

Abstract
This study evaluated the role of crystallins in retinal degeneration induced by chemical hypoxia. Wild type, αA-crystallin (−/−), and αB-crystallin (−/−) mice received intravitreal injection of 12 nmol (low dose), 33 nmol (intermediate dose) or 60 nmol (high dose) cobalt chloride (CoCl2). Hematoxylin and eosin and TdT–mediated dUTP nick-end labeling (TUNEL) stains were performed after 24 hours, 96 hours, and 1 week post-injection, while immunofluorescent stains for αA- and αB-crystallin were performed 1 week post-injection. The in vitro effects of CoCl2 on αB-crystallin expression in ARPE-19 cells were determined by real time RT-PCR, Western blot, and confocal microscopy and studies evaluating subcellular distribution of αB-crystallin in the mitochondria and cytosol were also performed. Histologic studies revealed progressive retinal degeneration with CoCl2 injection in wild type mice. Retinas of CoCl2 injected mice showed transient increased expression of HIF-1α which was maximal 24 hours after injection. Intermediate dose CoCl2 injection was associated with increased retinal immunofluorescence for both αA- and αB-crystallin; however, after high dose injection, increased retinal degeneration was associated with decreased levels of crystallin expression. Injection of CoCl2 at either intermediate or high dose in αA-crystallin (−/−) and αB-crystallin (−/−) mice resulted in much more severe retinal degeneration compared to wild type eyes. A decrease in ARPE-19 total and cytosolic αB-crystallin expression with increasing CoCl2 treatment and an increase in mitochondrial αB-crystallin were found. We conclude that lack of α-crystallins accentuates retinal degeneration in chemically-induced hypoxia in vivo.
Keywords: Oxidative stress, Crystallins, Retinal Pigment Epithelium, Retinal Degeneration, Hypoxia, Apoptosis
1. Introduction
The primary manifestations of age-related macular degeneration (AMD) are seen in the retinal pigment epithelium (RPE) layer, and dysfunction or loss of RPE will induce metabolic changes in the rest of the retina, especially the photoreceptors (Gehrs et al., 2006). Animal models of retinal degeneration have provided a better understanding of disease pathogenesis and have led to the development of novel therapeutic strategies (Delyfer et al., 2004). Many of these animal studies stem from genetically distinct mouse strains, such as the rd mouse, which possesses a mutation of rod-specific phosphodiesterase (Bowes et al., 1990; Jimenez et al., 1996). Other studies have focused attention on strain differences and metabolic consequences of light damage on retina during neonatal hyperoxia (Chan et al., 2005; Wu et al., 2006). However, much less is known about the environmental and metabolic contribution of tissue hypoxia to this process.
The retina is the most metabolically active tissue in the body, and it is highly sensitive to reduction in oxygen tension (Wangsa-Wirawan et al., 2003). Therefore, the role of the oxygen microenvironment in the retina may be of importance in understanding AMD and other retinal degenerative diseases. Manipulation of oxygen levels in animal models has been shown to modulate degeneration of photoreceptors (Maslim et al., 1997; Valter et al., 1998).
Cobalt treatment and hypoxia regulate a similar group of genes and cobalt chloride (CoCl2) is widely used as a hypoxia-mimicking agent (Lee et al., 2001; Vengellur et al., 2003). CoCl2 stimulates the hypoxia responsive pathways and has been shown to induce apoptosis by mitochondrial pathways and hypoxia inducible factor 1 alpha (HIF-1α) dependent and independent mechanisms (Badr et al., 1999; Guo et al., 2006); its ability to stabilize HIF1α allows HIF1α to translocate into the nucleus and enhance transcription of hypoxia-responsive genes. Furthermore, cobalt is essential for vitamin B12 synthesis, but excess exposure can lead to toxicity and can induce apoptosis (Karovic et al., 2007).
Crystallins are members of the small heat shock protein (sHSP) family (Derham and Harding, 1999). α-Crystallins have been studied extensively in the lens for their chaperone function, but it is now generally accepted that α-crystallins have additional different non-lens roles (Bhat, 2004; Andley, 2007) and are expressed in multiple tissues (Srinivasan et al., 1992). Crabb et al. (2002) using proteomic analysis, reported that crystallins were present in drusen preparations of AMD patients. Further, it was found that alpha crystallins accumulated in Bruch’s membrane, choroidal connective tissue, and RPE to a greater degree in AMD than in normal non-AMD controls (Nakata et al., 2005, De et al., 2007). Expression of stress related proteins apolipoprotein E and αB crystallin increased in the photoreceptors over Drusen (Johnson et al., 2005).
Analysis of the expression of crystallins in the mouse retina showed that αA, αB, β, and γ-crystallins were found in the inner and outer nuclear layers and the RPE layer (Xi et al., 2003). Our recent in vitro studies have shown that RPE cultured from α-crystallin knockout mice have an increased susceptibility to hydrogen peroxide-induced cell death compared to wild type RPE (Yaung et al., 2007). This finding is consistent with data from Alge et al. (2002) who found that RPE cells overexpressing αB-crystallin showed resistance to apoptosis, suggesting that α-crystallins may prevent stress-induced cell death. Others have found that αB crystallin protects retinal pericytes and endothelial cells from dicarbonyl stress (Nagaraj et al., 2005), and astrocytes from TNF-α induced stress (Ousman et al., 2007).
Inherited retinal degenerations are often associated with improper protein function due to aggregation or accumulation or misfolding of components crucial to the visual cascade (Garriga et al., 1996; Illing et al., 2002). Among the molecular chaperones, α-crystallins are utilized during protein synthesis and to prevent aggregation of misfolded or damaged proteins during oxidative stress (Horwitz, 1992). Loss of chaperone activity occurs in ocular tissues following a significant decrease in the crystallin content (Kapphahn et al., 2003). However, their chaperone activity is enhanced by stress and they can act at key regulatory steps in apoptosis. Hence, in the present study we examine differences in the level of retinal degeneration in α-crystallin deficient mice vs wild type mice with cobalt chloride, a hypoxia inducing agent. Hypoxia was imposed in vivo by intravitreal injections of CoCl2 in mice using modifications of a recently introduced technique (Hara et al., 2006). We propose that loss of protection by α-crystallin accelerates and augments the degeneration of the retina and RPE, which is supported by evidence of earlier and more severe degeneration in α-crystallin knockout retina. The study also examines the CoCl2 induced compartmental alterations in α-crystallin expression which showed accumulation of α-crystallin in mitochondria of RPE.
2. Materials and Methods
2.1. Intravitreal injection to mice
αA and αB crystallin knockout mice (αA crystallin (−/−) and αB crystallin (−/−)) were originally generated at the National Eye Institute by targeted gene disruption and were maintained in the 129 S6/SvEvTac background (Brady et al., 1997; 2001). The wild type mouse strain was also129 S6/SvEvTac (Taconic Farms, Germantown, NY). Six to eight-week old mice from the control group and αA crystallin (−/−) and αB crystallin (−/−) groups were maintained under 12 hour light: 12 hour dark cyclic lighting conditions and treated according to the ARVO Statement for the Use of Animals in Ophthalmic and Vision Research. For all surgical procedures, mice were anesthetized by intraperitoneal injection with 80mg/mL ketamine and 10mg/mL xylazine, and pupils were dilated with topical 1% tropicamide (Akorn, Buffalo Grove, IL) and 2.5% phenylephrine HCl (Akorn). Local anesthesia was also given to the eye (topical 0.5% Tetracaine HCl; Opthimal, Dublin, OH) as well as post-surgery antibiotics (Neo Poly Bac; Bausch and Lomb Inc., Tampa, FL).
To minimize injection outflow, viscous Gonak (Akorn) was applied to the injection site and surrounding external areas. An initial puncture through the sclera at the pars plana was performed with a 30 gauge needle with the top angled toward the posterior pole. A single injection of 3 microliters of CoCl2 solution (concentrations as indicated in figure legends) or PBS was made under microscopic visualization through the starter puncture and into the midvitreous cavity with a blunt-end 33 gauge needle. Caution was taken to avoid the retina and lens. Mice were euthanized with a lethal dose of pentobarbital after 24 hours, 96 hours or 1 week.
2.2. Histologic processing and immunostaining
Tissues were embedded in optimal cutting compound (Tissue-Tek; Miles, Elkhart, IN) and frozen in liquid nitrogen. Cryosections (8μm) were cut and stored at −80°C and then used for H&E staining, immunostaining, or terminal deoxynucleotidyl transferase biotin-dUTP nick end labeling (TUNEL) method. Sections for immunostaining were air-dried, fixed with 4% paraformaldehyde, and blocked with 1% bovine serum albumin (Sigma). Specimens were then incubated for 1 hour with the rabbit polyclonal antibody (αA-crystallin; αB-crystallin; Stressgen, Ann Arbor, MI; HIF1α, Affinity Bioreagents, Golden, CO), and 30 minutes with the FITC-conjugated secondary antibody (Vector Laboratories; Burlingame, CA).
Cell counts in the inner nuclear layer and the outer nuclear layer of retinas of wild type, αA-crystallin (−/−), and αB-crystallin (−/−) mice after intravitreal injection of CoCl2 were performed by counting the number of nuclei on images of H&E stained sections within a representative 150 micron wide column in each of 3 animals in each group.
2.3. TUNEL staining
The TdT–mediated dUTP nick-end labeling (TUNEL) staining method allowed direct detection of DNA fragmentation by fluorescence microscopy (In Situ Cell Death Detection Kit; Roche Applied Science, Indianapolis, IN). Air dried sections were fixed with 4% paraformaldehyde for 15 minutes and then permeabilized with 0.1% Triton X-100 and 0.1% sodium citrate on ice for 2 minutes. TUNEL reaction mixture was then added to each sample and the sample was incubated for 60 minutes at 37°C in the dark in a humidified chamber. Analysis was achieved by fluorescence microscopy at an excitation wavelength in the range of 520–560 nm for green and 570–620 nm for red as per manufacture’s protocol.
2.4. Cell culture and treatment
We have previously shown that absence of α-crystallin in murine RPE increases susceptibility to oxidative stress induced cell death (Yaung et al., 2007). Due to the limitation in the number of cells that can be isolated using the murine RPE isolation protocol, it would not be feasible to perform the in vitro studies proposed here using these cells. Therefore, the well characterized human RPE cell line ARPE-19 was used for all subsequent in vitro experiments. Cells were obtained from American Type Culture Collection (Manassas, VA), and cultured in a 1:1 ratio of Dulbecco’s modified Eagle’s medium (DMEM) and Ham’s F-12 medium (Mediatech; Herndon, VA) along with 10% fetal bovine serum (FBS), 100U/mL penicillin-streptomycin, and 2mM L-glutamine. Prior to CoCl2 treatment, culture media were changed from 10% FBS DMEM/F12 to 0% FBS for overnight. Time and dosage of CoCl2 are indicated in legends to figures.
2.5. Quantitative real-time RT-PCR
Quantitative expression of mRNA was examined using real-time reverse transcriptase PCR (Applied Biosystems). The following human primer sets were designed using Primer Express software (Applied Biosystems): β-actin 5′-TGACGGGGTCACCCACACTGTGCCCAT-3′ and 5′-CTAGAAGCATTTGCGG TGGACGATGGAGGG-3′; vascular endothelial growth factor (VEGF) 5′-CGCAAGAAATCCCGGTATAA-3′ and 5′-AAATGCTTTCTCCGCTCTGA-3′; and αB-crystallin 5′-TCCCCAGAGGAACTCAAAGTTAAG-3′ and 5′-GGCGCTCTTCATGTTTCCA-3′. Product formation detection was set in the center of the linear portion of PCR amplification, and the cycle at which each reaction reached the set threshold (CT) was determined. Relative change in mRNA expression was calculated to obtain the 2 − ΔΔCT values (Livak and Schmittgen, 2001). Four separate sets of RNA were isolated and examined, and each set was tested in triplicate. Levels were normalized relative to β-actin mRNA and reported as fold change over controls.
2.6. Isolation and fractionation of proteins
For preparation of ARPE-19 cell lysates, trypsinized cells were pelleted and washed once with PBS. Fifty microliters of Mammalian Protein Extraction buffer (Pierce, Rockford, IL), along with a 1:100 dilution of proteinase inhibitor mix (Sigma, St. Louis, MO), was added to each pellet. After incubation for one hour at 4°C, cell debris was pelleted at 14,000 rpm for 10 minutes. The remaining supernatant, containing the soluble cell proteins, was measured for protein concentration, using bovine serum albumin as a standard.
Separation of nuclear compartment from the cytoplasmic fraction of ARPE-19 cells was achieved by use of the Nuclear/Cytosol Fractionation Kit (BioVision, Mountain View, CA). Treated cells were first harvested by trypsin and then incubated with 200uL of Cytosol Extraction Buffer-A. After 10 minutes, 11uL of Cytosol Extraction Buffer-B was added, and supernatant was collected as the cytoplasmic portion. Forty microliters of Nuclear Extraction Buffer Mix was then added to the pellet and vortexed every 10 minutes for 40 minutes. Cells were centrifuged for 10 minutes, and supernatant was harvested as the nuclear extract. The purity of the fractions was checked by Western blot analysis with an antibody against histone H1 (Abcam, Cambridge, MA) and GW182, a cytoplasmic resident protein (Novus Biologicals, Littleton, CO). To separate mitochondrial proteins from cytosolic proteins, mitochondria were first isolated as previously described (Yaung et al., 2007).
2.7. Immunoblot analysis
Protein expression was examined using Western blot techniques. Concentration of harvested proteins was examined by the Bradford-Lowry assay. Equal amounts of protein lysate (10μg/uL) were resolved on 15% Tris-HCl polyacrylamide gels (Ready Gel; Bio-Rad, Hercules, CA) at 120 V and then transferred to a PVDF blotting membrane (Millipore, Bedford, MA). Each membrane was blocked with 5% milk for 30 min at room temperature, incubated with rabbit anti-αB crystallin antibody, rabbit anti-HSP27 antibody (Stressgen, Ann Arbor, MI ), or rabbit anti-HIF1α (Novus Biologicals, Littleton, CO) overnight at 4° C. After incubation with the corresponding secondary antibody (Vector Laboratories, Burlingame, CA), protein bands were detected by chemiluminescence (Amersham Pharmacia Biotech, Cleveland, OH). To verify equal loading of proteins, gels were stained with Coomassie stain (Bio-Safe Coomassie Stain, BioRad).
2.8. Confocal microscopy
ARPE-19 cells, grown to >90% confluency on chamber slides (Lab-Tek, Naperville, IL) and exposed to CoCl2, were used for determining α-crystallin expression and mitochondrial localization. Cells were permeabilized with 0.1% Triton-X100 (J. T. Baker Chemical Co, Phillipsburg, NJ) for 15 minutes, and blocked with 5% milk (Bio-Rad) in Tris-buffered saline plus 0.1% Tween for 15 minutes. Incubation conditions comprised of 1:100 dilution of αB-crystallin antibody overnight at 4°C prior to addition of Cy5-conjugated goat anti-rabbit secondary antibody (Jackson ImmunoResearch Laboratories, West Grove, PA) for 30 minutes the following day. Slides were examined using a Zeiss LSM510 (Zeiss, Thornwood, NY) confocal microscope.
2.9. Statistical analysis
The results are expressed as mean ± SEM. An unpaired, two-tailed Student’s t-test was used to determine the statistical difference between two group means. The differences were considered statistically significant at P<0.05.
3. Results
3.1. Histological and apoptotic evaluation of CoCl2-induced retinal degeneration
Morphological evaluation of retina after intravitreal injection of CoCl2 showed a time and dose-dependent retinal degeneration. H& E staining revealed that cell loss was most prominent in photoreceptors in the outer nuclear layer; higher concentrations of CoCl2 were associated with severe loss in the inner nuclear layer and with focal loss of RPE (Fig. 1A–C). Injection of CoCl2 revealed retinal degeneration, with mild, moderate, and severe damage with 12 nmol, 33 nmol, and 60 nmol, respectively. Apoptosis in the retina was examined by staining of nuclear DNA fragmentation by the TUNEL method (Fig. 1D–F). CoCl2 caused an increase in cell death with time which was most prominent in the outer nuclear layer and was maximal at 96h with a dose of 33 nmol (Fig. 1E). TUNEL positive RPE were also seen in the retinas of CoCl2 injected mice and were most consistently observed in wild type mice 1 week after the 33 nmol dose (Fig. 1F).
Fig. 1.
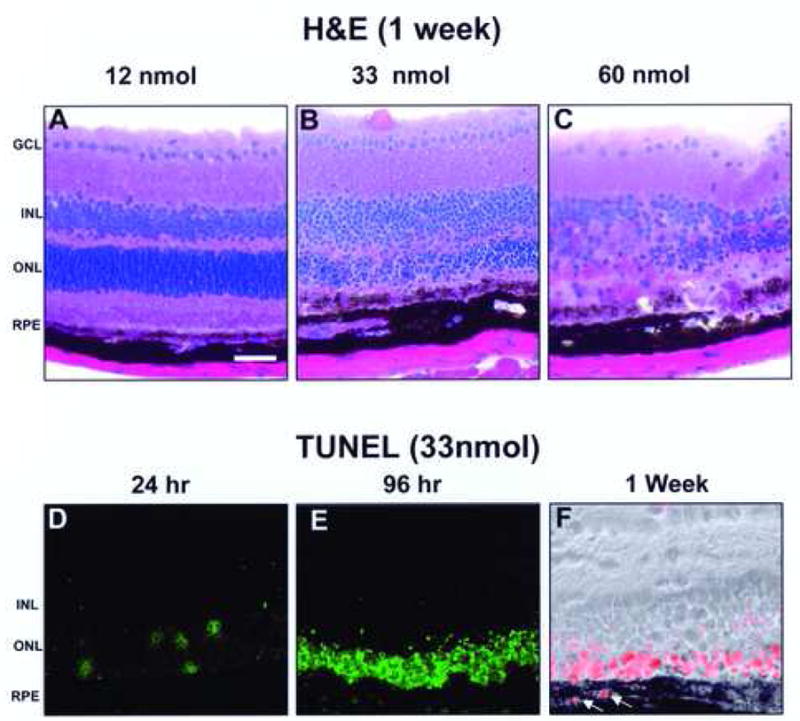
Time course and dose response of CoCl2 on degeneration in mouse retina by H&E staining and TUNEL analysis. H&E staining showed dose-dependent retinal degeneration 1-week post-injection in the wild type (WT) mouse retina. As the CoCl2 dosage increased, degeneration of the retina also increased (A–C). At 33 nmol, retinal layers show obvious signs of degeneration (B), while at 60 nmol, degeneration was more severe (C). TUNEL staining showed that the outer nuclear layer was susceptible to apoptosis in a time dependent manner with maximal staining at 96 h post injection (D–E). TUNEL positive cells continued to be present in the outer nuclear layer in 1 week CoCl2 treated wild type mice; however, TUNEL positive RPE were also evident (arrows; panel F; TUNEL stained with phase overlay). The images shown are representative from at least 3 experiments per group for both H&E staining and TUNEL staining. Bar indicates 75μm. GCL- Ganglion cell layer, INL- Inner nuclear layer, ONL- Outer nuclear layer, RPE- Retinal pigment epithelium.
Since CoCl2 stabilizes HIF1α, immunofluorescent staining for HIF1α was performed at early time points to assess metabolic effects of CoCl2 injections (Fig. 2B). There was prominent HIF1α immunopositivity in the outer aspect of the outer nuclear layer, the photoreceptor inner and outer segments and in the vicinity of the RPE layer at 24 h; by 72 h post-injection, HIF-1α expression had decreased (Fig. 2C).
Fig. 2.
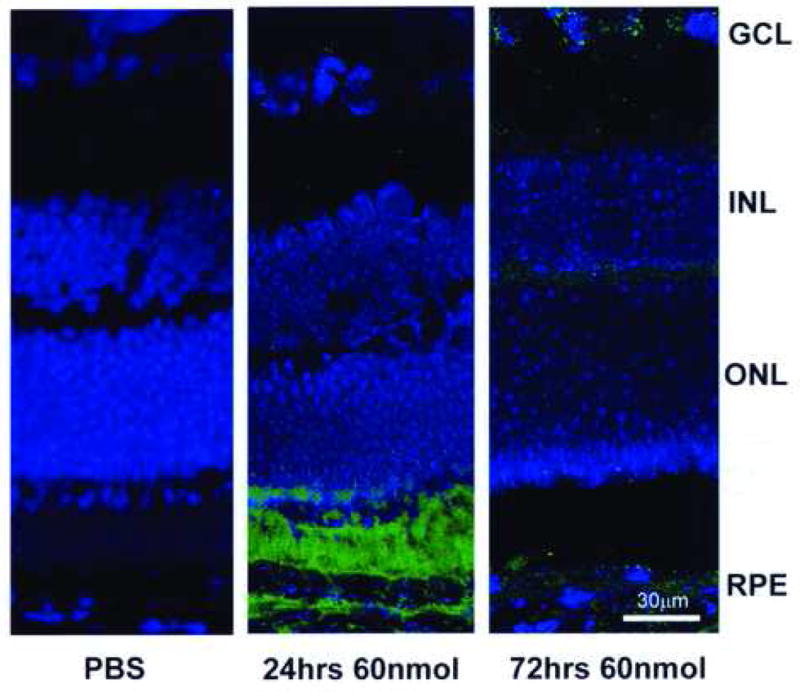
HIF-1α immunofluorescence of mouse retina following intravitreal injection of 60 nmol CoCl2. At 24 h post-injection, HIF-1α expression staining was strong in the RPE and photoreceptor layer. By 72 h, HIF-1α fluorescent intensity was greatly diminished. GCL-Ganglion cell layer, INL- Inner nuclear layer, ONL- Outer nuclear layer, RPE- Retinal pigment epithelium.
To determine whether the retinal degeneration associated with CoCl2 injection was associated with alterations in α-crystallin expression, we evaluated the pattern of expression of αA- and αB-crystallins by immunofluorescent confocal microscopy (Fig. 3). In control animals both αA- and αB-crystallins were most prominently expressed in the inner retina (ganglion cell layer and inner synaptic layer) and in photoreceptors. In animals treated with 33 nmol CoCl2 (1 week after injection) there was increased immunofluorescence for both αA- and αB-crystallin and expression was more diffusely distributed in the retina (Fig. 3). In contrast, high dose CoCl2 injections (60 nmol) were associated with increased retinal degeneration and decreased levels of immunofluorescence for αA- and αB-crystallin (Fig. 3).
Fig. 3.

Pattern of immunofluorescence for αA- and αB-crystallin in wild type mice injected with CoCl2 at intermediate dose (33 nmol), high dose (60 nmol) or with PBS alone. One week after intraocular injection of CoCl2, frozen sections of retina were stained immunohistochemically for αA- or αB-crystallin and evaluated by confocal microscopy. Both αA- and αB-crystallin showed increased and more diffuse retinal staining in animals injected with intermediate (33 nmol) dose CoCl2. GCL- Ganglion cell layer, INL- Inner nuclear layer, ONL- Outer nuclear layer, RPE-Retinal pigment epithelium.
3.2. Accelerated CoCl2-induced degeneration in α-Crystallin knockout retina
Our previous findings suggested an important role for α-crystallins in protecting retinal cells from cell death. RPE cultured from α-crystallin knockout mice revealed increased susceptibility to H2O2-induced apoptosis. Retinas from αA-crystallin knockout mice (Fig. 4D–F) and αB-crystallin knockout mice (Fig. 4G–I) revealed earlier and more severe degeneration from CoCl2 injection as compared to wild type retinas (Fig. 4A–C). At 33 nmol CoCl2 injection, degeneration in the crystallin knockout retina was most prominent in the inner and outer nuclear layers. At 60 nmol CoCl2 injection, the degeneration was prominent and in some cases nearly complete. An assessment of the extent of retinal damage with CoCl2 was made by counting the number of nuclei in the outer nuclear layer and inner nuclear layer of control and crystallin knockout retinas (4 J, K). This semiquantitaive analysis revealed that loss of nuclei increased with increasing CoCl2 dose at 96 h post injection in control and crystallin knockout retina. However, the decrease in the number of nuclei per unit area achieved significance (P<0.01 vs WT) only with 33 and 66 nmol CoCl2 injection in the outer nuclear layer and inner nuclear layer of crystallin knockout mice (Fig. 4J, K).
Fig. 4.

Accelerated CoCl2-induced retinal degeneration in αA(−/−) and αB(−/−) mice, 96 hour post-injection. With 12 nmol CoCl2 αA(−/−) and αB(−/−) retina revealed no apparent change compared to wild type retina (panels A,D,G). At 33 nmol, αA(−/−) and αB (−/−) retina showed selective degeneration in both inner nuclear layer and outer nuclear layers while the wild type retina appeared comparatively intact (panels B,E,H). At 60 nmol, retina from both αA(−/−) and αB(−/−) mice exhibited extensive degeneration in all layers, while the wild type retina showed early changes of retinal degeneration (panels C,F,I). The number of mice from which data were collected from wild type, αA (−/−) and αB(−/−) groups were 4, 4, and 3, respectively. Quantification of retinal damage of wild type, αA (−/−) and αB(−/−) groups (3–4/group) were presented in panels J,K, respectively for outer nuclear and inner nuclear layers. One and two asterisks indicate p<0.05 and p<0.01 respectively vs the PBS-treated controls marked C.
3.3. CoCl2 treatment decreases αB-crystallin transcript and expression in ARPE-19 cells
Since CoCl2 treatment at higher doses led to degeneration and loss of RPE in vivo, further work was undertaken to evaluate the effect of CoCl2 treatment on α-crystallin expression in ARPE-19 cells in vitro. The CoCl2 doses used for in vitro studies did not result in significant cell death as measured by cell counting and trypan blue exclusion (results not shown). We confined these in vitro studies to αB-crystallin since absolute levels of αB-crystallin are much greater than αA-crystallin in RPE cells (Yaung et al., 2007).
Downstream effects of CoCl2 include the stabilization and nuclear translocation of HIF1α, which was verified by HIF1α immunoblotting (Fig. 5A, B). Increasing CoCl2 treatment induced a significant (p<0.05 at 30 nmol and p<0.01 at 100, 250 and 350 μM) dose-dependent upregulation of HIF1α in the nucleus with expression at a level lower than the detection limit in the cytosol. Confocal microscopy (Fig. 6A) and Western blot analysis (Fig. 6B) were used to examine αB-crystallin expression after CoCl2 treatment. Confocal microscopy revealed that treatment with CoCl2 was associated with decreased cytoplasmic immunofluorescence for αB-crystallin which may in part be due to an apparent decrease in cell size. Western blot also revealed a decrease in protein expression with increasing doses of CoCl2 treatment with strongest effect at the 250 μM dose (p<0.05 vs untreated controls; Fig. 6. B–C, left panels). HSP27, another member of the sHSP family, showed a significant (p<0.05 vs control) dose dependent increase in expression with CoCl2 as previously described (Whitlock et al., 2005) (Fig. 6B,C; right panels). The decrease in αB crystallin expression was also seen at the transcriptional level after 24 h of 100 μM CoCl2, which showed a small but statistically significant decrease compared to untreated controls (Fig. 6D). As a positive control, we verified a significant linear increase in the gene expression of the angiogenic factor vascular endothelial growth factor (VEGF) which is known to be upregulated in hypoxia (Shweiki et al., 1992).
Fig. 5.
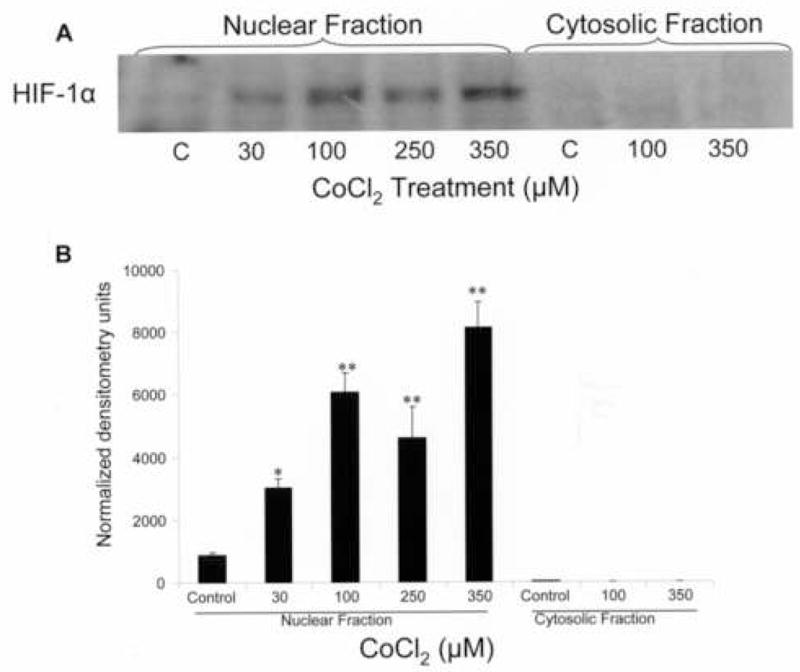
Nuclear localization of HIF-1α after CoCl2 treatment of ARPE-19 cells. Nuclear and cytosolic proteins were fractionated and examined for HIF-1α by immunoblotting. Increasing CoCl2 dose induced a dose-dependent translocation of HIF-1α to the nucleus. A representative gel from 3 independent experiments is shown. The one and two asterisks in the bar graph in B denote statistical difference at p<0.05 and p<0.01 respectively as compared to untreated control.
Fig. 6.

αB-Crystallin expression in ARPE-19 cells decreases with increasing CoCl2 stress. Confocal microscopy (A) and Western blotting (B, C) were used to examine protein expression after CoCl2 exposure. Cells were treated for 24 hours with 100 μM and 250 μM CoCl2. A dose dependent decrease in αB-crystallin immunoreactivity (A) along with preservation of cytoplasmic staining in the perinuclear region at 250 μM CoCl2 was found ( A, bottom right panel viewed at higher magnification, double stained with αB-crystallin [green] and nuclear stain DAPI [blue]). In contrast, HSP27 expression significantly (p<0.01) increased with CoCl2 treatment. αBCrystallin mRNA expression (D) showed a small increase at 4 hour and a significant (p< 0.05 vs control) decrease with 24 hour treatment of 100 μM CoCl2. VEGF, a downstream mediator of HIF-1α signaling, showed a time dependent linear increase in expression. Error bars represent SEM. Western blot analysis were performed in 4 independent cultures of ARPE-19 cells and the mRNA data represent mean ± SEM (n=4). One and two asterisks in the bar graphs indicate p<0.05, 0.01, respectively.
3.4. Changes in the mitochondrial distribution of αB-crystallin in CoCl2-treated ARPE-19 cells
A large portion of the αB-crystallin fraction resides in the cytosol as seen in the confocal micrograph in Figure 6A. Upon increasing CoCl2 dose, the cytosolic fraction of the RPE showed a decrease in αB-crystallin expression which was significant (p<0.05 vs control) at 250 μM CoCl2, whereas the αB-crystallin expression in the mitochondrial fraction increased significantly at 250 μM (Fig. 7A,B).
Fig. 7.
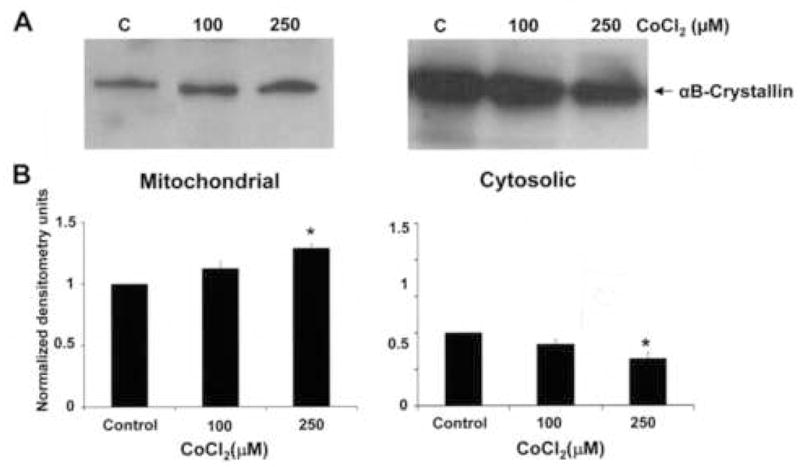
Mitochondrial and cytosolic distribution of αB-crystallin in CoCl2-treated RPE. A. Thecytosolic pools of αB-crystallin decreased with increasing doses of CoCl2 while the αB-crystallin expression in the mitochondria increased. B. Densitometry units indicate values normalized to untreated controls within the organelle compartment and are from 3 independent experiments. Asterisk indicates p<0.05 vs control.
4. Discussion
The role of α-crystallins in retinal degeneration induced by CoCl2 was examined in the present study and the hypoxic effects of CoCl2 on retina and RPE pathophysiology were elucidated. The methodology for intravitreal CoCl2 injection and its validation as an in vivo model of retinal degeneration were recently described (Hara et al., 2006). We have extensively modified the original technique to adapt to our studies. One main difference between our model versus the original procedure is the mode of injection technique itself. The injection site through the pars plana as opposed to the cornea allows us to bypass the iris and lens. Since crystallins are crucial to lenticular physiology, any manipulation of the lenticular tissue may skew our results on α-crystallin expression in the retina (Kamphuis et al., 2007). Subsequent adjustments to injection dose and volume, resulting from the difference in injection methodology and in mouse strain, have also been made.
Possible roles of the retina microenvironment in retinal degeneration have not been as yet fully clarified. Oxygen deprivation results in metabolic changes in the early phases of degeneration which may contribute to the further worsening and severity of photoreceptor cell loss. In this study, degeneration was induced by CoCl2 and the retina and RPE degenerated in atime and dose dependent manner. Similar results were reported by Hara et al. (2006) with CoCl2 in ddY mice and Wistar rats but with a different dosage regimen.
Two separate vascular systems, the retinal circulation and the choroid, supply oxygen to the retina. Unlike the inner retinal circulation which autoregulates under hypoxia, the choroidal blood flow is not regulated metabolically. Thus, hypoxia will lead to decreased choroidal oxygen levels, which subsequently decreases photoreceptor oxygen consumption (Wangsa-Wirawan et al., 2003). Photoreceptors are metabolically very active because they are enriched with mitochondria. Furthermore, the high oxygen consumption of photoreceptors contributes to their vulnerability to hypoxia, which may explain the selective photoreceptor cell degeneration with CoCl2 at earlier time points. However, with increasing dose or increased time of treatment, degeneration in other retinal layers was noted as well as apoptosis and cell loss in the RPE layer. Induction of cell death by in a number of other cell types like rat glioma cells, human alveolar macrophages, and neuronal PC12 cells has been reported (Zou et al., 2001; Araya et al., 2002; Yang et al., 2004). It has to be noted that the dosage and time required to induce significant cell death at the photoreceptor layers is different in different strains of mice. Hara et al. (2006) used 12 nmol CoCl2 in ddY mice and found significant cell death at the outer nuclear layer at 48 h post injection. However, in the present study no significant cell death was observed with the same dose in 129 SVE mice, suggesting dose and time response varies between mouse strains.
While CoCl2 has largely been used as a hypoxia-mimicking agent, our data provide evidence for it being an inducer of retinal degeneration; indeed, the pattern of retinal degeneration with lower doses of CoCl2 has similarities to that seen in murine models of inherited retinal degeneration such as the rd mouse. Our data further showed that at an intermediate dose, the apoptotic cell death was predominantly in the outer nuclear layer and was maximal at 96 h after CoCl2 injection. In addition, we also found RPE cells undergoing apoptosis in wild type mice one week after injection, suggesting that although RPE cells are affected, they may be more resistant to CoCl2 induced cytotoxicity than photoreceptors. We further investigated the role of α-crystallins in CoCl2-induced injury to the retina in light of our previous findings that absence of α-crystallins resulted in increased oxidant-induced RPE cell death (Yaung et al., 2007). We hypothesized that α-crystallins play a protective role in the retina and tested it using αA and αB-crystallin knockout mice. Retinas from αA and αB-crystallin knockout mice showed earlier and more severe degeneration from CoCl2 injection than retinas from wild type mice, suggesting that α-crystallins play an important function in maintaining retinal physiology in the face of hypoxic injury. An intriguing possibility for the observed protective role of crystallins in retinal degeneration is the potential differential regulation of hypoxia-inducible genes with neuroprotective function (eg Pigment epithelial derived factor, VEGF) or those that affect vascular permeability (eg. VEGF) in crystallin knockouts and controls (Nishijima et al., 2007; Takita et al., 2003).
It has been noted that an increase in alpha crystallin expression occurs in murine models of retinal degeneration and light mediated damage and the increase is proportional to the severity, type and onset of retinal degeneration (Jones et al., 1998; Organisciak et al., 2006). Our finding that intermediate dose CoCl2-induced retinal degeneration was associated with increased and more diffuse expression of αA- and αB-crystallin supports this view. Hence, the early photoreceptor degeneration observed in the present study supports the protective and chaperone function of alpha crystallins. However, the signal transduction mechanisms and pathways of α-crystallin action require further investigation. Overexpression of αB-crystallin confers protection against a large number of apoptotic stimuli while its suppression or silencing sensitizes human MDA-MB-231 breast carcinoma cells to apoptosis (Kamradt et al. 2001; 2005). Alpha crystallins have been shown to bind with pro-apoptotic molecules like Bax, Bcl-Xs and P53 and prevent their translocation to the mitochondria (Mao et al., 2004; Liu et al., 2007). Hence, the severe cell death observed in the present model of alpha crystallin knockouts could be mediated through these pathways (Gosh et al., 2007). The αB crystallin sequences have strong interactions with FGF-2, VEGF, insulin and beta catenin. It was also reported αB crystallin protects FGF-2 and VEGF from unfolding and aggregation through its surface interactive sequences (Gosh et al., 2007). Further it was found that laser photocoagulation in mice upregulated alpha crystallins significantly possibly for maintaining cytoskeletol network in order to protect the retina from further damage (Binz et al., 2005).
Since mitochondria are the chief organelle linked to reactive oxygen species generation and subsequent signaling of cell death, we used ARPE-19 cells in culture to study the effect of CoCl2 incubation of mitochondrial alpha crystallin expression. Our in vitro studies with ARPE-19 cells revealed a dose dependent decrease in αB-crystallin expression with increasing CoCl2 treatment in whole cell and cytosolic αB-crystallin protein. However, further subcellular fractionation techniques revealed that mitochondrial pool of αB-crystallin increased with increasing doses of CoCl2. Perhaps increase in mitochondrial expression offers more potent protection against CoCl2-induced insult compared to cytosolic αB-crystallin, a phenomenon analogous to the significant protective effect of mitochondrial GSH as compared to cytosolic GSH reported in hepatocytes (Lluis et al., 2005).
A recent article dealing with optic nerve head astrocytes after hypoxia and reperfusion injury found elevated levels of αB-crystallin mRNA and protein expression which suggests a protective role in damaged neural retina (Yu et al., 2007). Our present study examined the α-crystallin loss of function by use of knockout mice, in which αA(−/−)and αB(−/−) retinas degenerated earlier and to a greater extent than wild type retinas. In both these instances, mitochondria are likely to play an essential role. Studies using gene silencing techniques, in which crystallins can be silenced to varying degrees, could offer clues on the threshold of α-crystallins required for minimal protection. Future studies using over expression of crystallins will further examine the protective qualities of these proteins and associated mechanisms.
Acknowledgments
We thank Fernando Gallardo and Lina Flores for technical assistance, and Ernesto Barron for assistance with confocal microscopy. This work was supported in part by Grants EY02061 and EY03040 from the National Institutes of Health, an award from the Arnold and Mabel Beckman Foundation, and a grant to the Department of Ophthalmology from Research to Prevent Blindness, Inc. (New York, NY).
Footnotes
Presented at The Association for Research in Vision and Ophthalmology Meeting, Fort Lauderdale, FL; 2007; E-Abstract #48-2508.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Alge CS, Priglinger SG, Neubauer AS, Kampik A, Zillig M, Bloemendal H, Welge-Lussen U. Retinal pigment epithelium is protected against apoptosis by αB-crystallin. Invest Ophthalmol Vis Sci. 2002;43:3575–3582. [PubMed] [Google Scholar]
- Andley UP. Crystallin in the eye: function and pathology. Prog Retin Eye Res. 2007;26:78–98. doi: 10.1016/j.preteyeres.2006.10.003. [DOI] [PubMed] [Google Scholar]
- Araya J, Maruyama M, Inoue A, Fujita T, Kawahata J, Sassa K, Hayashi R, Kawagishi Y, Yamashita N, Sigiyama E, Kobayashi M. Inhibition of proteasome activity is involved in cobalt-induced apoptosis of human alveolar macrophages. Am J Physiol: Lung Cell Mol Physiol. 2002;282:L849–L858. doi: 10.1152/ajplung.00422.2001. [DOI] [PubMed] [Google Scholar]
- Badr GA, Zhang JZ, Tang T, Kern TS, Ismail-Beigi F. Glut1 and glut3 expression, but not capillary density, is increased by cobalt chloride in rat cerebrum and retina. Mol Brain Res. 1999;64:24–33. doi: 10.1016/s0169-328x(98)00301-5. [DOI] [PubMed] [Google Scholar]
- Bhat SP. Transparency and non-refractive functions of crystallins – a proposal. Exp Eye Res. 2004;79:809–816. doi: 10.1016/j.exer.2004.08.020. [DOI] [PubMed] [Google Scholar]
- Binz N, Graham CE, Simpson K, Lai YKY, Shen W, Lai C, Speed TP, Rakoczy PE. Long-term effect of therapeutic laser photocoagulation on gene expression in the eye. FASEB J. 2005;20:383–385. doi: 10.1096/fj.05-3890fje. [DOI] [PubMed] [Google Scholar]
- Bowes C, Li T, Danciger M, Baxter LC, Applebury ML, Farber DB. Retinal degeneration in the rd mouse is caused by a defect in the beta subunit of rod cGMP-phosphodiesterase. Nature. 1990;347:677–680. doi: 10.1038/347677a0. [DOI] [PubMed] [Google Scholar]
- Brady JP, Garland D, Duglas-Tabor Y, Robison WG, Jr, Groome A, Wawrousek EF. Targeted disruption of the mouse alpha A-crystallin gene induces cataract and cytoplasmic inclusion bodies containing the small heat shock protein alpha B-crystallin. Proc Natl Acad Sci U S A. 1997;94:884–889. doi: 10.1073/pnas.94.3.884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brady JP, Garland DL, Green DE, Tamm ER, Giblin FJ, Wawrousek EF. AlphaB-crystallin in lens development and muscle integrity: a gene knockout approach. Invest Ophthalmol Vis Sci. 2001;42:2924–2934. [PubMed] [Google Scholar]
- Chan CK, Pham LN, Zhou J, Spee C, Ryan SJ, Hinton DR. Differential expression of pro-and antiangiogenic factors in mouse strain-dependent hypoxia-induced retinal neovascularization. Lab Invest. 2005;85:721–733. doi: 10.1038/labinvest.3700277. [DOI] [PubMed] [Google Scholar]
- Crabb JW, Miyagi M, Gu X, Shadrach K, West KA, Sakaguchi H, Kamei M, Hasan A, Yan L, Rayborn ME, Salomon RG, Hollyfield JG. Drusen proteome analysis: an approach to the etiology of age-related macular degeneration. Proc Natl Acad Sci U S A. 2002;99:14682–14687. doi: 10.1073/pnas.222551899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De S, Rabin DM, Salero E, Lederman PL, Temple S, Stern JH. Human retinal pigment epithelium cell changes and expression of alphaB-crystallin: a biomarker for retinal pigment epithelium cell change in age-related macular degeneration. Arch Ophthalmol. 2007;125:641–645. doi: 10.1001/archopht.125.5.641. [DOI] [PubMed] [Google Scholar]
- Delyfer MN, Leveillard T, Mohand-Said S, Hicks D, Picaud S, Sahel JA. Inherited retinal degenerations: therapeutic prospects. Biol Cell. 2004;96:261–269. doi: 10.1016/j.biolcel.2004.01.006. [DOI] [PubMed] [Google Scholar]
- Derham BK, Harding JJ. Alpha-crystallin as a molecular chaperone. Prog Retin Eye Res. 1999;18:463–509. doi: 10.1016/s1350-9462(98)00030-5. [DOI] [PubMed] [Google Scholar]
- Garriga P, Liu X, Khorana HG. Structure and function in rhodopsin: Correct folding and misfolding in point mutants at and in proximity to the site of the retinitis pigmentosa mutation Leu-125-Arg in the transmembrane helix. Proc Natl Acad Sci USA. 1996;93:4560–4564. doi: 10.1073/pnas.93.10.4560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gehrs KM, Anderson DH, Johnson LV, Hageman GS. Age-related macular degeneration – emerging pathogenetic and therapeutic concepts. Ann Med. 2006;38:450–471. doi: 10.1080/07853890600946724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gosh JG, Shenoy AK, Jr, Clark JI. interactions between important regulatory proteins and human αB crystallin. Biochemistry. 2007;46:6308–6317. doi: 10.1021/bi700149h. [DOI] [PubMed] [Google Scholar]
- Guo M, Song LP, Jiang Y, Liu W, Yu Y, Chen GQ. Hypoxia-mimetic agents desferrioxamine and cobalt chloride induce leukemic cell apoptosis through different hypoxia-inducible factor-1α independent mechanisms. Apoptosis. 2006;11:67–77. doi: 10.1007/s10495-005-3085-3. [DOI] [PubMed] [Google Scholar]
- Hara A, Niwa M, Aoki H, Kumada M, Kunisada T, Oyama T, Yamamoto T, Kozawa O, Mori H. A new model of retinal photoreceptor cell degeneration induced by a chemical hypoxia-mimicking agent, cobalt chloride. Brain Res. 2006;1109:192–200. doi: 10.1016/j.brainres.2006.06.037. [DOI] [PubMed] [Google Scholar]
- Horwitz J. Alpha crystallin can function as a molecular chaperone. Proc Natl Acad Sci USA. 1992;89:10449–10453. doi: 10.1073/pnas.89.21.10449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Illing ME, Rajan RS, Bence NF, Kopito RR. A rhodopsin mutant linked to autosomal dominant retinitis pigmentosa is prone to aggregate and interacts with the ubiquitin proteosome system. J Biol Chem. 2002;277:34150–34160. doi: 10.1074/jbc.M204955200. [DOI] [PubMed] [Google Scholar]
- Jimenez AJ, Garcia-Fernandex JM, Gonzalez B, Foster RG. The spatio-temporal pattern of photoreceptor degeneration in the aged rd/rd mouse retina. Cell Tissue Res. 1996;284:193–202. doi: 10.1007/s004410050579. [DOI] [PubMed] [Google Scholar]
- Johnson PT, Brown MN, Pulliam BC, Anderson DH, Johnson LV. Synaptic pathology, altered gene expression, and degeneration in photoreceptors impacted by drusen. Invest Ophthalmol Vis Sci. 2005;46:4788–4795. doi: 10.1167/iovs.05-0767. [DOI] [PubMed] [Google Scholar]
- Jones SE, Jomary C, Grist J, Thomas MR, Neal MJ. Expression of alphaB-crystallin in a mouse model of inherited retinal degeneration. Neuroreport. 1998;21:4161–4165. doi: 10.1097/00001756-199812210-00030. [DOI] [PubMed] [Google Scholar]
- Kamphius W, Dijk F, Kraan W, Bergen AA. Transfer of lens-specific transcripts to retinal RNA samples may underlie observed changes in crystallin-gene transcript levels after ischemia. Mol Vis. 2007;13:220–228. [PMC free article] [PubMed] [Google Scholar]
- Kamradt MC, Chen F, Cryns VL. The small heat shock protein alpha B-crystallin negatively regulates cytochrome c- and caspase-8-dependent activation of caspase-3 by inhibiting its autoproteolytic maturation. J Biol Chem. 2001;276:16059–16063. doi: 10.1074/jbc.C100107200. [DOI] [PubMed] [Google Scholar]
- Kamradt MC, Lu M, Werner ME, Kwan T, Chen F, Strohecker A, Oshita S, Wilkinson JC, Yu C, Oliver PG, Duckett CS, Buchsbaum DJ, LoBuglio AF, Jordan VC, Cryns VL. The small heat shock protein alpha B-crystallin is a novel inhibitor of TRAIL-induced apoptosis that suppresses the activation of caspase-3. J Biol Chem. 2005;280:11059–11066. doi: 10.1074/jbc.M413382200. [DOI] [PubMed] [Google Scholar]
- Kapphahn RJ, Ethen EM, Peters EA, Higgins L, Ferrington DA. Modified αA crystallin in the retina: Altered expression and truncation with aging. Biochem. 2003;42:15313–15325. doi: 10.1021/bi034774e. [DOI] [PubMed] [Google Scholar]
- Karovic O, Tonazzini I, Rebola N, Edstrom E, Lovdahl C, Fredholm BB, Dare E. Toxic effects of cobalt in primary cultures of mouse astrocytes. Similarities with hypoxia and role of HIF-1alpha. Biochem Pharmacol. 2007;73:694–708. doi: 10.1016/j.bcp.2006.11.008. [DOI] [PubMed] [Google Scholar]
- Lee SG, Lee H, Rho HM. Transcriptional repression of the human p53 gene by cobalt chloride mimicking hypoxia. FEBS Lett. 2001;507:259–263. doi: 10.1016/s0014-5793(01)02989-1. [DOI] [PubMed] [Google Scholar]
- Liu S, Li J, Tao Y, Xiao X. Small heat shock protein alphaB-crystallin binds to p53 to sequester its translocation to mitochondria during hydrogen peroxide-induced apoptosis. Biochem Biophys Res Commun. 2007;354:109–114. doi: 10.1016/j.bbrc.2006.12.152. [DOI] [PubMed] [Google Scholar]
- Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2 − ΔΔCT method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- Lluis JM, Morales A, Blasco C, Colell A, Mari M, Garcia-Ruiz C, Fernandez-Checa JC. Critical Role of Mitochondrial Glutathione in the Survival of Hepatocytes during Hypoxia. J Biol Chem. 2005;280:3224 – 3232. doi: 10.1074/jbc.M408244200. [DOI] [PubMed] [Google Scholar]
- Mao YW, Liu JP, Xiang H, Li DW. Human alphaA- and alphaB-crystallins bind to Bax and Bcl-X(S) to sequester their translocation during staurosporine-induced apoptosis. Cell Death Differ. 2004;11:512–526. doi: 10.1038/sj.cdd.4401384. [DOI] [PubMed] [Google Scholar]
- Maslim J, Valter K, Egensperger R, Hollander H, Stone J. Tissue oxygen during a critical developmental period controls the death and survival of photoreceptors. Invest Ophthalmol Visual Sci. 1997;38:1667–1677. [PubMed] [Google Scholar]
- Nagaraj RH, Oya-Ito T, Bhat M, Liu B. Dicarbonyl stress an dapoptosis of vascular cells: Prevention by αB-crystallin. Ann N Y Acad Sci. 2005;1043:158–165. doi: 10.1196/annals.1333.020. [DOI] [PubMed] [Google Scholar]
- Nakata K, Crabb JW, Hollyfield JG. Crystallin distribution in Bruch’s membrane-choroid complex from AMD and age-matched donor eyes. Exp Eye Res. 2005;80:821–826. doi: 10.1016/j.exer.2004.12.011. [DOI] [PubMed] [Google Scholar]
- Nishijima K, Ng YS, Zhong L, Bradley J, Schubert W, Jo N, Akita J, Samuelsson SJ, Robinson GS, Adamis AP, Shima DT. Vascular endothelial growth factor-A is a survival factor for retinal neurons and a critical neuroprotectant during the adaptive response to ischemic injury. Am J Pathol. 2007;171:53–67. doi: 10.2353/ajpath.2007.061237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Organisciak D, Darrow R, Gu X, Barsalou L, Crabb JW. Genetic, age and light mediated effects on crystallin protein expression in the retina. Photochem Photobiol. 2006;82:1088–1096. doi: 10.1562/2005-06-30-RA-599. [DOI] [PubMed] [Google Scholar]
- Ousman SS, Tomooka BH, van Noort JM, Wawrousek EF, O’Conner K, Hafler DA, Sobel RA, Robinson WH, Steinman L. Protective and therapeutic role for alphaB-crystallin in autoimmune demyelination. Nature. 2007;448:474–479. doi: 10.1038/nature05935. [DOI] [PubMed] [Google Scholar]
- Shweiki D, Itin A, Soffer D, Keshet E. Vascular endothelial growth factor induced by hypoxia may mediate hypoxia-initiated angiogenesis. Nature. 1992;359:843–845. doi: 10.1038/359843a0. [DOI] [PubMed] [Google Scholar]
- Srinivasan AN, Nagineni CN, Bhat SP. αA-Crystallin is expressed in non-ocular tissues. J Biol Chem. 1992;267:23337–23341. [PubMed] [Google Scholar]
- Takita H, Yoneya S, Gehlbach PL, Duh EJ, Wei LL, Mori K. Retinal neuroprotection against ischemic injury mediated by intraocular gene transfer of pigment epithelium-derived factor. Invest Ophthalmol Vis Sci. 2003;44:4497–4504. doi: 10.1167/iovs.03-0052. [DOI] [PubMed] [Google Scholar]
- Valter K, Maslim J, Bowers F, Stone J. Photoreceptor dystrophy in the RCS rat: roles of oxygen, debris, and bFGF. Invest Ophthalmol Visual Sci. 1998;39:2427–2442. [PubMed] [Google Scholar]
- Vengellur A, Woods BJ, Ryan HE, Johnson RS, LaPres JJ. Gene expression profiling of the hypoxia signaling pathway in hypoxia-inducible factor 1alpha null mouse embryonic fibroblasts. Gene Exp. 2003;11:187–197. doi: 10.3727/000000003108749062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wangsa-Wirawan ND, Linsenmeier RA. Retinal oxygen: fundamental and clinical aspects. Arch Ophthalmol. 2003;121:547–557. doi: 10.1001/archopht.121.4.547. [DOI] [PubMed] [Google Scholar]
- Whitlock NA, Agarwal N, Ma JX, Crosson CE. Hsp27 upregulation by HIF-1 signaling offers protection against retinal ischemia in rats. Invest Ophthalmol Vis Sci. 2005;46:1092–1098. doi: 10.1167/iovs.04-0043. [DOI] [PubMed] [Google Scholar]
- Wu J, Seregard S, Algvere PV. Photochemical damage of the retina. Surv Ophthalmol. 2006;51:461–481. doi: 10.1016/j.survophthal.2006.06.009. [DOI] [PubMed] [Google Scholar]
- Xi J, Farjo S, Yoshida TS, Kern A, Swaroop UP, Andley A comprehensive analysis of the expression of crystallins in mouse retina. Mol Vis. 2003;9:410–419. [PubMed] [Google Scholar]
- Yang SJ, Pyen J, Lee I, Lee H, Kim Y, Kim T. Cobalt chloride-induced apoptosis and extracellular signal-regulated protein kinase1/2 activation in rat C6 glioma cells. J Biochem Mol Biol. 2004;37:480–486. doi: 10.5483/bmbrep.2004.37.4.480. [DOI] [PubMed] [Google Scholar]
- Yaung J, Jin ML, Barron E, Spee C, Wawrousek EF, Kannan R, Hinton DR. α-Crystallin distribution in retinal pigment epithelium and effect of gene knockouts on sensitivity to oxidative stress. Mol Vis. 2007;13:566–577. [PMC free article] [PubMed] [Google Scholar]
- Yu AL, Ruchshofer R, Birke M, Priglinger SG, Eibl KH, Kampik A, Bloemendal H, Welge-Lussen U. Hypoxia/Reoxygenation and TGF-β increase αB-crystallin expression in human optic nerve head astrocytes. Exp Eye Res. 2007;84:694–706. doi: 10.1016/j.exer.2006.12.008. [DOI] [PubMed] [Google Scholar]
- Zou W, Yan M, Xu W, Huo H, Sun L, Zheng Z, Liu X. Cobalt chloride induces PC12 cells apoptosis through reactive oxygen species and accompanied by AP-1 activation. J neurosci Res. 2001;64:646–653. doi: 10.1002/jnr.1118. [DOI] [PubMed] [Google Scholar]