levels of spontaneous or induced reactivation increased upon vPK induction. When vPK was induced by doxycycline, virion-associated DNA in the supernatant at 48hrs after induction was about ∼3-fold higher compared to the controls (Fig.6A-left-panel). Western blot analysis confirmed higher expression levels of K-Rta, K-bZIP and K8.1 in the vPK induced cells (Fig.6A-right-panel), further suggesting that vPK enhances viral reactivation. Next, we determined whether vPK could also augment chemically induced viral reactivation with either TPA or TSA. TPA functions through activation of the protein kinase C pathway, whereas TSA inhibits histone deacetylase. Interestingly, vPK overexpression synergizes with TPA, but not TSA, for induction of virus production (Fig.6B and C). The above observation suggests that vPK-mediated reactivation utilizes the histone acetylation pathway.
Figure 6. vPK enhances KSHV viral replication and lytic gene expression.
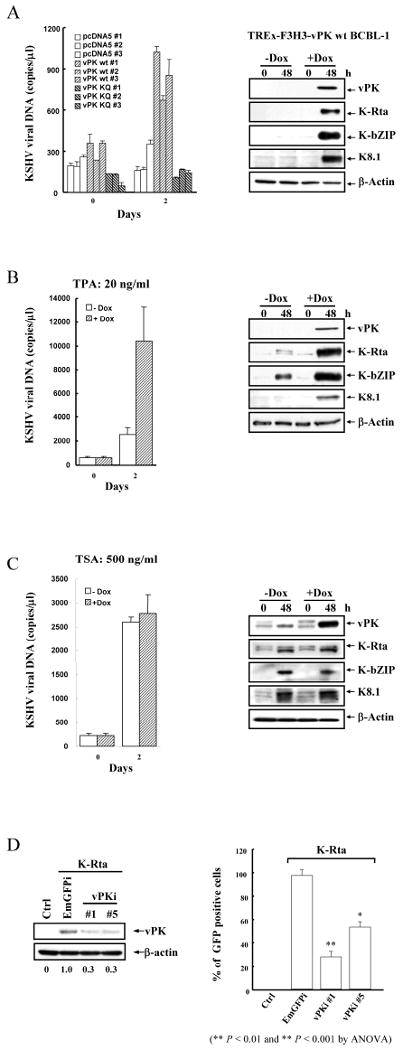
A, Supernatants of non-induced and doxycycline-induced TREx-F3H3-vPK BCBL-1 cells were collected and determined as described in Fig.1A (left-panel). TCL from the same cells were immunoblotted with the indicated antibodies (right-panel). B and C, The non-induced and doxycycline-induced TREx-F3H3-vPK BCBL-1 cells were incubated with TPA or TSA and subjected to detection of virion-associated KSHV DNA by qPCR amplification. TCL from the same cells were immunoblotted with specific antibodies. D, Vero-rKSHV.219 cells were cotransfected with two different vPK shRNA constructs and K-Rta. Eighteen hours post-transfection, sodium butyrate (1mM) was applied to stimulate KSHV reactivation of K-Rta treated cells. Forty-eight hours after transfection, TCLs were immunoblotted with anti-vPK and β-actin antibody. Filtered supernatants were collected at 72hrs post-transfection and used to infect 293T cells. The GFP positive cells were counted by flow cytometry at 48hrs post-infection. Data were normalized to % GFP positive cells transfected with shEmGFP.
Conversely, we determined whether knockdown of vPK would decrease viral production in the reactivation model. Using the Vero-rKSHV.219 cell line, which allows high efficiency of shRNA delivery, we tested whether vPK knockdown reduces virus production. In this experiment, K-Rta and shRNA-vPK were co-transfected, and levels of extracellular virion DNA were measured. Introduction of shRNA-vPK reduced viral titer to about 40∼60% of the K-Rta induced value in comparison with shRNA against GFP; these findings further support that vPK plays a role in facilitating viral reactivation (Fig.6D).
Discussion
A common feature of oncogenic viruses is their ability to enter latency. Understanding viral and cellular factors involved in establishment and maintenance of latency can provide insight into intervention strategies targeting malignancies associated with these viruses. Increasing evidence suggests that chromatinization and histone modification play important roles in regulating viral latency (20, 29). During latency, the herpesvirus genome is maintained as an episome that is organized in a compact chromatin conformation, with limited transcriptionally active regions within the viral genome (30-33). During reactivation, condensed viral chromosome gradually transitions into euchromatin state. Heterochromatin is characterized by trimethylation of H3K9 and assembly with HP1, which interacts with SUV39H1, a histone methylase (3). For KSHV, the viral latency protein LANA directly interacts with HP1 and links the latent viral episome to heterochromatin domains of the host cell (34-36). Beyond involvement of LANA and HP1, little is known about cellular factors involved in assembly of heterochromatin during KSHV latency, nor the disassembly during reactivation. Here, we provide the first evidence that KAP-1 is involved in regulating KSHV latency. We demonstrated that knockdown of KAP-1 significantly increases KSHV replication upon K-Rta induction. Even in the absence of induction, knockdown of KAP-1 increased spontaneous viral reactivation in Vero-rKSHV.219 cells which contain latent viral episomes.
KAP-1 has emerged as an important cellular factor in heterochromatin assembly. KAP-1 is a SUMO-ligase (12), and when itself sumoylated, it interacts with SETBD1, an H3K9 histone methylase (3). KAP-1 also recruits HDACs to remove acetyl residues from H3K9 and interacts with HP1 through its chromoshadow domain (37). One proposed model is that KAP-1 serves as an “enforcer” for heterochromatin formation by promoting H3K9m3 histone code through association with HDAC and SETDB1, thus stabilizing HP1 and chromatin interactions. HP1, in concert with KAP-1, spreads heterochromatin via the associated SUV39H1 histone methylase. Disruption of the interaction between KAP-1 and HP1 converts the heterochromatin-like conformation to an open form (28). Here, we show that in naturally infected BCBL-1 cells, KAP-1 occupancy on viral promoters was significantly higher during latency than in the lytic phase. In order to confirm that loss of KAP-1 binding was not due to encapsidation of new lytic viral genomes, we analyzed the expression profile of the virion protein K8.1 in KAP-1 knockdown cells. K8.1 was expressed at 24hrs after KAP-1 dissociated from the viral genome, indicating that loss of KAP-1 binding happened before virus encapsidation (Fig.1C). Consistently, decrease of KAP-1 occupancy during reactivation may dissociate HP1 from chromatin, resulting in demethylation of H3K9. Our data suggest that KAP-1 may regulate chromatin assembly and disassembly and thereby control viral latency. However, the present study does not address whether KAP-1 is the initiating factor or merely a facilitator of viral latency. KAP-1 has not previously been implicated in regulation of herpesvirus latency, although this cellular protein has been found to be associated with OriLyt (lytic replication origin) of EBV (38). KAP-1 is also involved in suppression of human papillomavirus transcription and replication mediated by viral gene E8-E2C (39), as well as silencing of murine leukemia virus proviral DNA in embryonic cells (40). Thus, it appears that multiple viruses exploit the KAP-1 pathway to silence genes and maintain a state of latency.
As a corepressor of KRAB-domain-containing zinc-finger protein transcriptional factors (KRAB-ZFP), KAP-1 is associated with a wide spectrum of chromosomal sites (27). Currently, only a few target DNA consensus sequences have been identified in the KRAB-ZFP family; these targets include ZBRK1, ZNF202 and KS1. Based on sequence alignments, we found several ZBRK1 and ZNF202 binding sites within our viral USR library (Fig.2A marked by *). Thus, it is likely that KAP-1 associates with the KSHV genome via interaction with the ZBRK1 and ZNF202 transcriptional factors, as well as other yet-to-be determined transcriptional factors. A more comprehensive examination of the involvement of ZBRK1 and ZNF202 sites in KAP-1 binding awaits the development of high-avidity antibodies to these proteins.
We note that the ability of KAP-1 to condense chromatin is regulated by both sumoylation and phosphorylation (9, 10, 12, 13). Interaction with the histone methylase SETDB1 is critical for KAP-1 to compact chromosome, and this interaction depends on KAP-1 sumoylation (12). The auto-sumoylation ability of KAP-1 depends on the extent of phosphorylation. Upon DNA damage, KAP-1(Ser824) phosphorylation by ATM decreases KAP-1 sumoylation and opens chromatin near DNA damage sites (11). KAP-1(Ser824) phosphorylation also decreases the ability to repress gene expression (9). These reports prompted us to determine whether phosphorylation of KAP-1 by vPK played a role in the regulation of viral latency. We found that viral reactivation was accompanied by increased KAP-1(Ser824) phosphorylation dependent on vPK. These studies demonstrated that this phosphorylation by vPK interferes with sumoylation of KAP-1. We and others have previously demonstrated that vPK is an early gene product and packaged into the virion (24, 41). The expression pattern of vPK correlates with the appearance of phospho-KAP-1(Ser824), and vPK levels affect KSHV reactivation. We also showed that vPK can further enhance chemically induced reactivation by TPA, but not TSA. These data, when taken together, support a model whereby vPK regulates viral latency by phosphorylating KAP-1 in order to ensure that the KAP-1 heterochromatin complex remains dissociated from the viral episome. It should be noted that vPK may not be absolutely required for KSHV replication under all conditions and in all cell types. EBV vPK (BGLF4) was shown to be required for viral replication (42); however, vPK/ORF36 of murine gamma-hepresvirus 68 (MHV-68) is critical for infection of primary macrophages, but not fibroblasts (43). Our current study also showed that vPK phosphorylated the same site of KAP-1 as ATM. Tarakanova et al. (43) reported that the viral kinases of MHV-68 and EBV both phosphorylate γH2AX, a substrate of ATM during DNA damage; these findings revealed overlapping phosphorylation specificity of vPK and certain cellular protein kinases. We also found that KSHV vPK phosphorylated γH2AX in vitro (data not shown). Thus, it is conceivable that vPK may play a diminished role in viral replication under conditions where ATM activity is high. In this regard, it is interesting that Shih et al. reported that the KSHV vIRF protein interacts with and attenuates ATM activity in infected cells (44), likely accentuating the importance of vPK in KSHV replication.
Using mass-spectroscopy, we found that KAP-1 is a major cellular protein associated with vPK. This was confirmed in cells overexpressing vPK as well as in naturally infected BCBL-1 cells. Taking advantage of the phosphorylation site mutant of KAP-1, we found that vPK contributes to the phosphorylation at Ser824. However, KAP-1-Ser824A remained slightly phosphorylated, suggesting that there are other vPK phosphorylation sites on KAP-1 which have yet to be mapped and characterized. Other than KAP-1, the predominant cellular proteins which interact with vPK are mostly chaperone proteins (HSP70, HSP90, cdc37, etc.) (45).
In summary, the present study provides new findings on the regulation of KSHV replication by analysis of viral and cellular proteins that control transcription by influencing chromatin dynamics. First and foremost, KAP-1 was shown to be a regulator of viral latency, and its association with transcriptional promoters in the viral genome correlated with the formation of heterochromatin complexes. Second, KAP-1 was modulated by vPK phosphorylation; this post-translational modification prevents KAP-1 from associating with chromatin, ensuring a fully relaxed viral chromatin state conducive to lytic replication.
Acknowledgments
This work was supported by NIH grants CA111185 and DE019085. L. Fitzgerald is a trainee of T32 CA108459 and CA114575-S1. We thank Dr. Cliff Tepper (UC Davis Cancer Center Gene Expression Resource) for v-Chip support, Dr. Jeffrey Vieira (University of Washington) for Vero-rKSHV.219 cells, and Dr. Frank Rausher 3rd (Wistar Institute) for KAP-1 plasmids and helpful discussions.
References
- 1.Friedman JR, Fredericks WJ, Jensen DE, et al. KAP-1, a novel corepressor for the highly conserved KRAB repression domain. Genes Dev. 1996;10:2067–78. doi: 10.1101/gad.10.16.2067. [DOI] [PubMed] [Google Scholar]
- 2.Ryan RF, Schultz DC, Ayyanathan K, et al. KAP-1 corepressor protein interacts and colocalizes with heterochromatic and euchromatic HP1 proteins: a potential role for Kruppel-associated box-zinc finger proteins in heterochromatin-mediated gene silencing. Mol Cell Biol. 1999;19:4366–78. doi: 10.1128/mcb.19.6.4366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Schultz DC, Ayyanathan K, Negorev D, Maul GG, Rauscher FJ., 3rd SETDB1: a novel KAP-1-associated histone H3, lysine 9-specific methyltransferase that contributes to HP1-mediated silencing of euchromatic genes by KRAB zinc-finger proteins. Genes Dev. 2002;16:919–32. doi: 10.1101/gad.973302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Schultz DC, Friedman JR, Rauscher FJ., 3rd Targeting histone deacetylase complexes via KRAB-zinc finger proteins: the PHD and bromodomains of KAP-1 form a cooperative unit that recruits a novel isoform of the Mi-2alpha subunit of NuRD. Genes Dev. 2001;15:428–43. doi: 10.1101/gad.869501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Underhill C, Qutob MS, Yee SP, Torchia J. A novel nuclear receptor corepressor complex, N-CoR, contains components of the mammalian SWI/SNF complex and the corepressor KAP-1. J Biol Chem. 2000;275:40463–70. doi: 10.1074/jbc.M007864200. [DOI] [PubMed] [Google Scholar]
- 6.Tsuruma R, Ohbayashi N, Kamitani S, et al. Physical and functional interactions between STAT3 and KAP1. Oncogene. 2008;27:3054–9. doi: 10.1038/sj.onc.1210952. [DOI] [PubMed] [Google Scholar]
- 7.Wang C, Ivanov A, Chen L, et al. MDM2 interaction with nuclear corepressor KAP1 contributes to p53 inactivation. EMBO J. 2005;24:3279–90. doi: 10.1038/sj.emboj.7600791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yang B, O'Herrin SM, Wu J, et al. MAGE-A, mMage-b, and MAGE-C proteins form complexes with KAP1 and suppress p53-dependent apoptosis in MAGE-positive cell lines. Cancer Res. 2007;67:9954–62. doi: 10.1158/0008-5472.CAN-07-1478. [DOI] [PubMed] [Google Scholar]
- 9.Li X, Lee YK, Jeng JC, et al. Role for KAP1 serine 824 phosphorylation and sumoylation/desumoylation switch in regulating KAP1-mediated transcriptional repression. J Biol Chem. 2007;282:36177–89. doi: 10.1074/jbc.M706912200. [DOI] [PubMed] [Google Scholar]
- 10.White DE, Negorev D, Peng H, Ivanov AV, Maul GG, Rauscher FJ., 3rd KAP1, a novel substrate for PIKK family members, colocalizes with numerous damage response factors at DNA lesions. Cancer Res. 2006;66:11594–9. doi: 10.1158/0008-5472.CAN-06-4138. [DOI] [PubMed] [Google Scholar]
- 11.Ziv Y, Bielopolski D, Galanty Y, et al. Chromatin relaxation in response to DNA double-strand breaks is modulated by a novel ATM- and KAP-1 dependent pathway. Nat Cell Biol. 2006;8:870–6. doi: 10.1038/ncb1446. [DOI] [PubMed] [Google Scholar]
- 12.Ivanov AV, Peng H, Yurchenko V, et al. PHD domain-mediated E3 ligase activity directs intramolecular sumoylation of an adjacent bromodomain required for gene silencing. Mol Cell. 2007;28:823–37. doi: 10.1016/j.molcel.2007.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lee YK, Thomas SN, Yang AJ, Ann DK. Doxorubicin down-regulates Kruppel-associated box domain-associated protein 1 sumoylation that relieves its transcription repression on p21WAF1/CIP1 in breast cancer MCF-7 cells. J Biol Chem. 2007;282:1595–606. doi: 10.1074/jbc.M606306200. [DOI] [PubMed] [Google Scholar]
- 14.Zeng L, Yap KL, Ivanov AV, et al. Structural insights into human KAP1 PHD finger-bromodomain and its role in gene silencing. Nat Struct Mol Biol. 2008;15:626–33. doi: 10.1038/nsmb.1416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dupin N, Fisher C, Kellam P, et al. Distribution of human herpesvirus-8 latently infected cells in Kaposi's sarcoma, multicentric Castleman's disease, and primary effusion lymphoma. Proc Natl Acad Sci U S A. 1999;96:4546–51. doi: 10.1073/pnas.96.8.4546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chang Y, Cesarman E, Pessin MS, et al. Identification of herpesvirus-like DNA sequences in AIDS-associated Kaposi's sarcoma. Science. 1994;266:1865–9. doi: 10.1126/science.7997879. [DOI] [PubMed] [Google Scholar]
- 17.Krishnan HH, Naranatt PP, Smith MS, Zeng L, Bloomer C, Chandran B. Concurrent expression of latent and a limited number of lytic genes with immune modulation and antiapoptotic function by Kaposi's sarcoma-associated herpesvirus early during infection of primary endothelial and fibroblast cells and subsequent decline of lytic gene expression. J Virol. 2004;78:3601–20. doi: 10.1128/JVI.78.7.3601-3620.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Haque M, Wang V, Davis DA, Zheng ZM, Yarchoan R. Genetic organization and hypoxic activation of the Kaposi's sarcoma-associated herpesvirus ORF34-37 gene cluster. J Virol. 2006;80:7037–51. doi: 10.1128/JVI.00553-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yu Y, Black JB, Goldsmith CS, Browning PJ, Bhalla K, Offermann MK. Induction of human herpesvirus-8 DNA replication and transcription by butyrate and TPA in BCBL-1 cells. J Gen Virol. 1999;80(Pt 1):83–90. doi: 10.1099/0022-1317-80-1-83. [DOI] [PubMed] [Google Scholar]
- 20.Knipe DM, Cliffe A. Chromatin control of herpes simplex virus lytic and latent infection. Nat Rev Microbiol. 2008;6:211–21. doi: 10.1038/nrmicro1794. [DOI] [PubMed] [Google Scholar]
- 21.Nakamura H, Lu M, Gwack Y, Souvlis J, Zeichner SL, Jung JU. Global changes in Kaposi's sarcoma-associated virus gene expression patterns following expression of a tetracycline-inducible Rta transactivator. J Virol. 2003;77:4205–20. doi: 10.1128/JVI.77.7.4205-4220.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Vieira J, O'Hearn PM. Use of the red fluorescent protein as a marker of Kaposi's sarcoma-associated herpesvirus lytic gene expression. Virology. 2004;325:225–40. doi: 10.1016/j.virol.2004.03.049. [DOI] [PubMed] [Google Scholar]
- 23.Hamza MS, Reyes RA, Izumiya Y, Wisdom R, Kung HJ, Luciw PA. ORF36 protein kinase of Kaposi's sarcoma herpesvirus activates the c-Jun N-terminal kinase signaling pathway. J Biol Chem. 2004;279:38325–30. doi: 10.1074/jbc.M400964200. [DOI] [PubMed] [Google Scholar]
- 24.Izumiya Y, Izumiya C, Van Geelen A, et al. Kaposi's sarcoma-associated herpesvirus-encoded protein kinase and its interaction with K-bZIP. J Virol. 2007;81:1072–82. doi: 10.1128/JVI.01473-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Krishnan HH, Sharma-Walia N, Zeng L, Gao SJ, Chandran B. Envelope glycoprotein gB of Kaposi's sarcoma-associated herpesvirus is essential for egress from infected cells. J Virol. 2005;79:10952–67. doi: 10.1128/JVI.79.17.10952-10967.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ellison TJ, Izumiya Y, Izumiya C, Luciw PA, Kung HJ. A comprehensive analysis of recruitment and transactivation potential of K-Rta and K-bZIP during reactivation of Kaposi's sarcoma-associated herpesvirus. Virology. 2009 doi: 10.1016/j.virol.2009.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.O'Geen H, Squazzo SL, Iyengar S, et al. Genome-wide analysis of KAP1 binding suggests autoregulation of KRAB-ZNFs. PLoS Genet. 2007;3:e89. doi: 10.1371/journal.pgen.0030089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Riclet R, Chendeb M, Vonesch JL, et al. Disruption of the Interaction between TIF1{beta} and HP1 Leads to a Switch from DNA Hyper- to Hypomethylation and H3K9 to H3K27 Trimethylation on the MEST Promoter Correlating with Gene Reactivation. Mol Biol Cell. 2008 doi: 10.1091/mbc.E08-05-0510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sinclair J, Sissons P. Latency and reactivation of human cytomegalovirus. J Gen Virol. 2006;87:1763–79. doi: 10.1099/vir.0.81891-0. [DOI] [PubMed] [Google Scholar]
- 30.Alberter B, Ensser A. Histone modification pattern of the T-cellular Herpesvirus saimiri genome in latency. J Virol. 2007;81:2524–30. doi: 10.1128/JVI.01931-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Day L, Chau CM, Nebozhyn M, Rennekamp AJ, Showe M, Lieberman PM. Chromatin profiling of Epstein-Barr virus latency control region. J Virol. 2007;81:6389–401. doi: 10.1128/JVI.02172-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Murphy JC, Fischle W, Verdin E, Sinclair JH. Control of cytomegalovirus lytic gene expression by histone acetylation. EMBO J. 2002;21:1112–20. doi: 10.1093/emboj/21.5.1112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wang QY, Zhou C, Johnson KE, Colgrove RC, Coen DM, Knipe DM. Herpesviral latency-associated transcript gene promotes assembly of heterochromatin on viral lytic-gene promoters in latent infection. Proc Natl Acad Sci U S A. 2005;102:16055–9. doi: 10.1073/pnas.0505850102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sakakibara S, Ueda K, Nishimura K, et al. Accumulation of heterochromatin components on the terminal repeat sequence of Kaposi's sarcoma-associated herpesvirus mediated by the latency-associated nuclear antigen. J Virol. 2004;78:7299–310. doi: 10.1128/JVI.78.14.7299-7310.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Stuber G, Mattsson K, Flaberg E, et al. HHV-8 encoded LANA-1 alters the higher organization of the cell nucleus. Mol Cancer. 2007;6:28. doi: 10.1186/1476-4598-6-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Viejo-Borbolla A, Schulz TF. Kaposi's sarcoma-associated herpesvirus (KSHV/HHV8): key aspects of epidemiology and pathogenesis. AIDS Rev. 2003;5:222–9. [PubMed] [Google Scholar]
- 37.Lechner MS, Schultz DC, Negorev D, Maul GG, Rauscher FJ., 3rd The mammalian heterochromatin protein 1 binds diverse nuclear proteins through a common motif that targets the chromoshadow domain. Biochem Biophys Res Commun. 2005;331:929–37. doi: 10.1016/j.bbrc.2005.04.016. [DOI] [PubMed] [Google Scholar]
- 38.Liao G, Huang J, Fixman ED, Hayward SD. The Epstein-Barr virus replication protein BBLF2/3 provides an origin-tethering function through interaction with the zinc finger DNA binding protein ZBRK1 and the KAP-1 corepressor. J Virol. 2005;79:245–56. doi: 10.1128/JVI.79.1.245-256.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ammermann I, Bruckner M, Matthes F, Iftner T, Stubenrauch F. Inhibition of transcription and DNA replication by the papillomavirus E8-E2C protein is mediated by interaction with corepressor molecules. J Virol. 2008;82:5127–36. doi: 10.1128/JVI.02647-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wolf D, Goff SP. TRIM28 mediates primer binding site-targeted silencing of murine leukemia virus in embryonic cells. Cell. 2007;131:46–57. doi: 10.1016/j.cell.2007.07.026. [DOI] [PubMed] [Google Scholar]
- 41.Lu M, Suen J, Frias C, et al. Dissection of the Kaposi's sarcoma-associated herpesvirus gene expression program by using the viral DNA replication inhibitor cidofovir. J Virol. 2004;78:13637–52. doi: 10.1128/JVI.78.24.13637-13652.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gershburg E, Raffa S, Torrisi MR, Pagano JS. Epstein-Barr virus-encoded protein kinase (BGLF4) is involved in production of infectious virus. J Virol. 2007;81:5407–12. doi: 10.1128/JVI.02398-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tarakanova VL, Leung-Pineda V, Hwang S, et al. Gamma-herpesvirus kinase actively initiates a DNA damage response by inducing phosphorylation of H2AX to foster viral replication. Cell Host Microbe. 2007;1:275–86. doi: 10.1016/j.chom.2007.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Shin YC, Nakamura H, Liang X, et al. Inhibition of the ATM/p53 signal transduction pathway by Kaposi's sarcoma-associated herpesvirus interferon regulatory factor 1. J Virol. 2006;80:2257–66. doi: 10.1128/JVI.80.5.2257-2266.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lee P, Shabbir A, Cardozo C, Caplan AJ. Sti1 and Cdc37 can stabilize Hsp90 in chaperone complexes with a protein kinase. Mol Biol Cell. 2004;15:1785–92. doi: 10.1091/mbc.E03-07-0480. [DOI] [PMC free article] [PubMed] [Google Scholar]