

Abstract
Platelet-derived growth factors (PDGFs) and their receptors (PDGFRs) have served as prototypes for growth factor and receptor tyrosine kinase function for more than 25 years. Studies of PDGFs and PDGFRs in animal development have revealed roles for PDGFR-α signaling in gastrulation and in the development of the cranial and cardiac neural crest, gonads, lung, intestine, skin, CNS, and skeleton. Similarly, roles for PDGFR-β signaling have been established in blood vessel formation and early hematopoiesis. PDGF signaling is implicated in a range of diseases. Autocrine activation of PDGF signaling pathways is involved in certain gliomas, sarcomas, and leukemias. Paracrine PDGF signaling is commonly observed in epithelial cancers, where it triggers stromal recruitment and may be involved in epithelial–mesenchymal transition, thereby affecting tumor growth, angiogenesis, invasion, and metastasis. PDGFs drive pathological mesenchymal responses in vascular disorders such as atherosclerosis, restenosis, pulmonary hypertension, and retinal diseases, as well as in fibrotic diseases, including pulmonary fibrosis, liver cirrhosis, scleroderma, glomerulosclerosis, and cardiac fibrosis. We review basic aspects of the PDGF ligands and receptors, their developmental and pathological functions, principles of their pharmacological inhibition, and results using PDGF pathway-inhibitory or stimulatory drugs in preclinical and clinical contexts.
Keywords: PDGF receptor, cancer, development, fibrosis, platelet-derived growth factor
Platelet-derived growth factor (PDGF) was identified more than three decades ago as a serum growth factor for fibroblasts, smooth muscle cells (SMCs), and glia cells (Kohler and Lipton 1974; Ross et al. 1974; Westermark and Wasteson 1976). Human PDGF was originally identified as a disulfide-linked dimer of two different polypeptide chains, A and B, separable using reversed phase chromatography (Johnsson et al. 1982). The B-chain (PDGF-B) was characterized by amino acid sequencing, revealing a close homology between PDGF-B and the product of the retroviral oncogene v-sis of simian sarcoma virus (SSV) (Doolittle et al. 1983; Waterfield et al. 1983). Subsequent studies confirmed that the human cellular counterpart (c-sis) was identical to PDGF-B and that autocrine PDGF activity was sufficient for SSV transformation in vitro. This was a paradigm-shifting discovery about the relationship between neoplastic cell transformation and normal growth control. For the first time, the importance of autocrine growth stimulation in neoplastic transformation was demonstrated. As discussed below, it is now well established that autocrine PDGF stimulation plays a role also in some human cancers.
PDGF-A was characterized by cDNA cloning (Betsholtz et al. 1986). This resolved a paradoxical lack of correlation between secretion of PDGF-like growth factors from tumor cell lines and their expression of c-sis; it turned out that most such cell lines express PDGF-A and secrete PDGF-AA homodimers (Heldin et al. 1986). Together with the demonstration that PDGF-BB homodimers are produced by SSV-transformed or PDGF-B-expressing cells, these results showed that the PDGF family consisted of three proteins—PDGF-AA, PDGF-AB, and PDGF-BB—encoded by two genes, PDGF-A and PDGF-B (for review, see Heldin and Westermark 1999). This view lasted for more than 15 years until combinations of genomic and biochemical efforts identified two additional PDGF genes and proteins—PDGF-C (Li et al. 2000) and PDGF-D (Bergsten et al. 2001; LaRochelle et al. 2001). The currently known PDGF genes and polypeptides belong to a family of structurally and functionally related growth factors including also the vascular endothelial growth factors (VEGFs) (Fredriksson et al. 2004a). PDGF/VEGF growth factors are conserved throughout the animal kingdom (Fig. 1) and form part of a large superfamily of proteins containing cystine knots (McDonald and Hendrickson 1993).
Figure 1.

The PDGF/VEGF family in mammals and invertebrates. Mammalian PDGFs fall into two classes (I and II) distinguished by the presence of basic retention motifs (A and B) or CUB domains (C and D). Mammalian VEGFs also fall into two classes (III and IV). C. elegans (Ce) and Drosophila (D) PVFs are most similar to VEGF-C and VEGF-D based on domain organization but may functionally be most similar to VEGF-A, VEGF-B, and PlGF.
The PDGFs have crucial roles during development, but there is limited evidence for normal physiological functions in the adult. Increased PDGF activity has been linked with several diseases and pathological conditions, however. Causal pathogenic roles of the PDGFs have been established for some diseases, providing prospects for therapy using PDGF antagonists. PDGF receptor-inhibiting substances are now extensively tested in preclinical models as well as in human clinical trials. In addition, recombinant human PDGF-BB has been introduced in the clinic as a wound-healing therapy.
The present review summarizes current knowledge about PDGF functions in health and disease. We provide a brief background to PDGF biochemistry and cell biology and discuss how some of the cellular responses to PDGFs relate to functions in mammalian development and disease. In this context, we also discuss the recently established roles of PDGF/VEGF-like growth factors (PVFs) in invertebrates. We summarize how different mechanisms contribute to the regulation of bioavailability and tissue distribution of the PDGFs, which are key parameters during development. For detailed information on particular aspects of PDGF biology, such as signal transduction and the many reported effects of PDGFs in cell culture, the reader is referred to other reviews and original literature, some of which are cited below.
The PDGF/VEGF family of ligands and receptors
Mammalian PDGF/VEGFs
All PDGFs and VEGFs are dimers of disulfide-linked polypeptide chains (for review, see Heldin and Westermark 1999). In mammals, a total of nine different genes encode four different PDGF chains (PDGF-A, PDGF-B, PDGF-C, and PDGF-D) and five different VEGF chains (VEGF-A, VEGF-B, VEGF-C, VEGF-D; and placenta growth factor, PlGF) (for review, see Ferrara et al. 2003; Fredriksson et al. 2004a). One heterodimer (PDGF-AB) has been demonstrated in human platelets. Although the PDGF-AB heterodimer is endowed with somewhat different signaling properties from the homodimers (Ekman et al. 1999), its physiological importance remains unclear. PDGF-AB occurrence in platelets may be specific to humans (Stroobant and Waterfield 1984). Also, the endogenous expression patterns of PDGF-A and PDGF-B are generally nonoverlapping (Hoch and Soriano 2003), suggesting that heterodimers are infrequent in vivo. Presently, evidence for genetic interactions between pdgfa and pdgfb is also lacking (Li et al. 2000). Thus, although there may be special cases for heterodimer formation and function within the PDGF ligand family, homodimers appear to dominate, at least during development.
Mammalian PDGFs and VEGFs separate into four distinguishable classes of proteins (Fig. 1). All members carry a growth factor core domain containing a conserved set of cysteine residues. The core domain is necessary and sufficient for receptor binding and activation. Classification into PDGFs or VEGFs is based on receptor binding. It has been generally assumed that PDGFs and VEGFs are selective for their own receptors. This view was recently challenged by the demonstration that VEGF-A may bind to and activate PDGF receptors in bone-marrow-derived mesenchymal stem cells (Ball et al. 2007). This study also challenges the general view that VEGFs target mainly endothelial cells, whereas mesenchymal cells are targeted by PDGFs. Further challenge to the functional distinctions between PDGFs and VEGFs comes from findings that VEGF-C and PDGF-A both regulate oligodendrocyte development, however, through distinct receptors. VEGFs and PDGFs also both appear to function in hematopoietic development, neurogenesis, and neuroprotection. These functions are further discussed below.
Mammalian PDGF receptors
PDGFs act via two RTKs (PDGFR-α and PDGFR-β) with common domain structures, including five extracellular immunoglobulin (Ig) loops and a split intracellular tyrosine kinase (TK) domain. This structure is shared with c-Fms, c-Kit, and Flt3, the receptors for CSF-1, SCF, and Flt3-ligand, respectively. The VEGFs act through a distinct but structurally related subfamily of RTKs— VEGFRs 1, 2, and 3. Ligand binding promotes receptor dimerization, which initiates signaling. Depending on ligand configuration and the pattern of receptor expression, different receptor dimers may form (Heldin and Westermark 1999). Theoretically (and generally based on cell culture experiments), the possible PDGF–PDGFR interactions are multiple and complex and include the formation of receptor heterodimers (Fig. 2). However, in vivo there is functional evidence only for a few interactions; i.e., those of PDGF-AA and PDGF-CC via PDGFR-α, and PDGF-BB via PDGFR-β (Fig. 2).It is likely that also PDGF-DD acts through PDGFR-β in vivo, but evidence for this is currently lacking. PDGFR heterodimer formation occurs in vivo as seen by crossing mice carrying PDGFR-α and PDGFR-β signaling mutants (Klinghoffer et al. 2002). The importance of the different PDGF–PDGFR interactions, and the signals elicited, are further discussed below. For a comprehensive overview of PDGF receptor signal transduction mechanisms, the reader is referred to previous reviews on this topic (Heldin and Westermark 1999; Rönnstrand and Heldin 2001; Tallquist and Kazlauskas 2004).
Figure 2.

PDGF–PDGFR interactions. Each chain of the PDGF dimer interacts with one receptor subunit. The active receptor configuration is therefore determined by the ligand dimer configuration. The top panel shows the interactions that have been demonstrated in cell culture. Hatched arrows indicate weak interactions or conflicting results. The bottom panel shows interactions proven to be of importance in vivo during mammalian development. Note that PDGF-D has not yet been investigated in this regard.
Invertebrate PDGF/VEGFs
PDGF/VEGF signaling is conserved throughout the animal kingdom. The fruit fly Drosophila melanogaster has three PVFs (PVF-1–3), and a single PDGF/VEGF receptor (PVR). The nematode Caenorhabditis elegans has four receptors (VER-1–4), and one putative ligand (PVF-1) (Hoch and Soriano 2003; Tarsitano et al. 2006), whose direct interaction with the VERs remains to be established. Invertebrate PVR/VERs resemble the mammalian VEGF receptors in that they have seven (rather than five) extracellular Ig loops. The PVFs also resemble the mammalian VEGFs in that they contain a C-terminal cysteine-rich motif, which is missing in the PDGFs (Fig. 1). Thus, it appears that an ancestral VEGF system has duplicated to generate the vertebrate VEGF and PDGF families through divergent evolution. C. elegans (Ce)PVF-1 was recently found to bind and activate human VEGFR-1 and VEGFR-2 and to induce angiogenic responses in human umbilical vein endothelial cells and chick chorioallantoic membranes. In contrast, CePVF-1 did not bind VEGFR-3 or PDGFR-β (Tarsitano et al. 2006). The invertebrate PFV-PVR/VER system therefore seems to be structurally and functionally orthologous to mammalian VEGF-A/B/PlGF–VEGFR1/2.
Expression, processing, and bioavailability of the PDGF ligands
PDGF ligand and receptor expression patterns
PDGFs act primarily as paracrine growth factors. PDGFs may also be engaged in autocrine loops in tumors (discussed below). Physiological autocrine roles, similar to those recently described for VEGF-A in endothelial cells (Lee et al. 2007), have not been demonstrated for the PDGFs. PDGFs are generally produced by discrete populations of cells and act locally to drive different cellular responses (for review, see Hoch and Soriano 2003; Betsholtz 2004). Both PDGF and PDGFR expression patterns are spatio-temporally regulated in vivo during development and in certain physiological hypertrophic responses. PDGF expression in cultured cells is dynamic and responsive to a variety of stimuli, including hypoxia, thrombin, cytokines, and growth factors, including PDGF itself (for review, see Heldin and Westermark 1999). Also, PDGFR expression is dynamic. General mesenchymal expression of PDGFRs is low in vivo, but increases dramatically during inflammation and in culture. Several factors induce PDGFR expression, including TGF-β, estrogen (probably linked to hypertrophic smooth muscle responses in the pregnant uterus), interleukin-1α (IL-1α), basic fibroblast growth factor-2 (FGF-2), tumor necrosis factor-α, and lipopolysaccharide (Heldin and Westermark 1999 and references therein).
The detailed expression patterns of the individual PDGF ligands and receptors are complex and have been reviewed elsewhere (Heldin and Westermark 1999; Hoch and Soriano 2003). There are some general patterns, however: PDGF-B is mainly expressed in vascular endothelial cells, megakaryocytes, and neurons. PDGF-A and PDGF-C are expressed in epithelial cells, muscle, and neuronal progenitors. PDGF-D expression is less well characterized, but it has been observed in fibroblasts and SMCs at certain locations (possibly suggesting autocrine functions via PDGFR-β). PDGFR-α is expressed in mesenchymal cells. Particularly strong expression of PDGFR-α has been noticed in subtypes of mesenchymal progenitors in lung, skin, and intestine and in oligodendrocyte progenitors (OPs) (discussed further below). PDGFR-β is expressed in mesenchyme, particularly in vascular SMCs (vSMCs) and pericytes.
The mammalian PDGF and PDGFR genes are located on different chromosomes, and their transcriptional regulation seems largely independent. It remains to be established if some of the overlapping expression patterns for PDGF-A and PDGF-C result from common transcription regulatory mechanisms. The transcriptional regulation of the PDGF-A and PDGF-B genes has been extensively studied and is reviewed elsewhere (Heldin and Westermark 1999; Kaetzel 2003). Little is still known about the transcriptional regulation of PDGF-C and PDGF-D and the PDGFRs.
PDGF biosynthesis and secretion
PDGF biosynthesis and processing are controlled at multiple levels and differ for the different PDGFs. There is currently no evidence for regulated secretion of the PDGFs, which instead appears to be constitutively released (Fruttiger et al. 2000). PDGF-A and PDGF-B become disulfide-linked into dimers already as propeptides. PDGF-C and PDGF-D have been less studied in this regard. PDGF-A and PDGF-B contain N-terminal pro-domains that are removed intracellularly by furin or related proprotein convertases (for review, see Fredriksson et al. 2004a). N-terminal processing is necessary for PDGF-A to acquire receptor-binding ability (for review, see Heldin and Westermark 1999; Fredriksson et al. 2004a). Likely, PDGF-B also requires N-terminal propeptide removal to become active.
In contrast, PDGF-C and PDGF-D are not processed intracellularly but are instead secreted as latent (conditionally inactive) ligands (for review, see Fredriksson et al. 2004a; Reigstad et al. 2005). Activation in the extracellular space requires dissociation of the growth factor domain from the CUB domain (Fig. 1). Plasmin and tissue plasminogen activator (tPA) have been demonstrated to proteolytically remove the CUB domain in PDGF-C, rendering it biologically active (Fredriksson et al. 2004b). Although the endogenous protease(s) responsible for PDGF-C activation in vivo remains to be identified, tPA endogenously expressed in cultured fibroblasts activates PDGF-CC expressed by the same cells. Plasmin can cleave and activate also PDGF-D, but tPA cannot (Fredriksson et al. 2004b). TPA needs to interact with both the CUB domain and the core domain in order for cleavage and activation of PDGF-C to occur, which likely explains this specificity.
Extracellular retention and distribution of PDGFs
Spatially uneven distribution (gradients and depots) of growth factors, cytokines, and morphogens defines their biological activity and action range. Diffusion of PDGF in the tissue interstitium is regulated by binding to extracellular matrix components (Fig. 3). For PDGF-A and PDGF-B, such binding is accomplished in part by the positively charged C-terminal motifs (referred to as retention motifs) containing a high proportion of basic amino acid residues (Fig. 1). PDGF-C and PDGF-D lack basic retention motifs, but CUB domains are implicated in protein–protein and protein–carbohydrate interactions in other contexts and may regulate extracellular distribution of latent PDGF-C and PDGF-D. The presence of the retention motif is determined by alternative splicing in PDGF-A and by alternative C-terminal proteolytic processing in PDGF-B (Fig. 3). Alternative splicing has also been demonstrated for several of the members of the VEGF family, leading to the formation of multiple isoforms that differ in extracellular matrix binding (Fig. 1; for review, see Ferrara et al. 2003). Alternative splicing of the PDGF-A transcript is cell type-specific and differs both among tumor cell lines (Afrakhte et al. 1996) and in different organs during development (J. Andrae, H. Boström, and C. Betscholtz, unpubl.).
Figure 3.

Processing and extracellular retention of the PDGFs. (Top panel) Through alternative splicing, PDGF-A may be translated into a protein with or without a retention motif (green). Both isoforms may bind and activate PDGFR-α (active). Heterodimers between the long and short forms of PDGF-A are not illustrated but can likely form since both splice isoforms are produced by the same cells in most situations. (Middle panel) PDGF-B is produced as a single precursor containing the retention motif. This protein may be secreted, in which case it gets trapped at the external side of the cell membrane or in pericellular matrix such as the basement membrane, where it is active and participates in pericyte recruitment. In platelets, PDGF-B is processed intracellularly into a soluble and active isoform lacking the retention motif. There is experimental evidence for trafficking of a proportion of synthesized PDGF-B toward degradation without prior secretion. (Bottom panel) PDGF-C and PDGF-D are produced and secreted as inactive growth factors containing a CUB domain (yellow). The CUB domain may help in localizing these PDGFs in the extracellular space. Active PDGF-C and PDGF-D are produced through extracellular proteolysis.
C-terminal proteolytic processing of PDGF-B may take place intracellularly or extracellularly. The endogenous protease(s) responsible for C-terminal cleavage of PDGF-B has not been identified, but thrombin is a putative candidate for extracellular proteolysis (Kelly et al. 1993). Certain cells transfected with PDGF-B expression vectors secrete soluble PDGF-BB into the conditioned medium. However, a major part of endogenously expressed PDGF-B becomes trapped on the cell surface or in the extracellular matrix, where it subsequently can be released by thrombin (Kelly et al. 1993; Soyombo and DiCorleto 1994).
Insights into the role of PDGF retention have come from studies of PDGF interaction with heparan sulfate proteoglycans (HSPGs) and phenotypic analysis of PDGF-B retention motif knockout mice. PDGFs bind to heparin and HSPGs similar to many other growth factors and morphogens with critical functions during development (e.g., hedgehogs, bone morphogenetic protieins [BMPs], and Wnts) (Feyzi et al. 1997; Lustig et al. 1999; Lin 2004; Häcker et al. 2005; Abramsson et al. 2007). Targeted deletion of the PDGF-B retention motif in mice leads to pericyte detachment from the microvessel wall (Abramsson et al. 2003; Lindblom et al. 2003). Reduced heparan sulfate (HS) N-sulfation (caused by lack of the enzyme N-deacetylase/N-sulfotransferase-1) similarly leads to pericyte detachment and delayed pericyte migration in vivo (Abramsson et al. 2007). This is probably caused by attenuated PDGF-BB binding to HS. PDGF-BB/HS interaction appears to depend on overall N- and O-sulfation of HS, whereas saccharide fine structure appears to be of lesser importance. Taken together, available evidence suggests a model in which PDGF-BB secreted from endothelial cells interacts with HS at the endothelial surface or in the periendothelial matrix. This would lead to local deposits of PDGF-BB, which, in turn, are critical for the correct investment of pericytes in the vessel wall. HS binding is also necessary for proper localization and function of VEGF-A (Ruhrberg et al. 2002). It was shown recently that HS may act in trans—i.e., from pericytes—to potentiate VEGF-receptor function in endothelial cells (Jakobsson et al. 2006). Similarly, HS expressed on endothelial cells may function to enhance PDGF-BB-mediated PDGFR-β signaling in neighboring pericytes.
PDGF binds also to certain non-HSPG extracellular proteins, but the physiological relevance of these interactions is unclear. Binding of PDGF-B has been demonstrated to α-2-macroglobulin (Bonner and Osornio-Vargas 1995), possibly acting as a scavenger for PDGF-B through low-density lipoprotein (LDL) receptor-related protein (LRP) receptors on macrophages and other cells (Bonner et al. 1995). PDGF-B also binds to SPARC and adiponectin, which may trap the growth factor in the extracellular space (Raines et al. 1992; Arita et al. 2002).
PDGF receptor signaling transduction and downstream events
Dimerization is the key event in PDGF receptor activation as it allows for receptor autophosphorylation on tyrosine residues in the intracellular domain (Kelly et al. 1991). Autophosphorylation activates the receptor kinase and provides docking sites for downstream signaling molecules (Kazlauskas and Cooper 1989). Docking of receptor substrates and further signal propagation involves protein–protein interactions through specific domains; e.g., Src homology 2 (SH2) and phosphotyrosine-binding (PTB) domains recognizing phosphorylated tyrosines, SH3 domains recognizing proline-rich regions, pleckstrin homology (PH) domains recognizing membrane phospholipids, and PDZ domains recognizing C-terminal-specific sequences (for review, see Heldin et al. 1998). Most of the PDGFR effectors bind to specific sites on the phosphorylated receptors through their SH2 domains.
PDGFR-induced signaling pathways
Both PDGFR-α and PDGFR-β engage several well-characterized signaling pathways—e.g. Ras-MAPK, PI3K, and PLC-γ—which are known to be involved in multiple cellular and developmental responses (Fig. 4). PDGFRs connect to Ras-MAPK mainly through the adaptor proteins Grb2 and Shc. Grb2 binds the activated PDGFR through its SH2 domain and complexes Sos1 through its SH3 domains. Sos1 in turn activates Ras, leading to downstream activation of Raf-1 and the MAPK cascade. MAPK signaling activates gene transcription, leading to stimulation of cell growth, differentiation, and migration (for review, see Bar-Sagi and Feramisco 1986; Seger and Krebs 1995).
Figure 4.

PDGFR signal transduction and links to the cytoskeleton. (A) The intracellular domains of PDGFR-α and PDGFR-β and some of their direct interactors are illustrated. Arrows imply links to major signal transduction pathways and secondary effectors. Negative feedback signaling is indicated in red. (B) Schematic illustration of how PDGFR-β may link to the cytoskeleton and to other signaling components of focal adhesions through the adapter NHERF, the merlin and ezrin/radixin/moezin family of cytoskeletal linkers (MERM), and focal adhesion kinase (FAK).
PI3K is a family of enzymes phosphorylating phosphoinositides. Effectors of PI3K signaling include serine/threonine kinases such as Akt/PKB (Burgering and Coffer 1995; Franke et al. 1995), some members of the PKC family including atypical isoforms (Nakanishi et al. 1993; Akimoto et al. 1996), p70 S6 kinase (Chung et al. 1994), JNK (Lopez-Ilasaca et al. 1997), and small GTPases of the Rho family (Hawkins et al. 1995). Activation of the PI3K pathway by PDGFRs promotes actin reorganization, directed cell movements, stimulation of cell growth, and inhibition of apoptosis (Hu et al. 1995).
PLC-γ binds PDGFRs, which results in its activation through phosphorylation (for review, see Tallquist and Kazlauskas 2004). PLC-γ activation leads to mobilization of intracellular calcium ions and the activation of PKC (Berridge 1993). The effects of PDGFR-mediated PLC-γ activation include stimulation of cell growth and motility (Kundra et al. 1994).
Several additional signaling molecules are engaged by PDGFRs, including enzymes, adaptors, and transcription factors. Activation of the Src TK promotes Myc transcription and mitogenic responses (for review, see Erpel and Courtneidge 1995). Also, members of the Fer and Fes TK family bind to PDGFRs (Kim and Wong 1995). PKC-δ is phosphorylated by PDGFR-β, leading to its activation and translocation to the cell membrane (Li et al. 1996). This signal may be involved in cell differentiation. The adaptors Nck and Crk bind to PDGFRs through their SH2 domain and are involved in activation of JNK (Nishimura et al. 1993; Su et al. 1997). The adaptor Grb7 contains a SH2 domain and binds PDGFR-β (Yokote et al. 1996). STAT transcription factors may bind to PDGF receptors, leading to their phosphorylation and activation (Darnell 1997).
PDGF receptors interact also with integrins, which enhances cell proliferation, migration, and survival (for review, see Assoian 1997; Frisch and Ruoslahti 1997). PDGFR interaction with integrins helps localizing PDGFRs and interacting molecules at focal adhesions, which are sites where several signaling pathways initiate and cross-talk (Clark and Brugge 1995). Recently, Na+/H+ exchanger regulatory factors (NHERFs) were shown to bind PDGFR-β and link it with focal adherence kinase and the cortical actin cytoskeleton (James et al. 2004), as well as to N-cadherin (Theisen et al. 2007) and the phosphatase PTEN (Fig. 4; Takahashi et al. 2006). Additional evidence for compartmentalization of PDGFRs and their downstream signals within cells comes from the intriguing observation that PDGFR-α (but not PDGFR-β) localizes to the primary cilium of fibroblasts (Schneider et al. 2005). Mutants that fail to form cilia do not activate PDGFR-α but maintain PDGFR-β signaling. Primary cilia are also essential for hedgehog signaling (Eggenschwiler and Anderson 2007). Hedgehog receptors (patched) and PDGFR-α show a conspicuous overlap in their expression pattern during development (Karlsson et al. 1999, 2000), and it is thus possible that this correlation extends to signaling at the subcellular level as well.
Negative control of PDGFR signaling
PDGF signaling is carefully regulated by feedback control mechanisms. Stimulatory and inhibitory signals arise in parallel, and the ultimate response depends on the balance between these signals (for review, see Heldin et al. 1998). The SHP-2 tyrosine phosphatase binds PDGFR through its SH2 domain and dephosphorylates the receptor and its substrates (Lechleider et al. 1993). Ras-GAP, which negatively regulates Ras, also binds PDGFR-β through its SH2 domain (Fantl et al. 1992).
Ligand occupancy of PDGFRs promotes endocytotic receptor internalization. The major destiny of internalized PDGFRs seems to be lysosomal degradation, thereby limiting the duration of PDGFR signaling (Heldin et al. 1982; Sorkin et al. 1991; Mori et al. 1995). Recycling of PDGFR-β, but not PDGFR-α, was recently observed in cells deficient for the phosphatase TC-PTP (Karlsson et al. 2006), which is a negative regulator of PDGFR-β phosphorylation (Persson et al. 2004). Trafficking toward lysosomal degradation of PDGFR-β depends on interactions with c-Cbl and receptor ubiquitination. The adapter protein Alix, which interacts with the C-terminal domain of PDGFR-β, facilitates ubiquitination and degradation of c-Cbl, thereby inhibiting PDGFR-β down-regulation (Lennartsson et al. 2006).
Cellular responses to PDGFR signaling
Some of the cellular responses to PDGFs take place within seconds to minutes after PDGFR activation and are independent of gene expression and protein synthesis. PDGFR-α and PDGFR-β mediate similar but not identical rapid responses. Both receptors stimulate rearrangement of actin filaments, but only PDGFR-β promotes formation of circular ruffles. PDGFR-β also mobilizes calcium ions more efficiently than PDGFR-α (Diliberto et al. 1992; Fatatis and Miller 1997). PDGFR-β inhibits gap junctional communication between cells through phosphorylation of the gap junction protein connexin 43 (Hossain et al. 1998). It is unclear whether this ability is shared with PDGFR-α.
In addition to the rapid post-transcriptional responses, PDGFRs (like other RTKs) induce fast transcriptional changes involving so-called immediate early genes (IEGs) (Cochran et al. 1983). IEGs are direct targets of the transcription factors that get activated (by post-translational modification) through various signaling pathways. The IEG responses are likely necessary for many of the long-term effects of PDGFs in vitro and in vivo. To what extent the IEG responses contribute specificity to PDGFR signaling is, however, unclear. Different RTKs induce virtually identical sets of IEGs (Fambrough et al. 1999). Also, different signaling pathways activated by the same receptor (PDGFR-β) induce largely overlapping sets of IEGs in vitro (Fambrough et al. 1999). From these studies, it would appear that quantitative rather than qualitative differences in the IEG responses mediate the specific responses to different RTKs and signaling pathways. This view is supported by in vivo analysis of an allelic series of pdgfrb tyrosine-to-phenylalanine mutations disrupting connection to different substrates and signaling pathways (Tallquist et al. 2003). Mutations in single or multiple tyrosine residues led to quantitatively different but qualitatively similar developmental effects (Tallquist et al. 2003). In contrast, similar analyses of PDGFR-α revealed strikingly different roles of the different downstream signaling pathways. For PDGFR-β, the disruption of signaling through PI3K alone has no, or only minor, developmental consequences (Heuchel et al. 1999; Tallquist et al. 2000a). In contrast, PI3K is indispensable for PDGFR-α function during development (Klinghoffer et al. 2002). Intriguingly (and in further contrast to PDGFR-β), deficient coupling to Src from PDGFR-α resulted in specific problems with the oligodendrocyte lineage, whereas other processes, such as skeletal development, remained normal.
The different IEGs appear to cooperate in their regulation of downstream cellular and developmental events. By analyzing gene trap mutants for >20 IEGs downstream from PDGFR signaling, a striking degree of phenotypic overlap and genetic interaction was revealed. Different mutated IEGs produced qualitatively similar responses. Moreover, the combination of mutations in several genes strengthened specific phenotypic outcomes known to depend on PDGFR signaling, such as craniofacial, cardiovascular, or kidney developmental processes (Schmahl et al. 2007). The large overlap between the signaling pathways, IEGs, and biological processes argue in favor of a model in which specificity is generated through a combination of quantitative differences in magnitude and duration of the responses occurring at different levels in the signaling cascade. One shall not forget, however, that a major part of the specificity of the PDGFRs in developmental functions depends on cell type- and context-specific PDGFR expression. The expression patterns of the PDGFRs correlate with the major sites of their functions; e.g., PDGFR-α is strongly expressed in OPs and PDGFR-β in pericyte progenitors. Lack of redundancy is therefore dependent in part on the different transcriptional regulation of the two pdgfr genes. By knocking in a PDGFR-β intracellular domain into the pdgfra gene, phenotypically normal animals were obtained, showing that PDGFR-β signaling can fully substitute for PDGFR-α signaling if it is expressed at the right place and time (Klinghoffer et al. 2001). The reverse replacement, however, indicates that PDGFR-α signaling can only partially compensate for the loss of PDGFR-β signaling. Targeted replacements in PDGFR-α with other RTK signaling domains have also demonstrated partial rescue, at best (Klinghoffer et al. 2001; Hamilton et al. 2003). Taken together, these data suggest that specificity of PDGFR signaling is achieved through a combination of cell type-specific expression and differential engagement of downstream signaling pathways.
Principles of genetic and pharmacological PDGF and PDGFR inhibition
Several strategies to block PDGF signaling have been applied to evaluate the role of PDGF signaling in biological and pathological processes. Targeted gene inactivation has been invaluable in the analysis of PDGF physiology in mice. Dominant-negative PDGFs or PDGFRs have been used in mammalian cell culture experiments and in Xenopus laevis. Since both PDGFs and PDGFRs require dimerization for functionality, any nonfunctional PDGF ligand or receptor retaining the ability to dimerize is a potential dominant-negative mutant. For example, N-terminal processing-deficient PDGF ligands, or PDGF receptors with inactivated or truncated kinase domains, act in this way. The PDGF/PDGFR complex is also amenable to pharmacological inhibition in several different ways (Fig. 5). Neutralizing antibodies exist for PDGF ligands and receptors and have been used extensively in experiments evaluating the importance of PDGF signaling in pathogenic processes. Aptamers (stabilized oligonucleotides that bind proteins with very high specificity) have been developed for the inhibition of PDGF-B and have been used to test the pathogenic role of this protein in various rodent disease models.
Figure 5.

Principles of pharmacological inhibition of PDGF signaling. PDGFs can be blocked by neutralizing antibodies, recombinant dimeric soluble PDGFR extracellular domains, or nucleic acids (aptamers), which all bind to the PDGFs with high affinity. PDGFRs can be blocked by antibodies or dominant-negative ligands. PDGFR function may be blocked by kinase inhibitors. The possibility of silencing the PDGF-A promoter by help of small-molecule pharmacologicals has emerged recently.
One of the most efficient ways to block PDGFR signaling is to inhibit the PDGFR kinase activity. Kinase inhibitors act by binding at or near the ATP-binding pocket of the kinase domain. Several kinase inhibitors have been developed that block PDGFRs, but the inhibitors available so far are not completely specific. One of them, imatinib mesylate (imatinib, STI571, or Gleevec), inhibits PDGFR-α and PDGFR-β, the bcr–abl fusion protein (toward which it was developed) (Carroll et al. 1997), c-kit, and Flt3. Imatinib has been approved by the US Food and Drug Administration for treatment of patients with Philadelphia chromosome-positive chronic myelogenous leukemia (carrying the bcl–abl fusion protein) (Druker et al. 2001) and gastrointestinal stromal tumors (GIST) carrying activating mutations in c-kit or PDGFR-α (Demetri et al. 2002). Imatinib has also demonstrated clinical efficacy against myeloproliferative diseases involving rearrangements of the PDGFRB gene (Apperley et al. 2002) and dermatofibrosarcoma protuberans (DP), a tumor carrying translocated and activated PDGF-B genes (McArthur et al. 2005). Several other PDGFR kinase inhibitors have been developed (for review, see Levitzki 2004) and tested in clinical trials (Homsi and Daud 2007). All PDGFR kinase inhibitors used so far also block c-Kit and Flt3, two of the other three receptors with similar domain organization as PDGFRs. This specificity problem must be kept in mind when kinase inhibitors are used in order to evaluate the role of PDGF signaling in (patho)physiological processes. Imatinib nevertheless shows promise as a drug for a number of clinical conditions in which PDGFR signaling is activated (discussed in subsequent sections). The side effects of imatinib also appear to be minor from a general perspective as well as in comparison with other PDGFR inhibitors tested so far (for review, see Homsi and Daud 2007).
Currently, other strategies for small-molecule-mediated inhibition of PDGFR signaling are scarce. A recent study identified a nuclease-hypersensitive element in the PDGF-A promoter as a putative target for small molecule pharmacologicals, however. The guanine-rich strand of the DNA in this region forms a dynamic G-quadruplex, which upon binding of the small molecule TMPyP4 becomes stabilized, silencing the PDGF-A promoter (Qin et al. 2007). How specific this inhibition is and whether this can be used to shut down PDGF-A expression in vivo remain to be established.
Developmental functions of PDGFs/PDGFRs
Developmental roles of PDGFs and PDGFRs have been unraveled mainly by way of gene-targeted mice. A large number of knockout, knock-in, or transgenic mutants have been used to identify specific cell types that are primary targets of PDGF signaling during development. The importance of these findings lies not only in the information achieved about the in vivo roles of PDGFs, but also in the new knowledge gained about some of the cell types regulated by PDGFs. For example, the critical roles of microvascular pericytes (Lindahl et al. 1997a), lung alveolar SMCs (Boström et al. 1996), and gastrointestinal villus cluster cells (Karlsson et al. 2000) were all pinpointed for the first time through the analysis of PDGF-B and PDGF-A knockout mice, respectively. Important developmental roles for PDGF/PDGFR-like proteins have also been demonstrated in nonmammalian vertebrates. As discussed below, some of these appear to be analogous to the roles of the mammalian PDGFs and VEGFs.
Role of PDGF signaling in gastrulation and formation of cranial and cardiac neural crest
Several observations suggest that PDGF-A and PDGFR-α have early developmental functions. Xenopus embryos injected with dominant-negative PDGF receptor mRNA show aberrant gastrulation, in which the involuting prospective head mesodermal cells fail to migrate beneath the overlying PDGF-A-positive blastocoel roof ectoderm (Ataliotis et al. 1995). Upon PDGFR inhibition, the mesodermal cells detach from the ectoderm and undergo apoptosis. Rescue from apoptosis did not rescue the migration defect, suggesting that PDGFR(s) independently controls both processes (Van Stry et al. 2004). In contrast to these effects, inhibition of PDGFR signaling did not affect the migration and survival of axial mesoderm in Xenopus. Axial mesoderm develops through a different morphogenetic process (mediolateral intercalation) compared with head mesoderm and at a distance from (and therefore probably independent of) the PDGF-A-expressing ectoderm (Van Stry et al. 2004).
A role for PDGF signaling during gastrulation is also suggested from experiments in sea urchins. PDGFR expression occurs in the developing mesoderm, and different strategies to inhibit PDGF or PDGFR function result in gastrulation defects (Ramachandran et al. 1995, 1997). A role for PDGF signaling in the mesoderm during zebrafish gastrulation has also been proposed (Montero and Heisenberg 2004). The PDGFR-specific kinase inhibitor AG1296 inhibited formation of mesendodermal cell protrusions and cell polarization in vivo in fish embryos. In vitro, PDGF-A stimulated these activities (Montero and Heisenberg 2004). Taken together, data from frog, sea urchin, and fish suggest an evolutionary conserved role for PDGF signaling during gastrulation in deuterostomes.
In mouse embryo development, PDGF-A expression onsets early and can be detected already in the preimplantation embryo (Rappolee et al. 1988; Mercola et al. 1990; Palmieri et al. 1992). At this stage, PDGF-A is coexpressed with PDGFR-α in the blastocyst inner cell mass. In spite of these early expression patterns, a role for PDGFs during mouse gastrulation is not apparent (Hoch and Soriano 2003). PDGF-A and PDGFR-α-null mouse embryos nevertheless show severe impairment of early mesenchymal derivatives in both embryo and extraembryonic tissues (Hamilton et al. 2003). A proportion of these embryos die before or at embryonic day 10.5 (E10.5), which is, however, variable and genetic background-dependent (Hoch and Soriano 2003).
Although early developmental routes may differ between vertebrate species, some of the later consequences of gastrulation defects in Xenopus resemble the phenotypes observed in certain PDGF/R mutant mice. PDGFR signaling-inhibited Xenopus embryos develop reduced anterior structures and defective vertebral arch closure (spina bifida). Similar defects are observed in mouse PDGFR-α knockouts and PDGF-A/C double knockouts due to problems with the developing cranial neural crest and axial skeleton. Neural crest mesenchyme expresses PDGFR-α strongly in mouse (Morrison-Graham et al. 1992), zebrafish (Liu et al. 2002a), and Xenopus (Ho et al. 1994). Complete or neural crest-specific PDGFR-α knockout in mice leads to severe developmental defects in neural crest mesenchyme derivatives, such as the cardiac outflow tract, the thymus, and the skeletal components of the facial region (Morrison-Graham et al. 1992; Soriano 1997; Tallquist and Soriano 2003). Both PDGF-A and PDGF-C are expressed at locations toward which neural crest cells migrate, such as the branchial arch epithelium (Orr-Urtreger and Lonai 1992; Ding et al. 2000; Aase et al. 2002; Liu et al. 2002b). For cardiac neural crest development, roles for both PDGFR-α and PDGFR-β have been demonstrated (Richarte et al. 2007). PDGFR-α is important mainly for nonneuronal cardiac neural crest development (Morrison-Graham et al. 1992). PDGFR-β and PDGF-B appear to play a role in neuronal cardiac neural crest development, as both PDGFR-β and PDGF-B knockouts display abnormal cardiac innervation (Van den Akker et al. 2008). Thus, PDGFR-α and PDGFR-β signaling appear to have complementary roles in cardiac neural crest development.
Directed cell migration—a conserved morphogenetic function of PDGFs
In addition to the mentioned anatomical similarities between the phenotypes resulting from PDGFR inhibition in Xenopus and PDGFR-α ablation in mice, the cellular processes that depend on PDGFR-α signaling as well as the critical signaling pathways engaged appear to be similar (Soriano 1997; Van Stry et al. 2004). For example, signaling through PI3K downstream from PDGFR-α appears to be critical during early embryonic development in mouse (Klinghoffer et al. 2002), Xenopus (Symes and Mercola 1996), and zebrafish (Montero et al. 2003).
Cell migration is a well-documented effect of PDGF stimulation in vitro (Heldin and Westermark 1999), but our understanding of PDGF-dependent cell movements in vivo is limited. PDGF signaling is undoubtedly required for the spreading of various populations of cells in developmental processes. This includes spreading of oligodendrocyte precursors in the spinal cord, of neural crest mesenchymal cells toward the branchial pouches and cardiac outflow tract, and of pericytes along newly formed angiogenic sprouts. The mechanisms by which the cells spread, and how PDGF regulates this process, remain obscure for most of the situations, however. Recent studies in Drosophila, Xenopus, and mouse have demonstrated a role for PDGF/VEGF family members in directed cell migration in at least some developmental processes in vivo.
The mechanism of PDGF-A/PDGFR-α-mediated mesodermal cell migration has been analyzed in some detail in Xenopus (Nagel et al. 2004). Using an in vitro explant model and a range of genetic tools, it was demonstrated that the blastocoel roof establishes a matrix-bound gradient of PDGF-A (long splice version) along which the PDGFR-α-positive mesodermal cells migrate directionally (Fig. 6). When graded signaling was disrupted experimentally, directional migration by mesodermal cells toward the animal pole was replaced by random migration (Nagel et al. 2004). This role for PDGF-A during Xenopus gastrulation resembles that of Drosophila PVF1 in the guidance of border cell migration in the egg chamber (Duchek et al. 2001). Also, there signaling is paracrine, with PVF1 expressed in the oocyte and PVR in the border cells. The border cells move as an aggregate from which a single cell takes a lead position by extending a long cellular protrusion in the direction of the migration of the cell aggregate. The directionality of this process depends on spatially graded signaling exerted by PVF1 (Fulga and Rørth 2002) in concert with the Drosophila EGFR ligands Keren and Spitz (McDonald et al. 2006). Specific misexpression of PVF1 misdirects the border cells to new locations (McDonald et al. 2003). However, uniform misexpression of PVF1 abolishes formation of the cellular protrusion and disrupts directionality of the migration, similar to the effect of uniform PDGF-A misexpression in the Xenopus blastocoel roof. These two examples of PDGF/PVF-induced cell migration are analogous to the role of VEGF-A in endothelial sprout guidance during angiogenesis. There, graded distribution of VEGF-A directs filopodial extensions and migratory behavior of endothelial tip cells, resulting in proper orientation of the angiogenic sprouts (Ruhrberg et al. 2002; Gerhardt et al. 2003). Thus, the ability to guide cell migration through the formation of growth factor gradients in the extracellular space appears to be common to several members of the PDGF/VEGF family, and conserved between vertebrates and invertebrates (Fig. 6).
Figure 6.
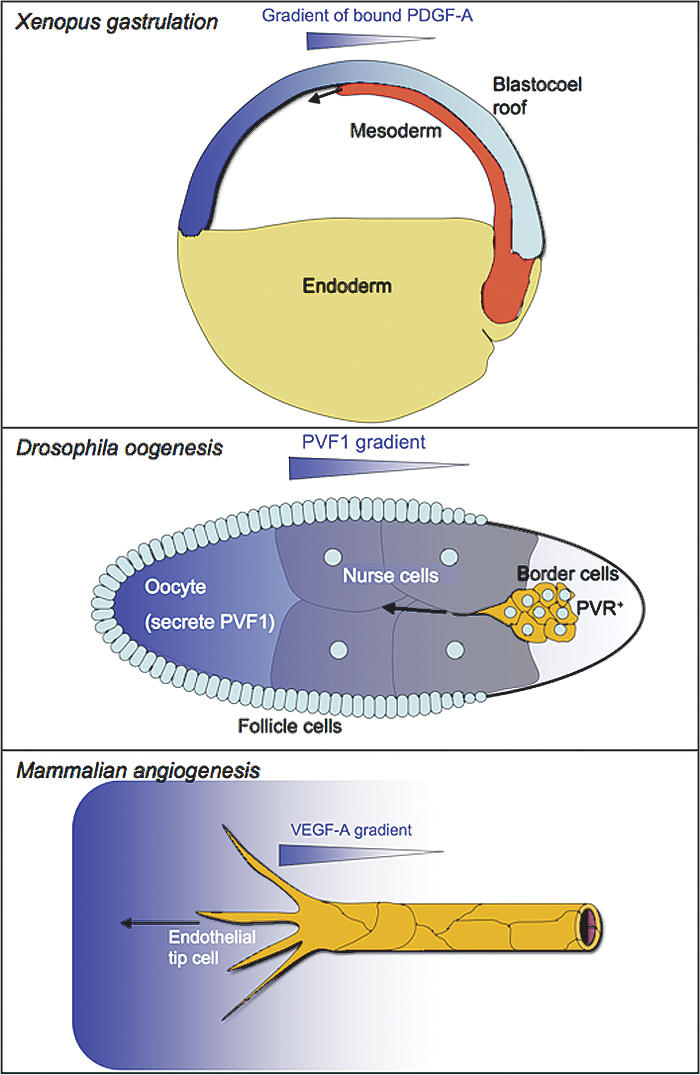
PDGFs, PVFs, and VEGFs direct cell migration in developmental processes. (Top panel) PDGF-A (long splice version) is expressed by the ectodermal cells of the blastocoel roof and gets deposited in a gradient that drives mesodermal cell movements along the blastocoel roof. (Middle panel) PVF1 is expressed by the oocyte and distributes in a graded fashion in the Drosophila egg chamber. This guides migration of the border cells toward the oocyte. (Bottom panel) Graded distribution of VEGF-A directs filopodial extensions and migration of the endothelial tip cells, thereby orienting the angiogenic sprout toward the highest VEGF-A concentration.
PDGF signaling in organogenesis
During post-implantation development, PDGF-A expression occurs in epithelia, nervous tissue, myotome, and vascular and visceral smooth muscle. PDGFR-α, in contrast, is expressed by most mesenchymal cell populations, and often in a reciprocal pattern compared with PDGF-A (for review, see Ataliotis and Mercola 1997; Hoch and Soriano 2003). A few sites of epithelial PDGFR-α expression have been noticed; e.g., lens epithelium (Reneker and Overbeek 1996), limb apical ectodermal ridge, and the early epithelial somite (Soriano 1997).
Analysis of various gene-targeted and transgenic models for PDGF-A and PDGFR-α demonstrates the importance of PDGF-A as an epithelium-derived factor promoting proliferation and spreading of nearby located PDGFR-α-positive mesenchymal cells. Thus, PDGF-A/PDGFR-α is a common signaling pair in epithelial–mesenchymal interaction, acting in concert with other signaling molecules such as hedgehogs, FGFs, BMPs, and Wnts. The complex reciprocal signaling involved in tissue induction should be kept in mind, as some of the phenotypes in PDGF-A or PDGFR-α mutants may reflect secondary consequences of the loss of mesenchymal cells that were primarily dependent on PDGF-A.
Lung and intestine—the formation of large specialized epithelial surfaces
Pdgfa−/− mice that survive birth develop a lung emphysema-like condition reflecting a complete failure of alveolar septum formation (Boström et al. 1996). Alveogenesis is a postnatal process in mouse, and it is driven by alveolar SMCs, which are missing in PDGF-A knockouts. These cells likely originate from clusters of PDGFR-α-positive mesenchymal cells that form near the epithelial buds during the pseudoglandular stage of lung development. Subsequently, the cells detach from the clusters to spread into the walls of the alveolar sacs, where they participate in (or initiate the onset of) alveogenesis (Fig. 7A). In PDGF-A knockouts, PDGFR-α-positive mesenchymal cell clusters develop normally, but subsequent spreading of the cells to the alveolar saccules fails (Lindahl et al. 1997b; Boström et al. 2002). This suggests that PDGF-AA, expressed by the lung epithelium, acts in paracrine mode to regulate the proliferation and migration of the PDGFR-α-positive alveolar SMC progenitors.
Figure 7.

Developmental roles of PDGFs. (A) During the saccular phase of lung development, PDGF-A secreted by the epithelium (yellow) drives spreading and proliferation of alveolar SMC progenitors (green). These cells differentiate into alveolar SMC and drive formation and maintain alveolar walls by producing deposits of elastin. (B) In intestinal development, the epithelium produces PDGF-A to drive proliferation of mesenchymal cells with a critical role in intestinal villus formation. (C) In the developing skin, PDGF-A secreted from keratinocytes promote proliferation of mesenchymal cells with functions in hair follicle morphogenesis. (D) In testicular development, PDGF-A (and PDGF-C) secreted from the epithelial tubes drives expansion of mesenchymal cells that subsequently differentiate into testosterone-producing Leydig cells. The testosterone production is in turn necessary for further testicular development and spermatogenesis. (E) In the developing CNS (the example illustrates the cerebellum), PDGF-A drives proliferative expansion of OPs, which subsequently myelinate nerve fibers throughout the CNS. Lack of PDGF-A leads to a complete absence of myelin in peripheral parts of the CNS, such as the optic nerve. (F) PDGF-C (and PDGF-A) is critical for development of the palatal shelves. PDGF-C is produced in the epithelium and acts on mesenchymal cells (green) in the shelves. (G) PDGF-B produced by endothelial cells (orange) drives proliferation and spreading of vSMC and pericytes in conjunction with angiogenesis and arteriogenesis. (H) In developing kidney glomeruli, PDGF-B expressed by glomerular endothelium drives the proliferation of mesangial cells (green).
Transgenic gain-of-function approaches have also been used to study the role of PDGFs in the lung. Ectopic transgenic expression of PDGF-A, PDGF-B, and PDGF-C in distal lung epithelium resulted in two different phenotypes. Mice expressing PDGF-A or PDGF-C died perinatally as a result of enlarged lungs with thickened mesenchyme and poorly differentiated cells (Li and Hoyle 2001; Zhuo et al. 2006). This phenotype likely reflects massive overproliferation of PDGFR-α-positive alveolar SMC progenitors. PDGF-B expression provided a different lung histology, comprising enlarged airspaces, inflammation, and fibrosis (Zhuo et al. 2006). This is somewhat surprising since PDGF-B should be able to activate PDGFR-α and thereby produce the same severe lung phenotype as the PDGF-A and PDGF-C transgenes. These differences may therefore reflect the different extracellular distribution and bioavailability of the different PDGFs, as discussed previously.
PDGF-A knockout mice also develop abnormally few and generally misshapen gastrointestinal villi. Villus formation is preceded by proliferation and clustering of PDGFR-α-positive cells subjacent to the epithelium (Fig. 7B). As the villus grows in length, the PDGFR-α-positive cells detach from the cluster and distribute as a single mesenchymal cell layer lining the basement membrane. The exact role of these cells in intestinal villus formation is not understood. However, PDGF-A/PDGFR-α signaling appears to secure renewal of this population of cells during the consecutive waves of villus formation that take place during intestinal development (Karlsson et al. 2000). Clustering of PDGFR-α-positive cells coincides with proliferative quiescence and onset of BMP2 and BMP4 expression by these cells (Karlsson et al. 2000). These BMPs likely signal to adjacent villus epithelial cells expressing BMPR1a. Transgenic expression of the BMP inhibitor noggin in intestinal epithelium led to ectopic formation of intestinal crypts (Haramis et al. 2004), as well as to increased epithelial expression of PDGF-A, and increased mesenchymal growth (Batts et al. 2006). This suggests that BMP expression by the villus cluster serves to restrict de novo crypt formation and mesenchymal expansion. However, the role of BMPs during gastrointestinal development is likely more complex. Transgenic models with early onset noggin expression in the intestinal epithelium show a different phenotype involving reduced villus numbers, resembling that in PDGF-A knockouts (Batts et al. 2006). Thus, BMPs may have different roles at different stages of gastrointestinal development.
Endodermal derivatives such as lung and gut are classical sites for inductive epithelial–mesenchymal interactions. The specific expression patterns for PDGF-A and PDGFR-α combined with the mouse knockout phenotypes suggest a general role for endodermal epithelium-derived PDGF-A to promote proliferation (and possibly also migration and differentiation) of specific populations of mesenchymal cells that drive morphogenesis of lung alveolar septa and intestinal villi. Both these processes involve folding of the epithelial surface and the formation of a vascularized mesenchymal core of each fold. Alveolar and intestinal surface areas are severely compromised in pdgfa−/− mice as a result of hypoplasia of the specialized PDGFR-α-positive mesenchymal cells. PDGF-A/PDGFR-α signaling is therefore instrumental in the formation of large surfaces for specialized interaction and molecular exchange with the environment; i.e., respiration and absorption (Fig. 7A,B).
Skin
PDGF-A and PDGFR-α have been implicated in development of the dermis, both in mice through genetic analyses (Betsholtz 2004) and in Xenopus using PDGFR-inhibiting substances (the TK inhibitor AG1296 and soluble PDGFR-α ectodomains) (Utoh et al. 2003). PDGF-A, PDGF-C, and PDGFR-α mouse knockouts all display dermal defects. PDGFR-α-null mice show severe dermal mesenchymal hypoplasia and skin blistering (Soriano 1997). A similar but milder phenotype is observed in PDGF-C knockouts (Ding et al. 2004). Postnatal PDGF-A-null mice show progressive loss of dermal mesenchyme and reduced hair development (Karlsson et al. 1999). Conversely, dermal injection of PDGF-AA or PDGF-BB in mice triggers resting hair follicles to enter the hair growth cycle (Tomita et al. 2006). The role of PDGF-A in the developing skin appears to be to secure the renewal of PDGFR-α-positive mesenchymal cells that are consumed as they are recruited to hair follicles to become dermal papillae and dermal sheaths (Fig. 7C; Karlsson et al. 1999).
Testis
During embryonic testicular development, tubular epithelial cells express PDGF-A, whereas interstitial mesenchymal cells express PDGFR-α. Postnatal pdgfa−/− mice display reduced testicular size and show loss of adult Leydig cells, reduced circulating testosterone, and disrupted spermatogenesis (Gnessi et al. 2000). This phenotype could be explained with the failure of development of the adult Leydig cells, the mayor testosterone-producing cells of the postnatal mammal (Fig. 7D; Gnessi et al. 2000). Studies of the PDGFR-α-null embryos revealed a more severe testicular phenotype involving a deficiency also in the fetal population of Leydig cells. This may be explained by an early overlapping function of PDGF-A and PDGF-C, both of which are expressed in the fetal male gonad (Brennan et al. 2003). It is not yet clear if lack of PDGF-A/C leads to Leydig cell deficiency entirely through proliferative arrest and progressive depletion of Leydig cell precursors, or if PDGFR signaling also controls Leydig cell differentiation.
Kidney
The interstitial kidney mesenchyme expresses PDGFR-α, and PDGFR-α-null mice show severe mesenchymal hypoplasia in the embryonic kidney. Formation of interstitial kidney mesenchyme seems to occur through the combined activity of PDGF-A and PDGF-C, which are expressed in different parts of the developing kidney (Li et al. 2000). Neither individual knockout for PDGF-A or PDGF-C showed any defect in the kidney mesenchyme, but the double PDGF-A/C knockout phenocopied the PDGFR-α-null mutant kidney phenotype (Li et al. 2000; Ding et al. 2004).
Lens
Autocrine PDGF-A–PDGFR-α expression occurs in lens epithelium and appears to play a role during lens development. Transgenic overexpression of PDGF-A in the lens leads to hyperproliferation and ectopic differentiation of the lens epithelium into lens fiber cells (Reneker and Overbeek 1996). In vitro studies confirmed that PDGF can induce proliferation in lens epithelium, but that the differentiation into fiber cells required the presence of FGF (Kok et al. 2002). PDGFR-β is also expressed in the lens, and PDGF-D was recently found to be expressed in the iris and ciliary body leading to accumulation of PDGF-D protein in aqueous humor. In eye anterior segment organ culture, anti-PDGF-D specifically and potently inhibited lens epithelial cells proliferation, suggesting that PDGF-D secreted from the iris/ciliary body regulates lens epithelial proliferation (Ray et al. 2005).
Role of PDGFs in glial cell development and neuroprotection
Oligodendrocytes are the myelin-forming glia cells of the CNS. PDGFR-α-positive OPs originate as bilateral rows of cells along the spinal cord central canal around E12 in the mouse (Pringle and Richardson 1993). PDGFR-α signaling is not required for the specification of OPs, but the further proliferation and spreading of OPs in the CNS depend on PDGF-A signaling through PDGFR-α (Fig. 7E; Calver et al. 1998; Fruttiger et al. 1999). Studies using PDGFR-α signaling mutants implicate both Src and PI3K as critical mediators of OP proliferation (Klinghoffer et al. 2002). Transgenic mice expressing different amounts of PDGF-A in the CNS have revealed that OPs proliferate until the growth factor becomes limiting; i.e., when OPs “consume” PDGF-A at a faster rate than it is produced (van Heyningen et al. 2001). When available PDGF is limiting (or withdrawn experimentally), OPs differentiate into myelinating oligodendrocytes. PDGF-A drives not only the proliferation of OPs in the embryo, but it also determines the number of OPs in the adult brain (Woodruff et al. 2004). Increased OP densities in the adult do not lead to enhanced remyelination following experimental demyelination, suggesting that the number of OPs is not limiting for myelin repair in adult mice. It is noteworthy that the normal number of OPs is also not limiting for myelination in wild-type mouse embryos. Instead, OPs are normally produced in excess numbers. pdgfa+/− mice become normally myelinated in spite of half the normal number of OPs. Only when the OP densities fall very low, as in the complete absence of PDGF-A, myelination deficiency becomes apparent (Fruttiger et al. 1999). Available genetic data suggest that PDGF-A is a critical ligand for PDGFR-α in oligodendrocyte development. PDGF-A is expressed by neurons and astrocytes throughout the CNS and is constitutively released from neural cell bodies (Fruttiger et al. 2000). PDGF-C is also expressed in the developing and adult CNS (Aase et al. 2002), but it is not known if it has a role for OP development.
PDGF-A and PDGFR-α appear to regulate also astrocyte recruitment in the developing retina. PDGF-A-null mice show retinal astrocyte hypoplasia (Gerhardt et al. 2003), and mice ectopically overexpressing PDGF-A in the retina develop astrocytic hyperplasia (Fruttiger et al. 1996; Gerhardt et al. 2003).
In vitro experiments have implicated PDGF signaling in neural development and function (Smits et al. 1991). The patterns of expression of PDGFR-α and PDGFR-β in various populations of embryonic neuronal cells or progenitors and the expression of PDGF-B in postnatal neurons are consistent with such roles (for review, see Heldin and Westermark 1999; Hoch and Soriano 2003). Neuron-specific ablation of PDGF-B was, however, not associated with apparent developmental abnormalities in the CNS or with changes in the astroglial response to local injuries (Enge et al. 2003). Neuron-specific ablation of PDGFR-β also did not produce apparent developmental defects. However, these mice showed exacerbated cerebral damage following certain brain injury experiments; e.g., cryogenic injury and NMDA-induced excitotoxicity (Ishii et al. 2006). Conversely, PDGF administration to the CNS protected against NMDA-induced injury (Egawa-Tsuzuki et al. 2004). These studies suggest that PDGFR-β signaling might exert a neuroprotective function in the adult mouse. PDGF signaling, possibly through PDGFR-α, has also been implicated in neuropathic pain following nerve injury (Narita et al. 2005).
Preliminary evidence suggests that invertebrate PVFs play a role in glial development in both C. elegans and Drosophila. C. elegans VERs are expressed by CNS cells analogous to glia (Popovici et al. 2002), and the Drosophila PVR is expressed by ventral midline glia (Cho et al. 2002). The possible developmental roles of invertebrate PVRs remain to be elucidated, but the expression patterns of VERs/PVR and PVFs in the invertebrate CNS may suggest an evolutionarily conserved role for PDGF family members in neuronal/glia development. Since the PVFs are more similar to VEGFs than to PDGFs, it is worth pointing out that also the mammalian VEGFs have been implicated as neurogenic and/or neuroprotective molecules (for review, see Storkebaum and Carmeliet 2004; Greenberg and Jin 2005). Targeted mutagenesis of the VEGF-A promoter region results in motoneuron death and the development of amyotrophic lateral sclerosis-like disease (Oosthuyse et al. 2001). Recently, VEGF-C signaling through VEGFR-3 was shown to play a role in oligodendrocyte development (Le Bras et al. 2006). Thus, an ancient role of a common PDGF/VEGF ancestor in neural development may have evolved into roles for both PDGFs and VEGFs in oligodendrocyte development and neuroprotection.
Development of the axial skeleton, palate, and teeth
PDGFR-α is expressed throughout the early (epithelial) somites but becomes progressively localized to the sclerotome and dermatome as differentiation of the somite proceeds (Soriano 1997). PDGF-A and PDGF-C are both expressed in the myotome (Orr-Urtreger and Lonai 1992; Ding et al. 2000; Aase et al. 2002). PDGFR-α-null embryos show abnormally shaped and sometimes fused somites, defects that correlate with later occurring abnormalities in ribs and vertebrae (Soriano 1997; Tallquist et al. 2000b). The expression patterns of PDGF-A/C and PDGFR-α suggest paracrine interactions between myotome, sclerotome, and dermatome, but the exact role of these signals in somite formation and differentiation is unclear. A function in chondrogenesis has been suggested based on experiments with mouse and chick micromass explant cultures (Ataliotis 2000; Tallquist et al. 2000b).
PDGFR-α-null or PI3K signaling mutants develop skeletal abnormalities, including defective vertebral neural arch formation, which results in spina bifida (Soriano 1997; Klinghoffer et al. 2002). PDGF-C appears to have a nonredundant role in vertebral development, since its ablation leads to spina bifida, although this defect is milder than that observed in PDGFR-α knockouts (Ding et al. 2004). Double knockout of PDGF-A and PDGF-C leads to severe spina bifida similar to the ablation of PDGFR-α. This suggests a role also for PDGF-A in vertebral development, but this role appears partially redundant with PDGF-C since PDGF-A-null mice have normal vertebrae (H. Boström and C. Betsholtz, 1996; unpubl.).
PDGF-C–PDGFR-α signaling plays a critical role in the formation of the palate (Fig. 7F). PDGFR-α-null embryos show cleft face and cleft palate, and PDGFR-α PI3K signaling mutants display a fully penetrant cleft palate (Soriano 1997; Klinghoffer et al. 2002). PDGF-C knockouts display cleft palate (fully penetrant in 129 sv genetic background) (Ding et al. 2004). PDGF-A-null mice show partially penetrant cleft palate, and the double PDGF-A/C knockout phenocopies the cleft face phenotype of PDGFR-α knockouts (Ding et al. 2004). Thus, as for vertebral development, palate formation appears to nonredundantly involve PDGF-C, whereas PDGF-A plays a role that is partially redundant with that of PDGF-C.
Why palatal shelves fail to fuse in PDGF-C-null embryos is not fully understood. PDGFR-α is expressed in the mesenchyme and PDGF-C in the palate epithelium, so the primary defect likely occurs in the mesenchyme. PDGFR-α-deficient palatal shelves may fuse if placed side by side in organ culture (Xu et al. 2005). This suggests that absence of PDGFR-α (or PDGF-C) impairs apposition of the shelves in vivo, which is necessary for their subsequent fusion. A conspicuous loss of expression of MMP2 in the PDGFR-α-deficient shelf mesenchyme may suggest that remodeling of extracellular matrix is a prerequisite for shelf apposition and fusion in vivo (Xu et al. 2005). Moreover, recent work in zebrafish suggests that PDGF-A mediates attraction of PDGFR-α-positive cranial neural crest cells to the oral ectoderm, where crest-derived signals induce expression of hedgehog and the transcription factor pitx2 (Eberhart et al. 2008). The same study identified a microRNA (Mirn140), which binds to the 3′ untranslated region of the PDGFR-α mRNA and negatively regulates its translation (Eberhart et al. 2008).
PDGF-A and PDGFR-α are reciprocally expressed during tooth morphogenesis. In the absence of PDGFR-α, odontoblast proliferation and differentiation take place, but ECM formation is perturbed, leading to abnormal growth of the dental cusp. The mechanisms are unclear, but as in palate development, PDGFR-α may regulate MMP2 and extracellular matrix remodeling (Xu et al. 2005).
Role of PDGF-B and PDGFR-β in the development of the cardiovascular system
In the mouse embryo, PDGF-B expression is concentrated to the developing vascular endothelium (Lindahl et al. 1997a). PDGFR-β is expressed by perivascular mesenchymal cells likely representing vascular mural cell (vSMC and pericyte) progenitors (Hellström et al. 1999). PDGF-B is particularly strongly expressed in tip cells of angiogenic sprouts and in the endothelium of growing arteries; i.e., at sites where pericytes are actively recruited and the vSMC population is expanding (Hellström et al. 1999; Gerhardt et al. 2003). The spatio-temporal expression patterns of PDGF-B and PDGFR-β together with the results of endothelium-specific knockout of PDGF-B (Enge et al. 2002; Bjarnegård et al. 2004) suggest that PDGF-BB released from endothelial cells promotes recruitment and proliferation of adjacent mural cell progenitors (Fig. 7G).
Pericyte and vSMC deficiency is seen in PDGF-B- and PDGFR-β-null embryos already at the onset of angiogenic sprouting (e.g., at E10 in the developing CNS). The degree of loss of these cells differs between different organs. This likely reflects the role of PDGF-B/PDGFR-β as a selective rather than inductive signal in pericytes/vSMC development. Induction of pericyte/vSMC differentiation from perivascular mesenchyme, as occurs, for example, around the early aorta, takes place independently of PDGF-B or PDGFR-β. Genetic analysis instead implicates TGFβ signaling in this step (for review, see Armulik et al. 2005). In contrast, the recruitment of pericytes from a pre-existing pool of such cells, by means of comigration and proliferation, is completely dependent on PDGF-B/PDGFR-β signaling. This would explain the near-complete loss of pericytes in organs such as the CNS in the absence of PDGF-B or PDGFR-β.
In spite of severe mural cell hypoplasia, PDGF-B and PDGFR-β knockout embryos continue to develop until E16–E19, at which time widespread hemorrhage and edema cause embryonic lethality. Histological analysis at E14.5 and later revealed abnormal kidney glomeruli, capillary microaneurysms, cardiac muscle hypotrophy, and placental defects (Levéen et al. 1994; Soriano 1994; Lindahl et al. 1997a, 1998; Hellström et al. 1999; Ohlsson et al. 1999). Lack of pericytes coincides with endothelial hyperplasia and an abnormally variable capillary diameter, suggesting that pericytes exert negative control of endothelial cell proliferation (Hellström et al. 2001). Lack of pericytes also leads to excessive membrane folding and cytoplasmic protrusions at the luminal surface of the endothelial cells. This suggests that pericytes control distribution of plasma membrane between the luminal and abluminal sides of the endothelium. The loss of a smooth luminal endothelial surface and the variable vessel diameter likely impairs perfusion, leading to hypoxia and secondary vascular defects (Hellström et al. 2001).
In kidney glomeruli and in the labyrinthine layer of mouse placenta, failure of recruitment of pericytes to the newly formed blood vessels generates specific and highly similar vascular defects. Normally, both organs develop complex networks of capillaries, which create the large surfaces needed for filtration/excretion in the kidney and excretion/absorption in the placenta. In PDGF-B and PDGFR-β knockout kidneys, mesangial cells are not properly recruited into the developing glomerular tufts, and as a consequence capillary branching fails (Levéen et al. 1994; Soriano 1994; Lindahl et al. 1998). These data identify PDGFR-β-positive mesangial cells as a primary target for endothelium-derived PDGF-BB in glomerular development (Fig. 7H). The pericyte loss in PDGF-B and PDGFR-β knockouts is less severe in the placenta than in the kidney, but nevertheless dramatically affects the shape of the blood vessels (Ohlsson et al. 1999). The vascular phenotypes of PDGF-B- or PDGFR-β-deficient kidney and placenta suggest that mesangial cells and placenta pericytes control capillary branching, possibly through intussusceptive vessel splitting.
Most of the defects of PDGF-B and PDGFR-β knockout mutants appear secondary to the mural cells’ hypoplasia and resulting vascular defects. However, recent analyses of the complex heart phenotype in PDGF-B and PDGFR-β knockout embryos suggest a general problem with epicardial derivatives. This leads to complex malformations including ventricular septum defects and abnormal development of the atrioventricular valves (Van den Akker et al. 2008). Combined with the PDGF-B/PDGFR-β expression patterns in the developing heart (Van Den Akker et al. 2005), these defects suggest that PDGF-B produced by developing coronary endothelium and endocardium may play a role in epicardial epithelial-to-mesenchymal transition (EMT) and recruitment of epicardium-derived mesenchymal cells into the myocardium.
PDGFs in hematopoietic development
In Drosophila embryos, PVR signaling is required for the spreading of hemocytes, precursors of the blood cell lineage. Lack of functional PVR causes stalling and clustering of hemocytes along their normal migratory route (Cho et al. 2002). This phenotype was initially interpreted as a migration defect, but later studies have shown that the hemocyte stalling and clustering are caused mainly by apoptotic cell death of hemocytes followed by phagocytic engulfment by macrophages (Brückner et al. 2004). These observations are relevant in light of the problems to interpret cell-spreading defects in mice lacking PDGF-A or PDGF-B. It is possible that the failed spreading of oligodendrocytes, alveolar SMC, and pericytes in PDGF-A and PDGF-B knockouts, as discussed above, does not result from lack of directed migration but rather from increased apoptosis, decreased proliferation, or premature differentiation (or even complex secondary phenomena), either of which may disrupt cell spreading along specific paths. Further analyses of the fate of the alveolar SMC and pericyte progenitors are needed to distinguish between these possibilities.
In addition to the major role of PVR in hemocyte survival, a role in migration was indeed indicated through survival rescue experiments (Brückner et al. 2004). Additional studies further demonstrated that PVF2 and PVF3 expressed in the CNS, and PVF2 expressed along the dorsal vessel, drive hemocyte migration along these structures (Wood et al. 2006). PVF1 appears unable to redirect hemocyte migration (Cho et al. 2002), and PVF2 (but not PVF1) drives hemocyte proliferation (Munier et al. 2002). The PVF selectivity in hemocyte development (PVF2 and PVF3) is thus opposite to that in border cell migration (discussed above), where PVF1 is the active ligand. How two different PDGF/VEGF ligands can produce cell type-specific responses via the same receptor is presently unclear, but a coreceptor involvement, similar to that of the neuropilins 1 and 2 for the mammalian VEGF receptors, seems plausible.
Whereas mammalian VEGF is clearly involved in hematopoietic development (Gerber and Ferrara 2003), the role for PDGF signaling is less obvious. PDGF-B and PDGFR-β knockout mice show hematological defects (Levéen et al. 1994; Soriano 1994), but lethally irradiated recipients of PDGF-B or PDGFR-β-deficient hematopoietic stem cells have normal hematopoiesis (Kaminski et al. 2001). This suggests that the hematological disturbances observed in the knockout embryos are secondary to the vascular problems. However, a role for PDGFR signaling in the early differentiation of hematopoietic/endothelial precursors is suggested from genetic and pharmacological gain-of-function experiments (Rolny et al. 2006). This study identified a transient PDGFR-β-positive hemangioprecursor cell, which responds to PDGF-B by differentiating toward an endothelial fate at the expense of hematopoietic differentiation. These data may also resolve some of the conflict results about PDGFR-β expression in endothelial cells (Heldin and Westermark 1999), since there may be instances when PDGFR-β expression ensues in endothelial cells (such as upon in vitro culturing), although it appears that differentiated endothelial cells do not normally express PDGFR-β in vivo.
PDGF and disease
PDGF functions have been implicated in a broad range of diseases. For a few of them—i.e., some cancers—there is strong evidence for a causative role of PDGF signaling in the human disease process. In these cases, genetic aberrations cause uncontrolled PDGF signaling in the tumor cells. There are also numerous examples of disease processes in animal models where a functional role for PDGF signaling has been demonstrated through genetic or pharmacological loss-of-function approaches. Typically, different PDGF pathway inhibitors have been used to reverse the disease process. The limitation with most of these studies is that the applied inhibitors are not completely specific. For many of the animal models, the relevance for the corresponding human diseases is also unclear. For yet a number of diseases, the evidence for involvement of PDGF signaling is purely correlative, or inferred from phenotypes caused in transgenic animals by ectopic PDGF overexpression. Keeping the limitations of these studies in mind, evidence for the involvement of PDGF signaling in several disease processes is growing rapidly. With potent and nontoxic PDGFR inhibitors at hand (such as imatinib), current ideas about causative roles of PDGF signaling in disease can now be put to the test also in humans.
Below, we tentatively divided diseases in which PDGFs are implicated into three groups—tumors, vascular diseases, and fibroses. These groupings are clearly not definitive and the PDGF-driven pathogenic processes in the various diseases are sometimes common; e.g., the formation of fibrotic reactions in conjunction with tumors and chronic inflammatory conditions. In general, two types of cells appear to respond in a pathological fashion to PDGFs—SMCs and fibroblasts—promoting vessel wall pathologies and fibrotic tissue scarring, respectively. Another general remark is that PDGFR-β appears to be the dominant PDGFR involved in vascular pathology, whereas growing evidence instead suggests a pivotal role for PDGFR-α signaling in various types of mesenchymal cell/fibroblast-driven pathologies. These differential pathological roles for the two PDGFRs resemble the developmental roles summarized above, in which PDGFR-β signaling has a key role in vascular mural cell formation, whereas PDGFR-α has both general and specific roles in the development of various mesenchymal and fibroblastic cell compartments.
PDGF signaling in tumor biology
In their review on the hallmarks of cancer, Hanahan and Weinberg assign six acquired capabilities to cancer cells—self-sufficiency in growth signals, insensitivity to anti-growth signals, escape from apoptosis, sustained angiogenesis, tissue invasion and metastasis, and limitless replicative potential (Hanahan and Weinberg 2000). Numerous studies have demonstrated that self-sufficiency in growth signals may be established for certain cell types through autocrine growth stimulatory loops involving PDGF-B/PDGFR-β signaling (Fig. 8; for review, see Heldin and Westermark 1999). Autocrine PDGF stimulation in vitro has the same effects as prolonged exogenous PDGF stimulation of the same cells (Johnsson et al. 1985) and therefore does not provide a cell with a full repertoire of malignant behaviors. By injecting PDGF-B-producing retroviruses into the mouse brain, it was recently established that PDGF stimulation cooperates with other genetic changes caused by retroviral insertions in order to induce a fully malignant tumor phenotype (Uhrbom et al. 1998). Thus, in the simplest scenario, autocrine PDGF signaling contributes to tumorigenesis by driving proliferative expansion of clones of preneoplastic and/or genetically unstable cells, which become fully malignant through further genetic alteration. However, a more multifaceted role of PDGFs in cancer biology is now emerging. In addition to providing a cell-autonomous proliferative stimulus, autocrine PDGF signaling seems to play a role also in the invasion and metastasis of certain epithelial cancers. Furthermore, paracrine PDGF signaling likely plays a role in the recruitment of different types of stromal cells. The importance of tumor angiogenesis in the supply of oxygen and nutrients to the expanding tumor mass is well established. Recent studies also highlight the possible influence of the vascular stroma on metastasis, as well as of nonvascular stroma cells on neoplastic cells growth, survival, and metastasis (Fig. 8). Moreover, by regulating stroma cell functions, PDGF signaling may limit drug delivery to tumors through effects on interstitial tissue pressure. Based on current experimental evidence, PDGF signaling may therefore be implicated causally (and hence constitute a putative drug target) in at least three of the six Hanahan/Weinberg’s cancer cell traits—self-sufficient growth, angiogenesis, and metastasis—and furthermore in resistance to cytotoxic therapy.
Figure 8.

PDGFs in tumor biology. PDGFs are frequently produced by tumor cells and may affect tumor growth and dissemination in several different ways. PDGFs may directly (thick arrows) stimulate tumor cell growth and EMT. PDGFs may further be involved in the recruitment of tumor fibroblasts and pericytes. Tumor fibroblasts may in turn produce factors that directly act on the tumor cells to promote their proliferation and migration (HGF, CXCL12, FGF2, and FGF7). Tumor fibroblasts may also secrete angiogenic factors that help in sustaining tumor angiogenesis. Tumor fibroblasts may also promote metastasis by secreting CCL5, which induces metastatic behavior of the tumor cells. Tumor cell-derived PDGF-B and PDGF-D may, if produced in excess (such that the PDGF-B gradient established by the endothelial cells is overwhelmed), promote detachment of pericytes, which may facilitate metastasis. See the text for references to the different examples. The symbols are adapted from Hanahan and Weinberg (2000).
Autocrine PDGF signaling in cancer—the case of glioblastoma
Autocrine PDGF loops have been proposed to play a role in different types of malignant brain tumor of glial origin (gliomas). Virtually all tested human glioma cell lines and fresh tumor isolates express multiple PDGFs and PDGFRs (Nistér et al. 1988; Hermanson et al. 1992), especially PDGF-A, PDGF-C, and PDGFR-α, which are almost ubiquitously found, but also PDGF-B, PDGF-D, and PDGFR-β (Lokker et al. 2002). In various experimental systems, overexpression of PDGFs has been shown to cause the formation of glioma-like tumors (Uhrbom et al. 1998; Dai et al. 2001). Inhibition of PDGF/PDGFRs has been shown to slow down glioma cell growth in experimental models (Uhrbom et al. 2000; Lokker et al. 2002; Servidei et al. 2006). Inhibitors of PDGFR signaling, such as imatinib, have recently entered clinical trials for high-grade glioma patients (Newton 2003).
The mechanism by which PDGF expression is activated in gliomas is poorly understood. TGFβ pathway activation was demonstrated to correlate with high proliferation and poor prognosis in gliomas. In search of mechanisms, it was found that TGFβ signaling through Smad2/3/4 activates PDGF-B transcription and secretion, and that the effect of TGFβ on glioma cell proliferation depends on PDGF-B/PDGFR signaling (Bruna et al. 2007). This confirms earlier indications that PDGF might be involved in the response to TGFβ in glioma (Jennings et al. 1997). The response to TGFβ correlated with the methylation status of the PDGF-B gene. These results demonstrate that the proliferative response to TGFβ in human glioma, and perhaps also the poor prognosis associated with TGFβ pathway activation in this disease, is mediated by PDGF-B expression and function. In part, this may be dictated by absence of epigenetic silencing of the PDGF-B gene.
Perhaps the most interesting scenario from both biological and therapeutic standpoints would be if glioma tumor stem cells respond to PDGFs. Data in support of such a scenario were recently published (Jackson et al. 2006). Adult neural stem cells in the subventricular zone, capable of generating both oligodendrocytes and neurons, were shown to express PDGFR-α and respond to PDGF by proliferation, leading to the formation of glioma-like hyperplasias. Neuroepithelial cells express PDGFR-α as early as E8.5 (Andrae et al. 2001), but the role of PDGFR-α at this time of development is not well understood. However, PDGFR-α-positive stem cells capable of generating oligodendrocytes develop later, and some of them are retained throughout life. It is therefore an intriguing possibility that adult PDGFR-α-positive stem cells constitute the cellular origin of gliomas and that the autocrine PDGF/PDGFR loops operating in such tumors reflect a normal role for PDGFs in regulating self-renewal and differentiation in this stem cell population.
Mutations in PDGF and PDGFR genes leading to autocrine PDGF signaling in cancer
There are several ways by which genetic alterations may lead to overexpression or altered function of the gene product in tumors; e.g., gene amplification, translocation, and activating mutations. All of these have been described in human tumors with high PDGF expression (Fig. 9).
Figure 9.

Genetic causes of PDGF/PDGFR deregulation. Translocation in the PDGF-B gene leads to an autocrine loop that sustains tumor cell proliferation in DP. Chromosomal translocations and deletions lead to the production of dimeric fusion proteins involving the intracellular PDGFR-β domain. Amplification of the PDGFR-A gene is frequently found in glioblastoma. GISTs may carry activating point mutations in the PDGFR-α kinase domain. See the text for details and references.
There are several reports on PDGFR-A amplifications, especially in glioblastomas (Fleming et al. 1992; Hermanson et al. 1996). Here, overexpression of PDGFR-α appears to precede amplification of the gene. Up-regulation of PDGFR-α protein is detected in most gliomas of all malignant grades, the more malignant the higher expression. In the most malignant form, gene amplification occurs in a subset of the tumors (Hermanson et al. 1996). PDGFR-A amplifications have also been described in anaplastic oligodendrogliomas (Smith et al. 2000), esophageal squamous cell carcinoma (Arai et al. 2003), and pulmonary artery intimal sarcomas (Zhao et al. 2002). It should be kept in mind that the amplification of a gene does not prove its involvement in the disease process—another gene within the amplicon might instead be critical. A high frequency of amplifications within a specific group of tumors implies a role in tumor development, however, suggesting that further studies of pulmonary sarcomas could be of interest (Zhao et al. 2002). This study comprised only eight patients, but PDGFR-A amplifications were found in five of them.
Most GISTs carry activating mutations in the Kit receptor tyrosine kinase, and imatinib is currently used to treat these patients. In a subset of GISTs with wild-type Kit, gain-of-function mutations in PDGFR-A have been detected (Heinrich et al. 2003; Hirota et al. 2003). Kit and PDGFR-α mutations are mutually exclusive, and 85%–90% of all GISTs carry one of the two mutations. PDGFR-α mutations are found either in the juxtamembrane region or in any of the two TK domains and lead to constitutive activation of the receptor. Signaling through PI3K and STAT3 is thought to be of most importance for this activation (for review, see Ali et al. 2007).
DP is a rare, slow-growing, infiltrative dermal sarcoma. More than 95% of all DPs have a unique translocation of the PDGF-B gene, which results in a fusion of collagen type 1 α1 chain (COL1A1) and PDGF-B genes, such that the PDGF-B exon 2 ends up under the control of the COL1A1 promoter (Simon et al. 1997). This results in overexpression of PDGF-B in skin fibroblasts (which normally strongly express COL1A1), creating an autocrine stimulatory loop that drives cell proliferation and fibrosis (for review, see Sirvent et al. 2003). The role for active PDGFR signaling in DP is supported by experiments in culture and animals. When transplanted onto mice, DP tumor growth is inhibited by imatinib (Sjöblom et al. 2001).
Most translocations involving PDGFRs result in a constitutively active receptor. In myeloid disorders and leukemias, at least six different translocations of the PDGFR-B gene have been reported (Sirvent et al. 2003). Most of these have only been found once, but the ETV6–PDGFR-B fusion gene has been identified >30 times and has also been more extensively characterized (Golub et al. 1994). The ETV6–PDGFR-B fusion results in a protein containing the transmembrane and catalytic intracellular part of the receptor, a protein that cannot bind to PDGF but is nevertheless constitutively active.
Fusion of the FIP1L1 and PDGFR-A genes is found in a subset of patients with hypereosinophilic syndrome (HES) (Cools et al. 2003a; Griffin et al. 2003). HES is characterized by high levels of eosinophils, and in some patients, HES develops into acute leukemia. The FIP1L1–PDGFR-A fusion gene has been identified also in systemic mastocytosis (Pardanani et al. 2003). The FIP1L1–PDGFR-A fusion protein is a constitutively active tyrosine kinase that can transform hematopoietic cells. Mice infected with retroviral FIP1L1–PDGFR-α develop myeloproliferative disease within a few weeks (Cools et al. 2003b). The paradigm for constitutive activation of PDGFs through translocation-mediated fusion with other proteins is enforced dimerization mediated by the fusion partner. The constitutive activity of the FIP1L1–PDGFR-α fusion protein, however, is independent of FIP1L1 and dimerization. Instead, it involves the deletion of an autoinhibitory PDGFR-α juxtamembrane domain (Stover et al. 2006).
Autocrine PDGF signaling in epithelial cancers
Autocrine PDGF signaling has been implicated in glioblastomas and sarcomas (tumors supposed to originate from PDGFR-positive cells). PDGF production in carcinomas, which is frequently observed (Heldin and Westermark 1999), is generally thought to act in a paracrine fashion to promote tumor stroma; i.e., mesenchymal cells and blood vessels (Pietras et al. 2003a; Furuhashi et al. 2004). However, the possibility also exists that autocrine signaling may play a role in carcinomas in conjunction with ectopic onset of PDGFR expression (Heldin et al. 1988). Expression of PDGF has been found to correlate with advanced tumor stages and unfavorable prognosis in human breast carcinoma (Seymour et al. 1993). Increased expression of PDGF pathway genes has also been correlated with EMT (Jechlinger et al. 2003). Using PDGF-neutralizing antibodies, dominant-negative PDGFR expression, or imatinib, it was recently demonstrated that autocrine PDGF signaling maintains EMT and promotes metastasis in mouse mammary carcinoma (Fig. 8; Jechlinger et al. 2006). Thus, PDGF expression in epithelial cancers may have an unexpected role in EMT and tumor dissemination, but it remains to be established if this phenomenon is common or rare.
Paracrine PDGF signaling and the recruitment of tumor stroma
Tumor stroma consists of nonneoplastic cells (and extracellular matrix) recruited into the tumor from the circulation or neighboring normal tissue. The stroma makes up a variable proportion of the tumor mass—certain carcinomas may consist of >90% of fibrous stroma—and the cellular composition of the stroma varies as well. Usually, the stroma is divided into a vascular part consisting of endothelial cells and associated mural cells (tumor pericytes) and a fibrous component consisting of mesenchymal cells (tumor fibroblasts).
PDGF expression is commonly found in the neoplastic component of different human tumors. PDGFR expression is regularly found in the stromal compartment of carcinomas, including skin, breast, colorectal, lung, pancreas, prostate, ovarian, and others (for review, see Heldin and Westermark 1999; Ostman 2004). Analyses of experimental tumors have confirmed the ability of tumor cell-derived PDGFs to contribute to the recruitment of both fibroblastic and vascular tumor stroma (Forsberg et al. 1993; Skobe and Fusenig 1998; Shao et al. 2000; Pietras et al. 2007). Through its effects on tumor stroma, PDGF signaling may directly or indirectly promote tumor growth, blood perfusion, metastatic dissemination, and drug resistance (Fig. 8).
Vascular stroma—PDGFR-β expression in solid tumors occurs mainly in vascular mural cells. PDGF-BB or PDGF-DD expression in experimental fibrosarcoma or melanoma has been shown to enhance pericyte recruitment to tumor vessels, correlating with tumor vessel stabilization, tumor cell survival, and tumor growth (Abramsson et al. 2003; Furuhashi et al. 2004). The importance of pericyte recruitment in tumor angiogenesis has also been indicated through studies of tumor growth in the RIP-Tag mice. In this model, it was demonstrated that pharmacological inhibition of PDGFR-β signaling in tumor pericytes synergized with the inhibition of VEGFR signaling in endothelial cells in suppressing tumor growth (Bergers et al. 2003).
While the quantity of recruited pericytes may affect tumor vessel function and tumor growth, there are also recent notions that the quality of the pericyte coating may affect hematogeneous tumor dissemination. Increased metastasis was reported in mouse mutants where tumor pericytes detach from the vessels as a result of loss of HSPG binding of PDGF-B or NCAM deficiency (Fig. 8; Xian et al. 2006). Low pericyte coverage is commonly observed in tumor vasculature (Schlingemann et al. 1991; Hashizume et al. 2000; Abramsson et al. 2002). Moreover, low expression of smooth muscle markers correlates positively with metastatic propensity in human solid tumors (Ramaswamy et al. 2003). Based on these findings, it would appear dangerous to inhibit pericytes as part of an anti-tumor angiogenesis strategy, as it might simultaneously impair tumor vessel function and increase metastasis (Gerhardt and Semb 2008). Further experiments are required to distinguish between possible benefits and drawbacks, with therapy aimed at inhibiting tumor pericyte recruitment.
Tumor fibroblasts—Tumor fibroblasts have for long been viewed as passive component of tumors. Emerging data suggest, however, that these cells may affect the malignant behavior of the neoplastic cells and contribute to tumor progression and resistance to therapy. It has been demonstrated that (PDGF-B-driven) recruitment of fibroblasts increases the progression of immortal human keratinocytes from a nontumorigenic to a tumorigenic phenotype (Skobe and Fusenig 1998). Recent studies pinpointed hepatocyte growth factor as the likely mediator of this effect (Fig. 8; Lederle et al. 2006). Recruited fibroblasts may also contribute to tumor angiogenesis. Using vegfa gene-inactivated transplantable tumors, it was demonstrated that tumor cell-derived PDGF-AA promoted recruitment of PDGFR-α-positive fibroblasts, which in turn produced VEGF-A and rescued tumor angiogenesis (Fig. 8; Dong et al. 2004). The production of angiogenic growth factors by tumor fibroblasts may also explain why experimental PDGF-B overexpression in melanoma leads to prominent recruitment of both fibroblastic and vascular stroma components (Forsberg et al. 1993). In preclinical models of human lung tumors, PDGF-A and PDGF-C were shown to recruit PDGFR-α-positive tumor fibroblasts, thereby promoting tumor growth (Tejada et al. 2006). PDGF-driven fibroblast recruitment was recently also demonstrated to drive tumor cell proliferation in a model of mouse cervical cancer, where the recruited fibroblasts secrete FGF2 and FGF7 (Fig. 8; Pietras et al. 2007). PDGFR inhibition by imatinib or PDGFR antibodies, or inhibition of the stroma-derived FGFs through ligand trapping, were both effective in reducing tumor growth in this model. Other studies have demonstrated the importance of cancer-associated fibroblasts in supporting tumor growth through secretion of SDF-1/CXCL12, which binds to CXCR4 on tumor cells (Orimo et al. 2005). Also, cancer fibroblast-derived CCL5/RANTES may enhance metastatic behavior of tumor cells (Fig. 8; Karnoub et al. 2007). It may be anticipated (but remains to be established) that SDF-1- and CCL5-positive stromal fibroblasts are recruited into the tumors by tumor cell-derived PDGFs.
Interstitial tumor pressure—Solid tumors commonly display high interstitial fluid pressure (IFP), which reduces fluid convection rate across the capillary wall. Increased IFP has been suggested to interfere with drug uptake in malignancies. Earlier work suggested that PDGFR-β signaling could regulate IFP in normal tissues (Heuchel et al. 1999). Consistent with a role of PDGF signaling also for tumor IFP, PDGFR inhibition by imatinib was shown to enhance uptake and/or anti-tumor effects of cytotoxic drugs (Pietras et al. 2001, 2002, 2003a, b; Hwang et al. 2003; for review, see Heldin et al. 2004).
PDGF signaling in vascular disorders
Atherosclerosis and restenosis
PDGF expression has been reported in virtually every cell type of the atherosclerotic arterial wall, as well as in infiltrating inflammatory cells (for review, see Raines 2004). All PDGFs (A–D) and, in particular, A and B have been found in atherosclerotic lesions at increased levels compared with the normal vessel wall (Raines 2004). PDGFR-α and PDGFR-β are also expressed at increased levels in atherosclerotic vessel wall vSMCs. The mechanisms leading to increased PDGF and PDGFR expression in conjunction with atherosclerosis are not known. Several conditions associated with cardiovascular disease affect PDGF or PDGFR gene expression, such as increased blood pressure and α-adrenergic stimulation of the vessel wall, which enhances vascular PDGF-A expression (Majesky et al. 1990; Negoro et al. 1995); reduced endothelial shear stress, which induces both PDGF-A and PDGF-B (Mondy et al. 1997); and hypercholesterolemia, which dramatically increases PDGF-A and PDGF-B expression in circulating mononuclear cells (Mondy et al. 1997).
In the response-to-injury hypothesis of atherosclerosis pathogenesis, PDGF released from aggregating platelets at sites of endothelial injury was assigned a key role in the migration of vSMC from the media into the intima, as well as in the subsequent proliferation of the vSMCs at this site (Ross and Glomset 1976; Ross 1993). The current view of atherogenesis is, however, that of a local chronic inflammation. Lipoprotein retention and the infiltration and activation of different types of immune cells are believed to play key roles (Figure 10; Hansson 2005). PDGF is one of many factors secreted in the vessel wall by activated immune cells. A number of different gain- and loss-of-function experiments suggest a role for PDGF in animal models of atherosclerosis. Specifically, in different animal models of acute arterial injury by balloon catheterization, neointimal SMC accumulation was reduced by the administration of various PDGF pathway inhibitors, including neutralizing PDGF (AB) antibodies (Ferns et al. 1991; Lewis et al. 2001), PDGF-B aptamers (Leppänen et al. 2000), PDGFR kinase inhibitors (Banai et al. 1998; Yamasaki et al. 2001), and PDGFR-neutralizing antibodies (Giese et al. 1999; Hart et al. 1999). Restenosis following angioplasty of atherosclerotic vessels in minipigs (Bilder et al. 1999) and chronic cardiac transplant rejection-induced atherosclerosis in rats (Sihvola et al. 1999) were also inhibited by PDGFR-blocking kinase inhibitors. In ApoE-deficient mice, atherosclerotic lesions were inhibited by neutralizing PDGFR-β antibodies (Sano et al. 2001) and a PDGFR blocking kinase inhibitor (Kozaki et al. 2002). Conversely, infusion of PDGF-BB (Jawien et al. 1992) or local transfection of a PDGF-B expression vector (Nabel et al. 1993) led to increased SMC proliferation and intima thickening in certain models of arterial injury. The mentioned results were all obtained using tools inhibiting or activating signaling through PDGFR-β. Evidence is currently lacking for a role of PDGFR-α in neointimal proliferative responses to acute or chronic arterial injury.
Figure 10.

PDGFs and atherosclerosis. (A) Schematic illustration of a section through an arterial wall with an atherosclerotic plaque containing vSMC (green), inflammatory and immune cells (blue), and lipids (dark yellow). Invading inflammatory cells release a large number of growth factors and cytokines that maintain a chronic inflammatory reaction. PDGFs released by the inflammatory cells (and perhaps also from the endothelial cells) help in attracting vSMC from the media to the intima (light yellow). In a stable plaque, the intimal vSMC forms a subendothelial fibrous cap. Red arrows indicate routes of cell recruitment from the circulation and the arterial media. (B) LRP1 is a critical negative regulator of PDGFR-β in vSMC. This effect appears to be enhanced by ApoE. LRP1 inhibits PDGFR-β transcription (via TGF-β) and promotes PDGFR-β degradation (via c-Cbl). The interplay between LRP1 and PDGFR-β is complex. PDGF-B may bind to LRP1, and PDGFR-β phosphorylates LRP1.
Support for a functional role of PDGF-B and PDGFR-β in different aspects of atherosclerosis or restenosis has been obtained also using genetic models of PDGF-B or PDGFR-β deficiency. Analysis of mouse chimeras composed of pdgfrb+/+ and pdgfrb−/− cells demonstrated limited pdgfrb−/− cell contribution to the vSMC lineage (Crosby et al. 1998), as expected given the critical role for PDGFR-β in vSMC development. Carotid ligation experiments in such chimeras demonstrated a low proportion of pdgfrb−/− cells in the neointima but an increased contribution to the media. This suggests impaired migration of vSMC from the media to the intima in the absence of functional PDGFR-β (Buetow et al. 2003). Another type of chimeric situation was obtained by bone marrow transplantations of PDGF-B-deficient marrow into lethally irradiated, ApoE-deficient recipients (Kozaki et al. 2002). The lack of PDGF-B production by infiltrating inflammatory cells led to a small and transient inhibition of neointimal fibrous cap formation. The reason for the relatively modest effect may be that PDGF-B has the opposing effects (on atherosclerosis) of being anti-inflammatory, on the one hand, and stimulating vSMC proliferation in the vessel wall, on the other (Tang et al. 2005).
One of the most striking results in support of a central role for PDGF-BB, PDGFR-β, and SMC migration and proliferation in atherogenesis comes from studies of LRP1. LRP1 is a multifunctional transmembrane receptor, which is expressed by many cell types and binds a biologically diverse array of ligands. LRP1 is tyrosine phosphorylated in response to PDGF-BB. LRP1 binds PDGF-BB and coimmunoprecipitates with PDGFR-β. Thus, biochemical data suggest a molecular interaction between PDGFR-β and LRP1, as well as PDGFR-β-dependent tyrosine phosphorylation of LRP1 (Boucher et al. 2002; Loukinova et al. 2002; Newton et al. 2005). Cell culture data also suggest that ApoE binding to LRP1 inhibits PDGF-BB-induced phosphorylation of LRP1 and PDGF-BB-dependent SMC migration and proliferation. SMC-specific knockout of LRP1 leads to a markedly enhanced atherosclerotic response in an LDL receptor-deficient background (Boucher et al. 2003). This response was preceded by a dramatic increase in PDGFR-β expression and autophosphorylation in SMC. Thus, LRP1 normally limits PDGFR-β expression and function in vascular SMC. This function appears to involve at least two independent and synergizing mechanisms: (1) LRP1 inhibition of thrombospondin-mediated TGFβ activation and signaling through Smad2/3, leading to decreased PDGFR-β expression (Boucher et al. 2007); and (2) LRP1-dependent endocytosis and c-Cbl-mediated ubiquitinylation of PDGFR-β (Takayama et al. 2005). Inhibition of PDGFR signaling by imatinib had a major protective effect on atherosclerosis development in SMC-specific LRP1 knockouts. These results demonstrate an atheroprotective role of LRP1, which appears to be mediated, fully or partially, through suppression of PDGFR-β expression and signaling in vSMC (Fig. 10B).
Pulmonary hypertension
Pulmonary arterial hypertension (PAH) is a severe condition that may lead to right cardiac ventricular failure and death. The etiology and pathogenesis of PAH is poorly understood. Pulmonary artery vSMC hyperplasia is a hallmark of the disease, and up-regulated expression of PDGFs and PDGFRs has been correlated with PAH in various experimental animal models (Humbert et al. 1998; Balasubramaniam et al. 2003) and in humans (Schermuly et al. 2005). The basis for increased PDGFR signaling in PAH is unknown. However, recent data may provide links to ApoE and adiponectin. The combined action of ApoE deficiency and insulin resistance (leading to lower adiponectin levels) promoted development of PAH in mice. This correlated with the ability of ApoE and adiponectin to suppress proliferation of pulmonary artery vSMC (Hansmann et al. 2007). Both these proteins inhibit PDGF signaling in pulmonary vSMC. ApoE promotes down-regulation of PDGFR-β (Boucher et al. 2003), perhaps through binding to LRP1, as suggested for aortic vSMC (see above and Fig. 10B). Adiponectin appears to bind PDGF-B and scavenge it (Arita et al. 2002; Wang et al. 2005).
The cellular origin of the proliferative pulmonary artery vSMC in PAH remains unclear. Increased expression of PDGFR-β in PAH has been observed in perivascular fibroblast-like cells, suggesting that these cells may differentiate into SMCs in conjunction with pulmonary hypertension (Jones et al. 2006).
Imatinib was recently shown to reverse experimental hypertension in two animal models of PAH (Schermuly et al. 2005). A promising preliminary result with imatinib has also been presented in a human case of PAH (Ghofrani et al. 2005). Imatinib reverted pulmonary arterial vSMC proliferation and neointima formation, processes linked to PDGF signaling in other vascular beds. The effects of imatinib on PAH may thus reflect inhibition of PDGFR signaling. As mentioned, imatinib is not a completely specific inhibitor of PDGFRs, but the other known targets of the drug (Abl, Kit, and Flt3) have not been implicated in vSMC growth. The therapeutic effects of imatinib in the animal models of PAH were better and the side effects smaller than those observed with previously attempted therapies (Schermuly et al. 2005).
Retinal vascular disease
A pathogenic role for PDGF-B has been implicated in ischemic retinopathies such as proliferative diabetic retinopathy, proliferative vitreoretinopathy, and choroidal neovascularization. Intraocular injection of PDGF-B-inhibiting aptamers was shown to protect against retinal detachment in a rabbit model of proliferative retinopathy (Akiyama et al. 2006). Inhibition of PDGFR signaling by antibodies or imatinib was also shown to enhance the therapeutic effect of anti-VEGF-A treatment in multiple mouse models of ocular neovascularization (Jo et al. 2006). Somewhat conflicting results have been obtained in other studies, however. Imatinib treatment of a rat ROP model increased VEGF and VEGFR levels and promoted pathological angiogenesis (Wilkinson-Berka et al. 2004). Focal neovascular retinal disease resembling proliferative diabetic retinopathy has also been reported in two different mouse mutants with PDGF-B deficiency and accompanying loss of retinal pericytes (Enge et al. 2002; Lindblom et al. 2003). Lack of one copy of the pdgfb gene also enhanced retinal pericyte loss and endothelial death in conjunction with diabetes in mice (Hammes et al. 2002). Taken together, these studies suggest that PDGF-B may play a role in the pathogenesis of vitreoretinopathy, while pericyte loss (caused by PDGF-B deficiency) may trigger the development of a diabetic retinopathy-like condition. Pericyte loss is a hallmark of human diabetic retinopathy (for review, see Betsholtz 2004) and may be causally involved in its pathogenesis, as suggested by the animal studies, but it is not known if diabetes-induced pericyte loss reflects changes in PDGF signaling in the retina.
Role of PDGF signaling in fibrotic diseases
Chronic fibrotic changes may occur in several different organs and organ systems. PDGF signaling has been implicated in several fibrotic conditions and is assumed to play a role in driving proliferation of cells with a myofibroblast phenotype. Tissue fibrosis also involves excessive deposition of extracellular matrix, which may in part reflect increased PDGF activity, but likely also increased TGFβ function. Together, excessive mesenchymal cell proliferation and matrix deposition lead to tissue scarring and progressive loss of organ function.
Evidence for a role of PDGF signaling in human fibrotic disease is mainly circumstantial and correlative. However, as discussed below, a causative role in several animal models of lung and cardiac fibrosis and glomerulosclerosis has been demonstrated by specific loss- and gain-of-function approaches. Also, provocative new results suggest that PDGFR-activating autoantibodies may participate in autoimmune fibroses. An emerging picture common to most fibrotic diseases is that inflammatory cells such as activated macrophages release PDGFs. The same cells also produce inflammatory cytokines that promote up-regulation of PDGFRs on mesenchymal cells (for review, see Bonner 2004). As a result, PDGF-mediated proliferation of mesenchymal cells may be a hallmark of all chronic inflammations (Fig. 11).
Figure 11.

PDGFs and fibrosis. (A) In lung fibrosis, PDGFs released from alveolar macrophages promote proliferation of alveolar (myo)fibroblasts and fibrogenesis. The latter express PDGFR-α, which is up-regulated by inflammatory cytokines, some of which are increased by air pollutants. (B) In liver fibrosis, hepatic stellate cells up-regulate PDGFR-β in response to TGF-β. PDGFs produced by resident and invading macrophages contribute to stellate cell proliferation and fibrogenesis. (C) In scleroderma, PDGFs released by macrophages promote proliferation of dermal (myo)fibroblasts. Fibroblast PDGFR-α and PDGF expression is up-regulated in response to Il-1α and TGF-β. Circulating autoantibodies that activate PDGF receptor have been described in scleroderma patients. (D) In a variety of renal diseases and disease models, PDGF-B released from invading macrophages and PDGF-D released from mesangial cells may drive mesangial cell proliferation and matrix deposition, leading to glomerulosclerosis. (E) In transgenic models, the expression of PDGFs in myocardial cells drives proliferative expansion of PDGFR-α-positive cardiac fibroblasts and collagen deposition, leading to severe cardiac fibrosis.
Pulmonary fibrosis (PF)
PF involves the proliferation of myofibroblasts in alveolar walls, pulmonary blood vessels, and bronchi. In humans, PF may occur as a result of different types of environmental exposures leading to acute toxic or chronic inflammatory responses in the lung. Chronic allergic reactions (asthma), chronic pulmonary transplant rejection, or idiopathic causes may also lead to PF. Most human PFs have been associated with increased PDGF levels in the lung tissue or bronchoalveolar lavage fluid. These results have been confirmed in a variety of rodent models of lung injury and fibrosis (Bonner 2004). Studies in animal models have also revealed that lung fibroblasts respond to PDGFs by increased proliferation and cell migration. Alveolar macrophages appear to be the principal source of PDGF (mainly B) in PF (Martinet et al. 1987; Antoniades et al. 1990; Bonner 2004). PDGF-A and PDGF-C expression has also been detected in other cell types in the injured or fibrotic lung (Homma et al. 1995; Zhuo et al. 2004). The mechanisms that mediate enhanced PDGF ligand expression in the process of PF are poorly understood. However, inflammatory cytokines such as IFN-γ, IL-1β, and IL-13 have been implicated (Bonner 2004). Both PDGFR-α- and PDGFR-β-mediated signals have been assigned roles in PF. PDGFR-α is strongly up-regulated in the lung mesenchyme in response to inflammatory mediators such as IL-1β and TGF-β (Fig. 11A). Up-regulated PDGFR-α has also been observed in the lung as a response to a number of environmental factors, including asbestos and air pollution particulates (Fig. 11A; Bonner et al. 1993). PDGFR-β expression in the lung appears constitutive and unaffected by inflammatory stimuli, but may nevertheless play a role in mediating fibroproliferative responses alone or in heterodimer configuration with PDGFR-α. These expression data suggest that PDGF-induced signals may constitute a target for treatment of PF. Support for such a concept has been obtained in animal models. Three distinct TK inhibitors, which all potently inhibit PDGFRs but have otherwise different selectivity profiles (SU9518, SU11657, and imatinib), were all shown to reduce lung fibrosis and increase life span in a model of radiation-induced lung injury (Abdollahi et al. 2005). In this model, all four PDGFs were up-regulated. In another study, mesenchymal cell proliferation and collagen accumulation were reduced by the PDGFR TK inhibitor AG1296 in a model of metal-induced lung fibrosis (Rice et al. 1999).
Liver fibrosis
Liver fibrosis (and its end stage cirrhosis) has a variety of causes, including drugs, alcohol, infections, metabolic changes, and idiopathic etiologies. The key fibrogenic cells are the stellate cells, which are sinusoidal vessel pericytes. Quiescent stellate cells are normally negative for PDGFR-β, and, unlike other pericytes, they do not depend on PDGF-B or PDGFR-β for their development (Hellström et al. 1999). PDGFR-β expression is, however, turned on in activated stellate cells both in vitro and in vivo in animals and humans (Fig. 11B; for review, see Bonner 2004). This transition renders stellate cells highly responsive to PDGF-B and PDGF-D. TGFβ appears to be a major inducer of PDGFR-β in stellate cells. Several different cell types in the reactive liver produce TGFβ, including the stellate cells themselves. When activated, resident liver macrophages (Kupffer cells) are a major source of PDGF-B, but infiltrating cells contribute too (Fig. 11B; Pinzani et al. 1996). Increased PDGF-D expression has been detected in the perisinusoidal and periportal areas in experimental liver fibrosis in rats, but the producer cell type has not been established (Borkham-Kamphorst et al. 2007).
Dermal fibrosis
Scleroderma (also known as systemic sclerosis) is an autoimmune disorder characterized primarily by progressive dermal and vascular fibrosis. Other organs are affected too, including lung, heart, esophagus, intestine, and kidney. Increased expression of both PDGFs and PDGFRs has been demonstrated in scleroderma skin (for review, see Bonner 2004). A role for PDGF signaling in bleomycin-induced experimental scleroderma in mice was also suggested by the therapeutic effect of imatinib in this model (Distler et al. 2007).
As for other fibroses, PDGF-B expression seems to occur in activated macrophages and infiltrating inflammatory cells (Fig. 11C; Gay et al. 1989). In addition, scleroderma myofibroblasts appear to be constitutively phenotypically changed in comparison with normal skin fibroblasts, perhaps indicating that they have a distinct origin. Possibly, they originate from pericytes since the increased PDGFR-β expression was mainly observed around blood vessels in the scleroderma skin (Klareskog et al. 1990). A pericyte origin has also been suggested for myofibroblasts in scarring tissue and tumors (Lindmark et al. 1993; Reuterdahl et al. 1993). One distinct feature of scleroderma myofibroblasts is that they constitutively express PDGF-A in an autocrine loop driven by Il-1α (Fig. 11C; Kawaguchi et al. 1999). This appears to contrast with normal fibroblasts in which transient PDGF-A expression occurs after exposure to mitogens (Paulsson et al. 1987). Another feature that distinguishes scleroderma fibroblasts from their normal counterparts is that TGFβ increases the expression of PDGFR-α and enhances the mitogenic effect of PDGF-A in the former (Fig. 11C) but seems to have opposite effects on the latter (Yamakage et al. 1992). Down-regulation of Smad7 and up-regulation of Smad3 in scleroderma fibroblasts may account for this difference. Also thrombin and Il-1α have been implicated in the enhanced responsiveness till PDGF stimulation in scleroderma fibroblasts (for review, see Bonner 2004).
It was recently reported that autoantibodies directed toward the PDGF receptors are present in the serum from patients with scleroderma (Fig. 11C; Baroni et al. 2006). Autoantibodies are commonly found in autoimmune disorders and sometimes play direct pathogenic roles. A well-known example is Grave’s disease, where agonistic autoantibodies for the thyroid-stimulating hormone receptor activate the thyroid gland, leading to hyperthyroidism. PDGFR autoantibodies were detected in serum from 46 out of 46 scleroderma patients but in no controls. The antibodies activated both PDGFR-α and PDGFR-β phosphorylation and downstream signaling through the Ras-MAPK pathway. Moreover, the antibodies were shown to convert normal human fibroblasts into collagen type-1-expressing myofibroblasts in vitro. This is an interesting finding with potential openings for new diagnostics. If these antibodies play a direct role in scleroderma pathogenesis, their elimination may also be considered as a putative therapy. Preliminary data suggest that PDGF autoantibodies are present also in patients with chronic graft-versus-host disease (Svegliati et al. 2007). The possibility of PDGFR activation through autoantibodies is a novel and provocative concept in autoimmune fibrotic diseases, which will most certainly be extensively explored over coming years.
Renal fibrosis
Renal fibrosis is obligatorily linked with the chronic renal failure caused by diabetes and other systemic diseases (such as hypertension), toxicity, autoimmune and Ig complex diseases, as well as idiopathic (often monogenic) diseases. Fibrosis may affect the glomeruli, in which case mesangial cells react. In tubulointerstitial fibrosis mesenchymal cells of less known identity and origin proliferate and deposit ECM. As discussed above, mesangial cell development depends on a PDGF-B–PDGFR-β paracrine loop. Tubulointerstitial mesenchyme development instead depends on combined PDGF-A and PDGF-C signaling over PDGFR-α. Several observations suggest involvement of the same signaling pathway during renal fibrosis. Glomerular disease is frequently accompanied by up-regulated expression of PDGFR-β (Fig. 11D; Fellström et al. 1989). In experimental animal models of mesangioproliferative disease, neutralizing PDGF antibodies, PDGF-B-binding aptamers, or imatinib all prevent mesangial cell proliferation and matrix accumulation (Johnson et al. 1992; Floege et al. 1999; Gilbert et al. 2001). Also, systemic administration of PDGF-BB promotes mesangial cell proliferation in the absence of other discernable effects, suggesting that these cells are able to respond to PDGF-B independent of prior inflammatory activation (Tang et al. 1996). A relatively modest (twofold) increase in circulating plasma PDGF-B levels also correlated with increased numbers of mesangial cells in mice with increased megakaryocytes and platelets due to overexpression of thrombopoietin (Shimoda et al. 2007). All PDGFs (A–D) have been shown to be up-regulated in conjunction with renal fibroses. Evidence for a role of PDGF-A and PDGF-C remains circumstantial, but PDGF-D has been shown to play a role in experimental mesangioproliferative glomerulonephritis in rats by use of a PDGF-D-specific neutralizing antibody (Ostendorf et al. 2006). The source(s) of PDGF-B during mesangioproliferative pathology is not entirely clear. Endothelial cells may contribute, as they do during development, but it is also possible that PDGF-B is released by invading mononuclear cells. PDGF-D appears to be strongly up-regulated in the mesangial cells themselves during their pathological expansion (Ostendorf et al. 2003) and might act in an autocrine loop (Fig. 11D).
Cardiac fibrosis
Cardiac fibrosis may occur as a result of cardiac infarction, hypertension, chronic rejection, or endocrine disorders. Cardiac fibrosis is believed to involve the proliferation and excessive collagen deposition by interstitial cardiac fibroblasts (Fig. 11E). Indications for a role of PDGF signaling in cardiac fibrosis mainly come from gain- and loss-of-function studies in experimental animals. Mice treated with neutralizing PDGFR antibodies following myocardial infarction show attenuated healing responses depending on antibody specificity. PDGFR-β blockade results in impaired maturation of cardiac neovessels, consistent with the roles of PDGFR-β signaling in mural cell recruitment and vascular development. PDGFR-α blockade, on the other hand, did not affect the vasculature but decreased collagen deposition, suggesting a role for this receptor in fibroblasts activation (Zymek et al. 2006). Transgenic studies also support for a role of PDGFR-α in cardiac fibroblast activation. Overexpression of PDGF-C in the heart of transgenic mice induced diffuse cardiac fibrosis with extensive hyperplasia of fibroblast-like cells and large collagen deposits (Pontén et al. 2003). This coincided with dilated cardiomyopathy in females, whereas males developed hypertrophic response. PDGF-D overexpression in the heart promoted diffuse cardiac fibrosis but also increased thickness of the vSMC coat in the coronary vessels (Pontén et al. 2005). Adeno-associated virus (AAV)-mediated transfer of PDGF-A, PDGF-C, or PDGF-D accelerated cardiac fibrosis and chronic rejection in heterotypic heart transplants in rats (Tuuminen et al. 2006). PDGF-AA is a potent inducer of proliferation in cardiac fibroblasts in vitro (Simm et al. 1998). AAV-derived PDGF-B lacked this effect, which may seem paradoxical since PDGF-B can activate both PDGFR-α and PDGFR-β. One possible explanation for this result is that PDGF-B bioavailability in the heart is limited due to interactions with ECM. Consistent with this idea, we found that heart-specific transgenic PDGF-A expression led to excessive cardiac fibrosis and death within a few weeks after birth, whereas PDGF-B expression from the same promoter resulted in a much weaker fibrotic response (P. Lindblom, C. Bondjers, and C. Betsholtz, unpubl.).
PDGFs have been implicated in the cardiac fibrotic response to angiotensin II (Nishioka et al. 2007). Angiotensin-II-induced cardiac fibrosis is reduced in mice lacking one copy of the transcription factor KLF5, correlating with reduced cardiac expression of PDGF-A. Evidence for direct binding of KLF5 to the PDGF-A promoter has been presented, and regulation of expression of PDGF-A by KLF5 occurs also in other organs (Shindo et al. 2002). Together, the mentioned results implicate PDGFR-α stimulation as a major fibrogenic process in the heart, whereas PDGFR-β stimulation in the heart affects mainly vascular mural cells leading to vessel wall pathology.
Polymorphisms in PDGF and PDGFR genes linked to diseases
Single nucleotide polymorphisms have been found in PDGF and PDGFR genes, but most of them currently lack significant association with disease. A possible link between neural tube defects (including spina bifida and anencephaly) and polymorphic sites in the human PDGFR-A promoter has been described (Joosten et al. 2001). Five different haplotypes were identified. Two of them were overrepresented in patients with sporadic spina bifida, and these were associated with increased transcriptional activity of PDGFR-A. Together with the results of PDGFR-α knockout or knock-in mutagenesis, these findings indicate that the correct level of signaling via PDGFR-α is crucial for proper development of neural arches and suggest that both too high or too low levels of PDGFR-α expression or signaling may result in spina bifida. Association between different combinations of PDGFR-A promoter polymorphisms and the severity of childhood asthma was recently suggested, but these data appear preliminary and in need of confirmation (Wu et al. 2006).
Clinical use of PDGF-BB to improve wound healing
Recombinant human PDGF-BB (becaplermin) has been introduced in the clinic as a topical treatment for chronic neuropathic lower-extremity diabetic ulcers (Robson et al. 1992). It has also been used against pressure ulcers and to speed up healing in various surgical procedures. The tissue repair mechanisms induced by PDGF-BB appear to involve fibroblast proliferation, collagen production, and neovessel formation (Pierce et al. 1994) and may involve direct effects on PDGFR-β-expressing mesenchymal cells, but also up-regulated expression of the long splice variant of PDGF-A (Pierce et al. 1995). Clinical efficacy has been demonstrated in several phase III studies (Smiell et al. 1999), and the combined results suggest that topical application of PDGF-BB is safe and well tolerated (for review, see Edmonds et al. 2000; Perry et al. 2002). A recent study involving >900 patients confirmed that topical treatment with PDGF improved healing of chronic full-thickness diabetic foot ulcers (Steed 2006). Local delivery of PDGF-BB has also been tested in patients with severe periodontal disease and found to increase biomarkers for bone metabolism and turnover (Sarment et al. 2006) and provide improved periodontal regeneration (Nevins et al. 2003).
Outlook
The small size of the PDGF/PDGFR family has been a great advantage for the elucidation of the numerous developmental and pathological roles of the PDGF/Rs by genetic and pharmacological methods. Thus far, developmental studies in mammals have mostly used complete knockouts (PDGF-A, PDGF-B, and PDGF-C, and PDGFR-α and PDGFR-β), which are all lethal at embryonic or early postnatal time points. The critical and nonredundant functions of the PDGFs and PDGFRs in organogenesis have on the one hand facilitated analysis of PDGF physiological functions, while on the other hand restricted information to developmental stages before onset of lethality. In addition to this limitation, the multiorgan effects and systemic consequences of some of the phenotypes may produce secondary effects that confound interpretations. Problems with systemic effects of cardiovascular failure (which are notorious in mice) may to some extent be avoided by analyzing fish. PDGFs and PDGFRs are expressed but have not yet been systematically functionally analyzed in zebrafish embryos. Such studies would clearly be of value.
However, genetic approaches in mice are far from exhausted. The phenotypic consequences of knockout of PDGF-D have still not been reported, but null mice are currently available and under study. So far, application of conditional mutagenesis approaches to PDGF biology has been limited, and further studies using inducible and tissue-specific knockouts of PDGF ligands and receptors will most certainly provide us with more correct and detailed information about the developmental roles of PDGF signaling. In addition, such studies will tell us to what extent PDGF signaling is of physiological importance in adult animals. PDGFs are indeed expressed in many normal cell types in adult mammals, often at significant levels, but the physiological role of this expression remains unclear. These questions are obviously important considering that potent PDGFR inhibitors, such as imatinib, are being introduced into the clinic. Imatinib is already in clinical use for certain uncommon neoplasias, but as exemplified in this review, there are clearly prospects for broad application of PDGF/R antagonists in more prevalent cancers, vascular disorders, and fibrotic conditions. Clinical trials with imatinib are already ongoing for some of these conditions.
Huge amounts of information on PDGFR signaling have accumulated over the past decade, but our understanding about how signaling specificity is obtained downstream from the individual PDGFRs, as well as in selected cell types at specific stages of development, is still limited. To what extent the cell systems commonly used for in vitro studies represent and reflect the critical PDGF signaling pathways in vivo remains largely unclear. A typical example is the PI3K pathway downstream from PDGFR-β, which appears relatively insignificant in vivo in contrast to what might have been predicted from in vitro analyses. In marked contrast, the same signaling pathway appears to be absolutely essential for PDGFR-α function during development. There are also obvious challenges in interpreting the biological significance of the intersection between signaling through PDGFR and other receptor systems. Cross-talk between PDGFR signaling and signaling through other RTKs, G protein-coupled receptors, and integrins (and more) adds immensely to signaling complexity. Again, most if not all of these effects have been analyzed in vitro, while the relevance in vivo remains unknown and a formidable challenge to tackle. Most likely, progress will come with technologies that allow signaling through distinct pathways to be monitored in intact organisms with cellular resolution and in real time. There is still a long way to go before this can be accomplished for PDGFRs, but progress is being made in several areas toward this goal.
As summarized in the present review, imatinib and other efficient PDGF pathway inhibitors have been invaluable in the search of physiological and pathological processes that require PDGFR signaling. Since available kinase inhibitors are not entirely specific, positive results must be followed up using more specific PDGF pathway inhibitors before firm conclusions can be made concerning the involvement of PDGF signaling for the particular process. Nevertheless, imatinib and similar substances are excellent research tools that allow efficient screening for putative PDGF-dependent pathophysiological processes in experimental animals. Clinical trials with imatinib will also reveal which human situation may benefit from PDGFR inhibition, and conversely, putative side effects will highlight physiological processes that depend on PDGF signaling.
Cancer research is an area where PDGF biology has gone through a recent revival. There are now numerous examples of the direct involvement of mutated PDGF signaling components in cancer, but the affected tumor types are relatively uncommon. Nevertheless, the progress is important, especially since some of these tumor types, particularly glioblastoma, are extremely aggressive and in urgent need for therapeutic options. Concerning the potential involvement of PDGF signaling in common cancers, it remains to be established if PDGF involvement in EMT goes beyond anecdotal examples in experimental tumors. However, this is clearly an important area worthy of further investigation. Perhaps more interesting is the emerging evidence for a broader role of PDGF signaling in tumor stroma recruitment, and, in turn, roles for tumor stroma in tumor growth, metastasis, and therapeutic resistance. The progress in this area is quite recent and based primarly on experimental tumor models. Clearly much more work is needed before we may conclude about any general role(s) of PDGF signaling in tumor stromogenesis. The possibility of finding therapeutic tagets in (putatively PDGF-dependent) fibroblastic or vascular tumor stroma is being developed also in humans, obviously reinforced by emerging positive results of anti-angiogenic therapy.
In summary, studies of PDGFs and PDGFRs in animal development and disease have revealed common principles for growth factor function in organogenesis, cancer, vascular inflammation, and tissue fibrosis. This review briefly summarizes the basic aspects of PDGF function and discusses in somewhat more detail the developmental roles of PDGFs in animals, focusing on gastrulation, neural crest, epithelial organogenesis, glial cell development, skeletal development, angiogenesis, and hematopoiesis. The numerous suggested roles of PDGF signaling in different diseases, ranging from cancer to vascular disorders and fibrotic diseases, are discussed in depth. We also review the current principles of pharmacological inhibition and stimulation of PDGF signaling and its applications in research and clinical practice.
Acknowledgments
The unpublished work we cited was supported by grants from the Ludwig Institute for Cancer Research, the Swedish Cancer Foundation, the Association for International Cancer Research (UK), and the Wallenberg, Söderberg, and Lundberg Foundations.
Footnotes
Article is online at http://www.genesdev.org/cgi/doi/10.1101/gad.1653708.
References
- Aase K., Abramsson A., Karlsson L., Betsholtz C., Eriksson U. Expression analysis of PDGF-C in adult and developing mouse tissues. Mech. Dev. 2002;110:187–191. doi: 10.1016/s0925-4773(01)00560-3. [DOI] [PubMed] [Google Scholar]
- Abdollahi A., Li M., Ping G., Plathow C., Domhan S., Kiessling F., Lee L.B., McMahon G., Gröne H.J., Lipson K.E., et al. Inhibition of platelet-derived growth factor signaling attenuates pulmonary fibrosis. J. Exp. Med. 2005;201:925–935. doi: 10.1084/jem.20041393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abramsson A., Berlin O., Papayan H., Paulin D., Shani M., Betsholtz C. Analysis of mural cell recruitment to tumor vessels. Circulation. 2002;105:112–117. doi: 10.1161/hc0102.101437. [DOI] [PubMed] [Google Scholar]
- Abramsson A., Lindblom P., Betsholtz C. Endothelial and nonendothelial sources of PDGF-B regulate pericyte recruitment and influence vascular pattern formation in tumors. J. Clin. Invest. 2003;112:1142–1151. doi: 10.1172/JCI18549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abramsson A., Kurup S., Busse M., Yamada S., Lindblom P., Schallmeiner E., Stenzel D., Sauvaget D., Ledin J., Ringvall M., et al. Defective N-sulfation of heparan sulfate proteoglycans limits PDGF-BB binding and pericyte recruitment in vascular development. Genes & Dev. 2007;21:316–331. doi: 10.1101/gad.398207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Afrakhte M., Nistér M., Ostman A., Westermark B., Paulsson Y. Production of cell-associated PDGF-AA by a human sarcoma cell line: Evidence for a latent autocrine effect. Int. J. Cancer. 1996;68:802–809. doi: 10.1002/(SICI)1097-0215(19961211)68:6<802::AID-IJC19>3.0.CO;2-1. [DOI] [PubMed] [Google Scholar]
- Akimoto K., Takahashi R., Moriya S., Nishioka N., Takayanagi J., Kimura K., Fukui Y., Osada S., Mizuno K., Hirai S., et al. EGF or PDGF receptors activate atypical PKCλ through phosphatidylinositol 3-kinase. EMBO J. 1996;15:788–798. [PMC free article] [PubMed] [Google Scholar]
- Akiyama H., Kachi S., Silva R.L., Umeda N., Hackett S.F., McCauley D., McCauley T., Zoltoski A., Epstein D.M., Campochiaro P.A. Intraocular injection of an aptamer that binds PDGF-B: A potential treatment for proliferative retinopathies. J. Cell. Physiol. 2006;207:407–412. doi: 10.1002/jcp.20583. [DOI] [PubMed] [Google Scholar]
- Ali Y., Lin Y., Gharibo M.M., Gounder M.K., Stein M.N., Lagattuta T.F., Egorin M.J., Rubin E.H., Poplin E.A. Phase I and pharmacokinetic study of imatinib mesylate (Gleevec) and gemcitabine in patients with refractory solid tumors. Clin. Cancer Res. 2007;13:5876–5882. doi: 10.1158/1078-0432.CCR-07-0883. [DOI] [PubMed] [Google Scholar]
- Andrae J., Hansson I., Afink G.B., Nistér M. Platelet-derived growth factor receptor-α in ventricular zone cells and in developing neurons. Mol. Cell. Neurosci. 2001;17:1001–1013. doi: 10.1006/mcne.2001.0989. [DOI] [PubMed] [Google Scholar]
- Antoniades H.N., Bravo M.A., Avila R.E., Galanopoulos T., Neville-Golden J., Maxwell M., Selman M. Platelet-derived growth factor in idiopathic pulmonary fibrosis. J. Clin. Invest. 1990;86:1055–1064. doi: 10.1172/JCI114808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Apperley J.F., Gardembas M., Melo J.V., Russell-Jones R., Bain B.J., Baxter E.J., Chase A., Chessells J.M., Colombat M., Dearden C.E., et al. Response to imatinib mesylate in patients with chronic myeloproliferative diseases with rearrangements of the platelet-derived growth factor receptor β. N. Engl. J. Med. 2002;347:481–487. doi: 10.1056/NEJMoa020150. [DOI] [PubMed] [Google Scholar]
- Arai H., Ueno T., Tangoku A., Yoshino S., Abe T., Kawauchi S., Oga A., Furuya T., Oka M., Sasaki K. Detection of amplified oncogenes by genome DNA microarrays in human primary esophageal squamous cell carcinoma: Comparison with conventional comparative genomic hybridization analysis. Cancer Genet. Cytogenet. 2003;146:16–21. doi: 10.1016/s0165-4608(03)00106-7. [DOI] [PubMed] [Google Scholar]
- Arita Y., Kihara S., Ouchi N., Maeda K., Kuriyama H., Okamoto Y., Kumada M., Hotta K., Nishida M., Takahashi M., et al. Adipocyte-derived plasma protein adiponectin acts as a platelet-derived growth factor-BB-binding protein and regulates growth factor-induced common postreceptor signal in vascular smooth muscle cell. Circulation. 2002;105:2893–2898. doi: 10.1161/01.cir.0000018622.84402.ff. [DOI] [PubMed] [Google Scholar]
- Armulik A., Abramsson A., Betsholtz C. Endothelial/pericyte interactions. Circ. Res. 2005;97:512–523. doi: 10.1161/01.RES.0000182903.16652.d7. [DOI] [PubMed] [Google Scholar]
- Assoian R.K. Anchorage-dependent cell cycle progression. J. Cell Biol. 1997;136:1–4. doi: 10.1083/jcb.136.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ataliotis P. Platelet-derived growth factor A modulates limb chondrogenesis both in vivo and in vitro. Mech. Dev. 2000;94:13–24. doi: 10.1016/s0925-4773(00)00321-x. [DOI] [PubMed] [Google Scholar]
- Ataliotis P., Mercola M. Distribution and functions of platelet-derived growth factors and their receptors during embryogenesis. Int. Rev. Cytol. 1997;172:95–127. doi: 10.1016/s0074-7696(08)62359-1. [DOI] [PubMed] [Google Scholar]
- Ataliotis P., Symes K., Chou M.M., Ho L., Mercola M. PDGF signalling is required for gastrulation of Xenopus laevis. Development. 1995;121:3099–3110. doi: 10.1242/dev.121.9.3099. [DOI] [PubMed] [Google Scholar]
- Balasubramaniam V., Le Cras T.D., Ivy D.D., Grover T.R., Kinsella J.P., Abman S.H. Role of platelet-derived growth factor in vascular remodeling during pulmonary hypertension in the ovine fetus. Am. J. Physiol. Lung Cell. Mol. Physiol. 2003;284:L826–L833. doi: 10.1152/ajplung.00199.2002. [DOI] [PubMed] [Google Scholar]
- Ball S.G., Shuttleworth C.A., Kielty C.M. Vascular endothelial growth factor can signal through platelet-derived growth factor receptors. J. Cell Biol. 2007;177:489–500. doi: 10.1083/jcb.200608093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banai S., Wolf Y., Golomb G., Pearle A., Waltenberger J., Fishbein I., Schneider A., Gazit A., Perez L., Huber R., et al. PDGF-receptor tyrosine kinase blocker AG1295 selectively attenuates smooth muscle cell growth in vitro and reduces neointimal formation after balloon angioplasty in swine. Circulation. 1998;97:1960–1969. doi: 10.1161/01.cir.97.19.1960. [DOI] [PubMed] [Google Scholar]
- Baroni S.S., Santillo M., Bevilacqua F., Luchetti M., Spadoni T., Mancini M., Fraticelli P., Sambo P., Funaro A., Kazlauskas A., et al. Stimulatory autoantibodies to the PDGF receptor in systemic sclerosis. N. Engl. J. Med. 2006;354:2667–2676. doi: 10.1056/NEJMoa052955. [DOI] [PubMed] [Google Scholar]
- Bar-Sagi D., Feramisco J.R. Induction of membrane ruffling and fluid-phase pinocytosis in quiescent fibroblasts by ras proteins. Science. 1986;233:1061–1068. doi: 10.1126/science.3090687. [DOI] [PubMed] [Google Scholar]
- Batts L.E., Polk D.B., Dubois R.N., Kulessa H. Bmp signaling is required for intestinal growth and morphogenesis. Dev. Dyn. 2006;235:1563–1570. doi: 10.1002/dvdy.20741. [DOI] [PubMed] [Google Scholar]
- Bergers G., Song S., Meyer-Morse N., Bergsland E., Hanahan D. Benefits of targeting both pericytes and endothelial cells in the tumor vasculature with kinase inhibitors. J. Clin. Invest. 2003;111:1287–1295. doi: 10.1172/JCI17929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergsten E., Uutela M., Li X., Pietras K., Ostman A., Heldin C.H., Alitalo K., Eriksson U. PDGF-D is a specific, protease-activated ligand for the PDGF β-receptor. Nat. Cell Biol. 2001;3:512–516. doi: 10.1038/35074588. [DOI] [PubMed] [Google Scholar]
- Berridge M.J. Inositol trisphosphate and calcium signalling. Nature. 1993;361:315–325. doi: 10.1038/361315a0. [DOI] [PubMed] [Google Scholar]
- Betsholtz C. Insight into the physiological functions of PDGF through genetic studies in mice. Cytokine Growth Factor Rev. 2004;15:215–228. doi: 10.1016/j.cytogfr.2004.03.005. [DOI] [PubMed] [Google Scholar]
- Betsholtz C., Johnsson A., Heldin C.H., Westermark B., Lind P., Urdea M.S., Eddy R., Shows T.B., Philpott K., Mellor A.L. cDNA sequence and chromosomal localization of human platelet-derived growth factor A-chain and its expression in tumour cell lines. Nature. 1986;320:695–699. doi: 10.1038/320695a0. [DOI] [PubMed] [Google Scholar]
- Bilder G., Wentz T., Leadley R., Amin D., Byan L., O’Conner B., Needle S., Galczenski H., Bostwick J., Kasiewski C., et al. Restenosis following angioplasty in the swine coronary artery is inhibited by an orally active PDGF-receptor tyrosine kinase inhibitor, RPR101511A. Circulation. 1999;99:3292–3299. doi: 10.1161/01.cir.99.25.3292. [DOI] [PubMed] [Google Scholar]
- Bjarnegård M., Enge M., Norlin J., Gustafsdottir S., Fredriksson S., Abramsson A., Takemoto M., Gustafsson E., Fässler R., Betsholtz C. Endothelium-specific ablation of PDGFB leads to pericyte loss and glomerular, cardiac and placental abnormalities. Development. 2004;131:1847–1857. doi: 10.1242/dev.01080. [DOI] [PubMed] [Google Scholar]
- Bonner J.C. Regulation of PDGF and its receptors in fibrotic diseases. Cytokine Growth Factor Rev. 2004;15:255–273. doi: 10.1016/j.cytogfr.2004.03.006. [DOI] [PubMed] [Google Scholar]
- Bonner J.C., Osornio-Vargas A.R. Differential binding and regulation of platelet-derived growth factor A and B chain isoforms by α 2-macroglobulin. J. Biol. Chem. 1995;270:16236–16242. doi: 10.1074/jbc.270.27.16236. [DOI] [PubMed] [Google Scholar]
- Bonner J.C., Goodell A.L., Coin P.G., Brody A.R. Chrysotile asbestos upregulates gene expression and production of α-receptors for platelet-derived growth factor (PDGF-AA) on rat lung fibroblasts. J. Clin. Invest. 1993;92:425–430. doi: 10.1172/JCI116584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonner J.C., Badgett A., Hoffman M., Lindroos P.M. Inhibition of platelet-derived growth factor-BB-induced fibroblast proliferation by plasmin-activated α 2-macroglobulin is mediated via an α 2-macroglobulin receptor/low density lipoprotein receptor-related protein-dependent mechanism. J. Biol. Chem. 1995;270:6389–6395. doi: 10.1074/jbc.270.11.6389. [DOI] [PubMed] [Google Scholar]
- Borkham-Kamphorst E., van Roeyen C.R., Ostendorf T., Floege J., Gressner A.M., Weiskirchen R. Pro-fibrogenic potential of PDGF-D in liver fibrosis. J. Hepatol. 2007;46:1064–1074. doi: 10.1016/j.jhep.2007.01.029. [DOI] [PubMed] [Google Scholar]
- Boström H., Willetts K., Pekny M., Levéen P., Lindahl P., Hedstrand H., Pekna M., Hellström M., Gebre-Medhin S., Schalling M., et al. PDGF-A signaling is a critical event in lung alveolar myofibroblast development and alveogenesis. Cell. 1996;85:863–873. doi: 10.1016/s0092-8674(00)81270-2. [DOI] [PubMed] [Google Scholar]
- Boström H., Gritli-Linde A., Betsholtz C. PDGF-A/PDGF α-receptor signaling is required for lung growth and the formation of alveoli but not for early lung branching morphogenesis. Dev. Dyn. 2002;223:155–162. doi: 10.1002/dvdy.1225. [DOI] [PubMed] [Google Scholar]
- Boucher P., Liu P., Gotthardt M., Hiesberger T., Anderson R.G., Herz J. Platelet-derived growth factor mediates tyrosine phosphorylation of the cytoplasmic domain of the low density lipoprotein receptor-related protein in caveolae. J. Biol. Chem. 2002;277:15507–15513. doi: 10.1074/jbc.M200428200. [DOI] [PubMed] [Google Scholar]
- Boucher P., Gotthardt M., Li W.P., Anderson R.G., Herz J. LRP: Role in vascular wall integrity and protection from atherosclerosis. Science. 2003;300:329–332. doi: 10.1126/science.1082095. [DOI] [PubMed] [Google Scholar]
- Boucher P., Li W.P., Matz R.L., Takayama Y., Auwerx J., Anderson R.G., Herz J. LRP1 functions as an atheroprotective integrator of TGFβ and PDFG signals in the vascular wall: Implications for Marfan syndrome. PLoS ONE. 2007;2:e448. doi: 10.1371/journal.pone.0000448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brennan J., Tilmann C., Capel B. Pdgfr-α mediates testis cord organization and fetal Leydig cell development in the XY gonad. Genes & Dev. 2003;17:800–810. doi: 10.1101/gad.1052503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brückner K., Kockel L., Duchek P., Luque C.M., Rørth P., Perrimon N. The PDGF/VEGF receptor controls blood cell survival in Drosophila. Dev. Cell. 2004;7:73–84. doi: 10.1016/j.devcel.2004.06.007. [DOI] [PubMed] [Google Scholar]
- Bruna A., Darken R.S., Rojo F., Ocaña A., Peñuelas S., Arias A., Paris R., Tortosa A., Mora J., Baselga J., et al. High TGFβ-Smad activity confers poor prognosis in glioma patients and promotes cell proliferation depending on the methylation of the PDGF-B gene. Cancer Cell. 2007;11:147–160. doi: 10.1016/j.ccr.2006.11.023. [DOI] [PubMed] [Google Scholar]
- Buetow B.S., Tappan K.A., Crosby J.R., Seifert R.A., Bowen-Pope D.F. Chimera analysis supports a predominant role of PDGFRβ in promoting smooth-muscle cell chemotaxis after arterial injury. Am. J. Pathol. 2003;163:979–984. doi: 10.1016/s0002-9440(10)63457-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgering B.M., Coffer P.J. Protein kinase B (c-Akt) in phosphatidylinositol-3-OH kinase signal transduction. Nature. 1995;376:599–602. doi: 10.1038/376599a0. [DOI] [PubMed] [Google Scholar]
- Calver A.R., Hall A.C., Yu W.P., Walsh F.S., Heath J.K., Betsholtz C., Richardson W.D. Oligodendrocyte population dynamics and the role of PDGF in vivo. Neuron. 1998;20:869–882. doi: 10.1016/s0896-6273(00)80469-9. [DOI] [PubMed] [Google Scholar]
- Carroll M., Ohno-Jones S., Tamura S., Buchdunger E., Zimmermann J., Lydon N.B., Gilliland D.G., Druker B.J. CGP 57148, a tyrosine kinase inhibitor, inhibits the growth of cells expressing BCR–ABL, TEL–ABL, and TEL–PDGFR fusion proteins. Blood. 1997;90:4947–4952. [PubMed] [Google Scholar]
- Cho N.K., Keyes L., Johnson E., Heller J., Ryner L., Karim F., Krasnow M.A. Developmental control of blood cell migration by the Drosophila VEGF pathway. Cell. 2002;108:865–876. doi: 10.1016/s0092-8674(02)00676-1. [DOI] [PubMed] [Google Scholar]
- Chung J., Grammer T.C., Lemon K.P., Kazlauskas A., Blenis J. PDGF- and insulin-dependent pp70S6k activation mediated by phosphatidylinositol-3-OH kinase. Nature. 1994;370:71–75. doi: 10.1038/370071a0. [DOI] [PubMed] [Google Scholar]
- Clark E.A., Brugge J.S. Integrins and signal transduction pathways: The road taken. Science. 1995;268:233–239. doi: 10.1126/science.7716514. [DOI] [PubMed] [Google Scholar]
- Cochran B.H., Reffel A.C., Stiles C.D. Molecular cloning of gene sequences regulated by platelet-derived growth factor. Cell. 1983;33:939–947. doi: 10.1016/0092-8674(83)90037-5. [DOI] [PubMed] [Google Scholar]
- Cools J., DeAngelo D.J., Gotlib J., Stover E.H., Legare R.D., Cortes J., Kutok J., Clark J., Galinsky I., Griffin J.D., et al. A tyrosine kinase created by fusion of the PDGFRA and FIP1L1 genes as a therapeutic target of imatinib in idiopathic hypereosinophilic syndrome. N. Engl. J. Med. 2003a;348:1201–1214. doi: 10.1056/NEJMoa025217. [DOI] [PubMed] [Google Scholar]
- Cools J., Stover E.H., Boulton C.L., Gotlib J., Legare R.D., Amaral S.M., Curley D.P., Duclos N., Rowan R., Kutok J.L., et al. PKC412 overcomes resistance to imatinib in a murine model of FIP1L1-PDGFRα-induced myeloproliferative disease. Cancer Cell. 2003b;3:459–469. doi: 10.1016/s1535-6108(03)00108-9. [DOI] [PubMed] [Google Scholar]
- Crosby J.R., Seifert R.A., Soriano P., Bowen-Pope D.F. Chimaeric analysis reveals role of Pdgf receptors in all muscle lineages. Nat. Genet. 1998;18:385–388. doi: 10.1038/ng0498-385. [DOI] [PubMed] [Google Scholar]
- Dai C., Celestino J.C., Okada Y., Louis D.N., Fuller G.N., Holland E.C. PDGF autocrine stimulation dedifferentiates cultured astrocytes and induces oligodendrogliomas and oligoastrocytomas from neural progenitors and astrocytes in vivo. Genes & Dev. 2001;15:1913–1925. doi: 10.1101/gad.903001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darnell J.E. STATs and gene regulation. Science. 1997;277:1630–1635. doi: 10.1126/science.277.5332.1630. [DOI] [PubMed] [Google Scholar]
- Demetri G.D., von Mehren M., Blanke C.D., Van den Abbeele A.D., Eisenberg B., Roberts P.J., Heinrich M.C., Tuveson D.A., Singer S., Janicek M., et al. Efficacy and safety of imatinib mesylate in advanced gastrointestinal stromal tumors. N. Engl. J. Med. 2002;347:472–480. doi: 10.1056/NEJMoa020461. [DOI] [PubMed] [Google Scholar]
- Diliberto P.A., Gordon G.W., Yu C.L., Earp H.S., Herman B. Platelet-derived growth factor (PDGF) α receptor activation modulates the calcium mobilizing activity of the PDGF β receptor in Balb/c3T3 fibroblasts. J. Biol. Chem. 1992;267:11888–11897. [PubMed] [Google Scholar]
- Ding H., Wu X., Kim I., Tam P.P., Koh G.Y., Nagy A. The mouse Pdgfc gene: Dynamic expression in embryonic tissues during organogenesis. Mech. Dev. 2000;96:209–213. doi: 10.1016/s0925-4773(00)00425-1. [DOI] [PubMed] [Google Scholar]
- Ding H., Wu X., Boström H., Kim I., Wong N., Tsoi B., O’Rourke M., Koh G.Y., Soriano P., Betsholtz C., et al. A specific requirement for PDGF-C in palate formation and PDGFR-α signaling. Nat. Genet. 2004;36:1111–1116. doi: 10.1038/ng1415. [DOI] [PubMed] [Google Scholar]
- Distler J.H., Jüngel A., Huber L.C., Schulze-Horsel U., Zwerina J., Gay R.E., Michel B.A., Hauser T., Schett G., Gay S., et al. Imatinib mesylate reduces production of extracellular matrix and prevents development of experimental dermal fibrosis. Arthritis Rheum. 2007;56:311–322. doi: 10.1002/art.22314. [DOI] [PubMed] [Google Scholar]
- Dong J., Grunstein J., Tejada M., Peale F., Frantz G., Liang W.C., Bai W., Yu L., Kowalski J., Liang X., et al. VEGF-null cells require PDGFR α signaling-mediated stromal fibroblast recruitment for tumorigenesis. EMBO J. 2004;23:2800–2810. doi: 10.1038/sj.emboj.7600289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doolittle R.F., Hunkapiller M.W., Hood L.E., Devare S.G., Robbins K.C., Aaronson S.A., Antoniades H.N. Simian sarcoma virus onc gene, v-sis, is derived from the gene (or genes) encoding a platelet-derived growth factor. Science. 1983;221:275–277. doi: 10.1126/science.6304883. [DOI] [PubMed] [Google Scholar]
- Druker B.J., Talpaz M., Resta D.J., Peng B., Buchdunger E., Ford J.M., Lydon N.B., Kantarjian H., Capdeville R., Ohno-Jones S., et al. Efficacy and safety of a specific inhibitor of the BCR-ABL tyrosine kinase in chronic myeloid leukemia. N. Engl. J. Med. 2001;344:1031–1037. doi: 10.1056/NEJM200104053441401. [DOI] [PubMed] [Google Scholar]
- Duchek P., Somogyi K., Jékely G., Beccari S., Rørth P. Guidance of cell migration by the Drosophila PDGF/VEGF receptor. Cell. 2001;107:17–26. doi: 10.1016/s0092-8674(01)00502-5. [DOI] [PubMed] [Google Scholar]
- Eberhart J.K., He X., Swartz M.E., Yan Y.L., Song H., Boling T.C., Kunerth A.K., Walker M.B., Kimmel C.B., Postlethwait J.H. MicroRNA Mirn140 modulates Pdgf signaling during palatogenesis. Nat. Genet. 2008;40:290–298. doi: 10.1038/ng.82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edmonds M., Bates M., Doxford M., Gough A., Foster A. New treatments in ulcer healing and wound infection. Diabetes Metab. Res. Rev. 2000;16 (Suppl. 1):S51–S54. doi: 10.1002/1520-7560(200009/10)16:1+<::aid-dmrr142>3.0.co;2-s. [DOI] [PubMed] [Google Scholar]
- Egawa-Tsuzuki T., Ohno M., Tanaka N., Takeuchi Y., Uramoto H., Faigle R., Funa K., Ishii Y., Sasahara M. The PDGF B-chain is involved in the ontogenic susceptibility of the developing rat brain to NMDA toxicity. Exp. Neurol. 2004;186:89–98. doi: 10.1016/j.expneurol.2003.11.001. [DOI] [PubMed] [Google Scholar]
- Eggenschwiler J.T., Anderson K.V. Cilia and developmental signaling. Annu. Rev. Cell Dev. Biol. 2007;23:345–373. doi: 10.1146/annurev.cellbio.23.090506.123249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ekman S., Thuresson E.R., Heldin C.H., Rönnstrand L. Increased mitogenicity of an αβ heterodimeric PDGF receptor complex correlates with lack of RasGAP binding. Oncogene. 1999;18:2481–2488. doi: 10.1038/sj.onc.1202606. [DOI] [PubMed] [Google Scholar]
- Enge M., Bjarnegård M., Gerhardt H., Gustafsson E., Kalén M., Asker N., Hammes H.P., Shani M., Fässler R., Betsholtz C. Endothelium-specific platelet-derived growth factor-B ablation mimics diabetic retinopathy. EMBO J. 2002;21:4307–4316. doi: 10.1093/emboj/cdf418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enge M., Wilhelmsson U., Abramsson A., Stakeberg J., Kühn R., Betsholtz C., Pekny M. Neuron-specific ablation of PDGF-B is compatible with normal central nervous system development and astroglial response to injury. Neurochem. Res. 2003;28:271–279. doi: 10.1023/a:1022421001288. [DOI] [PubMed] [Google Scholar]
- Erpel T., Courtneidge S.A. Src family protein tyrosine kinases and cellular signal transduction pathways. Curr. Opin. Cell Biol. 1995;7:176–182. doi: 10.1016/0955-0674(95)80025-5. [DOI] [PubMed] [Google Scholar]
- Fambrough D., McClure K., Kazlauskas A., Lander E.S. Diverse signaling pathways activated by growth factor receptors induce broadly overlapping, rather than independent, sets of genes. Cell. 1999;97:727–741. doi: 10.1016/s0092-8674(00)80785-0. [DOI] [PubMed] [Google Scholar]
- Fantl W.J., Escobedo J.A., Martin G.A., Turck C.W., del Rosario M., McCormick F., Williams L.T. Distinct phosphotyrosines on a growth factor receptor bind to specific molecules that mediate different signaling pathways. Cell. 1992;69:413–423. doi: 10.1016/0092-8674(92)90444-h. [DOI] [PubMed] [Google Scholar]
- Fatatis A., Miller R.J. Platelet-derived growth factor (PDGF)-induced Ca2+ signaling in the CG4 oligodendroglial cell line and in transformed oligodendrocytes expressing the β-PDGF receptor. J. Biol. Chem. 1997;272:4351–4358. doi: 10.1074/jbc.272.7.4351. [DOI] [PubMed] [Google Scholar]
- Fellström B., Klareskog L., Heldin C.H., Larsson E., Rönnstrand L., Terracio L., Tufveson G., Wahlberg J., Rubin K. Platelet-derived growth factor receptors in the kidney-upregulated expression in inflammation. Kidney Int. 1989;36:1099–1102. doi: 10.1038/ki.1989.306. [DOI] [PubMed] [Google Scholar]
- Ferns G.A., Raines E.W., Sprugel K.H., Motani A.S., Reidy M.A., Ross R. Inhibition of neointimal smooth muscle accumulation after angioplasty by an antibody to PDGF. Science. 1991;253:1129–1132. doi: 10.1126/science.1653454. [DOI] [PubMed] [Google Scholar]
- Ferrara N., Gerber H.P., LeCouter J. The biology of VEGF and its receptors. Nat. Med. 2003;9:669–676. doi: 10.1038/nm0603-669. [DOI] [PubMed] [Google Scholar]
- Feyzi E., Lustig F., Fager G., Spillmann D., Lindahl U., Salmivirta M. Characterization of heparin and heparan sulfate domains binding to the long splice variant of platelet-derived growth factor A chain. J. Biol. Chem. 1997;272:5518–5524. doi: 10.1074/jbc.272.9.5518. [DOI] [PubMed] [Google Scholar]
- Fleming T.P., Saxena A., Clark W.C., Robertson J.T., Oldfield E.H., Aaronson S.A., Ali I.U. Amplification and/or overexpression of platelet-derived growth factor receptors and epidermal growth factor receptor in human glial tumors. Cancer Res. 1992;52:4550–4553. [PubMed] [Google Scholar]
- Floege J., Ostendorf T., Janssen U., Burg M., Radeke H.H., Vargeese C., Gill S.C., Green L.S., Janjic N. Novel approach to specific growth factor inhibition in vivo: antagonism of platelet-derived growth factor in glomerulonephritis by aptamers. Am. J. Pathol. 1999;154:169–179. doi: 10.1016/S0002-9440(10)65263-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forsberg K., Valyi-Nagy I., Heldin C.H., Herlyn M., Westermark B. Platelet-derived growth factor (PDGF) in oncogenesis: Development of a vascular connective tissue stroma in xenotransplanted human melanoma producing PDGF-BB. Proc. Natl. Acad. Sci. 1993;90:393–397. doi: 10.1073/pnas.90.2.393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franke T.F., Yang S.I., Chan T.O., Datta K., Kazlauskas A., Morrison D.K., Kaplan D.R., Tsichlis P.N. The protein kinase encoded by the Akt proto-oncogene is a target of the PDGF-activated phosphatidylinositol 3-kinase. Cell. 1995;81:727–736. doi: 10.1016/0092-8674(95)90534-0. [DOI] [PubMed] [Google Scholar]
- Fredriksson L., Li H., Eriksson U. The PDGF family: Four gene products form five dimeric isoforms. Cytokine Growth Factor Rev. 2004a;15:197–204. doi: 10.1016/j.cytogfr.2004.03.007. [DOI] [PubMed] [Google Scholar]
- Fredriksson L., Li H., Fieber C., Li X., Eriksson U. Tissue plasminogen activator is a potent activator of PDGF-CC. EMBO J. 2004b;23:3793–3802. doi: 10.1038/sj.emboj.7600397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frisch S.M., Ruoslahti E. Integrins and anoikis. Curr. Opin. Cell Biol. 1997;9:701–706. doi: 10.1016/s0955-0674(97)80124-x. [DOI] [PubMed] [Google Scholar]
- Fruttiger M., Calver A.R., Krüger W.H., Mudhar H.S., Michalovich D., Takakura N., Nishikawa S., Richardson W.D. PDGF mediates a neuron–astrocyte interaction in the developing retina. Neuron. 1996;17:1117–1131. doi: 10.1016/s0896-6273(00)80244-5. [DOI] [PubMed] [Google Scholar]
- Fruttiger M., Karlsson L., Hall A.C., Abramsson A., Calver A.R., Boström H., Willetts K., Bertold C.H., Heath J.K., Betsholtz C., et al. Defective oligodendrocyte development and severe hypomyelination in PDGF-A knockout mice. Development. 1999;126:457–467. doi: 10.1242/dev.126.3.457. [DOI] [PubMed] [Google Scholar]
- Fruttiger M., Calver A.R., Richardson W.D. Platelet-derived growth factor is constitutively secreted from neuronal cell bodies but not from axons. Curr. Biol. 2000;10:1283–1286. doi: 10.1016/s0960-9822(00)00757-0. [DOI] [PubMed] [Google Scholar]
- Fulga T.A., Rørth P. Invasive cell migration is initiated by guided growth of long cellular extensions. Nat. Cell Biol. 2002;4:715–719. doi: 10.1038/ncb848. [DOI] [PubMed] [Google Scholar]
- Furuhashi M., Sjöblom T., Abramsson A., Ellingsen J., Micke P., Li H., Bergsten-Folestad E., Eriksson U., Heuchel R., Betsholtz C., et al. Platelet-derived growth factor production by B16 melanoma cells leads to increased pericyte abundance in tumors and an associated increase in tumor growth rate. Cancer Res. 2004;64:2725–2733. doi: 10.1158/0008-5472.can-03-1489. [DOI] [PubMed] [Google Scholar]
- Gay S., Jones R.E., Huang G.Q., Gay R.E. Immunohistologic demonstration of platelet-derived growth factor (PDGF) and sis-oncogene expression in scleroderma. J. Invest. Dermatol. 1989;92:301–303. doi: 10.1111/1523-1747.ep12276895. [DOI] [PubMed] [Google Scholar]
- Gerber H.P., Ferrara N. The role of VEGF in normal and neoplastic hematopoiesis. J. Mol. Med. 2003;81:20–31. doi: 10.1007/s00109-002-0397-4. [DOI] [PubMed] [Google Scholar]
- Gerhardt H., Semb H. Pericytes: Gatekeepers in tumour cell metastasis? J. Mol. Med. 2008;86:135–144. doi: 10.1007/s00109-007-0258-2. [DOI] [PubMed] [Google Scholar]
- Gerhardt H., Golding M., Fruttiger M., Ruhrberg C., Lundkvist A., Abramsson A., Jeltsch M., Mitchell C., Alitalo K., Shima D., et al. VEGF guides angiogenic sprouting utilizing endothelial tip cell filopodia. J. Cell Biol. 2003;161:1163–1177. doi: 10.1083/jcb.200302047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghofrani H.A., Seeger W., Grimminger F. Imatinib for the treatment of pulmonary arterial hypertension. N. Engl. J. Med. 2005;353:1412–1413. doi: 10.1056/NEJMc051946. [DOI] [PubMed] [Google Scholar]
- Giese N.A., Marijianowski M.M., McCook O., Hancock A., Ramakrishnan V., Fretto L.J., Chen C., Kelly A.B., Koziol J.A., Wilcox J.N., et al. The role of α and β platelet-derived growth factor receptor in the vascular response to injury in nonhuman primates. Arterioscler. Thromb. Vasc. Biol. 1999;19:900–909. doi: 10.1161/01.atv.19.4.900. [DOI] [PubMed] [Google Scholar]
- Gilbert R.E., Kelly D.J., McKay T., Chadban S., Hill P.A., Cooper M.E., Atkins R.C., Nikolic-Paterson D.J. PDGF signal transduction inhibition ameliorates experimental mesangial proliferative glomerulonephritis. Kidney Int. 2001;59:1324–1332. doi: 10.1046/j.1523-1755.2001.0590041324.x. [DOI] [PubMed] [Google Scholar]
- Gnessi L., Basciani S., Mariani S., Arizzi M., Spera G., Wang C., Bondjers C., Karlsson L., Betsholtz C. Leydig cell loss and spermatogenic arrest in platelet-derived growth factor (PDGF)-A-deficient mice. J. Cell Biol. 2000;149:1019–1026. doi: 10.1083/jcb.149.5.1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golub T.R., Barker G.F., Lovett M., Gilliland D.G. Fusion of PDGF receptor β to a novel ets-like gene, tel, in chronic myelomonocytic leukemia with t(5;12) chromosomal translocation. Cell. 1994;77:307–316. doi: 10.1016/0092-8674(94)90322-0. [DOI] [PubMed] [Google Scholar]
- Greenberg D.A., Jin K. From angiogenesis to neuropathology. Nature. 2005;438:954–959. doi: 10.1038/nature04481. [DOI] [PubMed] [Google Scholar]
- Griffin J.H., Leung J., Bruner R.J., Caligiuri M.A., Briesewitz R. Discovery of a fusion kinase in EOL-1 cells and idiopathic hypereosinophilic syndrome. Proc. Natl. Acad. Sci. 2003;100:7830–7835. doi: 10.1073/pnas.0932698100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Häcker U., Nybakken K., Perrimon N. Heparan sulphate proteoglycans: The sweet side of development. Nat. Rev. Mol. Cell Biol. 2005;6:530–541. doi: 10.1038/nrm1681. [DOI] [PubMed] [Google Scholar]
- Hamilton T.G., Klinghoffer R.A., Corrin P.D., Soriano P. Evolutionary divergence of platelet-derived growth factor α receptor signaling mechanisms. Mol. Cell. Biol. 2003;23:4013–4025. doi: 10.1128/MCB.23.11.4013-4025.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammes H.P., Lin J., Renner O., Shani M., Lundqvist A., Betsholtz C., Brownlee M., Deutsch U. Pericytes and the pathogenesis of diabetic retinopathy. Diabetes. 2002;51:3107–3112. doi: 10.2337/diabetes.51.10.3107. [DOI] [PubMed] [Google Scholar]
- Hanahan D., Weinberg R.A. The hallmarks of cancer. Cell. 2000;100:57–70. doi: 10.1016/s0092-8674(00)81683-9. [DOI] [PubMed] [Google Scholar]
- Hansmann G., Wagner R.A., Schellong S., Perez V.A., Urashima T., Wang L., Sheikh A.Y., Suen R.S., Stewart D.J., Rabinovitch M. Pulmonary arterial hypertension is linked to insulin resistance and reversed by peroxisome proliferator-activated receptor-γ activation. Circulation. 2007;115:1275–1284. doi: 10.1161/CIRCULATIONAHA.106.663120. [DOI] [PubMed] [Google Scholar]
- Hansson G.K. Inflammation, atherosclerosis, and coronary artery disease. N. Engl. J. Med. 2005;352:1685–1695. doi: 10.1056/NEJMra043430. [DOI] [PubMed] [Google Scholar]
- Haramis A.P., Begthel H., van den Born M., van Es J., Jonkheer S., Offerhaus G.J., Clevers H. De novo crypt formation and juvenile polyposis on BMP inhibition in mouse intestine. Science. 2004;303:1684–1686. doi: 10.1126/science.1093587. [DOI] [PubMed] [Google Scholar]
- Hart C.E., Kraiss L.W., Vergel S., Gilbertson D., Kenagy R., Kirkman T., Crandall D.L., Tickle S., Finney H., Yarranton G., et al. PDGFβ receptor blockade inhibits intimal hyperplasia in the baboon. Circulation. 1999;99:564–569. doi: 10.1161/01.cir.99.4.564. [DOI] [PubMed] [Google Scholar]
- Hashizume H., Baluk P., Morikawa S., McLean J.W., Thurston G., Roberge S., Jain R.K., McDonald D.M. Openings between defective endothelial cells explain tumor vessel leakiness. Am. J. Pathol. 2000;156:1363–1380. doi: 10.1016/S0002-9440(10)65006-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawkins P.T., Eguinoa A., Qiu R.G., Stokoe D., Cooke F.T., Walters R., Wennström S., Claesson-Welsh L., Evans T., Symons M. PDGF stimulates an increase in GTP-Rac via activation of phosphoinositide 3-kinase. Curr. Biol. 1995;5:393–403. doi: 10.1016/s0960-9822(95)00080-7. [DOI] [PubMed] [Google Scholar]
- Heinrich M.C., Corless C.L., Duensing A., McGreevey L., Chen C.J., Joseph N., Singer S., Griffith D.J., Haley A., Town A., et al. PDGFRA activating mutations in gastrointestinal stromal tumors. Science. 2003;299:708–710. doi: 10.1126/science.1079666. [DOI] [PubMed] [Google Scholar]
- Heldin C.H., Westermark B. Mechanism of action and in vivo role of platelet-derived growth factor. Physiol. Rev. 1999;79:1283–1316. doi: 10.1152/physrev.1999.79.4.1283. [DOI] [PubMed] [Google Scholar]
- Heldin C.H., Wasteson A., Westermark B. Interaction of platelet-derived growth factor with its fibroblast receptor. Demonstration of ligand degradation and receptor modulation. J. Biol. Chem. 1982;257:4216–4221. [PubMed] [Google Scholar]
- Heldin C.H., Johnsson A., Wennergren S., Wernstedt C., Betsholtz C., Westermark B. A human osteosarcoma cell line secretes a growth factor structurally related to a homodimer of PDGF A-chains. Nature. 1986;319:511–514. doi: 10.1038/319511a0. [DOI] [PubMed] [Google Scholar]
- Heldin N.E., Gustavsson B., Claesson-Welsh L., Hammacher A., Mark J., Heldin C.H., Westermark B. Aberrant expression of receptors for platelet-derived growth factor in an anaplastic thyroid carcinoma cell line. Proc. Natl. Acad. Sci. 1988;85:9302–9306. doi: 10.1073/pnas.85.23.9302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heldin C.H., Ostman A., Rönnstrand L. Signal transduction via platelet-derived growth factor receptors. Biochim. Biophys. Acta. 1998;1378:F79–F113. doi: 10.1016/S0304-419X(98)00015-8. [DOI] [PubMed] [Google Scholar]
- Heldin C.H., Rubin K., Pietras K., Ostman A. High interstitial fluid pressure—An obstacle in cancer therapy. Nat. Rev. Cancer. 2004;4:806–813. doi: 10.1038/nrc1456. [DOI] [PubMed] [Google Scholar]
- Hellström M., Kalén M., Lindahl P., Abramsson A., Betsholtz C. Role of PDGF-B and PDGFR-β in recruitment of vascular smooth muscle cells and pericytes during embryonic blood vessel formation in the mouse. Development. 1999;126:3047–3055. doi: 10.1242/dev.126.14.3047. [DOI] [PubMed] [Google Scholar]
- Hellström M., Gerhardt H., Kalén M., Li X., Eriksson U., Wolburg H., Betsholtz C. Lack of pericytes leads to endothelial hyperplasia and abnormal vascular morphogenesis. J. Cell Biol. 2001;153:543–553. doi: 10.1083/jcb.153.3.543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hermanson M., Funa K., Hartman M., Claesson-Welsh L., Heldin C.H., Westermark B., Nistér M. Platelet-derived growth factor and its receptors in human glioma tissue: Expression of messenger RNA and protein suggests the presence of autocrine and paracrine loops. Cancer Res. 1992;52:3213–3219. [PubMed] [Google Scholar]
- Hermanson M., Funa K., Koopmann J., Maintz D., Waha A., Westermark B., Heldin C.H., Wiestler O.D., Louis D.N., von Deimling A., et al. Association of loss of heterozygosity on chromosome 17p with high platelet-derived growth factor α receptor expression in human malignant gliomas. Cancer Res. 1996;56:164–171. [PubMed] [Google Scholar]
- Heuchel R., Berg A., Tallquist M., Ahlén K., Reed R.K., Rubin K., Claesson-Welsh L., Heldin C.H., Soriano P. Platelet-derived growth factor β receptor regulates interstitial fluid homeostasis through phosphatidylinositol-3′ kinase signaling. Proc. Natl. Acad. Sci. 1999;96:11410–11415. doi: 10.1073/pnas.96.20.11410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirota S., Ohashi A., Nishida T., Isozaki K., Kinoshita K., Shinomura Y., Kitamura Y. Gain-of-function mutations of platelet-derived growth factor receptor α gene in gastrointestinal stromal tumors. Gastroenterology. 2003;125:660–667. doi: 10.1016/s0016-5085(03)01046-1. [DOI] [PubMed] [Google Scholar]
- Ho L., Symes K., Yordán C., Gudas L.J., Mercola M. Localization of PDGF A and PDGFR α mRNA in Xenopus embryos suggests signalling from neural ectoderm and pharyngeal endoderm to neural crest cells. Mech. Dev. 1994;48:165–174. doi: 10.1016/0925-4773(94)90057-4. [DOI] [PubMed] [Google Scholar]
- Hoch R.V., Soriano P. Roles of PDGF in animal development. Development. 2003;130:4769–4784. doi: 10.1242/dev.00721. [DOI] [PubMed] [Google Scholar]
- Homma S., Nagaoka I., Abe H., Takahashi K., Seyama K., Nukiwa T., Kira S. Localization of platelet-derived growth factor and insulin-like growth factor I in the fibrotic lung. Am. J. Respir. Crit. Care Med. 1995;152:2084–2089. doi: 10.1164/ajrccm.152.6.8520779. [DOI] [PubMed] [Google Scholar]
- Homsi J., Daud A.I. Spectrum of activity and mechanism of action of VEGF/PDGF inhibitors. Cancer Control. 2007;14:285–294. doi: 10.1177/107327480701400312. [DOI] [PubMed] [Google Scholar]
- Hossain M.Z., Ao P., Boynton A.L. Platelet-derived growth factor-induced disruption of gap junctional communication and phosphorylation of connexin43 involves protein kinase C and mitogen-activated protein kinase. J. Cell. Physiol. 1998;176:332–341. doi: 10.1002/(SICI)1097-4652(199808)176:2<332::AID-JCP11>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- Hu Q., Klippel A., Muslin A.J., Fantl W.J., Williams L.T. Ras-dependent induction of cellular responses by constitutively active phosphatidylinositol-3 kinase. Science. 1995;268:100–102. doi: 10.1126/science.7701328. [DOI] [PubMed] [Google Scholar]
- Humbert M., Monti G., Fartoukh M., Magnan A., Brenot F., Rain B., Capron F., Galanaud P., Duroux P., Simonneau G., et al. Platelet-derived growth factor expression in primary pulmonary hypertension: Comparison of HIV seropositive and HIV seronegative patients. Eur. Respir. J. 1998;11:554–559. [PubMed] [Google Scholar]
- Hwang R.F., Yokoi K., Bucana C.D., Tsan R., Killion J.J., Evans D.B., Fidler I.J. Inhibition of platelet-derived growth factor receptor phosphorylation by STI571 (Gleevec) reduces growth and metastasis of human pancreatic carcinoma in an orthotopic nude mouse model. Clin. Cancer Res. 2003;9:6534–6544. [PubMed] [Google Scholar]
- Ishii Y., Oya T., Zheng L., Gao Z., Kawaguchi M., Sabit H., Matsushima T., Tokunaga A., Ishizawa S., Hori E., et al. Mouse brains deficient in neuronal PDGF receptor-β develop normally but are vulnerable to injury. J. Neurochem. 2006;98:588–600. doi: 10.1111/j.1471-4159.2006.03922.x. [DOI] [PubMed] [Google Scholar]
- Jackson E.L., Garcia-Verdugo J.M., Gil-Perotin S., Roy M., Quinones-Hinojosa A., VandenBerg S., Alvarez-Buylla A. PDGFR α-positive B cells are neural stem cells in the adult SVZ that form glioma-like growths in response to increased PDGF signaling. Neuron. 2006;51:187–199. doi: 10.1016/j.neuron.2006.06.012. [DOI] [PubMed] [Google Scholar]
- Jakobsson L., Kreuger J., Holmborn K., Lundin L., Eriksson I., Kjellén L., Claesson-Welsh L. Heparan sulfate in trans potentiates VEGFR-mediated angiogenesis. Dev. Cell. 2006;10:625–634. doi: 10.1016/j.devcel.2006.03.009. [DOI] [PubMed] [Google Scholar]
- James M.F., Beauchamp R.L., Manchanda N., Kazlauskas A., Ramesh V. A NHERF binding site links the βPDGFR to the cytoskeleton and regulates cell spreading and migration. J. Cell Sci. 2004;117:2951–2961. doi: 10.1242/jcs.01156. [DOI] [PubMed] [Google Scholar]
- Jawien A., Bowen-Pope D.F., Lindner V., Schwartz S.M., Clowes A.W. Platelet-derived growth factor promotes smooth muscle migration and intimal thickening in a rat model of balloon angioplasty. J. Clin. Invest. 1992;89:507–511. doi: 10.1172/JCI115613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jechlinger M., Grünert S., Tamir I.H., Janda E., Lüdemann S., Waerner T., Seither P., Weith A., Beug H., Kraut N. Expression profiling of epithelial plasticity in tumor progression. Oncogene. 2003;22:7155–7169. doi: 10.1038/sj.onc.1206887. [DOI] [PubMed] [Google Scholar]
- Jechlinger M., Sommer A., Moriggl R., Seither P., Kraut N., Capodiecci P., Donovan M., Cordon-Cardo C., Beug H., Grünert S. Autocrine PDGFR signaling promotes mammary cancer metastasis. J. Clin. Invest. 2006;116:1561–1570. doi: 10.1172/JCI24652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jennings M.T., Hart C.E., Commers P.A., Whitlock J.A., Martincic D., Maciunas R.J., Moots P.L., Shehab T.M. Transforming growth factor β as a potential tumor progression factor among hyperdiploid glioblastoma cultures: Evidence for the role of platelet-derived growth factor. J. Neurooncol. 1997;31:233–254. doi: 10.1023/a:1005767616500. [DOI] [PubMed] [Google Scholar]
- Jo N., Mailhos C., Ju M., Cheung E., Bradley J., Nishijima K., Robinson G.S., Adamis A.P., Shima D.T. Inhibition of platelet-derived growth factor B signaling enhances the efficacy of anti-vascular endothelial growth factor therapy in multiple models of ocular neovascularization. Am. J. Pathol. 2006;168:2036–2053. doi: 10.2353/ajpath.2006.050588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson R.J., Raines E.W., Floege J., Yoshimura A., Pritzl P., Alpers C., Ross R. Inhibition of mesangial cell proliferation and matrix expansion in glomerulonephritis in the rat by antibody to platelet-derived growth factor. J. Exp. Med. 1992;175:1413–1416. doi: 10.1084/jem.175.5.1413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnsson A., Heldin C.H., Westermark B., Wasteson A. Platelet-derived growth factor: Identification of constituent polypeptide chains. Biochem. Biophys. Res. Commun. 1982;104:66–74. doi: 10.1016/0006-291x(82)91941-6. [DOI] [PubMed] [Google Scholar]
- Johnsson A., Betsholtz C., Heldin C.H., Westermark B. Antibodies against platelet-derived growth factor inhibit acute transformation by simian sarcoma virus. Nature. 1985;317:438–440. doi: 10.1038/317438a0. [DOI] [PubMed] [Google Scholar]
- Jones R., Capen D., Jacobson M. PDGF and microvessel wall remodeling in adult lung: Imaging PDGF-Rβ and PDGF-BB molecules in progenitor smooth muscle cells developing in pulmonary hypertension. Ultrastruct. Pathol. 2006;30:267–281. doi: 10.1080/01913120600820336. [DOI] [PubMed] [Google Scholar]
- Joosten P.H., Toepoel M., Mariman E.C., Van Zoelen E.J. Promoter haplotype combinations of the platelet-derived growth factor α-receptor gene predispose to human neural tube defects. Nat. Genet. 2001;27:215–217. doi: 10.1038/84867. [DOI] [PubMed] [Google Scholar]
- Kaetzel D.M. Transcription of the platelet-derived growth factor A-chain gene. Cytokine Growth Factor Rev. 2003;14:427–446. doi: 10.1016/s1359-6101(03)00051-0. [DOI] [PubMed] [Google Scholar]
- Kaminski W.E., Lindahl P., Lin N.L., Broudy V.C., Crosby J.R., Hellström M., Swolin B., Bowen-Pope D.F., Martin P.J., Ross R., et al. Basis of hematopoietic defects in platelet-derived growth factor (PDGF)-B and PDGF β-receptor null mice. Blood. 2001;97:1990–1998. doi: 10.1182/blood.v97.7.1990. [DOI] [PubMed] [Google Scholar]
- Karlsson L., Bondjers C., Betsholtz C. Roles for PDGF-A and sonic hedgehog in development of mesenchymal components of the hair follicle. Development. 1999;126:2611–2621. doi: 10.1242/dev.126.12.2611. [DOI] [PubMed] [Google Scholar]
- Karlsson L., Lindahl P., Heath J.K., Betsholtz C. Abnormal gastrointestinal development in PDGF-A and PDGFR-α deficient mice implicates a novel mesenchymal structure with putative instructive properties in villus morphogenesis. Development. 2000;127:3457–3466. doi: 10.1242/dev.127.16.3457. [DOI] [PubMed] [Google Scholar]
- Karlsson S., Kowanetz K., Sandin A., Persson C., Ostman A., Heldin C.H., Hellberg C. Loss of T-cell protein tyrosine phosphatase induces recycling of the platelet-derived growth factor (PDGF) β-receptor but not the PDGF α-receptor. Mol. Biol. Cell. 2006;17:4846–4855. doi: 10.1091/mbc.E06-04-0306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karnoub A.E., Dash A.B., Vo A.P., Sullivan A., Brooks M.W., Bell G.W., Richardson A.L., Polyak K., Tubo R., Weinberg R.A. Mesenchymal stem cells within tumour stroma promote breast cancer metastasis. Nature. 2007;449:557–563. doi: 10.1038/nature06188. [DOI] [PubMed] [Google Scholar]
- Kawaguchi Y., Hara M., Wright T.M. Endogenous IL-1α from systemic sclerosis fibroblasts induces IL-6 and PDGF-A. J. Clin. Invest. 1999;103:1253–1260. doi: 10.1172/JCI4304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kazlauskas A., Cooper J.A. Autophosphorylation of the PDGF receptor in the kinase insert region regulates interactions with cell proteins. Cell. 1989;58:1121–1133. doi: 10.1016/0092-8674(89)90510-2. [DOI] [PubMed] [Google Scholar]
- Kelly J.D., Haldeman B.A., Grant F.J., Murray M.J., Seifert R.A., Bowen-Pope D.F., Cooper J.A., Kazlauskas A. Platelet-derived growth factor (PDGF) stimulates PDGF receptor subunit dimerization and intersubunit trans-phosphorylation. J. Biol. Chem. 1991;266:8987–8992. [PubMed] [Google Scholar]
- Kelly J.L., Sánchez A., Brown G.S., Chesterman C.N., Sleigh M.J. Accumulation of PDGF B and cell-binding forms of PDGF A in the extracellular matrix. J. Cell Biol. 1993;121:1153–1163. doi: 10.1083/jcb.121.5.1153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim L., Wong T.W. The cytoplasmic tyrosine kinase FER is associated with the catenin-like substrate pp120 and is activated by growth factors. Mol. Cell. Biol. 1995;15:4553–4561. doi: 10.1128/mcb.15.8.4553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klareskog L., Gustafsson R., Scheynius A., Hällgren R. Increased expression of platelet-derived growth factor type B receptors in the skin of patients with systemic sclerosis. Arthritis Rheum. 1990;33:1534–1541. doi: 10.1002/art.1780331011. [DOI] [PubMed] [Google Scholar]
- Klinghoffer R.A., Mueting-Nelsen P.F., Faerman A., Shani M., Soriano P. The two PDGF receptors maintain conserved signaling in vivo despite divergent embryological functions. Mol. Cell. 2001;7:343–354. doi: 10.1016/s1097-2765(01)00182-4. [DOI] [PubMed] [Google Scholar]
- Klinghoffer R.A., Hamilton T.G., Hoch R., Soriano P. An allelic series at the PDGFαR locus indicates unequal contributions of distinct signaling pathways during development. Dev. Cell. 2002;2:103–113. doi: 10.1016/s1534-5807(01)00103-4. [DOI] [PubMed] [Google Scholar]
- Kohler N., Lipton A. Platelets as a source of fibroblast growth-promoting activity. Exp. Cell Res. 1974;87:297–301. doi: 10.1016/0014-4827(74)90484-4. [DOI] [PubMed] [Google Scholar]
- Kok A., Lovicu F.J., Chamberlain C.G., McAvoy J.W. Influence of platelet-derived growth factor on lens epithelial cell proliferation and differentiation. Growth Factors. 2002;20:27–34. doi: 10.1080/08977190290022202. [DOI] [PubMed] [Google Scholar]
- Kozaki K., Kaminski W.E., Tang J., Hollenbach S., Lindahl P., Sullivan C., Yu J.C., Abe K., Martin P.J., Ross R., et al. Blockade of platelet-derived growth factor or its receptors transiently delays but does not prevent fibrous cap formation in ApoE null mice. Am. J. Pathol. 2002;161:1395–1407. doi: 10.1016/S0002-9440(10)64415-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kundra V., Escobedo J.A., Kazlauskas A., Kim H.K., Rhee S.G., Williams L.T., Zetter B.R. Regulation of chemotaxis by the platelet-derived growth factor receptor-β. Nature. 1994;367:474–476. doi: 10.1038/367474a0. [DOI] [PubMed] [Google Scholar]
- LaRochelle W.J., Jeffers M., McDonald W.F., Chillakuru R.A., Giese N.A., Lokker N.A., Sullivan C., Boldog F.L., Yang M., Vernet C., et al. PDGF-D, a new protease-activated growth factor. Nat. Cell Biol. 2001;3:517–521. doi: 10.1038/35074593. [DOI] [PubMed] [Google Scholar]
- Le Bras B., Barallobre M.J., Homman-Ludiye J., Ny A., Wyns S., Tammela T., Haiko P., Karkkainen M.J., Yuan L., Muriel M.P., et al. VEGF-C is a trophic factor for neural progenitors in the vertebrate embryonic brain. Nat. Neurosci. 2006;9:340–348. doi: 10.1038/nn1646. [DOI] [PubMed] [Google Scholar]
- Lechleider R.J., Sugimoto S., Bennett A.M., Kashishian A.S., Cooper J.A., Shoelson S.E., Walsh C.T., Neel B.G. Activation of the SH2-containing phosphotyrosine phosphatase SH-PTP2 by its binding site, phosphotyrosine 1009, on the human platelet-derived growth factor receptor. J. Biol. Chem. 1993;268:21478–21481. [PubMed] [Google Scholar]
- Lederle W., Stark H.J., Skobe M., Fusenig N.E., Mueller M.M. Platelet-derived growth factor-BB controls epithelial tumor phenotype by differential growth factor regulation in stromal cells. Am. J. Pathol. 2006;169:1767–1783. doi: 10.2353/ajpath.2006.060120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S., Chen T.T., Barber C.L., Jordan M.C., Murdock J., Desai S., Ferrara N., Nagy A., Roos K.P., Iruela-Arispe M.L. Autocrine VEGF signaling is required for vascular homeostasis. Cell. 2007;130:691–703. doi: 10.1016/j.cell.2007.06.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lennartsson J., Wardega P., Engström U., Hellman U., Heldin C.H. Alix facilitates the interaction between c-Cbl and platelet-derived growth factor β-receptor and thereby modulates receptor down-regulation. J. Biol. Chem. 2006;281:39152–39158. doi: 10.1074/jbc.M608489200. [DOI] [PubMed] [Google Scholar]
- Leppänen O., Janjic N., Carlsson M.A., Pietras K., Levin M., Vargeese C., Green L.S., Bergqvist D., Ostman A., Heldin C.H. Intimal hyperplasia recurs after removal of PDGF-AB and -BB inhibition in the rat carotid artery injury model. Arterioscler. Thromb. Vasc. Biol. 2000;20:E89–E95. doi: 10.1161/01.atv.20.11.e89. [DOI] [PubMed] [Google Scholar]
- Levéen P., Pekny M., Gebre-Medhin S., Swolin B., Larsson E., Betsholtz C. Mice deficient for PDGF B show renal, cardiovascular, and hematological abnormalities. Genes & Dev. 1994;8:1875–1887. doi: 10.1101/gad.8.16.1875. [DOI] [PubMed] [Google Scholar]
- Levitzki A. PDGF receptor kinase inhibitors for the treatment of PDGF driven diseases. Cytokine Growth Factor Rev. 2004;15:229–235. doi: 10.1016/j.cytogfr.2004.03.010. [DOI] [PubMed] [Google Scholar]
- Lewis C.D., Olson N.E., Raines E.W., Reidy M.A., Jackson C.L. Modulation of smooth muscle proliferation in rat carotid artery by platelet-derived mediators and fibroblast growth factor-2. Platelets. 2001;12:352–358. doi: 10.1080/09537100120071013. [DOI] [PubMed] [Google Scholar]
- Li J., Hoyle G.W. Overexpression of PDGF-A in the lung epithelium of transgenic mice produces a lethal phenotype associated with hyperplasia of mesenchymal cells. Dev. Biol. 2001;239:338–349. doi: 10.1006/dbio.2001.0441. [DOI] [PubMed] [Google Scholar]
- Li W., Michieli P., Alimandi M., Lorenzi M.V., Wu Y., Wang L.H., Heidaran M.A., Pierce J.H. Expression of an ATP binding mutant of PKC-δ inhibits Sis-induced transformation of NIH3T3 cells. Oncogene. 1996;13:731–737. [PubMed] [Google Scholar]
- Li X., Pontén A., Aase K., Karlsson L., Abramsson A., Uutela M., Bäckström G., Hellström M., Boström H., Li H., et al. PDGF-C is a new protease-activated ligand for the PDGF α-receptor. Nat. Cell Biol. 2000;2:302–309. doi: 10.1038/35010579. [DOI] [PubMed] [Google Scholar]
- Lin X. Functions of heparan sulfate proteoglycans in cell signaling during development. Development. 2004;131:6009–6021. doi: 10.1242/dev.01522. [DOI] [PubMed] [Google Scholar]
- Lindahl P., Johansson B.R., Levéen P., Betsholtz C. Pericyte loss and microaneurysm formation in PDGF-B-deficient mice. Science. 1997a;277:242–245. doi: 10.1126/science.277.5323.242. [DOI] [PubMed] [Google Scholar]
- Lindahl P., Karlsson L., Hellström M., Gebre-Medhin S., Willetts K., Heath J.K., Betsholtz C. Alveogenesis failure in PDGF-A-deficient mice is coupled to lack of distal spreading of alveolar smooth muscle cell progenitors during lung development. Development. 1997b;124:3943–3953. doi: 10.1242/dev.124.20.3943. [DOI] [PubMed] [Google Scholar]
- Lindahl P., Hellström M., Kalén M., Karlsson L., Pekny M., Pekna M., Soriano P., Betsholtz C. Paracrine PDGF-B/PDGF-Rβ signaling controls mesangial cell development in kidney glomeruli. Development. 1998;125:3313–3322. doi: 10.1242/dev.125.17.3313. [DOI] [PubMed] [Google Scholar]
- Lindblom P., Gerhardt H., Liebner S., Abramsson A., Enge M., Hellström M., Backstrom G., Fredriksson S., Landegren U., Nyström H.C., et al. Endothelial PDGF-B retention is required for proper investment of pericytes in the microvessel wall. Genes & Dev. 2003;17:1835–1840. doi: 10.1101/gad.266803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindmark G., Sundberg C., Glimelius B., Påhlman L., Rubin K., Gerdin B. Stromal expression of platelet-derived growth factor β-receptor and platelet-derived growth factor B-chain in colorectal cancer. Lab. Invest. 1993;69:682–689. [PubMed] [Google Scholar]
- Liu L., Chong S.W., Balasubramaniyan N.V., Korzh V., Ge R. Platelet-derived growth factor receptor α (pdgfr-α) gene in zebrafish embryonic development. Mech. Dev. 2002a;116:227–230. doi: 10.1016/s0925-4773(02)00142-9. [DOI] [PubMed] [Google Scholar]
- Liu L., Korzh V., Balasubramaniyan N.V., Ekker M., Ge R. Platelet-derived growth factor A (pdgf-α) expression during zebrafish embryonic development. Dev. Genes Evol. 2002b;212:298–301. doi: 10.1007/s00427-002-0234-3. [DOI] [PubMed] [Google Scholar]
- Lokker N.A., Sullivan C.M., Hollenbach S.J., Israel M.A., Giese N.A. Platelet-derived growth factor (PDGF) autocrine signaling regulates survival and mitogenic pathways in glioblastoma cells: Evidence that the novel PDGF-C and PDGF-D ligands may play a role in the development of brain tumors. Cancer Res. 2002;62:3729–3735. [PubMed] [Google Scholar]
- Lopez-Ilasaca M., Li W., Uren A., Yu J.C., Kazlauskas A., Gutkind J.S., Heidaran M.A. Requirement of phosphatidylinositol-3 kinase for activation of JNK/SAPKs by PDGF. Biochem. Biophys. Res. Commun. 1997;232:273–277. doi: 10.1006/bbrc.1997.6289. [DOI] [PubMed] [Google Scholar]
- Loukinova E., Ranganathan S., Kuznetsov S., Gorlatova N., Migliorini M.M., Loukinov D., Ulery P.G., Mikhailenko I., Lawrence D.A., Strickland D.K. Platelet-derived growth factor (PDGF)-induced tyrosine phosphorylation of the low density lipoprotein receptor-related protein (LRP). Evidence for integrated co-receptor function betwenn LRP and the PDGF. J. Biol. Chem. 2002;277:15499–15506. doi: 10.1074/jbc.M200427200. [DOI] [PubMed] [Google Scholar]
- Lustig F., Hoebeke J., Simonson C., Ostergren-Lundén G., Bondjers G., Rüetchi U., Fager G. Processing of PDGF gene products determines interactions with glycosaminoglycans. J. Mol. Recognit. 1999;12:112–120. doi: 10.1002/(SICI)1099-1352(199903/04)12:2<112::AID-JMR452>3.0.CO;2-R. [DOI] [PubMed] [Google Scholar]
- Majesky M.W., Daemen M.J., Schwartz S.M. α 1-adrenergic stimulation of platelet-derived growth factor A-chain gene expression in rat aorta. J. Biol. Chem. 1990;265:1082–1088. [PubMed] [Google Scholar]
- Martinet Y., Rom W.N., Grotendorst G.R., Martin G.R., Crystal R.G. Exaggerated spontaneous release of platelet-derived growth factor by alveolar macrophages from patients with idiopathic pulmonary fibrosis. N. Engl. J. Med. 1987;317:202–209. doi: 10.1056/NEJM198707233170404. [DOI] [PubMed] [Google Scholar]
- McArthur G.A., Demetri G.D., van Oosterom A., Heinrich M.C., Debiec-Rychter M., Corless C.L., Nikolova Z., Dimitrijevic S., Fletcher J.A. Molecular and clinical analysis of locally advanced dermatofibrosarcoma protuberans treated with imatinib: Imatinib Target Exploration Consortium Study B2225. J. Clin. Oncol. 2005;23:866–873. doi: 10.1200/JCO.2005.07.088. [DOI] [PubMed] [Google Scholar]
- McDonald N.Q., Hendrickson W.A. A structural superfamily of growth factors containing a cystine knot motif. Cell. 1993;73:421–424. doi: 10.1016/0092-8674(93)90127-c. [DOI] [PubMed] [Google Scholar]
- McDonald J.A., Pinheiro E.M., Montell D.J. PVF1, a PDGF/VEGF homolog, is sufficient to guide border cells and interacts genetically with Taiman. Development. 2003;130:3469–3478. doi: 10.1242/dev.00574. [DOI] [PubMed] [Google Scholar]
- McDonald J.A., Pinheiro E.M., Kadlec L., Schupbach T., Montell D.J. Multiple EGFR ligands participate in guiding migrating border cells. Dev. Biol. 2006;296:94–103. doi: 10.1016/j.ydbio.2006.04.438. [DOI] [PubMed] [Google Scholar]
- Mercola M., Wang C.Y., Kelly J., Brownlee C., Jackson-Grusby L., Stiles C., Bowen-Pope D. Selective expression of PDGF A and its receptor during early mouse embryogenesis. Dev. Biol. 1990;138:114–122. doi: 10.1016/0012-1606(90)90181-h. [DOI] [PubMed] [Google Scholar]
- Mondy J.S., Lindner V., Miyashiro J.K., Berk B.C., Dean R.H., Geary R.L. Platelet-derived growth factor ligand and receptor expression in response to altered blood flow in vivo. Circ. Res. 1997;81:320–327. doi: 10.1161/01.res.81.3.320. [DOI] [PubMed] [Google Scholar]
- Montero J.A., Heisenberg C.P. Gastrulation dynamics: Cells move into focus. Trends Cell Biol. 2004;14:620–627. doi: 10.1016/j.tcb.2004.09.008. [DOI] [PubMed] [Google Scholar]
- Montero J.A., Kilian B., Chan J., Bayliss P.E., Heisenberg C.P. Phosphoinositide 3-kinase is required for process outgrowth and cell polarization of gastrulating mesendodermal cells. Curr. Biol. 2003;13:1279–1289. doi: 10.1016/s0960-9822(03)00505-0. [DOI] [PubMed] [Google Scholar]
- Mori S., Tanaka K., Omura S., Saito Y. Degradation process of ligand-stimulated platelet-derived growth factor β-receptor involves ubiquitin-proteasome proteolytic pathway. J. Biol. Chem. 1995;270:29447–29452. doi: 10.1074/jbc.270.49.29447. [DOI] [PubMed] [Google Scholar]
- Morrison-Graham K., Schatteman G.C., Bork T., Bowen-Pope D.F., Weston J.A. A PDGF receptor mutation in the mouse (Patch) perturbs the development of a non-neuronal subset of neural crest-derived cells. Development. 1992;115:133–142. doi: 10.1242/dev.115.1.133. [DOI] [PubMed] [Google Scholar]
- Munier A.I., Doucet D., Perrodou E., Zachary D., Meister M., Hoffmann J.A., Janeway C.A., Lagueux M. PVF2, a PDGF/VEGF-like growth factor, induces hemocyte proliferation in Drosophila larvae. EMBO Rep. 2002;3:1195–1200. doi: 10.1093/embo-reports/kvf242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nabel E.G., Yang Z., Liptay S., San H., Gordon D., Haudenschild C.C., Nabel G.J. Recombinant platelet-derived growth factor B gene expression in porcine arteries induce intimal hyperplasia in vivo. J. Clin. Invest. 1993;91:1822–1829. doi: 10.1172/JCI116394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagel M., Tahinci E., Symes K., Winklbauer R. Guidance of mesoderm cell migration in the Xenopus gastrula requires PDGF signaling. Development. 2004;131:2727–2736. doi: 10.1242/dev.01141. [DOI] [PubMed] [Google Scholar]
- Nakanishi H., Brewer K.A., Exton J.H. Activation of the zeta isozyme of protein kinase C by phosphatidylinositol 3,4,5-trisphosphate. J. Biol. Chem. 1993;268:13–16. [PubMed] [Google Scholar]
- Narita M., Usui A., Narita M., Niikura K., Nozaki H., Khotib J., Nagumo Y., Yajima Y., Suzuki T. Protease-activated receptor-1 and platelet-derived growth factor in spinal cord neurons are implicated in neuropathic pain after nerve injury. J. Neurosci. 2005;25:10000–10009. doi: 10.1523/JNEUROSCI.2507-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Negoro N., Kanayama Y., Haraguchi M., Umetani N., Nishimura M., Konishi Y., Iwai J., Okamura M., Inoue T., Takeda T. Blood pressure regulates platelet-derived growth factor A-chain gene expression in vascular smooth muscle cells in vivo. An autocrine mechanism promoting hypertensive vascular hypertrophy. J. Clin. Invest. 1995;95:1140–1150. doi: 10.1172/JCI117762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nevins M., Camelo M., Nevins M.L., Schenk R.K., Lynch S.E. Periodontal regeneration in humans using recombinant human platelet-derived growth factor-BB (rhPDGF-BB) and allogenic bone. J. Periodontol. 2003;74:1282–1292. doi: 10.1902/jop.2003.74.9.1282. [DOI] [PubMed] [Google Scholar]
- Newton H.B. Molecular neuro-oncology and development of targeted therapeutic strategies for brain tumors. Part 1: Growth factor and Ras signaling pathways. Expert Rev. Anticancer Ther. 2003;3:595–614. doi: 10.1586/14737140.3.5.595. [DOI] [PubMed] [Google Scholar]
- Newton C.S., Loukinova E., Mikhailenko I., Ranganathan S., Gao Y., Haudenschild C., Strickland D.K. Platelet-derived growth factor receptor-β (PDGFR-β) activation promotes its association with the low density lipoprotein receptor-related protein (LRP). Evidence for co-receptor function. J. Biol. Chem. 2005;280:27872–27878. doi: 10.1074/jbc.M505410200. [DOI] [PubMed] [Google Scholar]
- Nishimura R., Li W., Kashishian A., Mondino A., Zhou M., Cooper J., Schlessinger J. Two signaling molecules share a phosphotyrosine-containing binding site in the platelet-derived growth factor receptor. Mol. Cell. Biol. 1993;13:6889–6896. doi: 10.1128/mcb.13.11.6889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishioka T., Suzuki M., Onishi K., Takakura N., Inada H., Yoshida T., Hiroe M., Imanaka-Yoshida K. Eplerenone attenuates myocardial fibrosis in the angiotensin II-induced hypertensive mouse: Involvement of tenascin-C induced by aldosterone-mediated inflammation. J. Cardiovasc. Pharmacol. 2007;49:261–268. doi: 10.1097/FJC.0b013e318033dfd4. [DOI] [PubMed] [Google Scholar]
- Nistér M., Libermann T.A., Betsholtz C., Pettersson M., Claesson-Welsh L., Heldin C.H., Schlessinger J., Westermark B. Expression of messenger RNAs for platelet-derived growth factor and transforming growth factor-α and their receptors in human malignant glioma cell lines. Cancer Res. 1988;48:3910–3918. [PubMed] [Google Scholar]
- Ohlsson R., Falck P., Hellström M., Lindahl P., Boström H., Franklin G., Ahrlund-Richter L., Pollard J., Soriano P., Betsholtz C. PDGFB regulates the development of the labyrinthine layer of the mouse fetal placenta. Dev. Biol. 1999;212:124–136. doi: 10.1006/dbio.1999.9306. [DOI] [PubMed] [Google Scholar]
- Oosthuyse B., Moons L., Storkebaum E., Beck H., Nuyens D., Brusselmans K., Van Dorpe J., Hellings P., Gorselink M., Heymans S., et al. Deletion of the hypoxia-response element in the vascular endothelial growth factor promoter causes motor neuron degeneration. Nat. Genet. 2001;28:131–138. doi: 10.1038/88842. [DOI] [PubMed] [Google Scholar]
- Orimo A., Gupta P.B., Sgroi D.C., Arenzana-Seisdedos F., Delaunay T., Naeem R., Carey V.J., Richardson A.L., Weinberg R.A. Stromal fibroblasts present in invasive human breast carcinomas promote tumor growth and angiogenesis through elevated SDF-1/CXCL12 secretion. Cell. 2005;121:335–348. doi: 10.1016/j.cell.2005.02.034. [DOI] [PubMed] [Google Scholar]
- Orr-Urtreger A., Lonai P. Platelet-derived growth factor-A and its receptor are expressed in separate, but adjacent cell layers of the mouse embryo. Development. 1992;115:1045–1058. doi: 10.1242/dev.115.4.1045. [DOI] [PubMed] [Google Scholar]
- Ostendorf T., van Roeyen C.R., Peterson J.D., Kunter U., Eitner F., Hamad A.J., Chan G., Jia X.C., Macaluso J., Gazit-Bornstein G., et al. A fully human monoclonal antibody (CR002) identifies PDGF-D as a novel mediator of mesangioproliferative glomerulonephritis. J. Am. Soc. Nephrol. 2003;14:2237–2247. doi: 10.1097/01.asn.0000083393.00959.02. [DOI] [PubMed] [Google Scholar]
- Ostendorf T., Rong S., Boor P., Wiedemann S., Kunter U., Haubold U., van Roeyen C.R., Eitner F., Kawachi H., Starling G., et al. Antagonism of PDGF-D by human antibody CR002 prevents renal scarring in experimental glomerulonephritis. J. Am. Soc. Nephrol. 2006;17:1054–1062. doi: 10.1681/ASN.2005070683. [DOI] [PubMed] [Google Scholar]
- Ostman A. PDGF receptors-mediators of autocrine tumor growth and regulators of tumor vasculature and stroma. Cytokine Growth Factor Rev. 2004;15:275–286. doi: 10.1016/j.cytogfr.2004.03.002. [DOI] [PubMed] [Google Scholar]
- Palmieri S.L., Payne J., Stiles C.D., Biggers J.D., Mercola M. Expression of mouse PDGF-A and PDGF α-receptor genes during pre- and post-implantation development: Evidence for a developmental shift from an autocrine to a paracrine mode of action. Mech. Dev. 1992;39:181–191. doi: 10.1016/0925-4773(92)90045-l. [DOI] [PubMed] [Google Scholar]
- Pardanani A., Ketterling R.P., Brockman S.R., Flynn H.C., Paternoster S.F., Shearer B.M., Reeder T.L., Li C.Y., Cross N.C., Cools J., et al. CHIC2 deletion, a surrogate for FIP1L1–PDGFRA fusion, occurs in systemic mastocytosis associated with eosinophilia and predicts response to imatinib mesylate therapy. Blood. 2003;102:3093–3096. doi: 10.1182/blood-2003-05-1627. [DOI] [PubMed] [Google Scholar]
- Paulsson Y., Hammacher A., Heldin C.H., Westermark B. Possible positive autocrine feedback in the prereplicative phase of human fibroblasts. Nature. 1987;328:715–717. doi: 10.1038/328715a0. [DOI] [PubMed] [Google Scholar]
- Perry B.H., Sampson A.R., Schwab B.H., Karim M.R., Smiell J.M. A meta-analytic approach to an integrated summary of efficacy: A case study of becaplermin gel. Control. Clin. Trials. 2002;23:389–408. doi: 10.1016/s0197-2456(02)00220-9. [DOI] [PubMed] [Google Scholar]
- Persson C., Sävenhed C., Bourdeau A., Tremblay M.L., Markova B., Böhmer F.D., Haj F.G., Neel B.G., Elson A., Heldin C.H., et al. Site-selective regulation of platelet-derived growth factor β receptor tyrosine phosphorylation by T-cell protein tyrosine phosphatase. Mol. Cell. Biol. 2004;24:2190–2201. doi: 10.1128/MCB.24.5.2190-2201.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pierce G.F., Tarpley J.E., Allman R.M., Goode P.S., Serdar C.M., Morris B., Mustoe T.A., Vande Berg J. Tissue repair processes in healing chronic pressure ulcers treated with recombinant platelet-derived growth factor BB. Am. J. Pathol. 1994;145:1399–1410. [PMC free article] [PubMed] [Google Scholar]
- Pierce G.F., Tarpley J.E., Tseng J., Bready J., Chang D., Kenney W.C., Rudolph R., Robson M.C., Vande Berg J., Reid P. Detection of platelet-derived growth factor (PDGF)-AA in actively healing human wounds treated with recombinant PDGF-BB and absence of PDGF in chronic nonhealing wounds. J. Clin. Invest. 1995;96:1336–1350. doi: 10.1172/JCI118169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pietras K., Ostman A., Sjöquist M., Buchdunger E., Reed R.K., Heldin C.H., Rubin K. Inhibition of platelet-derived growth factor receptors reduces interstitial hypertension and increases transcapillary transport in tumors. Cancer Res. 2001;61:2929–2934. [PubMed] [Google Scholar]
- Pietras K., Rubin K., Sjöblom T., Buchdunger E., Sjöquist M., Heldin C.H., Ostman A. Inhibition of PDGF receptor signaling in tumor stroma enhances antitumor effect of chemotherapy. Cancer Res. 2002;62:5476–5484. [PubMed] [Google Scholar]
- Pietras K., Sjöblom T., Rubin K., Heldin C.H., Ostman A. PDGF receptors as cancer drug targets. Cancer Cell. 2003a;3:439–443. doi: 10.1016/s1535-6108(03)00089-8. [DOI] [PubMed] [Google Scholar]
- Pietras K., Stumm M., Hubert M., Buchdunger E., Rubin K., Heldin C.H., McSheehy P., Wartmann M., Ostman A. STI571 enhances the therapeutic index of epothilone B by a tumor-selective increase of drug uptake. Clin. Cancer Res. 2003b;9:3779–3787. [PubMed] [Google Scholar]
- Pietras K., Pahler J., Bergers G., Hanahan D. Functions of paracrine PDGF signaling in the pro-angiogenic tumor stroma revealed by pharmacological targeting. PloS Med. 2007;5:e19. doi: 10.1371/journal.pmed.0050019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinzani M., Milani S., Herbst H., DeFranco R., Grappone C., Gentilini A., Caligiuri A., Pellegrini G., Ngo D.V., Romanelli R.G., et al. Expression of platelet-derived growth factor and its receptors in normal human liver and during active hepatic fibrogenesis. Am. J. Pathol. 1996;148:785–800. [PMC free article] [PubMed] [Google Scholar]
- Pontén A., Li X., Thorén P., Aase K., Sjöblom T., Ostman A., Eriksson U. Transgenic overexpression of platelet-derived growth factor-C in the mouse heart induces cardiac fibrosis, hypertrophy, and dilated cardiomyopathy. Am. J. Pathol. 2003;163:673–682. doi: 10.1016/S0002-9440(10)63694-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pontén A., Folestad E.B., Pietras K., Eriksson U. Platelet-derived growth factor D induces cardiac fibrosis and proliferation of vascular smooth muscle cells in heart-specific transgenic mice. Circ. Res. 2005;97:1036–1045. doi: 10.1161/01.RES.0000190590.31545.d4. [DOI] [PubMed] [Google Scholar]
- Popovici C., Isnardon D., Birnbaum D., Roubin R. Caenorhabditis elegans receptors related to mammalian vascular endothelial growth factor receptors are expressed in neural cells. Neurosci. Lett. 2002;329:116–120. doi: 10.1016/s0304-3940(02)00595-5. [DOI] [PubMed] [Google Scholar]
- Pringle N.P., Richardson W.D. A singularity of PDGF α-receptor expression in the dorsoventral axis of the neural tube may define the origin of the oligodendrocyte lineage. Development. 1993;117:525–533. doi: 10.1242/dev.117.2.525. [DOI] [PubMed] [Google Scholar]
- Qin Y., Rezler E.M., Gokhale V., Sun D., Hurley L.H. Characterization of the G-quadruplexes in the duplex nuclease hypersensitive element of the PDGF-A promoter and modulation of PDGF-A promoter activity by TMPyP4. Nucleic Acids Res. 2007;35:7698–7713. doi: 10.1093/nar/gkm538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raines E.W. PDGF and cardiovascular disease. Cytokine Growth Factor Rev. 2004;15:237–254. doi: 10.1016/j.cytogfr.2004.03.004. [DOI] [PubMed] [Google Scholar]
- Raines E.W., Lane T.F., Iruela-Arispe M.L., Ross R., Sage E.H. The extracellular glycoprotein SPARC interacts with platelet-derived growth factor (PDGF)-AB and -BB and inhibits the binding of PDGF to its receptors. Proc. Natl. Acad. Sci. 1992;89:1281–1285. doi: 10.1073/pnas.89.4.1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramachandran R.K., Govindarajan V., Seid C.A., Patil S., Tomlinson C.R. Role for platelet-derived growth factor-like and epidermal growth factor-like signaling pathways in gastrulation and spiculogenesis in the Lytechinus sea urchin embryo. Dev. Dyn. 1995;204:77–88. doi: 10.1002/aja.1002040110. [DOI] [PubMed] [Google Scholar]
- Ramachandran R.K., Wikramanayake A.H., Uzman J.A., Govindarajan V., Tomlinson C.R. Disruption of gastrulation and oral–aboral ectoderm differentiation in the Lytechinus pictus embryo by a dominant/negative PDGF receptor. Development. 1997;124:2355–2364. doi: 10.1242/dev.124.12.2355. [DOI] [PubMed] [Google Scholar]
- Ramaswamy S., Ross K.N., Lander E.S., Golub T.R. A molecular signature of metastasis in primary solid tumors. Nat. Genet. 2003;33:49–54. doi: 10.1038/ng1060. [DOI] [PubMed] [Google Scholar]
- Rappolee D.A., Brenner C.A., Schultz R., Mark D., Werb Z. Developmental expression of PDGF, TGF-α, and TGF-β genes in preimplantation mouse embryos. Science. 1988;241:1823–1825. doi: 10.1126/science.3175624. [DOI] [PubMed] [Google Scholar]
- Ray S., Gao C., Wyatt K., Fariss R.N., Bundek A., Zelenka P., Wistow G. Platelet-derived growth factor D, tissue-specific expression in the eye, and a key role in control of lens epithelial cell proliferation. J. Biol. Chem. 2005;280:8494–8502. doi: 10.1074/jbc.M413570200. [DOI] [PubMed] [Google Scholar]
- Reigstad L.J., Varhaug J.E., Lillehaug J.R. Structural and functional specificities of PDGF-C and PDGF-D, the novel members of the platelet-derived growth factors family. FEBS J. 2005;272:5723–5741. doi: 10.1111/j.1742-4658.2005.04989.x. [DOI] [PubMed] [Google Scholar]
- Reneker L.W., Overbeek P.A. Lens-specific expression of PDGF-A alters lens growth and development. Dev. Biol. 1996;180:554–565. doi: 10.1006/dbio.1996.0328. [DOI] [PubMed] [Google Scholar]
- Reuterdahl C., Sundberg C., Rubin K., Funa K., Gerdin B. Tissue localization of β receptors for platelet-derived growth factor and platelet-derived growth factor B chain during wound repair in humans. J. Clin. Invest. 1993;91:2065–2075. doi: 10.1172/JCI116429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rice A.B., Moomaw C.R., Morgan D.L., Bonner J.C. Specific inhibitors of platelet-derived growth factor or epidermal growth factor receptor tyrosine kinase reduce pulmonary fibrosis in rats. Am. J. Pathol. 1999;155:213–221. doi: 10.1016/S0002-9440(10)65115-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richarte A.M., Mead H.B., Tallquist M.D. Cooperation between the PDGF receptors in cardiac neural crest cell migration. Dev. Biol. 2007;306:785–796. doi: 10.1016/j.ydbio.2007.04.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robson M.C., Phillips L.G., Thomason A., Robson L.E., Pierce G.F. Platelet-derived growth factor BB for the treatment of chronic pressure ulcers. Lancet. 1992;339:23–25. doi: 10.1016/0140-6736(92)90143-q. [DOI] [PubMed] [Google Scholar]
- Rolny C., Nilsson I., Magnusson P., Armulik A., Jakobsson L., Wentzel P., Lindblom P., Norlin J., Betsholtz C., Heuchel R., et al. Platelet-derived growth factor receptor-β promotes early endothelial cell differentiation. Blood. 2006;108:1877–1886. doi: 10.1182/blood-2006-04-014894. [DOI] [PubMed] [Google Scholar]
- Rönnstrand L., Heldin C.H. Mechanisms of platelet-derived growth factor-induced chemotaxis. Int. J. Cancer. 2001;91:757–762. doi: 10.1002/1097-0215(200002)9999:9999<::aid-ijc1136>3.0.co;2-j. [DOI] [PubMed] [Google Scholar]
- Ross R. The pathogenesis of atherosclerosis: A perspective for the 1990s. Nature. 1993;362:801–809. doi: 10.1038/362801a0. [DOI] [PubMed] [Google Scholar]
- Ross R., Glomset J.A. The pathogenesis of atherosclerosis (first of two parts) N. Engl. J. Med. 1976;295:369–377. doi: 10.1056/NEJM197608122950707. [DOI] [PubMed] [Google Scholar]
- Ross R., Glomset J., Kariya B., Harker L. A platelet-dependent serum factor that stimulates the proliferation of arterial smooth muscle cells. Proc. Natl. Acad. Sci. 1974;71:1207–1210. doi: 10.1073/pnas.71.4.1207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruhrberg C., Gerhardt H., Golding M., Watson R., Ioannidou S., Fujisawa H., Betsholtz C., Shima D.T. Spatially restricted patterning cues provided by heparin-binding VEGF-A control blood vessel branching morphogenesis. Genes & Dev. 2002;16:2684–2698. doi: 10.1101/gad.242002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sano H., Sudo T., Yokode M., Murayama T., Kataoka H., Takakura N., Nishikawa S., Nishikawa S.I., Kita T. Functional blockade of platelet-derived growth factor receptor-β but not of receptor-α prevents vascular smooth muscle cell accumulation in fibrous cap lesions in apolipoprotein E-deficient mice. Circulation. 2001;103:2955–2960. doi: 10.1161/01.cir.103.24.2955. [DOI] [PubMed] [Google Scholar]
- Sarment D.P., Cooke J.W., Miller S.E., Jin Q., McGuire M.K., Kao R.T., McClain P.K., McAllister B.S., Lynch S.E., Giannobile W.V. Effect of rhPDGF-BB on bone turnover during periodontal repair. J. Clin. Periodontol. 2006;33:135–140. doi: 10.1111/j.1600-051X.2005.00870.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schermuly R.T., Dony E., Ghofrani H.A., Pullamsetti S., Savai R., Roth M., Sydykov A., Lai Y.J., Weissmann N., Seeger W., et al. Reversal of experimental pulmonary hypertension by PDGF inhibition. J. Clin. Invest. 2005;115:2811–2821. doi: 10.1172/JCI24838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlingemann R.O., Rietveld F.J., Kwaspen F., de van Kerkhof P.C., de Waal R.M., Ruiter D.J. Differential expression of markers for endothelial cells, pericytes, and basal lamina in the microvasculature of tumors and granulation tissue. Am. J. Pathol. 1991;138:1335–1347. [PMC free article] [PubMed] [Google Scholar]
- Schmahl J., Raymond C.S., Soriano P. PDGF signaling specificity is mediated through multiple immediate early genes. Nat. Genet. 2007;39:52–60. doi: 10.1038/ng1922. [DOI] [PubMed] [Google Scholar]
- Schneider L., Clement C.A., Teilmann S.C., Pazour G.J., Hoffmann E.K., Satir P., Christensen S.T. PDGFRαα signaling is regulated through the primary cilium in fibroblasts. Curr. Biol. 2005;15:1861–1866. doi: 10.1016/j.cub.2005.09.012. [DOI] [PubMed] [Google Scholar]
- Seger R., Krebs E.G. The MAPK signaling cascade. FASEB J. 1995;9:726–735. [PubMed] [Google Scholar]
- Servidei T., Riccardi A., Sanguinetti M., Dominici C., Riccardi R. Increased sensitivity to the platelet-derived growth factor (PDGF) receptor inhibitor STI571 in chemoresistant glioma cells is associated with enhanced PDGF-BB-mediated signaling and STI571-induced Akt inactivation. J. Cell. Physiol. 2006;208:220–228. doi: 10.1002/jcp.20659. [DOI] [PubMed] [Google Scholar]
- Seymour L., Dajee D., Bezwoda W.R. Tissue platelet derived-growth factor (PDGF) predicts for shortened survival and treatment failure in advanced breast cancer. Breast Cancer Res. Treat. 1993;26:247–252. doi: 10.1007/BF00665802. [DOI] [PubMed] [Google Scholar]
- Shao Z.M., Nguyen M., Barsky S.H. Human breast carcinoma desmoplasia is PDGF initiated. Oncogene. 2000;19:4337–4345. doi: 10.1038/sj.onc.1203785. [DOI] [PubMed] [Google Scholar]
- Shimoda H.K., Yamamoto M., Shide K., Kamezaki K., Matsuda T., Ogawa K., Harada M., Shimoda K. Chronic thrombopoietin overexpression induces mesangioproliferative glomerulopathy in mice. Am. J. Hematol. 2007;82:802–806. doi: 10.1002/ajh.20970. [DOI] [PubMed] [Google Scholar]
- Shindo T., Manabe I., Fukushima Y., Tobe K., Aizawa K., Miyamoto S., Kawai-Kowase K., Moriyama N., Imai Y., Kawakami H., et al. Krüppel-like zinc-finger transcription factor KLF5/BTEB2 is a target for angiotensin II signaling and an essential regulator of cardiovascular remodeling. Nat. Med. 2002;8:856–863. doi: 10.1038/nm738. [DOI] [PubMed] [Google Scholar]
- Sihvola R., Koskinen P., Myllärniemi M., Loubtchenkov M., Häyry P., Buchdunger E., Lemström K. Prevention of cardiac allograft arteriosclerosis by protein tyrosine kinase inhibitor selective for platelet-derived growth factor receptor. Circulation. 1999;99:2295–2301. doi: 10.1161/01.cir.99.17.2295. [DOI] [PubMed] [Google Scholar]
- Simm A., Nestler M., Hoppe V. Mitogenic effect of PDGF-AA on cardiac fibroblasts. Basic Res. Cardiol. 1998;93 (Suppl. 3):40–43. doi: 10.1007/s003950050209. [DOI] [PubMed] [Google Scholar]
- Simon M.P., Pedeutour F., Sirvent N., Grosgeorge J., Minoletti F., Coindre J.M., Terrier-Lacombe M.J., Mandahl N., Craver R.D., Blin N., et al. Deregulation of the platelet-derived growth factor B-chain gene via fusion with collagen gene COL1A1 in dermatofibrosarcoma protuberans and giant-cell fibroblastoma. Nat. Genet. 1997;15:95–98. doi: 10.1038/ng0197-95. [DOI] [PubMed] [Google Scholar]
- Sirvent N., Maire G., Pedeutour F. Genetics of dermatofibrosarcoma protuberans family of tumors: From ring chromosomes to tyrosine kinase inhibitor treatment. Genes Chromosomes Cancer. 2003;37:1–19. doi: 10.1002/gcc.10202. [DOI] [PubMed] [Google Scholar]
- Sjöblom T., Shimizu A., O’Brien K.P., Pietras K., Dal Cin P., Buchdunger E., Dumanski J.P., Ostman A., Heldin C.H. Growth inhibition of dermatofibrosarcoma protuberans tumors by the platelet-derived growth factor receptor antagonist STI571 through induction of apoptosis. Cancer Res. 2001;61:5778–5783. [PubMed] [Google Scholar]
- Skobe M., Fusenig N.E. Tumorigenic conversion of immortal human keratinocytes through stromal cell activation. Proc. Natl. Acad. Sci. 1998;95:1050–1055. doi: 10.1073/pnas.95.3.1050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smiell J.M., Wieman T.J., Steed D.L., Perry B.H., Sampson A.R., Schwab B.H. Efficacy and safety of becaplermin (recombinant human platelet-derived growth factor-BB) in patients with nonhealing, lower extremity diabetic ulcers: A combined analysis of four randomized studies. Wound Repair Regen. 1999;7:335–346. doi: 10.1046/j.1524-475x.1999.00335.x. [DOI] [PubMed] [Google Scholar]
- Smith J.S., Wang X.Y., Qian J., Hosek S.M., Scheithauer B.W., Jenkins R.B., James C.D. Amplification of the platelet-derived growth factor receptor-A (PDGFRA) gene occurs in oligodendrogliomas with grade IV anaplastic features. J. Neuropathol. Exp. Neurol. 2000;59:495–503. doi: 10.1093/jnen/59.6.495. [DOI] [PubMed] [Google Scholar]
- Smits A., Kato M., Westermark B., Nistér M., Heldin C.H., Funa K. Neurotrophic activity of platelet-derived growth factor (PDGF): Rat neuronal cells possess functional PDGF β-type receptors and respond to PDGF. Proc. Natl. Acad. Sci. 1991;88:8159–8163. doi: 10.1073/pnas.88.18.8159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soriano P. Abnormal kidney development and hematological disorders in PDGF β-receptor mutant mice. Genes & Dev. 1994;8:1888–1896. doi: 10.1101/gad.8.16.1888. [DOI] [PubMed] [Google Scholar]
- Soriano P. The PDGF α receptor is required for neural crest cell development and for normal patterning of the somites. Development. 1997;124:2691–2700. doi: 10.1242/dev.124.14.2691. [DOI] [PubMed] [Google Scholar]
- Sorkin A., Westermark B., Heldin C.H., Claesson-Welsh L. Effect of receptor kinase inactivation on the rate of internalization and degradation of PDGF and the PDGF β-receptor. J. Cell Biol. 1991;112:469–478. doi: 10.1083/jcb.112.3.469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soyombo A.A., DiCorleto P.E. Stable expression of human platelet-derived growth factor B chain by bovine aortic endothelial cells. Matrix association and selective proteolytic cleavage by thrombin. J. Biol. Chem. 1994;269:17734–17740. [PubMed] [Google Scholar]
- Steed D.L. Clinical evaluation of recombinant human platelet-derived growth factor for the treatment of lower extremity ulcers. Plast. Reconstr. Surg. 2006;117 (7 Suppl.):143S–149S. doi: 10.1097/01.prs.0000222526.21512.4c. [DOI] [PubMed] [Google Scholar]
- Storkebaum E., Carmeliet P. VEGF: A critical player in neurodegeneration. J. Clin. Invest. 2004;113:14–18. doi: 10.1172/JCI200420682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stover E.H., Chen J., Folens C., Lee B.H., Mentens N., Marynen P., Williams I.R., Gilliland D.G., Cools J. Activation of FIP1L1-PDGFRα requires disruption of the juxtamembrane domain of PDGFRα and is FIP1L1-independent. Proc. Natl. Acad. Sci. 2006;103:8078–8083. doi: 10.1073/pnas.0601192103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stroobant P., Waterfield M.D. Purification and properties of porcine platelet-derived growth factor. EMBO J. 1984;3:2963–2967. doi: 10.1002/j.1460-2075.1984.tb02241.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su Y.C., Han J., Xu S., Cobb M., Skolnik E.Y. NIK is a new Ste20-related kinase that binds NCK and MEKK1 and activates the SAPK/JNK cascade via a conserved regulatory domain. EMBO J. 1997;16:1279–1290. doi: 10.1093/emboj/16.6.1279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svegliati S., Olivieri A., Campelli N., Luchetti M., Poloni A., Trappolini S., Moroncini G., Bacigalupo A., Leoni P., Avvedimento E.V., et al. Stimulatory autoantibodies to PDGF receptor in patients with extensive chronic graft-versus-host disease. Blood. 2007;110:237–241. doi: 10.1182/blood-2007-01-071043. [DOI] [PubMed] [Google Scholar]
- Symes K., Mercola M. Embryonic mesoderm cells spread in response to platelet-derived growth factor and signaling by phosphatidylinositol 3-kinase. Proc. Natl. Acad. Sci. 1996;93:9641–9644. doi: 10.1073/pnas.93.18.9641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi Y., Morales F.C., Kreimann E.L., Georgescu M.M. PTEN tumor suppressor associates with NHERF proteins to attenuate PDGF receptor signaling. EMBO J. 2006;25:910–920. doi: 10.1038/sj.emboj.7600979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takayama Y., May P., Anderson R.G., Herz J. Low density lipoprotein receptor-related protein 1 (LRP1) controls endocytosis and c-CBL-mediated ubiquitination of the platelet-derived growth factor receptor β (PDGFR β) J. Biol. Chem. 2005;280:18504–18510. doi: 10.1074/jbc.M410265200. [DOI] [PubMed] [Google Scholar]
- Tallquist M., Kazlauskas A. PDGF signaling in cells and mice. Cytokine Growth Factor Rev. 2004;15:205–213. doi: 10.1016/j.cytogfr.2004.03.003. [DOI] [PubMed] [Google Scholar]
- Tallquist M.D., Soriano P. Cell autonomous requirement for PDGFRα in populations of cranial and cardiac neural crest cells. Development. 2003;130:507–518. doi: 10.1242/dev.00241. [DOI] [PubMed] [Google Scholar]
- Tallquist M.D., Klinghoffer R.A., Heuchel R., Mueting-Nelsen P.F., Corrin P.D., Heldin C.H., Johnson R.J., Soriano P. Retention of PDGFR-β function in mice in the absence of phosphatidylinositol 3′-kinase and phospholipase Cγ signaling pathways. Genes & Dev. 2000a;14:3179–3190. doi: 10.1101/gad.844700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tallquist M.D., Weismann K.E., Hellström M., Soriano P. Early myotome specification regulates PDGFA expression and axial skeleton development. Development. 2000b;127:5059–5070. doi: 10.1242/dev.127.23.5059. [DOI] [PubMed] [Google Scholar]
- Tallquist M.D., French W.J., Soriano P. Additive effects of PDGF receptor β signaling pathways in vascular smooth muscle cell development. PLoS Biol. 2003;1:E52. doi: 10.1371/journal.pbio.0000052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang W.W., Ulich T.R., Lacey D.L., Hill D.C., Qi M., Kaufman S.A., Van G.Y., Tarpley J.E., Yee J.S. Platelet-derived growth factor-BB induces renal tubulointerstitial myofibroblast formation and tubulointerstitial fibrosis. Am. J. Pathol. 1996;148:1169–1180. [PMC free article] [PubMed] [Google Scholar]
- Tang J., Kozaki K., Farr A.G., Martin P.J., Lindahl P., Betsholtz C., Raines E.W. The absence of platelet-derived growth factor-B in circulating cells promotes immune and inflammatory responses in atherosclerosis-prone ApoE−/− mice. Am. J. Pathol. 2005;167:901–912. doi: 10.1016/S0002-9440(10)62061-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarsitano M., De Falco S., Colonna V., McGhee J.D., Persico M.G. The C. elegans pvf-1 gene encodes a PDGF/VEGF-like factor able to bind mammalian VEGF receptors and to induce angiogenesis. FASEB J. 2006;20:227–233. doi: 10.1096/fj.05-4147com. [DOI] [PubMed] [Google Scholar]
- Tejada M.L., Yu L., Dong J., Jung K., Meng G., Peale F.V., Frantz G.D., Hall L., Liang X., Gerber H.P., et al. Tumor-driven paracrine platelet-derived growth factor receptor α signaling is a key determinant of stromal cell recruitment in a model of human lung carcinoma. Clin. Cancer Res. 2006;12:2676–2688. doi: 10.1158/1078-0432.CCR-05-1770. [DOI] [PubMed] [Google Scholar]
- Theisen C.S., Wahl J.K., Johnson K.R., Wheelock M.J. NHERF links the N-cadherin/catenin complex to the platelet-derived growth factor receptor to modulate the actin cytoskeleton and regulate cell motility. Mol. Biol. Cell. 2007;18:1220–1232. doi: 10.1091/mbc.E06-10-0960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomita Y., Akiyama M., Shimizu H. PDGF isoforms induce and maintain anagen phase of murine hair follicles. J. Dermatol. Sci. 2006;43:105–115. doi: 10.1016/j.jdermsci.2006.03.012. [DOI] [PubMed] [Google Scholar]
- Tuuminen R., Nykänen A., Keränen M.A., Krebs R., Alitalo K., Koskinen P.K., Lemström K.B. The effect of platelet-derived growth factor ligands in rat cardiac allograft vasculopathy and fibrosis. Transplant. Proc. 2006;38:3271–3273. doi: 10.1016/j.transproceed.2006.10.056. [DOI] [PubMed] [Google Scholar]
- Uhrbom L., Hesselager G., Nistér M., Westermark B. Induction of brain tumors in mice using a recombinant platelet-derived growth factor B-chain retrovirus. Cancer Res. 1998;58:5275–5279. [PubMed] [Google Scholar]
- Uhrbom L., Hesselager G., Ostman A., Nistér M., Westermark B. Dependence of autocrine growth factor stimulation in platelet-derived growth factor-B-induced mouse brain tumor cells. Int. J. Cancer. 2000;85:398–406. doi: 10.1002/(sici)1097-0215(20000201)85:3<398::aid-ijc17>3.0.co;2-l. [DOI] [PubMed] [Google Scholar]
- Utoh R., Shigenaga S., Watanabe Y., Yoshizato K. Platelet-derived growth factor signaling as a cue of the epithelial–mesenchymal interaction required for anuran skin metamorphosis. Dev. Dyn. 2003;227:157–169. doi: 10.1002/dvdy.10302. [DOI] [PubMed] [Google Scholar]
- Van Den Akker N.M., Lie-Venema H., Maas S., Eralp I., DeRuiter M.C., Poelmann R.E., Gittenberger-De Groot A.C. Platelet-derived growth factors in the developing avian heart and maturating coronary vasculature. Dev. Dyn. 2005;233:1579–1588. doi: 10.1002/dvdy.20476. [DOI] [PubMed] [Google Scholar]
- Van den Akker N.M.S., Winkel L.C.J., Nisancioglu M.H., Maas S., Wisse L.J., Armulik A., Poelmann R.E., Lie-Venema H., Betsholtz C., Gittenberger-de Groot A.C. PDGF-B signaling is important for murine cardiac development: Its role in developing atrioventricular valves, coronaries, and cardiac innervation. Dev. Dyn. 2008;237:494–503. doi: 10.1002/dvdy.21436. [DOI] [PubMed] [Google Scholar]
- van Heyningen P., Calver A.R., Richardson W.D. Control of progenitor cell number by mitogen supply and demand. Curr. Biol. 2001;11:232–241. doi: 10.1016/s0960-9822(01)00075-6. [DOI] [PubMed] [Google Scholar]
- Van Stry M., McLaughlin K.A., Ataliotis P., Symes K. The mitochondrial-apoptotic pathway is triggered in Xenopus mesoderm cells deprived of PDGF receptor signaling during gastrulation. Dev. Biol. 2004;268:232–242. doi: 10.1016/j.ydbio.2003.12.030. [DOI] [PubMed] [Google Scholar]
- Wang Y., Lam K.S., Xu J.Y., Lu G., Xu L.Y., Cooper G.J., Xu A. Adiponectin inhibits cell proliferation by interacting with several growth factors in an oligomerization-dependent manner. J. Biol. Chem. 2005;280:18341–18347. doi: 10.1074/jbc.M501149200. [DOI] [PubMed] [Google Scholar]
- Waterfield M.D., Scrace G.T., Whittle N., Stroobant P., Johnsson A., Wasteson A., Westermark B., Heldin C.H., Huang J.S., Deuel T.F. Platelet-derived growth factor is structurally related to the putative transforming protein p28sis of simian sarcoma virus. Nature. 1983;304:35–39. doi: 10.1038/304035a0. [DOI] [PubMed] [Google Scholar]
- Westermark B., Wasteson A. A platelet factor stimulating human normal glial cells. Exp. Cell Res. 1976;98:170–174. doi: 10.1016/0014-4827(76)90476-6. [DOI] [PubMed] [Google Scholar]
- Wilkinson-Berka J.L., Babic S., De Gooyer T., Stitt A.W., Jaworski K., Ong L.G., Kelly D.J., Gilbert R.E. Inhibition of platelet-derived growth factor promotes pericyte loss and angiogenesis in ischemic retinopathy. Am. J. Pathol. 2004;164:1263–1273. doi: 10.1016/s0002-9440(10)63214-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood W., Faria C., Jacinto A. Distinct mechanisms regulate hemocyte chemotaxis during development and wound healing in Drosophila melanogaster. J. Cell Biol. 2006;173:405–416. doi: 10.1083/jcb.200508161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodruff R.H., Fruttiger M., Richardson W.D., Franklin R.J. Platelet-derived growth factor regulates oligodendrocyte progenitor numbers in adult CNS and their response following CNS demyelination. Mol. Cell. Neurosci. 2004;25:252–262. doi: 10.1016/j.mcn.2003.10.014. [DOI] [PubMed] [Google Scholar]
- Wu L.S., Tan C.Y., Wang L.M., Lin C.G., Wang J.Y. Variant in promoter region of platelet-derived growth factor receptor-α (PDGFRα) gene is associated with the severity and allergic status of childhood asthma. Int. Arch. Allergy Immunol. 2006;141:37–46. doi: 10.1159/000094180. [DOI] [PubMed] [Google Scholar]
- Xian X., Håkansson J., Ståhlberg A., Lindblom P., Betsholtz C., Gerhardt H., Semb H. Pericytes limit tumor cell metastasis. J. Clin. Invest. 2006;116:642–651. doi: 10.1172/JCI25705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu X., Bringas P., Soriano P., Chai Y. PDGFR-α signaling is critical for tooth cusp and palate morphogenesis. Dev. Dyn. 2005;232:75–84. doi: 10.1002/dvdy.20197. [DOI] [PubMed] [Google Scholar]
- Yamakage A., Kikuchi K., Smith E.A., LeRoy E.C., Trojanowska M. Selective upregulation of platelet-derived growth factor α receptors by transforming growth factor β in scleroderma fibroblasts. J. Exp. Med. 1992;175:1227–1234. doi: 10.1084/jem.175.5.1227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamasaki Y., Miyoshi K., Oda N., Watanabe M., Miyake H., Chan J., Wang X., Sun L., Tang C., McMahon G., et al. Weekly dosing with the platelet-derived growth factor receptor tyrosine kinase inhibitor SU9518 significantly inhibits arterial stenosis. Circ. Res. 2001;88:630–636. doi: 10.1161/01.res.88.6.630. [DOI] [PubMed] [Google Scholar]
- Yokote K., Margolis B., Heldin C.H., Claesson-Welsh L. Grb7 is a downstream signaling component of platelet-derived growth factor α- and β-receptors. J. Biol. Chem. 1996;271:30942–30949. doi: 10.1074/jbc.271.48.30942. [DOI] [PubMed] [Google Scholar]
- Zhao J., Roth J., Bode-Lesniewska B., Pfaltz M., Heitz P.U., Komminoth P. Combined comparative genomic hybridization and genomic microarray for detection of gene amplifications in pulmonary artery intimal sarcomas and adrenocortical tumors. Genes Chromosomes Cancer. 2002;34:48–57. doi: 10.1002/gcc.10035. [DOI] [PubMed] [Google Scholar]
- Zhuo Y., Zhang J., Laboy M., Lasky J.A. Modulation of PDGF-C and PDGF-D expression during bleomycin-induced lung fibrosis. Am. J. Physiol. Lung Cell. Mol. Physiol. 2004;286:L182–L188. doi: 10.1152/ajplung.00083.2003. [DOI] [PubMed] [Google Scholar]
- Zhuo Y., Hoyle G.W., Shan B., Levy D.R., Lasky J.A. Over-expression of PDGF-C using a lung specific promoter results in abnormal lung development. Transgenic Res. 2006;15:543–555. doi: 10.1007/s11248-006-9007-5. [DOI] [PubMed] [Google Scholar]
- Zymek P., Bujak M., Chatila K., Cieslak A., Thakker G., Entman M.L., Frangogiannis N.G. The role of platelet-derived growth factor signaling in healing myocardial infarcts. J. Am. Coll. Cardiol. 2006;48:2315–2323. doi: 10.1016/j.jacc.2006.07.060. [DOI] [PubMed] [Google Scholar]