

Abstract
This review addresses the localized regulation of voltage-gated ion channels by phosphorylation. Comprehensive data on channel regulation by associated protein kinases, phosphatases, and related regulatory proteins are mainly available for voltage-gated Ca2+ channels, which form the main focus of this review. Other voltage-gated ion channels and especially Kv7.1-3 (KCNQ1-3), the large- and small-conductance Ca2+-activated K+ channels BK and SK2, and the inward-rectifying K+ channels Kir3 have also been studied to quite some extent and will be included. Regulation of the L-type Ca2+ channel Cav1.2 by PKA has been studied most thoroughly as it underlies the cardiac fight-or-flight response. A prototypical Cav1.2 signaling complex containing the β2 adrenergic receptor, the heterotrimeric G protein Gs, adenylyl cyclase, and PKA has been identified that supports highly localized via cAMP. The type 2 ryanodine receptor as well as AMPA- and NMDA-type glutamate receptors are in close proximity to Cav1.2 in cardiomyocytes and neurons, respectively, yet independently anchor PKA, CaMKII, and the serine/threonine phosphatases PP1, PP2A, and PP2B, as is discussed in detail. Descriptions of the structural and functional aspects of the interactions of PKA, PKC, CaMKII, Src, and various phosphatases with Cav1.2 will include comparisons with analogous interactions with other channels such as the ryanodine receptor or ionotropic glutamate receptors. Regulation of Na+ and K+ channel phosphorylation complexes will be discussed in separate papers. This review is thus intended for readers interested in ion channel regulation or in localization of kinases, phosphatases, and their upstream regulators.
I. Introduction
The past decade has revealed an unanticipated number of protein-protein interactions that fundamentally changed our view of the localization and functional interactions of proteins inside cells. Signaling pathways are no exception. Proximity of the relevant control elements including protein kinases and phosphatases is critical for fast, efficient, and specific signaling by many different pathways (310). These targeting mechanisms are especially prevalent at the plasma membrane, where incoming signals may be relayed and integrated with high specificity. Spatial restriction is not only limited to kinases. Second messengers and especially cAMP can also act in a highly localized manner (78, 155, 329, 455). This article reviews mechanisms that promote localized and thereby selective regulation of voltage-gated ion channels by kinases and phosphatases. The localization of the protein kinases cAMP-dependent protein kinase (PKA); protein kinase C (PKC); Ca2+/calmodulin-dependent protein kinase II (CaMKII); Src; the phosphatases PP1, PP2A, and PP2B; their adaptor proteins [e.g., A kinase anchor proteins (AKAPs)]; and their regulators [e.g., G protein-coupled receptors (GPCRs) and G proteins] near or at ion channels are discussed in depth. Relevant aspects of the structures and anchoring mechanisms of the different kinases and phosphatases are included. This review largely focuses on those mechanisms for which interactions between ion channels and their regulators have been identified and ideally verified on a molecular level. Even with this limitation, it is not possible to discuss every contribution to this field. We apologize to those colleagues whose work could not be mentioned.
The most studied example of ion channel regulation by phosphorylation is the stimulation of the L-type Ca2+ channel Cav1.2 in the heart by signaling via cAMP and PKA. β-Adrenergic stimulation increases heart rate and contractility as part of the fight-or-flight response. Although many questions remain, various aspects of the control mechanisms of this channel have emerged over the last few years. Cav1.2 assembles the β2 adrenergic receptor (AR), the heterotrimeric G protein Gs, adenylyl cyclase (AC), PKA, and the counteracting phosphatase PP2A into a prototypical signaling complex (11, 78, 81). Similar complexes are formed by inward-rectifying Kir3 channels (231, 325) and the AMPAR GluR1 subunit (M. Joiner, D. Hall, Z. Malik, M. Lise, Y. Chen, A. Burette, R. Weinberg, A. El-Husseini, and J. Hell, unpublished data). The pioneering work on these complexes indicates that signaling from the β2 AR via cAMP to Cav1.2 is locally restricted (78). The discussion of the Cav1.2 complex is combined with an overview of findings that support the notion of spatially restricted cAMP pools and of stimulus-independent preassembly of G proteins with their cognate GPCRs or their downstream effectors for selective and effective signaling.
The review of Ca2+ channel regulation is interwoven with examples of analogous localized control of other ion channels. The type 2 cardiac ryanodine receptors (RyR2) in the sarcoplasmic reticulum will be discussed because it anchors PKA, PP1, and PP2A independently of Cav1.2, although it is in close proximity and functionally linked to Cav1.2. AMPARs and NMDARs constitute another group of ion channels that are regulated by anchored kinases and phosphatases. They are colocalized with Cav1.2 at postsynaptic sites and will be discussed in that context. Molecular and functional aspects of interactions of Na+ and K+ channels and other Ca2+ channels with kinases and phosphatases will be compared with those of Cav1.2. These channels include Kv7.1-3 (KCNQ1-3), the large-conductance Ca2+-activated K+ channel BK, the small-conductance Ca2+-activated K+ channel SK2, and the inward-rectifying K+ channels of the Kir3 family.
II. Structure and Function of Ion Channels
A. Ca2+ Channels
Ca2+ is a potent second messenger that controls a variety of cellular functions (44, 66, 137). As a major source of Ca2+ influx, voltage-gated Ca2+ channels fulfill critical roles in Ca2+ signaling. L-type Ca2+ channels regulate muscle contraction, hormone secretion, neuronal excitability, and gene expression. P/Q-, N-, and to some degree R- and L-type Ca2+ channels trigger neurotransmitter release at nerve terminals and other locations. T-type channels support neuronal burst firing and relaxation in coronary smooth muscle (56, 98, 134, 143, 248, 264, 383, 447) (see Refs. 52, 313 for most recent reviews). Ca2+ channels consist of a central α1 subunit, which forms the ion-conducting pore and defines the channel type (see below). The α1 subunit has four homologous domains, I-IV, each consisting of six transmembrane segments and a P-loop between segments 5 and 6 (Fig. 1). The auxiliary subunits α2-δ, β, and γ directly interact with α1 They modulate surface expression and biophysical properties such as channel activation and inactivation (6, 52, 83). The α2-δ subunit is created from a single transcript by proteolytic cleavage of the original polypeptide into two fragments. Four distinct genes encode α2-δ-1 through α2-δ-4, which are further diversified by differential splicing (83). The δ subunit consists of a short cytosolic COOH terminus, a single transmembrane segment, and a short extracellular domain, which is linked via a disulfide bridge to the heavily glycosylated and much larger (∼200 kDa) α2 polypeptide. The intracellular COOH terminus of δ is 1–15 residues long (83) and is unlikely to be phosphorylated by protein kinases. Coexpression of α2-δ generally increases surface expression of Ca2+ channels and influences to some degree their biophysical properties (83).
FIG. 1.

Membrane topology of Ca2+ channels. The central pore-forming subunit α1 (dark blue) consists of the four homologous domains I–IV that are linked to each other by the intracellular loops I/II, II/III, and III/IV, each containing six transmembrane segments and a P-loop between segments 5 and 6. The auxiliary subunits α2-δ (light blue) and β (magenta; Refs. 62, 308, 402) directly interact with α1 [the precise interaction sites of α1 with α2-δ and γ (medium blue) have not been defined]. Magenta: β subunits generally bind with their GK domains to loop I/II connecting domains I and II (AID); black X: calpain cleavage region.
In contrast to α2-δ, β is localized exclusively at the cytosolic face of the channel. The existence of four different β genes (β1-β4) and extensive differential splicing, especially of β1 and β2 transcripts, give rise to multiple isoforms (120). Recent structural studies demonstrate that β subunits consist of two protein-protein interaction domains, an SH3 domain, and a GK domain (62, 308, 402). Five sequential β strands constitute the core of the β SH3 domain analogous to canonical SH3 domains. However, the loops between strands 1 and 2 and strands 4 and 5 are much longer than in classic SH3 domains, in which the first loop contains several residues that form contacts with proline-rich domains. This arrangement is similar to the SH3-HOOK-GK motif in PSD-95 and its homologs (272, 389). The HOOK domain in PSD-95 corresponds to the large loop between strands 4 and 5 of the SH3 domain of β and has been suggested to obstruct access of proline-rich sequences to the unconventional SH3 domain (272).
The main interaction site on α1 for β subunits is a sequence of 18 residues in the loop between domain I and II (loop I/II) called the α interaction domain or AID, which binds to a hydrophobic grove in the GK domain of β (62, 308, 402). Additional interaction sites for β subunits have been identified in the NH2- and COOH-terminal regions of different α1 subunits (87, 407). The GK domain is important for Ca2+ channel trafficking to the cell surface likely by masking an ER retention signal in loop I/II (26, 238). Recent evidence, however, indicates that the SH3 domain mediates other functional effects of β subunits on channel activity including channel gating. The SH3 domain can act independently of the GK domain by binding to loop I/II (residues 520-532 in α11.2) downstream of AID (residues 458–475 in α11.2) (69, 238, 259, 273). Splice variants that mainly consist of the SH3 domain and lack the GK domain have been described for all four β isoforms (120, 174, 191, 286). The respective SH3 splice variant of the β1 subunit (β1d) does not support surface trafficking of α11.2 but increases mean open probability of the limited number of channels that is present at the surface in the absence of a GK-containing β subunit (69). PKA, PKC, and CaMKII can regulate Ca2+ channel activity at least in part via mechanisms that involve β subunits (see sects. iiiB2d, ivB, and vA).
Eight genes encode γ1-γ8, which share four putative transmembrane segments (NH2 and COOH termini are intracellular) and a signature motif (GLWXXC) as well as a pair of conserved cysteine residues in the first extracellular loop (reviewed in Ref. 213). These features are also characteristic for the otherwise more distantly related claudin family members, which are critical for formation of tight junctions (213, 397). The γ2, γ3, γ4, and γ8 subunits are more closely related to themselves than to the other family members, including the original γ subunit, γ1. In contrast to the other γ isoforms, γ2-γ4 and γ8 (as well as claudins) possess a PDZ domain binding consensus sequence at their very COOH termini that mediates interaction with PSD-95 and its homologs. Although interactions between the γ2 subfamily members and Ca2+ channels have been observed (213), their most prominent role is to support surface expression of AMPARs (see sect. iiiB4b and Fig. 10).
FIG. 10.
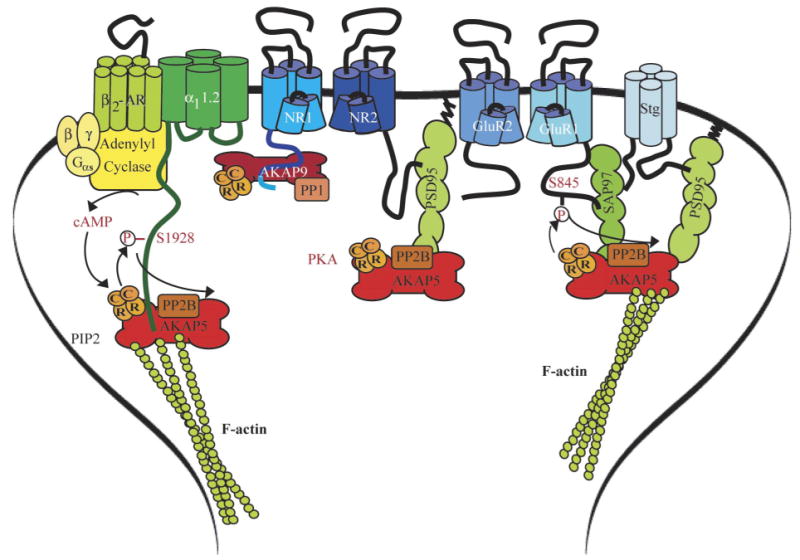
Postsynaptic A kinase anchor protein (AKAP)/PKA complexes. Left: the Cav1.2 signaling complex containing the β2-AR, adenylyl cyclase (AC), Gs, and the PKA holoenzyme (2C plus 2R), which is linked to the complex via AKAP150 (AKAP5). The PKA phosphorylation site serine-1928 is indicated. Middle: the NMDAR complex with Yotiao (AKAP9; binds to the C1 segment in the NR1 COOH terminus) and AKAP150. Yotiao functionally links not only PKA but also the counteractive phosphatase PP1 to the NMDAR. AKAP150 interacts with PSD-95 (or its homologs), which in turn bind to the very COOH-terminal ESDL-COO− motif of NR2A and 2B. A potential function of AKAP150 in NMDAR regulation is not known. Right: anchoring of PKA and PP2B by AKAP150 via SAP97 and via PSD-95/stargazin (stg) to the AMPAR GluR1 subunit. SAP97 and PSD-95 bind AKAP150 with their COOH-terminal portion containing SH3 and GK (the exact interaction sites have proven difficult to dissect). PKA and PP2B phosphorylation and dephosphorylation are indicated at serine-845 on GluR1.
As γ2-γ4 and γ8 are critical for postsynaptic targeting of AMPARs, they may also steer Ca2+ channels to this location, thereby fostering the colocalization and perhaps functional interaction of glutamate receptors and Ca2+ channels (215). Like glutamate receptors, L-type Ca2+ channels are clustered at dendritic spines, which constitute the postsynaptic sites of excitatory synapses (78, 163, 303). In fact, inhibition of L-type channels reduces maintenance, though not necessarily initial induction, of LTP (Lim and Hell, unpublished results). LTP refers to a stable increase in synaptic transmission that is at least in part mediated by a lasting elevation of glutamate receptor activity (31, 256–258). Back-propagating and locally generated dendritic action potentials contribute to LTP induction when occurring shortly after (10–50 ms) an excitatory postsynaptic potential (EPSP) at a given synapse. They do so by promoting Ca2+ influx through voltage-gated Ca2+ channels, including L-type channels, into dendritic spines (139, 262, 449). However, no γ subunits have been detected yet in the neuronal L-type channel complex (6, 52), although γ2 has been observed to coimmunoprecipitate with neuronal Cav2.1 and Cav2.2 and to affect channel activity in heterologous expression systems (213, 214).
Ca2+ channels are divided into high- and low-voltage-activated channels (HVA and LVA, respectively). LVA channels require less depolarization for activation and subsequent inactivation than HVA channels. L-type channels are HVA channels and are pharmacologically defined by their sensitivity to dihydropyridines and other so-called organic Ca2+ channel blockers. The four L-type channels Cav1.1-1.4 incorporate α11.1-1.4 (previously α1S, α1C, α1D, and α1F; for nomenclature, see Refs. 54, 55, 110). The other HVA family contains the P/Q-, N-, and R-type channels (Cav2.1-2.3 consisting of α12.1–2.3 also known as α1A, α1B, and α1E). P- and Q-type channels are created from α12.1 transcripts by differential splicing (35). They are selectively, but with different potency, inhibited by the funnel web spider toxin ω-AgaIVA and the cone snail toxin ω-CTx-MVIIC (35). N-type currents are quasi-irreversibly blocked by the cone snail toxin ω-CTx-GVIA and some, though not all, R-type currents are inhibited by the tarantula toxin SNX482. Cav2.3, which is selectively affected by SNX482, underlies a portion of the R-type current (295, 317, 430). LVA currents are mediated by the three T-type channels Cav3.1-3.3, which are formed by the related α13.1–3.3 subunits, (α1G, α1H, and α1I).
B. Na+ Channels
The structure of the pore-forming α subunit of voltage-gated Na+ channels mirrors that of Ca2+ channels with four homologous domains each consisting of six transmembrane segments and a reentry P loop (Fig. 2) (51). The auxiliary β1 and β2 subunits consist of an extracellular immunoglobulin-like domain, a single transmembrane segment, and a short intracellular domain. The β1 subunit binds to the extracellular segment of the α subunit that precedes transmembrane segment IVS6 (Fig. 2). Their coexpression with the α subunit accelerates activation and inactivation of the resulting Na+ currents (51). As suggested by their structural relationship to the large family of cell adhesion molecules, β subunits also regulate the subcellular distribution of Na+ channels (e.g., Ref. 271). Nine different α subunit genes encode Nav1.1–1.9 (53). Na+ channels contain one α subunit and either no β subunit, β1, β2, or both β subunits.
FIG. 2.

Membrane topology of Na+ channels. The central pore-forming subunit α (yellow) consists of the four homologous domains I–IV that are linked to each other by the intracellular loops I/II, II/III, and III/IV, each containing six transmembrane segments and a P-loop between segments 5 and 6. Some complexes contain the auxiliary subunits β1 and β2, which span the plasma membrane once (green). The extracellular interaction of β1 with the exctracellular segment preceding IVS6 is indicated (bracket).
C. K+ Channels
1. Kv7/KCNQ K+ channels
Kv7 channels are part of the large voltage-gated K+ channel family. Each of the Kv channels consists of four subunits that are homologous to each other and also to the individual four domains of Na+ and Ca2+ channels (Fig. 3A) (223, 453). In addition, Kv7.1/KCNQ1 assembles with the single transmembrane segment protein KCNE1/ MinK to form the slow K+ current IKs in the heart (12, 150, 346). This current is critical in repolarization of the cardiac action potential. Loss of function mutations prolong the Q-T interval, which leads to arrhythmias. Upregulation of IKs in the heart is important during sympathetic stimulation of the heart rate to ensure faster repolarization.
FIG. 3.

Membrane topology of K+ channels. The pore is formed by four homologous α subunits (purple), which interact with each other via their NH2 termini. α Subunits of Kv (A), BK (B), and SK (C) are formed by six transmembrane segments and a P-loop between segments 5 and 6, whereas Kir (D) lacks S1–S4. BK channel subunits contain an additional transmembrane segment (S0) NH2 terminal to the conserved S1 segment. Kv7/KCNQ channels (A) are typically associated with the auxiliary single transmembrane MinK/KCNE subunit (magenta), BK channels bind Ca2+ with their COOH termini (B), and SK channel α subunits dimerize through binding two CaM molecules (C).
Kv7.2 and Kv7.3 (KCNQ2/3) are mainly found in the nervous system, where they combine to form heteromeric channels that mediate the M-current (150, 408). This current received its name because it inactivates upon stimulation of muscarinic receptors. Muscarinic activation of phospholipase C (PLC) leads to depletion of phosphatidylinositol 4,5-bisphosphate (PIP2), which otherwise binds directly to Kv7.2/3 to activate the channel (184, 377).
2. Large-conductance Ca2+-activated K+ channel BK
Large-conductance Ca2+-activated K+ channels (BK, or “big-K”; also called KCa1.1, maxi-K, or slo) are activated by depolarization; intracellular Ca2+ reduces the degree of depolarization required for channel opening (37). The pore is formed by 4 α subunits encoded by a single gene (slo) first cloned from the Drosophila Slowpoke locus (9). The α subunit sequence is basically homologous to other K+ channel α subunits with six transmembrane segments but contains an additional transmembrane segment towards the NH2 terminus, which places the NH2 terminus on the extracellular side (Fig. 3B). Four homologous genes encode the auxiliary β1–4 subunits, which modulate Ca2+ sensitivity, voltage dependency, and gating kinetics to various degrees (37). BK critically contributes to the rapid phase of the afterhyperpolarization that follows action potentials.
BK channel activation can be induced by Ca2+ influx through L-type channels (24, 319, 379), P/Q-type channels (24, 319), N-type channels (264, 379), and NMDAR (199) but not R-type Cav2.3 Ca2+ channels (24). Biochemical analyses indicate that BK can form physical complexes with L-type Cav1.2 and Cav1.3, P/Q-type Cav2.1, and N-type Cav2.2 channels (24, 148, 249). The interaction between Cav1.2 and BK is stabilized in HEK293 cells by the β2 AR, which binds to both ion channels (249) as further discussed below [but see the interaction of BK with Cav1.2 in CHO cells transfected only with the α11.2 and BK α subunit, which appeared to be β2 AR-independent (24)]. These interactions place BK channels within a few nanometers of the Ca2+ channel pores (“nanodomains”) as required for their highly localized stimulation by Ca2+ influx due to their relatively low affinity for Ca2+ (10 μm and higher is required for effective BK activation under physiological conditions; Refs. 37, 293). Because of this proximity, it would be conceivable that the Cav1.2-associated signaling molecules including β2 AR, Gs, AC, and PKA can regulate BK not only indirectly by affecting Cav1.2 activity but also by directly phosphorylating BK. Interestingly, BK independently assembles itself a similar signaling complex as discussed in the next section.
3. Small-conductance Ca2+-activated K+ channel SK
Small-conductance Ca2+-activated K+ channels (SK) are activated by intracellular Ca2+ but not voltage (34). The pore-forming subunits are encoded by four different genes. SK1-3 are found in various brain regions, but the related IK1 (SK4) isoform is mainly expressed in peripheral tissue. Similar to other K+ channels, SK channels consist of four homologous domains, each containing six transmembrane segments and a P loop between segments 5 and 6 (Fig. 3C). The four SK subunits form pairs, each of which firmly binds two CaM molecules (355). During periods of depolarization, voltage-gated Ca2+ channels and NMDARs mediate influx of Ca2+, which binds to CaM in the SK complex, thereby causing conformational changes that open the SK channel pore. SK channels thereby mediate slow afterhyperpolarization and spike-frequency adaptation during trains of action potentials in neurons (34).
SK channels form functional units with various Ca2+ channels including L-type channels (264) and NMDARs (114, 296). SK channels can also be activated by Ca2+ influx through P-, N-, and T-type Ca2+ channels (106, 431). Recent biochemical and physiological experiments provide circumstantial evidence for α-actinin physically linking SK channels in general and specifically SK2 to Cav1.2 and Cav1.3 (250; see also Ref. 343). We also observed earlier that SK2 channels coimmunoprecipitate with α-actinin using two different SK2 antibodies (Fig. 4). However, more rigorous studies are required to firmly establish these α-actinin interactions. If SK channels are linked to L-type Ca2+ channels, signaling molecules associated with Cav1.2 including the β2 AR, Gs, AC, and PKA might be shared. Although there is no evidence that this signaling pathway regulates directly SK channels, regulation of SK-coupled Cav1.2 obviously will translate in altered SK channel activity.
FIG. 4.
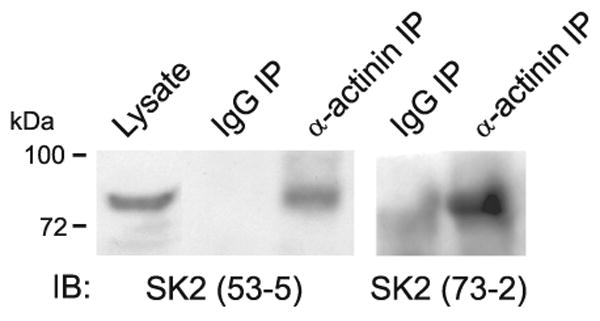
α-Actinin coimmunoprecipitates with SK2 from rat brain. Rat forebrains were homogenized in 1% Triton X-100, and nonsolubilized proteins were removed by ultracentrifugation before immunoprecipitation with anti-α-actinin or control IgG and immunoblotting with two different antibodies against SK2 (53–5 and 73–2) (see Refs. 153, 154 for more technical details). Both SK2 antibodies detected a single band of the expected molecular mass in anti-α-actinin but not control precipitates and in total lysate.
4. Inward rectifying Kir3 K+ channels
The pore-forming α subunits of G protein-gated inward rectifying K+ channels of the Kir3 family are encoded by four different genes, Kir3.1-3.4 (formerly GIRK1–4) (223, 453). As for other K+ channels, the gene products can assemble into tetramers in various combinations. However, Kir3 and in general Kirα subunits only contain two transmembrane segments, which flank the P-loop, similar to the S5/S6 region of the other K+ channel α subunits (Fig. 3D). These channels thus lack the voltage-sensing S4 transmembrane segment. Kir3 channels are largely activated by direct interactions of Gβγ with the channel (185, 328, 427). Several studies indicate that G proteins are preassociated with Kir3 channels and undergo conformational changes upon activation of G proteins (325, 331, 332). Although Gαi does not mediate regulation of Kir3 by G proteins, it directly binds to their NH2 and COOH termini. These findings further support the model that trimeric G proteins are preassociated with Kir3. Especially prominent is the function of the KACh channel in the heart, which is formed by Kir3.1 and 3.4. Parasympathetic release of acetylcholine leads to activation of the muscarinic m2 receptor Gi, and ultimately, via Gβγ released from Gi, KACh. Activation of this channel reduces cardiac excitability and thereby the heart rate.
III. Regulation of Ion Channels by Protein Kinase A
A. Regulation and Targeting of PKA
PKA is a tetramer consisting of two regulatory (R) and two catalytic (C) subunits. Distinct genes encode four R (RIα,β, RIIα,β) and three C subunits (Cα, β, and γ). C subunit catalytic activity is suppressed when associated with a homodimeric R core until release by cAMP binding to R (36). RII and to some degree RI dimers are recruited to certain substrates by AKAPs (46, 340, 433). AKAPs are a structurally diverse family of proteins, which share an amphipathic α helix that binds the R dimer. The Ht31 peptide derived from the RII binding site on the AKAP Ht31 disrupts all tested AKAP-RII interactions and is a powerful tool in delineating the functional importance of PKA anchoring by these AKAPs. Because some AKAPs are lipid-modified or can directly bind to certain phospholipids, earlier thinking assumed that AKAPs are recruited to defined subcellular compartments by their interactions with lipid membranes (52, 90, 126). However, it is a now well-established notion that AKAPs are more precisely targeted by binding to specific proteins (for early work, see Refs. 79, 426).
B. Regulation of Ca2+ Channels by PKA
1. Cav1.1 regulation by PKA
Cav1.1 is specifically expressed in skeletal muscle. Upon depolarization, Cav1.1 induces Ca2+ release from the sarcoplasmic reticulum (Ca2+-induced Ca2+ release) by the type 1 RyR (RyR1) and thereby contraction (excitation-contraction coupling). Cav1.1 likely activates the RyR1 (green structure in background in Fig. 5) via direct physical interaction by a Ca2+-independent mechanism (276, 322, 330). However, the increase in contraction force by epinephrine via ARs and cAMP signaling depends on extracellular Ca2+ and is, therefore, at least in part due to elevated Ca2+ entry through Cav1.1. Supporting this notion, PKA phosphorylates α11.1 and its β subunit and upregulates the activity of Cav1.1 (52, 74, 119). Phosphorylation of full-length α11.1 (1873 residues) by PKA occurs mostly at serine-1757 and serine 1854 in vitro and in myotubes (281, 339). The Ca2+-activated protease calpain removes these sites by cleaving α11.1 between residues 1685 and 1699 (84, 86, 193). The main phosphorylation site of the truncated α11.1 is serine-687 in the loop between domains II and III (337) though it is unclear whether this site is actually effectively regulated in intact cells (339). The prevailing β subunit in skeletal muscle is β1, which is phosphorylated by PKA in vitro on serine-182 and threonine-205 (84, 342). The phosphorylation sites that regulate channel activity of Cav1.1 have not been defined on a functional level.
FIG. 5.

The Cav1.1-AKAP15-PKA complex. Shown are the subunits α11.1 (dark green) and β1 (magenta). Dark red, AKAP15 and AKAP15 leucine zipper binding site on α11.1 (LZ); light red, PKA and identified phosphorylation sites for PKA on α1 and β1 (arrows). The main PKA sites in full-length α11.1 (serines 1757 and 1854) are removed by calpain (black X, calpain cleavage region). Magenta, β2 and its interaction with α11.1. The main in vitro PKA site of the truncated α11.1 is serine 687. Green, RyR1 (for simplicity only 2 of the 4 subunits that form one pore complex in the sarcoplasmic reticulum are shown). The large cytosolic foot structure of RyR1 directly interacts with α11.1.
PKA-mediated potentiation of Cav1.1 activity upon depolarization occurs quickly (in the range of tens of milliseconds) (e.g., Ref. 208). It likely reflects very fast phosphorylation events, which would best be accomplished if PKA would be anchored near Cav1.1. In fact, the Ht31 peptide, which universally disrupts AKAP-PKA RII interactions (see sect. iiiA), inhibits this rapid potentiation of Cav1.1 channel activity by PKA in skeletal myotubes (208). AKAP15 [also named AKAP18 (126) or AKAP7 (433)] was identified and cloned as Cav1.1-associated protein (144, 145). AKAP15 interacts with Cav1.1 by binding to a leucine zipper (LZ) -like motif close to the very COOH terminus of α11.1 (Table 1). A peptide derived from the LZ-like motif on AKAP15 inhibits depolarization-induced potentiation of Cav1.1 in skeletal myotubes (192). Collectively these observations indicate that AKAP15 mediates PKA binding to Cav1.1 and that this interaction is important for fast and effective regulation of the channel activity.
Table 1. Identified PKA phosphorylation sites and AKAP binding sites.
| Ion Channel | Phosphorylation Site(s) | AKAP Binding Region | AKAP | Reference Nos. |
|---|---|---|---|---|
| Cav1.1 | S1757/S1854 | L1786–L1814 | AKAP15 | 192, 339 |
| Cav1.2 | S1928 | I2073–L2094 | AKAP15 | 85, 194 |
| RyR type 1 | S2843 | I3039–L3075 | mAKAP | 266, 378 |
| RyR type 2 | S2808 | V3003–L3039 | mAKAP | 266, 336 |
| IP3R | S1589/S1755 | N1251–I1287 | Yotiao | 117, 399 |
PKA, cAMP-dependent protein kinase; AKAP, A kinase anchor protein; RyR, ryanodine receptor; IP3R, inositol 1,4,5-trisphosphate receptor.
In vitro binding of cAMP to the R-subunit dimer releases and thereby activates the C subunits, which are inhibited when tightly complexed with the R subunits under basal conditions. Unless there are additional anchoring mechanisms for C subunits, this mechanism should lead to a loss of C subunits from the AKAP-RII complexes and phosphorylation of the ultimate target proteins. In fact, there is some evidence that continued stimulation of PKA-dependent phosphorylation ultimately results in a reduction of the potentiation of Cav1.1 by PKA consistent with the possibility that the PKA C subunit becomes ultimately displaced from the channel-AKAP15-R subunit complex (208). However, C subunits can phosphorylate substrates without complete dissociation from RII subunits upon cAMP addition (404, 446). Such an incomplete dissociation mechanism might contribute to anchoring C near its substrates more permanently. RII seems to be anchored by AKAPs to a larger degree than RI isoforms, which releases its C subunit during substrate phosphorylation in vitro (404). Additional work is required to better understand C-subunit behavior upon stimulation by cAMP.
2. Cav1.2 regulation by PKA
A) Physiological Role of Cav1.2 and its Regulation by PKA
The regulation of Cav1.2 has been extensively studied because of its central role in cardiac function. Ca2+ influx through Cav1.2 sparks Ca2+ release from the sarcoplasmic reticulum by the type 2 RyR (RyR2) and subsequent contraction in the heart. The increase in heart rate and contractility during the fight-or-flight response is mediated to a substantial degree by β adrenergic stimulation of L-type Ca2+ channels (20, 309, 327) and the RyR2 (263, 267). Upon catecholamine binding, β ARs activate the stimulatory heterotrimeric G protein Gs by inducing the exchange of GDP for GTP on Gαs. Gαs dissociates from its Gβγ partners to stimulate AC and cAMP production, which in turn activates PKA. Dysregulation of Cav1.2, RyR2, and thereby Ca2+-induced Ca2+ release contributes to the contractile dysfunction in heart failure (140, 160, 353, 395).
Biochemical and functional studies indicate that Cav1.2 accounts for at least 75–80% of L-type channels in the brain (165, 167, 221, 364, 376). In neurons, Cav1.2 is concentrated at postsynaptic sites of asymmetric dendritic, presumably glutamatergic synapses and at somatic synapses, which are likely GABAergic (78, 163, 303). The postsynaptic localization of L-type channels is also supported by functional Ca2+ imaging studies of dendritic spines (32, 178, 449).
B) Function and Structure of the Cytosolic Cooh Terminus of α 11.2
Like α11.1, α11.2 exists in a long and a short form of ∼250 and 220 kDa (164, 167). The short form is created in intact neurons by proteolytic cleavage of the COOH terminal region by the Ca2+-activated protease calpain upon Ca2+ influx through NMDARs (163) (Fig. 6, black X in COOH terminus). Similar processing of α11.2 occurs in heart (85, 128, 129). In vitro phosphorylation of α11.2 isolated from rat brain is solely detectable in the long but not short form (164, 167). This observation indicates that the main phosphorylation site of α11.2 is in its COOH-terminal region, analogous to α11.1. In fact, at this point, the only observable phosphorylation site of α11.2 is serine-1928 (85, 281, 312), which is downstream of the calpain cleavage site (163). This COOH-terminal fragment can remain associated and functionally interacts with the rest of the channel after cleavage (128, 135, 196). Currents through α11.2 expressed in HEK293 cells are severalfold higher when the COOH-terminal region is truncated by insertion of stop codons around the predicted calpain cleavage site (128, 135, 196, 278, 417). This effect is at least in part due to an increase in coupling efficiency between the voltage sensor and the opening of the channel pore.
FIG. 6.

The Cav1.2-AKAP150-PKA complex. Blue, α11.2; magenta, β2 and its interactions with α11.2; yellow, CaM binding sites on α11.2 [there is one binding site in the NH2 terminus and three binding sites in tandem in the COOH terminus; the latter region also interacts with β subunits (magenta bracket and segment)]; red, AKAP79/150 binding sites (LZ, brackets, and segments), PKA, and PKA phosphorylation sites on α1 and β2 (arrows); gray, PP2A binding site; black X, calpain cleavage region; green, β2 AR; yellow-green, heterotrimeric G protein complex; yellow-orange, adenylyl cyclase.
Coexpression of COOH terminally truncated α11.2 with the COOH-terminal 350 residues as an independent polypeptide reverses the disinhibition caused by the truncation of α11.2; it actually overcompensates, leading to sixfold stronger inhibition compared with full-length α11.2 (196). Three negatively charged residues near the very COOH terminus (E2103, E2106, D2110) are critical for the COOH-terminal effect (196). These three residues interact with two positively charged residues in the membrane-proximal portion (R1696 and R1697), which are also required for the inhibitory effect of the distal COOH terminus (196). Similarly, injection of a polypeptide covering the very COOH-terminal 144 residues of α11.2, which contain E2103, E2106, D2110, also reversed the disinhibition otherwise observed when α11.2 was truncated 147 residues upstream of its COOH terminus (128). Additional evidence for an interaction between the NH2- and COOH-terminal portion of the full-length COOH terminus of L-type channels comes from the finding that the unique extension of the COOH terminus of α11.4 by 60 residues abrogates the Ca2+- and CaM-dependent inactivation of Cav1.4 and also Cav1.3 if added to the α11.3 COOH terminus (363, 406). This is an important mechanism for ensuring long-lasting dark currents by Cav1.4, which is only known to be present in photoreceptors and mediates the tonic neurotransmitter release of photoreceptors. Why the mechanism for Ca2+- and CaM-dependent inactivation of α11.4 has not been directly abrogated remains a puzzle. Perhaps the machinery mediating Ca2+- and CaM-dependent inactivation has additional yet to be discovered functions.
An attractive though currently speculative model is that the interaction of the very COOH terminus with the rest of the channel reduces ion conduction activity and that phosphorylation of serine-1928 (see sect. iiiB2c), which is 249 residues upstream of the COOH terminus of full-length α11.2, releases this inhibitory interaction. In support of this model, both COOH-terminal truncation of α11.2 and phosphorylation by PKA, lead to a left shift in the current-voltage curve. Functionally, such a left shift means that the channel opens more effectively at lower depolarization levels when the driving force for Ca2+ influx is higher, resulting in currents through the individual channels (128, 135, 195, 196, 278, 417). Like COOH-terminal truncation, PKA also increases the coupling efficiency between gating and pore opening as indicated by single-channel recording. These recordings show that Cav1.2 exists in three main modes. Cav1.2 is not available for activation in mode 0, exhibits short frequent openings in mode 1, and long-lasting openings with brief closings in between in mode 2 (173). β AR stimulation leads to transition from mode 0 to mode 1 or mode 2 in frog ventricular cells (20). Thus modifications by PKA and COOH-terminal truncation have comparable effects. It should be noted that earlier reports indicate that the majority of α11.2 in heart may exist in the cleaved state (85, 129). However, when high concentrations of calpain inhibitor I and II and of EGTA are present and all solutions and instruments are precooled to 0°C during rapid extraction procedures, immunoblotting typically shows that ∼50% and sometimes more of the detectable α11.2 is in its long form in extracts from brain (79, 81, 163, 166) and heart (Fig. 7).
FIG. 7.
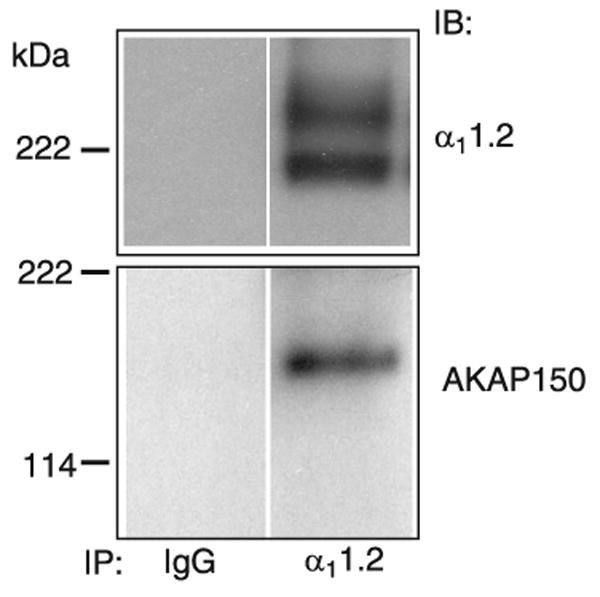
Cardiac α11.2 and its association with AKAP150. Cav1.2 was solubilized from rat heart extracts with 1% Triton X-100 before ultracentrifugation to remove nonsolublized material, immunoprecipitation (IP) with anti-α11.2 or control antibody (IgG), and immunoblotting (IB) with antibodies against α11.2 and AKAP150. Top: α11.2 long and short forms are present in a ratio of ∼1:1. Note the rather diffuse appearance of the two α11.2 bands, which suggests heterogeneity likely due to minor variations by differential splicing and other factors. Bottom: AKAP150 is prominent in total rat heart extract (data not shown) and coprecipitates with Cav1.2.
C) Role of α11.2 Serine-1928 in Cav1.2 Regulation by PKA
How PKA regulates Cav1.2 is not unequivocally established as it proved difficult if not impossible for a number of experienced investigators to reliably and reproducibly reconstitute regulation of Cav1.2 in heterologous cell lines without injection of exogenous PKA (52, 171, 278, 464). These difficulties proved also true even if AKAP79/150, which recruits PKA to Cav1.2 (see below) was coexpressed (76) [AKAP79 is the human homolog of AKAP150 but is missing 36 imperfect octapeptide repeats of unknown function that are present in the rodent AKAP150 (340)]. Perhaps additional but largely untested factors such as ahnak, a 700-kDa protein that can associate with Cav1.2, are important as well (151). In our hands, in 7 of 14 recordings from HEK293 cells transfected to transiently express Cav1.2 application of the cell-permeable phosphodiesterase resistant cAMP analog Sp-5,6-dichloro-1-β-d-ribofurano-sylbenzimidazole-3′,5′-cyclic monophosphorothioate (DClcBIMPS) lead to a >20% increase in Ca2+ currents (Fig. 8). Others made similar observations (M. J. Davis, Univ. of Missouri, Columbia, MO; personal communication). In some cases sequential recording of two cells within the same culture found one cell to be responsive to DClcBIMPS while another one was not. We do not know which factors determine whether DClcBMPS upregulates Cav1.2 currents. We can only speculate that the cell cycle status or other signaling mechanisms set the level of Cav1.2 phosphorylation before DClcBIMPS application in our system so that some but not other cells can respond possibly due to near-maximal basal phosphorylation of the relevant PKA site(s).
FIG. 8.
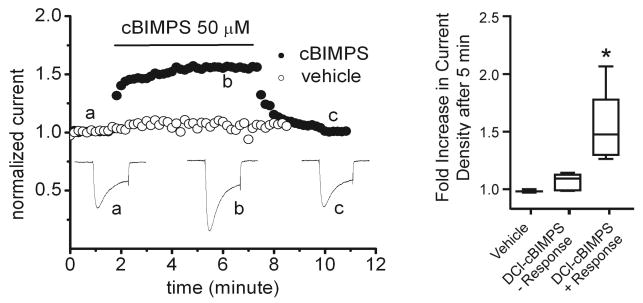
Exemplary Cav1.2 currents and their regulation by cAMP/PKA in HEK293 cells. HEK293 cells were grown on poly-d-lysine-coated coverslips in Dulbecco's modified Eagle's medium plus 10% fetal bovine serum at 37°C under 5% CO2 and transfected with full-length cardiac α11.2, β2a with FuGENE 6. Whole cell patch Ca2+ currents were elicited by depolarization from a holding potential of −70 to 0 mV for 200 ms at 24°C (leak-subtracted using p/4; extracellular: 125 mM NaCl, 10 mM tetraethylammonium chloride, 5 mM CaCl2, 5.4 mM CsCl, 1 mM 4-aminopyridine, 1 mM MgCl2, 10 mM HEPES-NaOH, 10 mM glucose, pH 7.4; intracellular: 120 mM CsCl, 10 mM tetraethylammonium chloride, 10 mM EGTA, 1 mM MgCl2, 3 mM MgATP, 0.5 mM Na3GTP, 10 mM HEPES-CsOH, pH 7.3). DClcBIMPS (closed circles) but not vehicle (open) induced in 7 of 14 experiments (see box blots) an increase in current of >20% (compare a and b), which readily reversed upon wash-out (c).
Nevertheless, it has been reported that currents through α11.2 coexpressed with β1b and α2δ in the HEK293-derived tsA-201 cells were increased by the membrane-permeable cAMP analog 8-Br-cAMP for wild type but not serine-1928 to alanine mutant α11.2 (149) (serine-1928 corresponds to serine-1901 in rat neuronal α11.2 investigated in Ref. 149). Others using BHK6 cells stably transfected with β1a and α2δ for transient expression of wild-type or serine-1928 to alanine mutant α11.2 (288) did not observe an increase in currents through wild-type Cav1.2 upon stimulation of AC with forskolin but observed a left-shift in the current-voltage curve, which allows Cav1.2 to open in response to smaller depolarization. This shift is another hallmark of the effect of PKA and was absent in the serine-1928 to alanine mutant in support of a role of serine-1928 in regulation of Cav1.2 by PKA.
Others expressed α11.2 and β2a together with AKAP150 (AKAP5, Ref. 433) in tsA-201 cells and described a potentiation of resulting currents by PKA; this potentiation was absent when serine-1928 in α11.2 was mutated to alanine, AKAP150 omitted, or binding of PKA to AKAPs inhibited by the Ht31 peptide (130). In more recent work, HEK293 cells expressing α11.2, β2b, and α2δ1 were pretreated with forskolin for maximal phosphorylation of Cav1.2 by PKA to provide equalized starting points before Ca2+ currents through these channels were monitored (304). Over time, the Ca2+ currents decreased mainly because the incoming Ca2+ stimulated the Ca2+-activated serine-threonine phosphatase PP2B, which, like PKA, is also anchored by AKAP79/150 (see sect. viiC). No rundown was observed if serine-1928 had been mutated to alanine or the membrane-permeable stearylated Ht31 peptide was present (which disrupts anchoring of PKA but not PP2B by AKAPs) likely because either manipulation prevented any initial upregulation of Cav1.2 channel activity by forskolin and PKA. These results suggested that serine-1928 as well as PKA anchoring by AKAP79/150 are critical for potentiation of the channel activity by PKA. However, neither Gui et al. (149) nor Naguro et al. (288) (see previous paragraph) ectopically expressed AKAP150. Perhaps the level of endogenous AKAP79 in tsA-201 and other cells varies and was sufficient in the latter two studies. A recent publication reports substantial amounts of endogenous AKAP79 in HEK293 cells used by these authors (133). Whether AKAP150 is present in BHK6 cells is unknown.
It is, therefore, still not firmly established that serine-1928 phosphorylation is largely responsible for the PKA-dependent increase in channel activity. In fact, Ganesan et al. (127a) provided evidence that serine-1928 is not absolutely critical for PKA-mediated upregulation of Cav1.2 activity. The authors used cardiomyocytes as an endogenous system to express α11.2 with two point mutations (T1066Y/E1089M) to make it insensitive to dihydropyridines. This elegant strategy allowed them to inhibit endogenous wild-type Cav1.2 with dihydropyridines and selectively measure currents through the ectopically expressed channel. Stimulation of β ARs with isoproterenol increased the activity of the ectopically expressed T1066Y/E1089M α11.2 by 50%. When serine-1928 was mutated to alanine in this α11.2 construct, the increase was 35%. Thus, in this system, serine-1928 may only play a secondary role in β-adrenergic stimulation of Cav1.2. However, isoproterenol increased the activity of the endogenous L-type channel (measured in the absence of dihydropyridines and without ectopic expression of α11.2) by >300% rather than 50%. Accordingly, only a small fraction of Cav1.2 regulation is reconstituted in this system. Serine-1928 phosphorylation might be responsible for a sizable portion of the missing 250% of Cav1.2 regulation.
Be that as it may, there is no question that serine-1928 is phosphorylated in vivo as a phosphorylation-state specific antibody against the phosphorylated serine-1928 site reacted with the α11.2 long but not short form isolated from heart (85) and brain (79–81) and showed increased staining of dissociated cardiomyocytes upon stimulation of either β1 or β2 AR (195). Quantitative analysis indicates that 16.7 ± 1.8% of α11.2 is phosphorylated on serine-1928 under basal conditions in rat brain hippocampi (80). This phosphorylation increases to ∼40– 60% upon activation of PKA by forskolin application in acutely prepared hippocampal slices (166) or in vivo by administration of isoproterenol (153). This upregulation of serine-1928 phosphorylation is blocked by coadministration of the β AR antagonist propranolol and absent in AKAP150 KO mice in vivo (153) (see note added in proof and Ref. 234a).
D) Role of β2 Serines-478 and -479 in Cav1.2 Regulation by PKA
When α11.2 is expressed alone in COS cells, Ba2+ currents through this channel are increased severalfold upon application of the active catalytic subunit of PKA in whole cell or excised inside-out patch recordings. These results show that α11.2 can be regulated by PKA independent of α2δ, β, or γ (147, 356). However, other evidence suggests that PKA also phosphorylates at least one of the cardiac Ca2+ channel β subunits in vitro and in vivo upon β-adrenergic stimulation (152). PKA can phosphorylate serines-459, -478, and -479 in the cardiac β2a subunit (136). These sites are conserved in most other β2 subunit splice forms but not in β2c or in β1, β3, or β4. PKA increases Ba2+ currents through a mutant α11.2 form that is truncated 265 residues upstream of its natural COOH terminus to eliminate serine-1928 and is expressed together with β2a in tsA-201 cells. Mutating serines-478 and -479, but not -459, to alanines in β2a eliminates this increase. These results suggest that phosphorylation of serine-478 or serine-479 but not serine-459 contributes to PKA-mediated regulation of Cav1.2 (39) (Fig. 6). As several different β subunits can interact with α11.2 (24, 49, 50, 99, 292, 316, 418) and other β subunits do not have analogous phosphorylation sites, it appears likely that other phosphorylation sites in Cav1.2 can mediate PKA-induced upregulation of current activity. Furthermore, given the difficulties with regard to reproducibility of the regulation of Cav1.2 after ectopic expression in cell lines (see above), additional evidence for the role of serine-478 and -479 phosphorylation in Cav1.2 regulation is necessary especially in the context of full-length α11.2 (perhaps with serine-1928 mutated to alanine to accentuate effects that are independent of serine-1928 phosphorylation).
3. The β2-adrenergic receptor-Cav1.2 signaling complex
PKA and cAMP had been assumed to diffuse with minimal hindrance throughout the cell. PKA would thereby gain access to most of its substrates. However, the past 10 years revealed that various kinases and phosphatases are anchored at or, upon activation, recruited to many of their substrates for fast, effective, and selective signaling (310). Furthermore, the coexistence of multiple GPCRs that are positively coupled to cAMP production prompted earlier considerations that signaling by these GPCRs may not be purely redundant but rather linked to different signaling pathways (for review, see Ref. 324). Such selectivity would require spatially restricted cAMP signals. Stimulation of either β1 or β2 ARs leads to increased Ca2+ influx through Cav1.2 into ventricular cardiomyocytes (5, 20, 441). However, only β1 but not β2 AR activation effectively stimulates PKA throughout the myocyte resulting in a global phosphorylation of phospholamban to foster Ca2+ sequestration in the sarcoplasmic reticulum, glycogen phosphorylase kinase to regulate glycogen hydrolysis, and troponins I and C to control contraction and relaxation (441). These observations suggest that PKA acts locally at the plasma membrane and specifically near Cav1.2 upon β2-AR stimulation but globally upon β1-AR stimulation. Activation of the glucagon receptor or the prostaglandin E1 receptor also results in locally restricted cAMP production (334, 371). In contrast to β2 AR activation, which leads to more prominent cAMP/PKA responses at the sarcolemma including a positive inotropic response, prostaglandin E1 preferably stimulates cytosolic PKA (159, 348, 416).
Assembly of critical signaling molecules into macromolecular signaling complexes may foster pathway selectivity and localized signaling by cAMP and PKA. Cav1.2 forms the core of such a complex or signalosome (11, 78, 79, 81, 153). Coimmunoprecipitation of functionally active PKA and an AKAP with Cav1.2 provided initial clues for the existence of a signaling complex assembled around Cav1.2 (79). A systematic search for additional signaling components upstream of PKA in this complex was further inspired by earlier indications for β2 AR-regulated localized cAMP signaling in heart (see above; Refs. 371, 441). β-AR stimulation with isoproterenol increased serine-1928 phosphorylation of Cav1.2 in vivo in the rat and mouse brain by more than twofold; this effect is prevented by co-administration of the β-AR antagonist propranolol (153). In dissociated cardiomyocytes, stimulation of either the β1 AR or β2 AR increased serine-1928 phosphorylation (195). Coimmunoprecipitation studies from rat brain and heart revealed that the β2 AR, Gs, and AC are constitutively associated with Cav1.2 (11, 78). The existence of a β2 AR-Cav1.2 signaling complex was further supported by immunofluorescence colocalization of the β2 AR with Cav1.2 at postsynaptic sites in the brain (78). In vitro pull-down experiments with bacterially expressed fusion proteins indicate that the COOH terminus of β2 AR directly binds to α11.2 (78) (Fig. 6). How Gs and AC are linked to Cav1.2 is unclear. However, AKAP150 selectively associates with AC V and VI (17). This interaction could recruit one or both AC isoforms to Cav1.2.
A) Localized Signaling from the β2 AR to Cav1.2
The assembly of the β2 AR-Cav1.2 signaling complex might foster localized cAMP signaling. During cell-attached patch-clamp recording from somata of hippocampal pyramidal neurons in culture with Ba2+ as charge carrier, application of the β2 AR-selective agonist albuterol resulted in a more than twofold increase in L-type-mediated current when applied inside, but not outside, the recording pipette electrode (78). Analogous findings were obtained for localized Cav1.2 regulation by β2-AR stimulation in cardiomyocytes (63). This stimulation is mediated by PKA as PKA inhibitors block β2-adrenergic upregulation of L-type currents in cardiomyocytes (5, 440, 441, 462) and neurons (178). The requirement for localized β2-AR stimulation suggests that cAMP signaling can be restricted to submicrometer dimensions. Synthesis of cAMP occurs presumably in the immediate surrounding outside the pipette when albuterol is added, as the β2 AR-Cav1.2 complex is distributed throughout the plasma membrane in these neurons (79). The wall of the pipette used for the cell-attached patch recordings is typically several hundred nanometers thick. The plasma membrane usually forms an Ω-shaped structure inside the tip of the patch pipette with the narrow neck of the Ω-structure closely attached to the glass wall inside the pipette for a few hundred nanometers. The lack of channel potentiation by albuterol applied outside the pipette suggests that cAMP does not reach concentrations that are high enough to stimulate PKA-mediated phosphorylation of Cav1.2 channels inside the patch, which are only several hundred nanometers away from the β2 AR outside the electrode. Accordingly, cAMP signaling is restricted to an area of <1 μm around the β2 AR.
Electrophysiological studies also provide evidence that signaling by the β1 AR in cardiomyocytes and neurons is much less locally restricted than β2 AR signaling (63, 78). These findings support the earlier biochemical studies mentioned above (371, 441) that demonstrated a more local signaling by β2 AR versus a more global one by β1-AR stimulation. Fluorescence resonance energy transfer imaging provided further support for the idea that cAMP is not freely distributing throughout cardiomyocytes but rather concentrating at the Z-line level upon β-adrenergic stimulation (455). The observed gradient, however, was exacerbated in these studies by the overexpression of the fluorescent RII-derived cAMP sensor, as PKA itself can buffer cAMP concentrations (348), and this cAMP sensor itself was preferably localized to membrane-associated AKAPs (416).
B) Spatial Restriction of Camp Signaling
I) Spatial restriction of cAMP by Gi
The β2 AR can couple not only to Gs but also to Gi, especially upon prolonged stimulation (8, 371, 440, 441). At least in fibroblast cell lines, this switch can be induced by PKA-mediated phosphorylation of the β2 AR (75, 456). The β2 AR can thus limit cAMP production by inhibiting AC after switching from coupling to Gs to coupling to Gi. This mechanism appears as an effective means to temporally restrict cAMP production.
II) Spatial restriction of cAMP by AC
A second such mechanism is based on the inhibition of AC V and VI by PKA (60, 204). AC V and VI themselves can associate with AKAP150 (17). When AC V and the tonically active Gαs mutant Q227L are coexpressed in HEK293 cells, coexpression of AKAP79 induces AC V phosphorylation by PKA and reduces cAMP production (17). S676 in AC V is homologous to S674 in AC VI, which is critical for inhibition by PKA (60). Mutating S676 to alanine prevents the reduction in cAMP production induced by AKAP79 overexpression in this system (17). This regulation creates a negative feedback for cAMP production. Although it is unknown which ACs are present in the β2 AR-Cav1.2 complex, AC V and VI could be associated with the Cav1.2 complex via AKAP150 (17) (see sect. iiiB4a), and their phosphorylation by PKA could provide such inhibitor feedback.
III) Spatial restriction of cAMP by phosphodiesterase
A third mechanism that can restrict the effective radius of cAMP is its hydrolysis by phosphodiesterases (PDEs) that compartmentalize cAMP in cardiomyocytes (159, 210, 234, 334, 335, 348, 371, 440, 441). More specifically, PDE inhibitors delocalize spatially restricted β2-adrenergic regulation of Cav1.2 (176, 210). Although evenly distributed PDEs would be sufficient to reduce the lifetime of cAMP and thereby its effective radius, some PDEs associate with AKAPs for localized reduction of cAMP (22) (see next paragraph). Recruitment of different PDE4D isoforms to activated β2 ARs via β-arrestin is yet another mechanism that can contribute to reduced cAMP signaling (10, 314). In general, β-arrestin binds to the β2 AR upon receptor stimulation. It is not known whether (β-arrestin also associates with the β2 AR in the Cav1.2 complex, but if so, this mechanism would further contribute to the spatiotemporal restriction of cAMP signaling in the vicinity of such a complex.
Both cGMP-inhibited cAMP PDE3 and cAMP-specific PDE4 isoforms can contribute to spatially restricted cAMP signaling (334). Certain GPCRs including the α1 AR regulate cAMP degradation by stimulating specific PDEs (371) for localized cAMP signaling. Furthermore, PDE4D selectively counteracts upregulation of the cardiac contraction rate by the β2 but not β1 AR via PKA (437). The abundant PDE4D3 isoform is recruited to various subcellular compartments by directly or indirectly binding to mAKAP (95, 96), AKAP350 (AKAP9) (386), and gravin [also designated AKAP250 and AKAP12 (433)] (429). These AKAPs act in part to assemble PKA and PDE4D into a complex in which localized phosphorylation of PDE4D by PKA results in its stimulation (71, 96). Gravin can directly bind to the COOH terminus of the β2 AR, which is important for agonist-induced internalization of the β2 AR as well as recovery of the β2 AR from this desensitization (116, 244, 360). This binding requires phosphorylation of the PKA binding domain of gravin itself by the anchored PKA (385). How the gravin-PKA complex promotes recycling of internalized β2 ARs to the plasma membrane is unclear. This mechanism might be analogous to the β1 AR, which requires association of the AKAP150-PKA complex with its COOH terminus for recycling. In this latter case, AKAP150-anchored PKA phosphorylates S312, which is required for receptor recycling (132, 133). The analogy between gravin and AKAP150 is especially noticeable as these two AKAPs share a number of features including anchoring of PKC and PP2B in addition to PKA and binding to negatively charged phospholipids via three positively charged segments in their NH2-terminal regions, which is antagonized by CaM in the presence of Ca2+ (90, 141, 384, 409). Whether PDE4D or another PDE binds to the β2 AR-Cav1.2 complex via gravin, AKAP150, or, upon β2-AR activation, β-arrestin is untested.
mAKAP also binds PDE4D (95) (see sect. iiiB4d). More specifically, it interacts with PDE4D3 but not PDE4D5, two of nine splice variants encoded by the PDE4D gene. In PDE4D knockout mice, isoproterenol-induced cAMP accumulation is increased in the heart with no change in total cAMP under resting conditions. Furthermore, PKA phosphorylates serine-54 of PDE4D3, which increases its hydrolytic activity, thereby reducing cAMP in the vicinity of this complex (358). PKA also phosphorylates serine-13 in the binding site of PDE4D3 for mAKAP. This phosphorylation increases the PDE4D3-mAKAP interaction, thereby increasing the presence of PDE4D3 in mAKAP complexes (45). The extracellular signal-regulated kinase ERK5 and potentially ERK2 are associated with the mAKAP complex via binding to a KIM and a FQF docking site upstream and downstream of serine-579 on PDE4D3, respectively (95, 253). Both ERKs phosphorylate PDE4D3 on serine-579, which reduces its PDE activity (95, 253). ERK5 in turn is downregulated by cAMP via the cAMP effector Epac1 but not PKA (95).
It is unclear how cAMP is targeted to the Cav1.2-associated PKA rather than diffusing away from the complex. Analogous to substrate channeling in certain metabolic enzyme complexes, we therefore proposed that analogous channeling could occur for cAMP from the Cav1.2-associated AC to PKA (155). This is the more conceivable as AC V or VI might be rather closely localized to PKA in the Cav1.2 complex due to a direct link by AKAP150. It is also worth considering in this context that one of the three AKAP150 attachment sites on α11.2 is ∼150 residues downstream of the main PKA phosphorylation site, serine-1928 (153, 304). Accordingly, the COOH-terminal portion of α11.2 together with AKAP150 could bring AC V or VI and PKA into close proximity to each other and to the PKA substrate site.
C) Association of Trimeric G Proteins with Signaling Complexes
Rodbell and co-workers provided early evidence for G proteins being part of large signaling complexes based on radiation inactivation and other methods (e.g., Ref. 350). Kinetic studies also argue that G proteins may remain associated with their immediate effectors (e.g., AC, Ref. 239) or their receptors (e.g., Gq interactions with the muscarinic M1 receptor, perhaps in conjunction with PLCβ1, Ref. 28) beyond the brief encounters that were postulated by the original collision-coupling model. Live interaction studies using bioluminescence resonance energy transfer (BRET) studies provide further evidence for the notion that G protein-GPCR interactions exist for an extended time period (127, 300), including G protein interactions with the β2 AR. Also, an increasing body of evidence indicates that GPCRs associate with trimeric G proteins in the early secretory pathway (103, 104).
Many cells have a variety of Gβγ heterodimers, which act as pathway-selective signal transducers in conjunction with specific Gα subunits (see Refs. 294, 324 and citations therein). For example, muscarinic M4 and somatostatin receptors inhibit voltage-gated Ca2+ channels in GH3 cells through Gαo1β3γ4 and Gαo2β1γ3, respectively (see Ref. 220 and citations therein). It is difficult to envision how this selectivity could be maintained if Gα and Gβγ completely dissociate from each other and from their cognate receptors upon stimulus-induced GDP/GTP exchange of Gα find all their binding partners including their cognate receptors and targets, and, after hydrolysis of GTP on Gα to GDP find each other again by random collision. Because different Gα subunits have similar affinities for most Gβγ dimers in vitro, there must be mechanisms in vivo that ensure reassociation of the original combinations. Analogously, different Gβγ complexes can activate the inward-rectifying muscarinic K+ channel (Kir3/GIRK; activated by m2 muscarinic receptors) (185, 225, 328, 427). Biochemical and biophysical evidence indicate that not only Gβγ but also Gα can stably interact with Kir3, further supporting the notion that heterotrimeric Gi can form a quite steady complex with Kir3 (185, 203, 298, 325). It should be noted, however, that binding of Gαi to Kir3 reflects at least in part an additional direct regulatory mechanism of Kir3 channel activity by Gαi (311).
Kinetic and biochemical evidence indicates that both Gαs and Gβγ are rather stably associated with its effector AC independent of their activation status (239, 261; see also Ref. 419). More recent FRET and BRET studies suggest that activation of Gi by coexpressed α2A AR upon agonist application does not lead to dissociation of Gαi from Gβγ but rather to a rearrangement of the trimeric complex (38, 124, 127). Similarly, total internal reflection fluorescence combined with FRET indicate that heterotrimeric G proteins are preassociated with Kir3.1/3.4 channels at the cell surface and undergo a conformational change upon activation by an upstream Go/i-linked GPCR (331, 332).
The members of the K+ channel Kir3 family are activated by direct binding of Gβγ to the NH2 and COOH termini of their subunits (27, 67, 202, 225). Selectivity for Gβγ appears to be minimal, but Gαi2 and Gαi3 seem to be the preferred donors of Gβγ. This selectivity for the Gα subunit may be in part due to direct binding of Gαi2 and Gαi3 to Kir3 subunits (203, 311), which may lead to preassociation of trimeric G proteins with these two subunits and thereby to effective and selective signal transduction by those G proteins. It is tempting to speculate that a similar direct interaction recruits Gs to Cav1.2, although the stable association of Gs could equally well be due to constitutive binding of Gs to other proteins in the Cav1.2 complex including the β2 AR or AC.
D) Caveolae and Membrane Rafts as Platforms for Signaling Complexes
An alternative for the assembly of signaling complexes at the plasma membrane by direct protein-protein interactions is the colocalization within distinct membrane domains such as Triton X-100-insoluble rafts, caveolae, or postsynaptic density fractions. Caveolae and postsynaptic densities may not be a structural requirement for the β2 AR-Cav1.2 complex as respective marker proteins are undetectable in this complex after Triton X-100 solubilization and immunopurification from rodent brain (78). However, cardiac β2 AR and Cav1.2 colocalize by immunofluorescence microscopy and co-fractionate during sucrose density gradient centrifugation with caveolin-3 (11). In fact, caveolin-3 coimmunoprecipitates with the β2 AR-Gs-AC-PKA-Cav1.2 complex from heart. Furthermore, disruption of caveolae by depletion of cholesterol with methyl β-cyclodextrin and siRNA knockdown of caveolin-3 abrogated β2-adrenergic upregulation of L-type current in mouse ventricular myocytes without affecting β1-adrenergic upregulation (11). These observations indicate that caveolin-3 and more generally caveolae fulfill in some cells a critical supportive role in either the formation of the β2 AR-Cav1.2 complex or its colocalization with other signaling or structural components.
4. Role of AKAPs in regulating Cav1.2 and colocalized ion channels
A) Role of Akap in the Regulation of Cav1.2 by Pka
The first AKAP that was found to be associated with Cav1.2 was MAP2B (79), a microtubule-associated protein known to bind PKA (400). It directly binds to three different site of the α11.2 subunit (153). However, the question of whether MAP2B mediates PKA binding to Cav1.2 proved difficult to answer because heterologous overexpression of MAP2B is detrimental to cells due to its strong microtubule bundling effect, because Cav1.2 and MAP2B are large proteins with at least three interaction sites, and because of other technical issues (Davare, Dong, Rubin, and Hell, unpublished observations). Like Cav1.2, MAP2B had been detected in dendritic spines (40), but it is mainly localized in dendritic shafts and is not a prevalent AKAP in the spines.
In contrast to MAP2B, AKAP150 is enriched at postsynaptic sites, perhaps in part due to its interactions with the structural postsynaptic protein PSD-95 (which interacts with AMPA receptors via stargazing/γ2; see sect. iiA) and its homolog SAP97 (which directly binds to the AMPAR GluR1 subunit) (41, 70, 141, 236, 388). Furthermore, according to an earlier report (130) (see sect. iiiB2c), PKA-mediated potentiation of Cav1.2 expressed in heterologous tsA-201 cells also required coexpression of AKAP150 (see also Ref. 304). Although AKAP79/150 was initially not detectable in coimmunoprecipitation experiments with Cav1.2 despite repeated efforts (79; Davare and Hell, data not shown), AKAP150 reproducibly coprecipitates with Cav1.2 from brain when 150 mM NaCl is added to the solubilization buffer (153; see also Ref. 304). Furthermore, coimmunoprecipitation of PKA with Cav1.2 from brain extracts is drastically reduced but not absent in AKAP150 knockout mice. Isoproterenol-induced phosphorylation of serine-1928 is eliminated in AKAP150 knockout mice in vivo (153). These findings indicate that AKAP150 is the major but not only AKAP that recruits PKA to neuronal Cav1.2 complexes.
MAP2B shows little if any expression outside the nervous system. In contrast, AKAP15 (see set. iiiB1) is abundant in heart, where it coimmunoprecipitates and colocalizes with Cav1.2 (194). Furthermore, Ca2+ currents through Cav1.2 are increased upon PKA activation when Cav1.2 is coexpressed with AKAP15 in tsA-201 cells, and this increase is not observed when AKAP15 is omitted (126). Finally, a peptide derived from the leucine-like zipper motif in AKAP15 that mediates its binding to Cav1.1 and inhibits upregulation of Cav1.1 by PKA (see sect. iiiB1) also blocks β-adrenergic stimulation of L-type Ca2+ currents in cardiomyocytes (194). Although this peptide may prevent not only AKAP15 but also other AKAPs from binding to Cav1.2 and the AKAP15-dependent increase in Cav1.2 activity in tsA-201 cells is rather modest (18%) (126), collectively these data suggested that AKAP15 recruits PKA to at least a sizeable Cav1.2 population for fast and efficient signaling in the heart. Because AKAP15 is present in brain and specifically in dendrites, it may help recruit PKA to a certain neuronal Cav1.2 population as well (194).
AKAP79/150 was thought to be absent or of low abundance in the heart. However, there is a detectable AKAP150 pool in cardiac tissue, and AKAP150 coimmunoprecipitates with Cav1.2 from cardiac extracts (Fig. 7). It thus could contribute to PKA anchoring in the heart. Furthermore, one of the three AKAP150 binding sites on α11.2 lies within the last 125 residues (153), which contain the LZ-like motif that anchors AKAP15 (194). Disrupting this motif with point mutations reduces the interaction between α11.2 and AKAP150 and inhibits regulation of Cav1.2 by PKA (304). The peptide derived from the LZ-like motif in AKAP15 as described in Reference 194 might thus also affect the AKAP150-α11.2 interaction.
AKAP150 binds not only to α11.2 but also the cytosolic COOH terminus and to a lesser degree the third intracellular loop of the β2 AR (Fig. 9) (125). AKAP150 could, therefore, fulfill an auxiliary role in the association of the β2 AR with Cav1.2. However, earlier evidence clearly shows also a direct interaction between the COOH termini of the β2 AR and Cav1.2 (78).
FIG. 9.
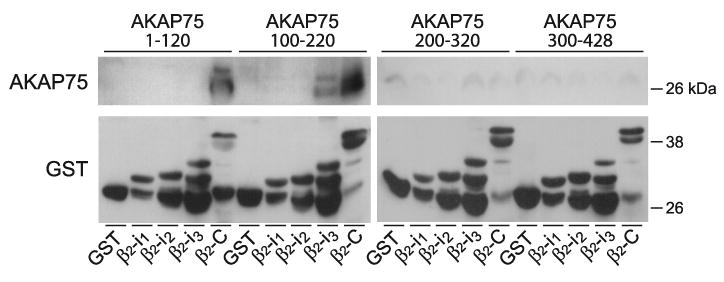
Direct interactions between the NH2-terminal half of AKAP150 and the β2 AR. Fusion proteins were expressed in E. coli and extracted using sarcosyl as described (81, 153, 236). GST fusion proteins of the first, second, and third intracellular loops of the β2 AR (β2-i1, -i2, -i3, respectively) or its cytosolic COOH terminus (β2-C), or GST alone (GST) were immobilized on glutathione Sepharose. Resins were incubated with E. coli lysates of 6xHis- and V5-tagged AKAP79 fragments encoding residues 1–120, 100–220, 200–320, and 300–428 (these constructs cover the full length of AKAP75). Top: immunoblotting with antibodies against V5 showed specific binding of the first two AKAP75 fragments to the COOH terminus of the β2 AR. Weak binding of the second fragment to the third intracellular loop of the β2 AR (β2-i3) was also detectable to variable degrees. Bottom: reprobing with anti-GST demonstrated that comparable amounts of the full-length GST fusion proteins were present, although loop fragments were also substantially degraded.
Finally, AKAP79 promotes cell surface expression of Cav1.2 in HEK293 cells (3). Although loop II/III, the intracellular connection between domains II and III, of α11.2 was implicated in this effect, there is actually no evidence that AKAP150 binds directly to this loop. Rather, AKAP150 interacts with the relatively short NH2 terminus, the loop I/II, and the distal COOH terminus of α11.2 (153) (Fig. 6). Intriguingly, MAP2B shows the same interaction pattern (153). It is unclear whether AKAP150 regulates Cav1.2 surface expression reported in Reference 3 by signaling via its binding partners PKA, PKC, or PP2B or by other means. AKAP150 binds with its NH2-terminal region not only PKC but also PIP2, F-actin, and cadherin, and might through these interactions foster Cav1.2 surface expression (90, 141, 142). It is also conceivable that it stabilizes a certain conformation of α11.2 by binding to its three different attachment sites. Although it is unclear whether AKAP150 simultaneously binds to two or three sites, such interactions could regulate the overall structure of α11.2. If AKAP150 concurrently interacts with the distal COOH terminus and one of the other two sites, it could stabilize the association of the distal COOH terminus with the rest of the channel after cleavage of the COOH terminus as discussed in section iiiB2b.
B) Role of Akap79/150 in Regulating the Ampar Glur1 Subunit
Cav1.2, AMPAR, and NMDAR are coclustered if not intermingled at postsynaptic sites yet contain their own AKAPs (Fig. 10). This molecular organization suggests once more that PKA is specifically targeted to individual channel complexes for phosphorylating defined substrates with high specificity and spatial restriction well below the dimensions of postsynanptic sites, which are formed by dendritic spines that are ∼1 μm diameter, reaching dimensions of the size and distances of individual channel complexes (a few nanometers). For this reason, postsynaptic PKA anchoring to AMPAR and NMDAR will be described in more detail.
AMPARs consist of four homologous subunits encoded by four different genes (GluR1-4), with GluR1/R2 and GluR2/R3 being the prevalent combinations in adult rodent cortex and hippocampus. Each subunit contains a large extracellular NH2-terminal domain, a transmembrane segment M1 that is followed by a reentry loop M2 and two other transmembrane segments M3 and M4, and the intracellular COOH terminus (Fig. 10). The NH2 terminus forms the clamshell-like glutamate binding site together with the extracellular loop between M2 and M3. In addition, AMPARs associate with stargazing/γ2 and its homologs γ3, γ4, and γ8 (see sect. iiA), which are necessary for surface expression and synaptic clustering (57), the latter depending on PSD-95 (351). They also promote and prolong ligand-induced opening of the receptor channel (280, 297, 321, 396) and have been named TARPs (397). Recently, γ7 was also shown to act as TARP (217). γ2 was originally identified as stargazin, which is defective in the stargazer mouse. γ2 is the only TARP expressed in the cerebellum, and the resulting cerebellar ataxia is due to the absence of surface expression of AMPARs in the cerebellum. Other brain regions express other members of the γ2 subfamily facilitating proper synaptic AMPAR expression (397).
AKAP150 binds to the SH3 domains of PSD-95 and SAP97 (70, 388). PSD-95 and its homologs SAP97, SAP102, and PSD-93/CHAPSYN110 consist of three PDZ domains, which typically bind to the very COOH termini of certain proteins, followed by an atypical SH3 domain and a GK domain, the latter two resembling Ca2+ channels β subunits (see sect. iiA). PSD-95 interacts with the COOH termini of TARPs including stargazing/γ2. As mentioned above, SAP97 directly binds to the COOH terminus of the AMPAR GluR1 subunit (41, 70, 141, 236, 388).
Reconstitution of GluR1 regulation by PKA in HEK293 cells requires coexpression of AKAP79 (388). Accordingly, AKAP79/150 targets individually PKA to AMPAR and Cav1.2 even if the different channel complexes are in close proximity to each other. AKAP79/150 also interacts with PKC and PP2B. AKAP79/150-anchored PP2B counterbalances PKA-mediated stimulation of GluR1 channel activity (68, 388) (see also sect. viiA). Anchoring of PKC via AKAP79/150 (219) is required for effective phosphorylation of GluR1 at serine-831 by PKC (387), which can otherwise also be phosphorylated by CaMKII (260).
C) Role of Yotiao in Regulating NMDAR
NMDARs consist of four subunits that are homologous to AMPAR subunits (see sect. iiiB4b) with an extracellular NH2 terminus, four membrane segments including the M2 reentry loop, and an intracellular COOH terminus. All NMDAR contain two NR1 subunits, which bind the coagonist glycine rather than glutamate, and two glutamate-binding NR2 subunits encoded by four different genes (NR2A-D, with NR2A and -2B being most common in the cortex and hippocampus). Yotiao, a ∼230-kDa splice form of AKAP350/450 (AKAP9), binds to the C1 cassette in the NR1 COOH terminus and links PKA and also the counteracting phosphatase PP1 physically and functionally to the NMDAR at postsynaptic sites (426). Cav1.2, which is localized in close proximity to postsynaptic NMDARs, does not coimmunoprecipitate Yotiao (153). The COOH termini of NR2 subunits bind to the first two PDZ domains of PSD-95, which can interact with AKAP150 (see sect. iiiB4b). However, it is unclear whether PSD-95 recruits AKAP150 to the NMDAR.
D) Role of Makap in Regulating the Ryanodine Receptor RyR2
The cardiac RyR (RyR2) is in close proximity and functionally coupled to Cav1.2 in cardiomyocytes but associates with PKA via its own AKAP. Localized regulation of the RyR2 versus Cav1.2 will therefore be discussed in more detail. Three genes encode RyR1–3. RyR1 is mainly expressed in skeletal muscle, where it is linked to Cav1.1 (see sect. iiiB1 and Fig. 5). RyR2 is the RyR in the heart, which releases Ca2+ upon a modest amount of Ca2+ influx through juxtaposed Cav1.2 (Ca2+-induced Ca2+ release). All three RyR are ∼5,000 residues long and consist of a very large cytosolic foot domain, four transmembrane segments, and a short cytosolic COOH terminus (Fig. 5).
RyR2 associates with mAKAP via a LZ-like motif in the foot domain (residues 3003–3039; Table 1) (266). mAKAP was originally named AKAP100 (269) but later renamed mAKAP after full-length cDNA was obtained due to its expression in muscle (216). More recently, it was also designated as AKAP6 (433). Like RyR2, mAKAP is enriched at the Z-line level at the t-tubule/junctional sarcoplasmic reticulum (443). Because RyRs and Cav1.2 are juxtaposed, PKA anchored via mAKAP to RyR2 would be in close proximity to Cav1.2; however, mAKAP does not coimmunoprecipitate with Cav1.2, at least not from brain, where RyR2 and mAKAP are also expressed at substantial levels (153) (Davare and Hell, data not shown). That PKA is linked to the RyR2 and Cav1.2 by different AKAPs suggests once more that within a radius that is likely substantially <100 nm, AKAP anchored PKA cannot effectively reach neighboring protein complexes. In fact, the identified AKAP binding sites on Cav1.1, Cav1.2, RyR1, RyR2, and type 1 IP3 receptors (IP3R1) are quite close to identified PKA phosphorylation sites on these different ion channels (Table 1), suggesting the requirement for a rather limited interaction range for PKA, its AKAPs, and its targets.
mAKAP also binds the PDE4D splice isoform PDE4D3 (see sect. iiiB3bIII) (95). As mAKAP binds to the cardiac RyR2, it might recruit PDE4D3 to the RyR2 (234). In PDE4D knockout mice phosphorylation of serine-2808, the PKA site of the RyR2, is elevated. PDE4D association with the RyR2 is reduced and serine-2808 phosphorylation increased in human heart failure. Serine-2808 phosphorylation increases the open probability of RyR2 possibly because it destabilizes binding of FKBP12.6 (calstabin2) to the RyR2, which renders the RyR leaky, thereby contributing to heart failure (266, 267; but see Ref. 438). Binding of PDE4D3 to RyR2 brings this PDE in close proximity of Cav1.2 in cardiomyocytes. RyR2-anchored PDE4D3 thus could contribute to locally restricting cAMP in a manner that affects not only the RyR2 but also Cav1.2.
C. Regulation of Na+ Channels by PKA
PKA and PKC synergistically regulate neuronal Na+ channel activity by reducing peak current and enhancing slow inactivation (48, 61, 77, 241). PKA phosphorylates four serine residues in loop I/II of the NaV1.2 α1.2 subunit (Fig. 11), the predominant Na+ channel in forebrain (287). PKC phosphorylates serine-1506 of α1.2 near the inactivation gate in loop III/IV (241, 423). Na+ channels interact with PKA via AKAP15 (394). Regulation by PKA requires anchoring via AKAP15 (42). AKAP15 in turn binds to the NH2-terminal segment of loop I/II in α1.2 (residues 446– 453) via a modified LZ motif (Fig. 11) (43, 118). This motif is shorter than established for other LZ motifs and contains a phenylalanine in the first position, which is canonically occupied by leucine. This motif is well conserved among all nine Na+ channel α subunits. A peptide derived from the LZ of AKAP15 prevents downregulation of Na+ channel peak currents upon stimulation of the dopamine D1/5 receptor in hippocampal neurons (118).
FIG. 11.

The Nav1.2-AKAP15-PKA complex. Yellow, sodium channel α1.2 subunit; dark red, AKAP15 binding site (LZ, bracket and segment); light red, PKA, four phosphorylation sites for PKA within loop I/II (arrows); orange, PKC, PKC phosphorylation site serine-1506, and the inactivation gate sequence within loop III/IV.
D. The Yotiao-Kv7.1 Complex and β2-AR Regulation
PKA is recruited to Kv7.1 via the AKAP Yotiao (265). Yotiao binds to the LZ motif in the COOH terminus of Kv7.1 that is formed by residues 588–616 (Fig. 12). PKA anchoring by Yotiao is critical for phosphorylation of serine-27 in Kv7.1, which leads to increased channel activity (265). The PP1/PP2A blocker okadaic acid enhances this increase presumably because in its absence Yotiao-anchored PP1 rapidly reverses serine-27 phosphorylation.
FIG. 12.

The Kv7.1-Yotiao-PKA/PP2A and Kv7.2-AKAP150-PKC complexes. Purple, α7.1/7.2; blue, Yotiao binding site on α7.1 (LZ); magenta, AKAP150 binding site on α7.2; bright red, PKA, identified phosphorylation site for PKA in NH2 terminus of α7.1; dark red, AKAP150 and AKAP150 binding sites on α7.1; orange, PKC binding to AKAP150.
In addition to its role in PKA anchoring, binding of Yotiao itself increases Kv7.1 currents, but only if serine-27 is phosphorylated or replaced with the phosphomimetic aspartate (58, 227). Although Yotiao binding per se mediates such an increase independent of PKA or PKC activity (227), phosphorylation of serine-43 of Yotiao magnified the increase without altering binding of Yotiao to the channel or its phosphorylation on serine-27 by Yotiao-anchored PKA (58).
Two different binding sites in Yotiao for Kv7.1 have been identified, one near its NH2 terminus (residues 29–46) and the other one near its COOH terminus (residues 1574–1643) (59). A missense mutation in the COOH-terminal binding site (S1570L) in a subpopulation of humans with long Q-T syndrome reduces binding of Yotiao to Kv7.1 and cAMP-induced phosphorylation and regulation of Kv7.1 (59). AKAP150 also interacts with Kv7 channels but appear mainly to function in anchoring PKC rather than PKA to these channels (see sect. ivD).
IKs slow K+ conductance currents are upregulated upon β-adrenergic stimulation. FRET between secondary antibodies labeling Kv7.1 and the β2 AR indicates close proximity of those two proteins in cardiomyocytes (94). Interestingly, overexpression of the β2 AR increases phosphorylation and channel activity of Kv7.1 independent of agonist stimulation of the β2 AR (94). Overexpression of the β2 AR did not increase L-type channel Cav1.2 activity under basal conditions. It appears that the β2 AR is especially intimately associated with Kv7.1 so that signaling from the receptor to the channel is spatially restricted and does not translate into stimulation of Cav1.2. A lack of effect on Cav1.2 is puzzling, however, as the β2 AR is structurally and functionally closely associated with Cav1.2 (see sect. iiiB3). Perhaps the β2 AR behaves differently when associated with the Kv7.1 versus Cav1.2 complex.
E. The PKA-β2 AR-BK Complex
1. PKA binding to BK
PKA activity can either increase or decrease BK channel activity depending on the splice isoform of BK. PKA increases the activity of BK channels (224) that lack the exon STREX-1 at splice site 2 by direct phosphorylation of serine-869 (serine-899 in some isoforms) in the COOH terminus of the α subunit (Fig. 13) (290, 393).
FIG. 13.

The BK-PKA complex. Purple, α subunit of BK; red, PKA, identified PKA phosphorylation sites; blue, leucine zipper motifs (LZ) within BK, unidentified AKAP that recruits PKA for serine-869 phosphorylation; green, β2 AR and its interactions with the BK α subunit; dark red, AKAP150.
PKA-mediated phosphorylation of the serine in position 4 of STREX-1 leads to BK channel inhibition independent of the presence of serine-869 (393). Whereas an increase in channel activity requires phosphorylation of all four serine-869 residues in the tetrameric channel, phopshorylation of a single serine residue in position 4 of the STREX-1 insert is sufficient to reduce channel activity (392).
Functional studies of native BK channels reconstituted into lipid bilayers or in membrane patches excised from BK-transfected Xenopus oocytes and HEK293 cells provided initial evidence for a tight association of PKA with BK (65, 112, 393). Subsequent biochemical studies indicate that the catalytic C subunit of PKA by itself but not when associated with regulatory R subunits or with the inhibitory PKI peptide can directly bind to the COOH terminus of the Drosophila BK α subunit dSlo (410, 461). Residues 922–956 in the BK COOH terminus are required for this binding and contain the PKA phosphorylation site serine-942, which is homologous to serine-869 in mammalian BK. Several experiments indicate that the association of the PKA C subunit with the BK COOH terminus is not simply mediated by a catalytic site-substrate interaction (461).
Direct binding of free PKA C subunit to dSlo is different from PKA targeting via AKAPs. In mammalian BK channels, one of two LZ-like motifs in the COOH terminus of BK (LZ1 but not LZ2 further COOH terminal) is required for coimmunoprecipitation of PKA with BK (391). LZ1 is also necessary for PKA-mediated upregulation of channel activity of STREX-1-lacking BK as well as for downregulation of STREX-1-containing BK (391). Although LZ-like motifs can anchor a number of different AKAPs (see above), regulation of BK by PKA is not inhibited by the Ht31 peptide, which blocks signaling by AKAP-anchored PKA by displacing PKA from the different AKAPs in numerous other systems (391). In contrast to dSlo (see previous paragraph), no interaction of the region surrounding serine-869/899 of mammalian BK with PKA C subunit was detectable by pull-down experiments (391). It appears that an unknown adaptor protein that interacts with LZ1 is required for functionally recruiting PKA holoenzyme to the channel complex.
2. The interaction of the β2 AR with BK
The β2 AR and BK coimmunoprecipitate from rat brain and colocalize in neurons (249). The β2 AR directly binds with its third intracellular loop (i3) to the rat BK α subunit (Fig. 13) (249). AKAP150, which can directly interact with the β2 AR (125), is recruited to the β2 AR-BK complex through its interaction with the β2 AR (249). At first glance, the β2 AR/AKAP150 complex could be the missing link that connects PKA to BK, but in this case, BK regulation by PKA should be Ht31 sensitive. Although this interaction could contribute to BK regulation, an alternative PKA anchoring mechanism remains sufficient.
The main binding region for AKAP150 in the β2 AR is its cytosolic COOH terminus, which binds to the NH2-terminal half of AKAP150 (Fig. 9). This arrangement can easily accommodate simultaneous binding of the β2 AR loop i3 to BK (Fig. 13). In cell-attached recordings, the β2-selective agonist salbutamol increased BK channel activity largely only if both the β2 AR and the AKAP-150 homolog AKAP79 were coexpressed in Xenopus oocytes (249). Importantly, bath application of salbutamol increased BK activity to a significantly lesser degree. These findings indicate that signaling from the β2 AR to BK via cAMP/PKA is restricted to domains in the range of several hundred nanometers and possibly less, similar to signaling from the β2 AR to Cav1.2 (see above and Refs. 63, 78).
IV. Regulation of Ion Channels by Protein Kinase C
A. Regulation and Targeting of PKC
Stimulation of Gq-coupled receptors leads to activation of PLCγ. This enzyme cleavages PIP2, generating inositol 1,4,5-trisphosphate (IP3) and diacylglycerol. IP3 activates IP3Rs, thereby inducing Ca2+ release from internal stores. Diacylglycerol can either alone or in conjunction with Ca2+ stimulate most PKC isoforms. Ten different PKC genes have been identified and their protein products divided into three different classes (282, 299). Conventional PKC isoforms (cPKC: PKCα, PKCβI and its splice isoform PKCβII, PKCγ) are activated when diacylglycerol or Ca2+ bind to the conserved C1 and C2 domains, respectively. Diacylglycerol and Ca2+ are required for full cPKC activity. Novel PKC isoforms (nPKC: PKCδ, PKCε, PKCη, PKCθ) contain only a C1 domain and require solely diacylglycerol for stimulation. Atypical PKC isoforms (aPKC: PKCι, PKCλ, PKCζ; human PKCι and mouse PKCλ may be orthologs) lack C1 and C2 domains. Their regulation is not well understood but may be in part mediated by arachidonic acid, ceramide, and other lipids.
Most PKC isoforms redistribute upon activation from one specific subcellular location including the cytosol and nucleus to another, depending on the PKC isoforms and cell type (101, 282). Ca2+ binding to the C2 domain induces its interaction with acidic phospholipids such as phosphatidylserine, thereby fostering recruitment of cPKC isoforms to membrane surfaces, especially the plasma membrane, although activation of cPKC can also lead to their localization at intracellular structures such as the nucleus or perinuclear region (101, 282). Subsequent binding of their C1 domains to diacylglycerol causes further activation and may stabilize membrane anchoring via the V1 region. The V1 region is upstream of the C1 domain and varies among PKC isoforms. It interacts with the catalytic domain, thereby acting as pseudosubstrate inhibitor. Diacylglycerol binding to C1 releases V1 from the catalytic domain. After dissociation from the catalytic domain, the V1 region itself may mediate membrane interactions, possibly fostering membrane association and targeting of PKC (302).
Differential recruitment of the various PKC isoforms to certain subcellular compartments is further fine-tuned by their interactions with specific PKC adaptor proteins named RACKs upon their activation (366). This mechanism is best characterized for PKCβ and PKCε. Subtype-specific sequences in the C2 domain and the V5 domain at the very COOH terminus (PKCβ) or the V1 domain (PKCε) become accessible for RACK interaction upon enzyme activation (366, 369). PKCβ selectively binds to RACK1 (338). Two different peptides derived from the C2 region of PKCβ inhibit this interaction. Both peptides antagonized L-type channel inhibition by phorbol esters in ventricular myocytes (459). Of the cPKC isoforms in the heart, only activated PKCβII is prominent at the surface of cardiomyocytes together with its PKCβ-specific RACK, RACK1. PKCβI and PKCα, which could also be affected by C2 domain-derived peptides, accumulate at other locations. PKCγ is not detectable in heart. It appears that PKCβII downregulates Cav1.2 upon its activation and subsequent recruitment by RACK1 to Cav1.2 complexes that are present in the plasma membrane (101). Stimulation of PKCε induces its translocation to Z lines in ventricular myocytes, placing it near L-type channels in t tubules (186, 333). Although this spatial proximity does not necessarily have to translate into a functional interaction between L-type channels and PKCε, it is tempting to speculate that different PKC isoforms may regulate Cav1.2 in opposite ways (see sect. ivB) that may in part depend on the exact localization of the Cav1.2-PKC complex, in this case the plasma membrane versus t tubules.
Various adaptor proteins that can constitutively recruit PKC to defined protein complexes have been described. For example, InaD consists of five PDZ domains and assembles TRP ion channels, PLCβ, and PKC into a signaling complex that is a critical part of the light-sensing system of Drosophila (398). Interaction of the Drosophila eye PKC with its PDZ domain binding site at its very COOH terminus to the fourth PDZ domain of InaD appears activation independent. Mammalian PKCα binds in a similar way to the multi-PDZ domain proteins PSD-95 and SAP102 (242) and to the PDZ domain in the scaffolding protein PICK1, which can form homo-oligomers (367). PICK1 in turn interacts with AMPAR GluR2 and GluR3 subunits (436) and the acid-sensing ion channels ASIC1 and -2 (181). Furthermore, PSD-95 and SAP102 bind to NMDAR NR2 subunits (283) and to stargazin and its homologs, which might thus recruit PSD-95-PKC complexes to AMPAR (see sects. iiA and iiiB4b). It is therefore conceivable that PICK1, PSD-95, or SAP102 can target PKCα to AMPAR or NMDAR. Cav1.2 also possesses a PDZ domain binding consensus sequence at its very COOH terminus that interacts with PDZ domain-containing proteins including NIL-16 (228). However, it is unknown whether PKC associates with the Cav1.2 via adaptor proteins similar to NIL-16 or, after activation, RACK1.
B. Regulation of Cav1.2 by PKC and Its Anchoring by AKAP150
In vitro experiments indicate that α11.2 and at least one β subunit can be phosphorylated by PKC (164, 167, 205). Activation of PKC via the Gq-linked α1AR, endothelin receptor, or angiotensin II receptor or by direct activators such as diacylglycerols or phorbol esters leads to upregulation, downregulation, or biphasic effects on currents through cardiac Cav1.2 channels (reviewed in Refs. 52, 212). The complexity of these results may in part be due to effects not mediated by PKC. For example, extracellular application of dioctanoylglycerol inhibited the Ca2+ currents by a PKC-independent mechanism, whereas intracellular photorelease of caged dioctanoylglycerol yielded a robust PKC-dependent stimulation (161).
Two alternatively spliced exons encode the initial segment of the NH2 terminus of α11.2, which is either 46 (exon 1a) or 16 (exon 1b) residues long and had originally been cloned from rodent heart and brain, respectively, (279, 365); both splice forms have been described in both organs (33, 361). PKC decreased Ba2+ currents through Cav1.2 expressed in HEK 293-derived tsA-201 cells by phosphorylating threonines-27 and -31 of the α11.2 isoform with the longer NH2 terminus (Fig. 14) (274). These residues are missing in isoforms with the shorter NH2 terminus, which does not show inhibition by PKC. Alanine but not aspartate substitution of either threonine prevented the PKC effect. These results indicate that phosphorylation of both threonines is necessary for the PKC effect, with aspartate effectively mimicking the negative phosphate charge. The first 20 residues of the long NH2 terminus of α11.2 are also important for the increase in channel activity by PKC as observed upon expression in Xenopus oocytes (361). However, this effect does not seem to involve direct phosphorylation of this segment by PKC but perhaps entails phosphorylation of another site in the Cav1.2 complex that functionally interacts with the very NH2 terminus (361). Similar to the decrease in channel activity of the α11.2 isoform with the longer NH2 terminus described above, phosphorylation of serine-81 in the NH2 terminus of α11.3 by PKC decreases the activity of Cav1.3 in tsA-201 cells ectopically expressing this channel (14). It is thus possible that analogous inhibitory effects by PKC on Cav1.2 and Cav1.3 exist.
FIG. 14.

The Cav1.2(-AKAP150-)PKC complex. Blue, α11.2; red, AKAP79/150 binding sites (brackets and segments); orange, identified direct binding sites independent of AKAP150 (segments) and phosphorylation sites (arrows, residues followed by number) for PKC on α1. Two separate fragments of the α1 COOH terminus (1509–1905, 1906–2170; Ref. 445) bind PKC. Magenta, β2 and its interactions with α11.2.
It is unknown how PKC increases Cav1.2. PKC is capable of phosphorylating serine-1928 in vitro and in intact HEK293 cells (445), which could upregulate channel activity (see sect. iiiB2c). However, phosphorylation by PKC appears to be much less prominent than by PKA (79).
As mentioned in section ivA, inactive PKC can be cytosolic or preassociate with defined subcellular structures (101, 282). In fact, cPKCα, cPKCβ, and cPKCγ but not nPKCδ or nPKCε coimmunoprecipitate from brain with Cav1.2 (445). PKCβ also coimmunoprecipitates with Cav1.2 from heart. Two different GST fusion proteins covering the membrane proximal and distal part of α11.2 COOH terminus (residues 1509–1905 and 1906–2170, respectively) pull down recombinant PKCα (445). Accordingly, cPKC can directly bind to α11.2 (Fig. 14). PKC also binds to the NH2-terminal region of AKAP150 (219). Because AKAP150 is associated with Cav1.2 (153, 304), it could help to recruit PKC to the Cav1.2 complex. In arterial smooth muscle cells, PKCα increases Ca2+ influx through Cav1.2 and thereby Ca2+ sparklets, leading to increased blood pressure. This regulation requires recruitment of PKCα but not of PKA by AKAP150 to Cav1.2 (291).
C. Antagonistic Regulation of Cav2.1 and Cav2.2 by Gβγ and PKC
N- and P/Q-type channels fulfill various functions, but perhaps most prominent is the presynaptic Ca2+ influx that triggers neurotransmitter release (383). Regulation of their activity has profound effects on synaptic transmission (382). α12.1 (P/Q-type), α12.2 (N-type), and α12.3 (R-type) are phosphorylated by PKA, PKC, CaMKII, and PKG in vitro and in intact neurons (162, 164, 166, 344, 452). However, little is known about the physiological functions of these different in vitro phosphorylations except that PKC can increase activity of N-type channels (368, 444), which potentiates fast synaptic transmission (382), and of R-type channels (368).
PKC acts by antagonizing the downregulation of channel activity by G proteins (381, 382). This downregulation by G proteins is membrane-delimited, i.e., it does not involve a freely diffusible second messenger but rather direct interactions with the Gβγ dimer upon its release from G proteins (88, 121, 169, 175, 197, 457). The respective G proteins must be in close proximity to the channels because the membrane-delimited signaling prevents access to the channels for G proteins outside a cell-attached recording electrode. The upregulation of N- and P/Q-type channel activity by PKC is tightly interwoven with its downregulation by Gβγ (recently reviewed in Refs. 100, 113). Because channels are to some degree tonically inhibited by Gβγ, PKC activation can lead to an increase in channel activity even under basal conditions, i.e., without previous pharmacological activation of trimeric G proteins.
Loop I/II of the Ca2+ channel α1 subunits is the major binding site for not only the auxiliary β subunits (see sect. iiA) but also for the βγ dimer upon its release from Gα (Fig. 15). β Subunits bind to a stretch of 18 residues that is 16 residues downstream of IS6, the last transmembrane segment of domain I. Gβγ dimers bind to two different segments in loop I/II. Each segment is ∼20 residues long. One partially overlaps with the NH2-terminal portion of the β-subunit binding site and one is ∼40 residues downstream of the β-subunit binding site (88, 457). Regions in the NH2 and COOH termini of these channels have also been implicated in Gβγ-mediated regulation (52, 100, 323), but details are less established than for loop I/II, which clearly plays a central role in these regulatory mechanisms. Gβγ binding results in a slowing of ion current activation kinetics, which likely mirrors the voltage-dependent release from inhibition. Strong depolarization to positive potentials release Gβγ from the channel, thereby increasing channel activity.
FIG. 15.

Cav2.2 PKC complexes. PKC increases channel activity by antagonizing the donwregulation by binding of Gβγ to loop I/II and of syntaxin to the synprint region in loop II/III. Blue, α12.2; orange, PKC phosphorylation site threonine-422 in loop I/II; this site is present in rat but not rabbit α12.2 and specifically antagonizes inhibition by Gβ1, which carries two unique aspartates, but not other trimeric Gβ subunits in rat. Other unidentified phosphorylation sites are responsible for the antagonistic action of PKC with respect to other Gβγ interactions. Grey, β1 and γ subunits of G proteins; black, segments within loop I/II that bind G protein β1 and γ subunits. Also shown in orange are PKC phosphorylation sites in and near the synprint region in loop II/III. Yellow, synprint region within loop II/III and inhibitory interacting protein Syntaxin 1A (Syn-1A); red, CaMKII, identified CaMKII phosphorylation sites in and near the synprint region in loop II/III; green, ENH binds with its LIM domains to both the COOH terminus of Cav2.2 (but not Cav2.1) and to PKC-ε (orange), thereby recruiting PKC-ε to the channel complex.
PKC phosphorylates threonine-422 in the second Gβγ binding site in loop I/II of rat α12.2, thereby antagonizing binding of Gβγ to this site and its inhibitory effect on channel activity (157, 457). It appears that all five Gβ isoforms Gβ1–5 can functionally interact with Cav2.1 and Cav2.2 but may differentially affect channel activity (7, 131, 341). PKC-mediated phosphorylation antagonizes Cav2.2 channel inhibition specifically through Gβ1 but not other Gβ isoforms (72, 97). This functional difference between Gβ1 and other Gβ subunits is due to a pair of aspartate residues that is only present in Gβ1.
The homologous position of threonine-422 in rabbit α12.2 is alanine and cannot be phosphorylated, yet PKC still antagonizes channel inhibition by Gβγ (25). Thus additional PKC phosphorylation sites can control channel inhibition by Gβγ (100). Furthermore, PKC as well as CaMKII can phosphoryalte loop II/III of α12.2 in the so-called synprint region. This region mediates binding of different presynaptic proteins involved in synaptic vesicle exocytosis including syntaxin 1A. This interaction reduces Cav2.2 channel activity. Phosphorylation of synprint prevents syntaxin 1A binding, thereby counteracting the inhibitory effect of syntaxin 1A on channel activity (450, 451).
Enigma homolog (ENH) is an adaptor protein that consists of a PDZ domain followed by three LIM domains, which are cysteine-rich double zinc finger motifs. It interacts through one or more of its LIM domains with the COOH terminus of α12.2 but not α12.1 (254). More precisely, ENH binds to a region within 150 residues upstream of the alternative splice site that creates the two main splice isoforms of α12.2 (254). Different LIM domains can interact with PKC, and all three LIM domains of ENH bind to PKCβI (226). PKCα, -βI, and -ζ but not PKCγ, -δ, and -ε also showed binding to ENH in vitro (226). However, only PKCε but not PKCα, PKCβII, and PKCγ coimmunoprecipitated with ENH from brain extracts (254). The differences in isoform binding to ENH versus coimmunoprecipitation with ENH might be due to variable assay sensitivity or brain-specific mechanisms that lead to different selectivity for ENH-PKC isoform interactions. PKCε also coimmunoprecipitated with Cav2.2 (254). Coexpression of ENH with Cav2.2 in Xenopus oocytes accelerated the increase in channel current as observed upon stimulation of endogenous PKC with the phorbol ester phorbol 12,13-dibutyrate (PDBu) (254). This effect depended on the COOH terminus of α12.2 consistent with the model that an ENH-PKCε complex binds to this region. A peptide derived from the NH2 terminus of PKCε that inhibits translocation of PKCε from the cytosol to the periphery blocked coimmunoprecipitation of ENH with PKCε. This peptide also inhibited phorbol ester-induced potentiation of N-type currents in neurons (254). Indirect evidence suggests that PKCδ can also be recruited to α12.2 upon activation by Gq-coupled receptors (362), although this interaction might be independent of ENH as ENH does not bind PKCδ in vitro (but see above for PKCε) (226).
An interesting twist with respect to membrane-delimited inhibition of N-type channels by Gβγ is the formation of a physical complex between the nociceptin receptor ORL1 and Cav2.2 (21). ORL1 suppressed constitutive N-type activity in a Gαi/q/Gβγ-dependent manner (21). This mechanism appears at least in part to be kinase independent. However, prolonged stimulation of ORL1 with nociceptin induced internalization of Cav2.2 in a PKC-dependent manner (4).
D. Regulation of Kv7.2/3 by AKAP150-Anchored PKC
The NH2-terminal region of AKAP150 (residues 1–143), which interacts with PKC, also binds to the membrane proximal region of the COOH terminus of Kv7.2 (residues 321–499), i.e., immediately downstream of the sixth (last) transmembrane segment (180). Coexpression of the Kv7.2 fragment spanning residues 321–499 or of mutant AKAP150 constructs lacking its PKC binding site with full-length Kv7.2 in CHO cells strongly reduces inhibition of currents induced by stimulation of the stably expressed muscarinic m1 receptor, which acts via Gq and PKC. Inhibitors that act on the diacylglycerol binding site of PKC (calphostin C and safingol) reduced muscarinic inhibition as well as phosphorylation of Kv7.2, but catalytic site inhibitors of PKC (bisindolylmaleimide and chelerythrine) did not (180). Perhaps AKAP150-anchored PKC is only accessible for the former but not latter inhibitors. It should be emphasized that depletion of PIP2, which can lead to PKC stimulation via IP3-mediated intracellular Ca2+ release, is primarily responsible for inactivation of the M current in native cells independent of PKC, with PKC only playing a modulatory role for this current (184, 377). In native superior cervical ganglion cells, however, knockdown of AKAP150 reduces muscarinic but not bradykinin-induced inhibition of endogenous M currents (179). The m1 receptor but not the bradykinin B2 receptor coimmunoprecipitated with AKAP150 from HEK293 cells when cotransfected (179). AKAP150 might thus be important for recruiting m1 to the endogenous Kv7.2 complex.
E. PKC Anchoring at the BK Signaling Complex
PKC inhibits BK channel activity at least in part by counteracting PKA-induced channel activation (13, 230). Recordings of BK channel activity in excised membrane patches or after reconstitution into planar lipid membranes imply that PKC is constitutively associated with the channel (29, 326). The PKC adaptor protein RACK1 binds to the COOH terminus of BK; this interaction might be direct or potentially mediated by other proteins present in the cell extract used by Isacson et al. (200). However, these authors were unable to detect coimmunoprecipitation of PKC with BK. Nevertheless, overexpression of RACK1 with BK in Xenopus oocytes shifted its activation curve towards more positive potentials in the absence but not presence of ectopically expressed channel β subunits. The activation time constant was decreased by RACK1 overexpression in the presence but not absence of β1. Although it is unclear whether and how PKC is associated with BK, these data suggest RACK1 as a likely link.
F. GPCR-Kir3 Complexes
Dopaminergic D2/3/4 receptors act via Gi/o and activate Kir3 channels (198, 422). Although regulation of Kir3 by Gβγ is typically initiated by Gi/o-coupled receptors, β2 AR can also stimulate Kir (243, 420), possibly in part because it can switch from Gs to Gi/o (8, 371, 420, 440, 441). D2 and D4 receptors and the β2 AR coimmunoprecipitate with different Kir3 subunits but not Kir2.1 upon coexpression in HEK293 and COS7 cells (231). Elegant BRET experiments confirmed the close proximity of these proteins with each other and with Gαs, Gβ1, and Gγ2 (231, 325). Kir3.2 coimmunoprecipitates with D2 receptor and β2 AR from brain detergent extracts after partial clearance of nonsoluble material by intermediate speed centrifugation (231). These complexes also incorporate AC II when coexpressed in cell lines; AC V or VI was detected in immunoprecipitates from brain extracts. These interactions were not sensitive to agonist treatment as determined by BRET, indicating that they are constitutive rather than transient (231, 325).
Gβ coimmunoprecipitates with Kir3.1 from solubilized atrial membranes after rigorous ultracentrifugation (298). Contrasting to some degree the findings described in the previous paragraphs, exposure of atrial membranes to GDP, which will promote reassociation of Gβγ with Gα, prevented Gβ coimmunoprecipitation with Kir3.1. This Kir3.1 complex also contained PKA, PP1, PP2A/C, and RACK1. RACK1 directly binds Gβγ (89) and PKC (338). Addition of PKC to inside-out patches of rat atrial myocytes inhibited KACh currents (298). However, the role of RACK1 was not tested in this system. PKC phosphorylates Kir3.1 in vitro (275). PKA increases KACh currents (284, 285) and PP2A decreases them (275, 285).
Activation of Kir3 channels by Gi/o-coupled GABAB receptors is well established (222, 252). FRET in combination with total internal reflection fluorescence indicates that GABA B1 and B2 subunits both are in close proximity with and likely directly linked to Kir3.2/3.4 channels at or near the cell surface (122). This interaction is independent of the activation status of the GABAB receptor and thus constitutive. Similarly, BRET, supplemented by coimmunoprecipitation experiments, suggests close proximity of GABA B1 and Kir3.1 (82). This interaction was observed independent of GABA B2 (82).
V. Regulation of Ion Channels by Calcium/Calmodulin-Dependent Protein Kinase II
A. Regulation of Ca2+ Channels by CaM and CaMKII
Ca2+ influx through Cav1.2 regulates channel activity via CaM in several different ways. During long depolarizing test pulses, Ca2+ currents through Cav1.2 decrease due to distinct voltage- and Ca2+-dependent inactivation processes starting within 50 ms and extending over a period of up to several seconds. Ca2+-dependent inactivation is usually measured as the difference in inactivation observed with Ca2+ versus Ba2+ as charge carriers. Inactivation observed with Ba2+ is largely voltage-dependent inactivation. Ca2+ binding to CaM likely induces a conformational change in the complex preformed between Ca2+-free apo-CaM and the membrane proximal region of the COOH terminus of α11.2 (Fig. 16) (109, 218, 315; for review, see Ref. 156). CaM binding to the NH2 terminus provides a further modest and typically masked contribution to Ca2+-dependent inactivation (92, 201).
FIG. 16.

Interaction of CaMKII with Cav1.2 and related targets. Blue, α11.2; magenta, β2 and its interactions with α11.2; red, CaMKII binding sites (brackets and segments) and defined phosphorylation sites (arrows and residues followed by number). Evidence for functional importance is available for CaMKII binding inside the CaM binding region (TVGKFY) and the COOH terminus of β2. Yellow, one CaM binding site has been identified in the NH2 terminus and a cluster of three sites in the COOH terminus; the cluster also interacts with β subunits (magenta arrow). Gray, PP2A binding site; black X, calpain cleavage region. Top: sequence alignment of the autoinhibitory domain of CaMKII with CaMKII binding sites on ion channel subunits. Residues at the top refer to interactions between residues in the segment of the CaMKII autoinhibitory domain that interacts with the P site under unstimulated conditions and residues in the large lobe of CaMKII. I205 is also important for binding of CaMKII to the NMDA receptor NR2B subunit (18). Boxes indicate residues that interact (or are homologous to other proteins) with CaMKII residues above.
Multiple short depolarizing pulses can lead to facilitation of certain Ca2+ channels, i.e., the peak current amplitude increases with pulse numbers (91, 232). This facilitation is especially prominent for Cav2.1 and in this case also mediated by CaM. Ca2+-dependent inactivation of Cav2.1 requires the two EF hands in the N domain of CaM, whereas Ca2+-dependent facilitation may depend either on the two EF hands in the C domain or on all four EF hands (91, 232). Ca2+-dependent facilitation is rather small, if at all detectable, for Cav1.2 because it is overshadowed by Ca2+-dependent inactivation; it can be unmasked by mutating isoleucine-1654 in α11.2, one of the residues important for Ca2+-dependent inactivation, to alanine (465). The small facilitation of Cav1.2 observed in cardiomyocytes and smooth muscle can be inhibited by CaMKII-specific blockers (105, 268, 439; see also Ref. 233). Addition of constitutively active CaMKII to excised inside-out patches prolonged single-channel open times and thereby overall current conduction by Cav1.2 (105). Ca2+-dependent facilitation of Cav1.2 is thus mediated mainly by CaMKII.
1. Regulation of Cav1.2 by CaMKII
CaMKII phosphorylates α11.2 and at least one of the β-subunit isoforms in vitro (164, 167, 205). To observe Ca2+-dependent facilitation by whole cell patch recording from HEK293 cells transfected with α11.2 and β2a, coexpression of CaMKII is necessary (233). Facilitation was prevented by KN93, which inhibits CaMKII as well as several other targets. In this system, mutating serine-1512 or serine-1570 to alanine, either individually or together, abolished the CaMKII-dependent facilitation, suggesting that these two residues are important for CaMKII-mediated facilitation. Mutating both sites reduced overall phosphorylation of full-length α11.2 and nearly abolished phosphorylation of a fusion protein spanning this region by CaMKII (233; see also Ref. 111). Similarly, inhibition of calcineurin leads to an increase in L-type currents by increasing mode 2. This increase is likely due to CaMKII as it is prevented by KN62, a KN93 analog, and after mutating serine-1512 to alanine (111). Collectively, these findings indicate that serine-1512 and -1570 are phosphorylated by CaMKII and contribute to increased channel activity by facilitation and transition to mode 2 gating. It is noteworthy that serine-1512 and -1570 flank the EF-hand motif located between the last transmembrane segment and the CaM binding region of α11.2 (Fig. 16). This motif does not appear to bind Ca2+, which is in agreement with general findings that single EF-hand motifs typically do not bind Ca2+. It rather is thought to play a structural role in mediating Ca2+-dependent inactivation that is triggered by Ca2+ binding to CaM associated with α11.2 downstream of the EF hand (e.g., Ref. 466).
To observe upregulation of the open probability of Cav1.2 by CaMKII in excised patch recordings from tsA293 cells, coexpression of β2a (or perhaps another β subunit) with α11.2 is necessary (147). In these experiments, constitutive active CaMKII was applied to the cytosolic face of the patches. Mutating threonine-498 in β2a (Fig. 16) to alanine blocked the CaMKII effect (147). It also reduced (but did not abolish) phosphorylation of β2a by CaMKII. Furthermore, ectopic expression of this mutant β2a subunit in rat cardiomyocytes abolished facilitation of L-type currents (147). These results indicate an important role of threonine-498 in β2a in the excised patch recordings and in intact cardiomyocytes. It is unclear at this point whether phosphorylation of serine-1512 and -1570 in α11.2 and threonine-498 in β2a (or on the homologous sites in β1 and β3) all have to occur for effective regulation of Cav1.2 by CaMKII as suggested by these two studies. Alternatively, the different expression and recording systems might reveal two different regulatory mechanisms of Cav1.2 by CaMKII.
2. CaMKII targeting to Cav1.2 and the NMDAR
Four closely related genes encode CaMKIIα, -β, -γ, and -δ (189). CaMKIIα and -β are the main isoforms in neurons and CaMKIIδ in heart. Immunofluorescence staining indicates that CaMKII (presumably δ) is enriched along Z-lines in cardiomyocytes (301, 435). This observation suggests the existence of anchoring mechanisms for CaMKII. The first identified anchoring protein that recruits CaMKII to a defined subcellular site is αKAP in skeletal muscle (19). αKAP is identical to the COOH-terminal portion of CaMKIIα called the association domain, which mediates multimerization of full-length CaMKII subunits to dodecamers. It is generated from an alternative promoter in the CaMKII coding region. Alternative splicing leads to addition of a hydrophobic segment at the NH2 terminus of αKAP. This segment targets αKAP-CaMKII complexes to the endoplasmic reticulum in skeletal muscle, where CaMKII may contribute to the regulation of RyRs (19).
Whether αKAP is critical for CaMKII anchoring in the vicinity of Cav1.2 is unclear. However, CaMKII and Cav1.2 coimmunoprecipitate from heart extracts (190, 233) and from HEK cells transfected with α11.2 plus β2a (233). In vitro interaction studies indicate that CaMKIIα can directly bind to multiple sites on α11.2 (188) and to residues 410–505 in β2a downstream of its GK domain (Fig. 16) (147). One of the α11.2 sites is immediately upstream of and partially overlapping with the third CaM binding site in the membrane-proximal region of the α11.2 COOH-terminal region, which contains the IQ motif (residues 1644–1649 in cardiac α11.2 from rabbit; Fig. 16) (188). The functional relevance of this site was tested in α11.2 with isoleucine at position 1654 in the IQ motif mutated to alanine. This α11.2 mutant shows clear Ca2+-dependent facilitation, which is abolished by additional mutation of the CaMKII binding site formed by residues 1644–1649 (188). However, whether this mutation abrogated Ca2+-dependent facilitation by preventing CaMKII binding to this site or by disrupting the precise interactions between CaM and α11.2 is unclear because this CaMKII binding site overlaps with residues critical for CaM binding including phenylalanine 1648 (115, 401) (Fig. 16). Furthermore, this region of α11.2 also binds to the NH2-terminal half of the β-subunit SH3 domain and the mutation might affect this interaction (458). Finally, overall binding of CaMKII to Cav1.2 and phosphorylation of α11.2 by CaMKII in vitro were not affected by mutating residues 1644–1649 (188).
Whether binding to residues 410–505 of β2a is necessary for CaMKII anchoring at Cav1.2 is untested. However, threonine-498 is clearly critical for CaMKII-mediated effects on Cav1.2 (147). The region surrounding threonine-498 exhibits striking similarity to the autoinhibitory domain of CaMKII and also to the CaMKII binding sites in the COOH terminus of the NMDAR NR2B subunit (18, 235, 237, 373, 374) and the Drosophila K+ channel Eag (dEag) (380) (Fig. 16, top). Serine-1303 in NR2B and threonine-286 in the autoinhibitory CaMKII segment are phosphorylated by CaMKII to inhibit their binding to the so-called T site of CaMKII, which is formed in part by tryptophan-237 and valine-298 (Fig. 16, top). Disruption of the interaction of the autoinhibitory CaMKII segment with the T site either by threonine-286 phosphorylation or by binding of the NR2B or the homologous Eag site allows CaMKII to maintain its activity after Ca2+/CaM has been removed (18, 247, 380). Finally, CaMKII binding to NR2B depends on induction of an active CaMKII conformation by either the presence of Ca2+/CaM or by T286 autophosphorylation (18, 235, 237, 373, 374). Therefore, β2a may similarly regulate CaMKII activity at Cav1.2.
3. CaMKII interactions with Cav2.1
When expressed in the HEK293-derived tsA-201 cells, Cav2.1 shows faster voltage-dependent inactivation in the presence of two different CaMKII inhibitors (the organic compund KN93 and ectopic expression of the endogenously occurring inhibitory polypeptide CaM-KIIN) than under control conditions (207). This effect is independent of interactions between CaM and either the IQ-like CaM binding motif (IQ domain) or the downstream CaM binding domain (CBD). The IQ-like motif is homologous to the third CaM binding site in the COOH terminus of α11.2, but a CBD that would functionally be equivalent to the CBD in α12.1 has not been identified in α11.2. However, a peptide derived from the α12.1 region that is homologous to the CaMKII binding region of α11.2 located upstream and partially overlapping with the IQ-like motif (residues 1897–1912 in α12.1) blocked pull-down of CaMKII by the COOH terminus of α12.1. The peptide also accelerated voltage-dependent inactivation of α12.1, similar to the effects of KN93 and CaM-KIIN (207). Because CaMKII coexpression increases inactivation when Ba2+ was used as charge carrier instead of Ca2+, which could otherwise activate CaMKII, the authors hypothesize that CaMKII might mainly act via binding to α12.1 and not by phosphorylating it. In fact, the AIP peptide, which is derived from the autoinhibitory domain of CaMKII and typically also a very effective and specific CaMKII blocker, had no effect on Cav2.1 currents obtained in the presence of ectopically expressed CaMKII. However, neither KN93 nor CaM-KIIN inhibited binding of CaMKII to the α12.1 COOH terminus, although they blocked the CaMKII effect. Although it is possible that CaMKII has to be in a certain conformation that is similar to the active one to exert its effect on Cav2.1 and that this conformation is prevented by either inhibitor, it is equally possible that AIP did not effectively gain access to the α12.1-CaMKII complex, whereas KN93 and CaM-KIIN did and thus might have acted by actually blocking basal CaMKII activity. This latter possibility is quite conceivable because CaMKII binding to NMDAR and dEAG has been found to induce autonomous activity in CaMKII (see sect. v, A2 and B) (18, 247, 380).
4. Regulation of Cav3.2 by CaMKII
T-type channels are activated by relatively small depolarizations with optimal activity when membrane potentials are around −50 mV. At such low potentials, the driving force for Ca2+ influx into cells is high. T-type channels can, therefore, mediate a substantial amount of Ca2+ influx. Three different genes encoding α1 subunits that form T-type channels exist: α13.1, α13.2, and α13.3. CaMKII selectively increases the activity of Cav3.2 but not Cav3.1 by shifting the half-activation potential to hyperpolarized potentials (421, 432). In other words, CaMKII lowers the magnitude of depolarization required to open Cav3.2, thus increasing the number of open channels at more negatively polarized potentials, which in turn provide a higher driving force for Ca2+ influx. This effect is mediated by phosphorylation of serine-1198 in the intracellular loop that connects domains II and III of the pore-forming channel portion (421, 448). Endogenous CaMKII also regulates Cav3.2 during excised membrane patch recordings, suggesting that CaMKII is anchored near or on the channel complex (15). Biochemical evidence indicates that CaMKIIγ can directly bind to loop II/III of α13.2 but not α13.1 (448). This interaction is not increased by activation of CaMKII in contrast to CaMKII binding to the NMDAR (18, 235, 237, 372, 373) and the eag K+ channel in Drosophila (see Fig. 4) (380). It is unclear whether CaMKII has to be associated with Cav3.2 for its regulation. In fact, activation of CaMKII by Ca2+/CaM and ATP leads to phosphorylation of serine-1198, which in turn results in the release of CaMKII from loop II/III. Accordingly, CaMKII can associate with Cav3.2 under basal conditions when Ca2+ concentrations are low but dissociates from the channel after stimulation and phosphorylation of serine-1198.
B. CaMKII Targeting to Drosophila K+ Channel Eag
dEag was first cloned from the Drosophila genetic ether-a-go-go locus (414). It is homologous to mammalian Kv10 channels, which belong to a superfamily of K+ channels that includes Erg (Kv11) and Elk (Kv12) channels (415). Its overall structure is similar to Shaker-type K+ channels (see also Fig. 3A). CaMKII phosphorylates dEag on threonine-787 in its COOH terminus (413). Inhibition of CaMKII and mutation of threonine-787 to alanine reduced dEag current amplitude and accelerated inactivation (413). CaMKII binds to a segment in the COOH terminus of dEag that is homologous to its binding site on NR2B (residues 773–794 in Eag; Fig. 16, top). As for CaMKII binding to NR2B (see sect. vA2), dEag binding requires active CaMKII as induced either by the presence of Ca2+/ CaM or by T286 autophosphorylation (380). Furthermore, binding of CaMKII to dEag keeps CaMKII in a constitutively active conformation (380) analogous to its binding to NR2B (see sect. vA2) (18).
Given that dEag also binds Slob (for Slowpoke binding protein) (352), Eag forms an intriguing signaling complex that could be regulated by phosphorylation-induced recruitment of 14-3-3ζ to Slob, although no further evidence supporting this hypothesis is currently available. Slob was originally identified as a protein associated with the Drosophila BK channel dSlo. Slob directly binds and recruits 14-3-3ζ to dSlo (460). As with most other 14-3-3 interactions, this interaction is mediated by phosphorylated serine residues. There are two different sites in Slob that can independently bind 14-3-3ζ: serine-54 and serine-79. Both sites are phosphorylated by CaMKII. Overexpression of constitutive active CaMKII leads to increased coimmunoprecipitation of Slob with 14-3-3ζ, and ectopic expression of a CaMKII inhibitory peptide causes decreased coimmunoprecipitation (460). 14-3-3ζ decreases dSlo channel activity by shifting the current-voltage curve to the right. Accordingly, channel activity will be lower in the presence of 14-3-3ζ at a given depolarization level (460).
VI. Regulation of Ion Channels by Src
A. Exemplary Regulation of the NMDAR by Src
Src-mediated stimulation of NMDAR activity is quite well characterized. This stimulation depends on the association of Src and its upstream activator Pyk2 with the NMDAR complex (187, 251, 345, 357, 454). Src associates with the NMDAR via two different proteins. The NADH dehydrogenase subunit ND2 is a mitochondrial protein that exists outside mitochondria at postsynaptic sites where it links Src to the NMDAR (138). Src binds with its unique domain located between the NH2-terminal SH4 and the SH3 domain to residues 239–321 of ND2. Src also interacts with its SH2 domain with the NH2 terminus of PSD-95 in an unusual phosphotyrosine-independent manner (211). This interaction inhibits Src activity but localizes it next to the tyrosine kinase Pyk2, which binds to the SH3 domain of PSD-95 (357). Autophosphorylation of Pyk2, which is also targeted to the NMDAR via PSD-95 (357), on tyrosine-402 creates a Src binding site, which recruits Src and activates it upon binding in other systems (93). Upon its activation, Pyk2 could reposition Src from its preassociated site on the NH2 terminus of PSD-95 under resting conditions to phosphorylated tyrosine 402 on Pyk2.
B. Regulation of Cav1.2 by Src
Regulation of voltage-gated ion channels by tyrosine phosphorylation has received more modest attention. Src increases current activity of L-type channels in smooth muscle and other cells including neurons (108, 183, 375, 434). Src-mediated upregulation of L-type channel currents can be triggered by integrin signaling (149, 434), perhaps via the Pyk2-related focal adhesion kinase FAK, although no direct experimental evidence that supports the latter notion is available. Tyrosine-2122 in the COOH-terminal region of rat α11.2 has been identified as a critical residue for phosphorylation by Src and the ensuing upregulation in Cav1.2 activity (23, 149). Similar to the main PKA site serine-1928 (see sect. iiB2a), tyrosine-2122 is cleaved off in the short form of α11.2 (23), although the COOH-terminal fragment may still functionally interact with Cav1.2 (see sect. iiiB2b). Of interest with respect to kinase targeting is the finding that Src specifically bound in vitro to a fusion protein of the COOH-terminal region of α11.2 downstream of the cleavage site (23). This COOH-terminal region contains two sites that match the consensus binding sequence for the SH3 domain of Src (RPLPXXP). Cav1.2 might coimmunoprecipitate with Src from colonic smooth muscle cells, but it is unclear whether the coimmunoprecipitation was specific because no negative control was exhibited (183). Whether the reported Src-Cav1.2 interactions reflect constitutive recruitment of Src to the channel complex perhaps via its SH3 domain or a catalytic site-substrate interaction and whether this interaction is necessary for effective phosphorylation and regulation of Cav1.2 by Src remains to be demonstrated. It should be noted that tyrosine-2122 is not conserved in rabbit α11.2 and might thus not be the only Src phosphorylation site in α11.2.
C. Binding of Src to BK and Kv1.5
Src coimmunoprecipitates with the Drosophila BK channel dSlo when expressed in HEK293 cells (410). Whether Src binds directly or through adaptor proteins is unknown. It is unclear how Src regulates dSlo, but initial preliminary data suggest that it regulates the subcellular distribution of BK (410). Overexpression of Src with mammalian BK in HEK293 cells increases channel activity by shifting the current-voltage curve to the left but only in the presence of 1–120 μM Ca2+ (246). Furthermore, Src phosphorylates BK, likely on tyrosine-766, because mutation of this tyrosine to phenylalanine prevented the Src-induced phosphorylation and increase in channel activity (246). Pyk2 might be upstream of Src activation in this system (245).
Src also directly binds a proline-rich domain in the NH2 terminus of human Kv1.5 (177). This domain is not conserved in rat Kv1.5, which does not bind Src. Src decreases channel activity of human Kv1.5 (177).
VII. Regulation of Ion Channels by Anchored Serine/Threonine Phosphatases
A. Serine/Threonine Phosphatase Targeting to Ion Channels Exemplified by AMPAR and RyR
Known mammalian phosphatases that dephosphorylate serine and threonine residues are designated as PP1, PP2A, PP2B, PP2C, PP4, PP5, PP6, and PP7. The catalytic subunits of these phosphatases are structurally related and form the PPP family (for “protein phosphatase of phosphorylase”). The exception is PP2C and related nuclear phosphatases, which form the PPM family (for “protein phosphatase requiring magensium”). Similar to kinases, phosphatases are associated with some if not many of their substrates (206, 405), often via scaffolding proteins that also anchor kinases (16). For example, AKAP79/ 150 does not only anchor PKA but also PP2B (calcineurin) (68, 305). The association of AKAP75/150 with the AMPAR via SAP97 and PSD-95/stargazin (see sect. iiiB4b) regulates dephosphorylation of serine-845, the PKA phosphorylation site on the GluR1 subunit of AMPAR (70, 388). The AKAP Yotiao constitutes another example that recruits both PKA and, in this case, the protein phosphatase PP1 to the NMDAR (426).
Monomeric PP1 consists of a catalytic subunit that exists in four isoforms encoded by three different genes (PP1 α, β, γ1, and γ2; the latter two arise by differential splicing from the same gene; PP1β had also been named PP1δ). PP1 binds mainly with a hydrophobic groove opposite its catalytic site to (R/K)(V/I)XF motifs present in anchoring proteins that recruit PP1 to other substrates, although additional interactions provide further stabilization and selectivity for one versus the other PP1 isoforms (47, 107, 390). For example, neurabin and the related spinophilin/neurabin 2 (2, 289, 347) recruit PP1 to various targets including ion channels. Spinophilin is critical for regulation of dynamic phosphorylation and dephosphorylation of AMPAR and therefore postsynaptic AMPAR function (182, 442). Spinophilin has an LZ motif that mediates binding to other targets including RyR2 (266). The (R/K)(V/I)XF motif also mediates PP1's inhibitory interaction with its endogenous blockers, inhibitor 1, DARPP-32, and perhaps also inhibitor 2 (229). Yotiao, which docks PKA and PP1 at the NMDAR, contains a KVXF sequence, but this sequence is apparently not necessary for its interaction with PP1 (426). However, PP1 binding to AKAP149 (also known as d-AKAP1 or AKAP1, Ref. 433) and AKAP220 (AKAP11) is mediated by this or a similar motif (349, 370). Although evidence indicates that PP1 regulates Ca2+ channels including Cav1.2, none of the PP1 adaptor proteins discussed in this paragraph has been reported to interact with a Ca2+ channel, and it is unclear whether and how PP1 is targeted to those channels.
PP2A is intimately associated with several of its substrates including RyR2 (266), β ARs (318), and CaMKIV (424). PP2A forms complexes with other kinases such as casein kinase II (168) and the p70 S6 kinase (425). PP2A is a heterotrimer composed of catalytic (C), structural (A), and regulatory (B) subunits (171, 206, 320, 359). To date, over 15 PP2A B subunits have been identified that are subdivided into three classes (B, B′, and B″) (73, 206, 270, 320). The substrate specificity of PP2A is at least in part determined by the B subunit present in the holoenzyme (320). The B” subunit PR130 (463) mediates PP2A binding to RyR2 by forming a leucine/isoleucine zipper interaction with the second leucine/isoleucine zipper segment of RyR2 (266). The first leucine/isoleucine segment of this receptor binds to PP1 (see previous paragraph) and the third to mAKAP (see sect. iiiB4d) (266). The first and third but not second leucine/isoleucine segments are present in the sekletal muscle RyR1, which also interacts with PP1 and PKA but not PP2A (266).
B. Regulation of Cav1.2 by PP1 and PP2A
Rundown of L-type channel currents in inside-out patches after excision from rabbit ventricular myocytes is reversed by endogenous and also purified PKA and inhibited by the phosphatase inhibitor okadaic acid (306). Okadaic acid also strongly increases currents through porcine ventricular L-type channels when reconstituted into lipid bilayers, although it is unclear how phosphatase inhibition can actually lead to such an increase in the absence of ATP (411). These results suggest that, like PKA, a phosphatase must be docked near or at the channel that antagonizes PKA-mediated regulation of Cav1.2 (81). Several pieces of evidence collectively indicate that PP2A and in some systems PP1 are involved in reversing PKA-mediated phosphorylation (171). For instance, application of purified PP1 and PP2A inhibits whole cell L-type currents in guinea pig and rat heart cells that have been upregulated by β-adrenergic stimulation; neither phosphatase affected basal currents in these cases (172). These findings suggest that in guinea pig heart PP1 and PP2A antagonize PKA-mediated upregulation of L-type currents because the rise in channel activity upon β-adrenergic stimulation is PKA dependent (see sect. iiiB3). In frog heart, PP1/PP2A inhibitors also reverse or occlude PKA-mediated increases in L-type currents (123, 158). In mouse ventricular myocytes, it is mainly PP1 that counteracts the stimulatory action of PKC on L-type currents (102).
A more detailed analysis indicates that PP1 and PP2A differentially regulate channel availability and length of openings. As discussed in section iiiB2b, Cav1.2 exists in three major modes. It shows no openings in mode 0, shows brief frequent openings in mode 1, and long openings and short closings in mode 2 (173). β-Adrenergic stimulation induces a switch from mode 0 to mode 1 or mode 2 in frog ventricular cells (20). Okadaic acid inhibits switching from mode 2 to mode 1 in cell-attached patch recordings from cardiac and smooth muscle cells while PP2A application to inside-out patches from human umbilical vein smooth muscle cells promotes such a switch (146, 307, 428). In guinea pig ventricular myocytes, the transition from mode 1 to 0 is less sensitive to okadaic acid. Calyculin A, on the other hand, inhibits the switch from mode 1 to 0 more effectively than the switch from mode 2 to 1 (428). Okadaic acid inhibits PP2A with higher potency than PP1, but calyculin A is about equipotent towards both phosphatases. Those results, therefore, indicate that PP2A plays a predominant role in the reversal of mode 2 to mode 1 and PP1 in that of mode 1 to mode 0. In rat heart, the situation may be reversed, with PP2A being more critical for switching Cav1.2 from mode 1 to 0 and PP1 for the mode 2 to 1 transition (307).
Initial experiments to determine the phosphorylation status of α11.2 in intact neurons before and after PKA stimulation revealed that α11.2 phosphorylation was unstable even during advanced purification steps unless a cocktail of serine/threonine phosphatase inhibitors including 2–4 μM microcystin LR was continuously present (Hell, unpublished observations; Ref. 166). Furthermore, okadaic acid prevents rundown of L-type channel currents in inside-out patches (306) (see above). These observations suggest that a phosphatase is associated and copurifies with Cav1.2. In fact, a more careful analysis shows that PP2A but not the γ isoforms of PP1 coimmunoprecipitates with Cav1.2 (81). This endogenous phosphatase reverses PKA-mediated phosphorylation of serine-1928 on α11.2 (81). Bacterially expressed fusion proteins of the catalytic subunit of PP2A (PP2A/C) binds directly to fusion proteins of the COOH terminus of α11.2 (81). This association is not due to a rather long-lasting catalytic site interaction as microcystin, which prevents access to the PP2A/C catalytic site, does not inhibit the PP2A/C-α11.2 COOH-terminal interaction. More recent studies show that PP2A/C binds ∼30–50 residues downstream of serine-1928 (154), nesting PP2A between this PKA phosphorylation site and the COOH-terminal AKAP150 binding site (see sect. iiiB2c; Fig. 6). These studies also indicate that binding to that site is critical for efficient dephosphorylation of serine-1928 in intact HEK293 cells. In general, B-type subunits are thought to dock PP2A at its targets. However, the work on the PP2A/C-α11.2 interaction unraveled an unexpected way of PP2A/C anchoring that is completely independent of the scaffolding A subunit or a B-type subunit (81, 154).
C. Regulation of Cav1.2 by PP2B
Although earlier work did not detect any effects of PP2B inhibition on L-type currents in the heart (123, 170) or neurons (403), more recent work indicates otherwise (304, 412). Furthermore, there is also evidence that Cav1.2 in smooth muscle cells is downregulated by PP2B (354). Especially well worked out are details for PP2B targeting to and regulation of Cav1.2 in neurons (304). PP2B interacts with AKAP79/150 (68, 305). AKAP79/150 is associated with Cav1.2 (153) (see sect. iiiB4a) and recruits PP2B to the Cav1.2 complex (304). Manipulation of the binding site for PP2B on AKAP79/150 and for AKAP79/150 on α11.2 indicates that tethering of PP2B to α11.2 by AKAP79/150 is important for highly dynamic and localized reversal of PKA-mediated upregulation of Cav1.2. However, it is somewhat surprising that Ca2+ influx through Cav1.2 activates PP2B without causing a more pronounced Ca2+-dependent inactivation via Cav1.2-associated CaM as seen by other investigators (see sect. vA).
D. Reversal of PKC-Mediated Cav2.2 Phosphorylation by Cav2.2-Anchored PP2C
The phosphatase PP2C shares little sequence similarity with other main serine/threonine phosphatases. Because no effective inhibitor is established for this phosphatase, little is known about its function. Recent work showed that PP2C directly binds to the COOH termini of α11.2, α12.1, and α12.2 (240). PP2C binds with its own COOH-terminal region, which is downstream of its catalytic domain, to a site that is likely within residues 1708–1864 in α12.2, right after the IVS6 segment (240). Among the four main phosphatases (i.e., PP1, 2A, 2B, and 2C), PP2C is most effective in dephosphorylation of PKC-phosphorylated α12.2 loop II/III (240). However, PKC-phosphorylated loop I/II was only partially dephosphorylated by PP2C. Overexpression of PP2C diminishes the phorbol ester-induced potentiation of Cav2.2 ectopically expressed in tsA201 cells. Ectopic expression of the COOH-terminal PP2C fragment acts as a dominant negative construct by increasing potentiation of Cav2.2 in neurons upon treatment with phorbol ester, which likely acts through PKC in this case (240). These data indicate that PP2C counteracts the stimulatory effect of PKC by dephosphorylation of loop II/III residues and perhaps one or more specific sites in loop I/II while other loop I/II phosphorylation sites are dephosphorylated by other phosphatases. As discussed above, PKC can prevent the inhibitory binding of syntaxin 1A to loop II/III and of Gβγ to loop I to the channel. Either of these mechanisms could be antagonized by Cav2.2-anchored PP2C.
E. Association of Casein Kinase 2 and PP2A With SK2
1. Association of casein kinase 2 with SK2
SK2 directly recruits casein kinase 2 (CK2), along with PP2A, which in turn regulate CaM phosphorylation and thereby its Ca2+ sensitivity in the SK2 complex (1, 30, 255). CK2 directly binds to three different SK2 segments, one in the NH2 terminus and two in the COOH terminus (1). The interaction between a positively charged segment in the NH2-terminal domain (specifically lysine-121) and CK2 promotes activation of CK2, which is otherwise accomplished by small basic molecules such as spermine. CK2 phosphorylates CaM on threonine-80. This phosphorylation occurs only when SK2 is in the closed state. It reduces the affinity of CaM in the SK2 complex for Ca2+, thereby reducing activation of SK2. It also leads to faster deactivation of SK2. SK2 also mediates the reduction of SK2 activity upon noradrenergic stimulation (255).
SK2 is in close proximity and tightly functionally coupled to the NMDAR at postsynaptic sites (114, 296). Although there is no evidence that CK2 is associated with the NMDAR complex, CK2 can phosphorylate serine-1480 of the NR2B subunit (64). This residue is critical for binding of NR2B to PDZ domains of PSD-95 and its homologs and its phosphorylation disrupts these interactions, thereby reducing NR2B surface expression (64). In this signaling complex, CK2 is directly or indirectly activated by CaMKII. CaMKII is directly associated with the NMDAR NR1 and NR2B subunits (237, 277, 373). It is conceivable that SK2 shares CK2 with the NMDAR complex.
2. Association of PP2A with SK2
PP2A binding to SK2 is mediated by a direct interaction of the structural PP2A A subunit with the COOH terminus of SK2 (30). As discussed in section viiA, PP2A targeting is typically mediated by one of more than a dozen B subunits. The A subunit functions to support the interaction of the catalytic C subunit with a B-type subunit. A direct interaction of the A subunit with a PP2A target is yet another example of targeting of PP2A to an ion channel independent of the B subunit (see the direct binding of the C subunit to Cav1.2; see sect. viiB). PP2A binding to SK2 requires the sequence EQRK (residues 469–472) in the SK2 COOH terminus (1, 255). Mutating this sequence to AEAA not only inhibited binding of PP2A to SK2 but also functionally prevented PP2A from counteracting CK2-mediated acceleration of channel deactivation. Although the catalytic CK2α subunit can also directly bind PP2A (168), these findings clearly show that PP2A has to be directly bound to SK2 for dephosphorylation of CaM in the SK2 complex. Consistent with this notion is that SK2 binds both the CK2α and the regulatory CK2β subunit which form heterotetrameric CK2 complexes that do not by themselves bind PP2A (168).
VIII. Conclusion
Only those structural and functional interactions of kinases and phosphatases with voltage-gated ion channels that are well established by coimmunopreciptiation and in vitro interaction assays could be discussed in this review. These interactions likely constitute only the tip of the iceberg, and many more such interactions will turn out to be critical for a variety of physiological functions. Similar complexes exist for other ion channels and, if at all, could only be mentioned in passing. Examples include the Trp-InaD complex in the Drosophila eye (see sect. ivA) and the signaling complex assembled by the NMDAR (see sects. iiiB4c, vA2, and viA).
Acknowledgments
We thank Dr. Mei Shi for the performance of coimmunoprecipitation experiments of SK2 with α-actinin and Dr. J. Adelman (Vollum Institute, Portland, OR) for the SK2 antibodies.
GRANTS: The work by the authors described in this review was supported by National Institute of Neurological Disorders and Stroke Grants R01-NS-35563 and R01-NS-46450 (to J. W. Hell), National Institute on Aging Grant R01-AG-17502 (to J. W. Hell), American Heart Association Established Investigator Grant 0040151N (to J. W. Hell), and the American Heart Association Scientist Development Grant 0040151N (to D. D. Hall). The content of this publication is solely the responsibility of the authors and does not necessarily represent the official views of the National Institute on Health or the American Heart Association.
Footnotes
Note Added in Proof: After acceptance of this manuscript, Lemke et al. (234a) reported on the functional relevance of Cav1.2 serine-1928 phosphorylation in response to β-adrenergic signaling within cardiac myocytes. Mutating serine-1928 to alanine in knock-in mice did not affect basal L-type calcium currents, β-adrenergic-dependent upregulation with isoproterenol, or PKA-dependent stimulation with forskolin in isolated ventricular myocytes. Furthermore, neither basal nor isoproterenol-stimulated cardiac contractility was affected in the knock-in animals. These results indicate that serine-1928 is not the main PKA phosphorylation site that mediates the strong upregulation in Cav1.2 current. However, more modest contributions of serine-1928 phosphorylation to Cav1.2 regulation cannot be ruled out at this point. For instance, a recent abstract by Rankovic et al. (323a) suggests that PKA regulates calcium-dependent inactivation of L-type channels in thalamocortical relay neurons in response to β-adrenergic stimulation.
References
- 1.Allen D, Fakler B, Maylie J, Adelman JP. Organization and regulation of small conductance Ca2+-activated K+ channel multi-protein complexes. J Neurosci. 2007;27:2369–2376. doi: 10.1523/JNEUROSCI.3565-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Allen PB, Ouimet CC, Greengard P. Spinophilin, a novel protein phosphatase 1 binding protein localized to dendritic spines. Proc Natl Acad Sci USA. 1997;94:9956–9961. doi: 10.1073/pnas.94.18.9956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Altier C, Dubel SJ, Barrere C, Jarvis SE, Stotz SC, Spaetgens RL, Scott JD, Cornet V, De Waard M, Zamponi GW, Nargeot J, Bourinet E. Trafficking of L-type calcium channels mediated by the postsynaptic scaffolding protein AKAP79. J Biol Chem. 2002;277:33598–33603. doi: 10.1074/jbc.M202476200. [DOI] [PubMed] [Google Scholar]
- 4.Altier C, Khosravani H, Evans RM, Hameed S, Peloquin JB, Vartian BA, Chen L, Beedle AM, Ferguson SS, Mezghrani A, Dubel SJ, Bourinet E, McRory JE, Zamponi GW. ORL1 receptor-mediated internalization of N-type calcium channels. Nat Neurosci. 2006;9:31–40. doi: 10.1038/nn1605. [DOI] [PubMed] [Google Scholar]
- 5.An R, Heath BM, Higgins JP, Koch WJ, Lefkowitz RJ, Kass RS. Beta2-adrenergic receptor overexpression in the developing mouse heart: evidence for targeted modulation of ion channels. J Physiol. 1999;516:19–30. doi: 10.1111/j.1469-7793.1999.019aa.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Arikkath J, Campbell KP. Auxiliary subunits: essential components of the voltage-gated calcium channel complex. Curr Opin Neurobiol. 2003;13:298–307. doi: 10.1016/s0959-4388(03)00066-7. [DOI] [PubMed] [Google Scholar]
- 7.Arnot MI, Stotz SC, Jarvis SE, Zamponi GW. Differential modulation of N-type 1B and P/Q-type 1A calcium channels by different G protein subunit isoforms. J Physiol. 2000;527:203–212. doi: 10.1111/j.1469-7793.2000.00203.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Asano T, Katada T, Gilman AG, Ross EM. Activation of the inhibitory GTP-binding protein of adenylate cyclase, Gi, by beta-adrenergic receptors in reconstituted phospholipid vesicles. J Biol Chem. 1984;259:9351–9354. [PubMed] [Google Scholar]
- 9.Atkinson NS, Robertson GA, Ganetzky B. A component of calcium-activated potassium channels encoded by the Drosophila slo locus. Science. 1991;253:551–555. doi: 10.1126/science.1857984. [DOI] [PubMed] [Google Scholar]
- 10.Baillie GS, Sood A, McPhee I, Gall I, Perry SJ, Lefkowitz RJ, Houslay MD. β-Arrestin-mediated PDE4 cAMP phosphodiesterase recruitment regulates β-adrenoceptor switching from Gs to Gi. Proc Natl Acad Sci USA. 2003;100:940–945. doi: 10.1073/pnas.262787199. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 11.Balijepalli RC, Foell JD, Hall DD, Hell JW, Kamp TJ. From the cover: localization of cardiac L-type Ca2+ channels to a caveolar macromolecular signaling complex is required for beta2-adrenergic regulation. Proc Natl Acad Sci USA. 2006;103:7500–7505. doi: 10.1073/pnas.0503465103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Barhanin J, Lesage F, Guillemare E, Fink M, Lazdunski M, Romey G. K(V)LQT1 and lsK (minK) proteins associate to form the I(Ks) cardiac potassium current. Nature. 1996;384:78–80. doi: 10.1038/384078a0. [DOI] [PubMed] [Google Scholar]
- 13.Barman SA, Zhu S, White RE. Protein kinase C inhibits BKCa channel activity in pulmonary arterial smooth muscle. Am J Physiol Lung Cell Mol Physiol. 2004;286:L149–L155. doi: 10.1152/ajplung.00207.2003. [DOI] [PubMed] [Google Scholar]
- 14.Baroudi G, Qu Y, Ramadan O, Chahine M, Boutjdir M. Protein kinase C activation inhibits Cav1.3 calcium channel at NH2-terminal serine 81 phosphorylation site. Am J Physiol Heart Circ Physiol. 2006;291:H1614–H1622. doi: 10.1152/ajpheart.00095.2006. [DOI] [PubMed] [Google Scholar]
- 15.Barrett PQ, Lu HK, Colbran R, Czernik A, Pancrazio JJ. Stimulation of unitary T-type Ca2+ channel currents by calmodulin-dependent protein kinase II. Am J Physiol Cell Physiol. 2000;279:C1694–C1703. doi: 10.1152/ajpcell.2000.279.6.C1694. [DOI] [PubMed] [Google Scholar]
- 16.Bauman AL, Scott JD. Kinase- and phosphatase-anchoring proteins: harnessing the dynamic duo. Nature Cell Biol. 2002;4:E203–E206. doi: 10.1038/ncb0802-e203. [DOI] [PubMed] [Google Scholar]
- 17.Bauman AL, Soughayer J, Nguyen BT, Willoughby D, Carnegie GK, Wong W, Hoshi N, Langeberg LK, Cooper DM, Dessauer CW, Scott JD. Dynamic regulation of cAMP synthesis through anchored PKA-adenylyl cyclase V/VI complexes. Mol Cell. 2006;23:925–931. doi: 10.1016/j.molcel.2006.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bayer KU, De Koninck P, Leonard AS, Hell JW, Schulman H. Interaction with the NMDA receptor locks CaMKII in an active conformation. Nature. 2001;411:801–805. doi: 10.1038/35081080. [DOI] [PubMed] [Google Scholar]
- 19.Bayer KU, Harbers K, Schulman H. alphaKAP is an anchoring protein for a novel CaM kinase II isoform in skeletal muscle. EMBO J. 1998;17:5598–5605. doi: 10.1093/emboj/17.19.5598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bean BP, Nowycky MC, Tsien RW. β-Adrenergic modulation of calcium channels in frog ventricular heart cells. Nature. 1984;307:371–375. doi: 10.1038/307371a0. [DOI] [PubMed] [Google Scholar]
- 21.Beedle AM, McRory JE, Poirot O, Doering CJ, Altier C, Barrere C, Hamid J, Nargeot J, Bourinet E, Zamponi GW. Agonist-independent modulation of N-type calcium channels by ORL1 receptors. Nat Neurosci. 2004;7:118–125. doi: 10.1038/nn1180. [DOI] [PubMed] [Google Scholar]
- 22.Beene DL, Scott JD. A-kinase anchoring proteins take shape. Curr Opin Cell Biol. 2007;19:192–198. doi: 10.1016/j.ceb.2007.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bence-Hanulec KK, Marshall J, Blair LA. Potentiation of neuronal L calcium channels by IGF-1 requires phosphorylation of the alpha1 subunit on a specific tyrosine residue. Neuron. 2000;27:121–131. doi: 10.1016/s0896-6273(00)00014-3. [DOI] [PubMed] [Google Scholar]
- 24.Berkefeld H, Sailer CA, Bildl W, Rohde V, Thumfart JO, Eble S, Klugbauer N, Reisinger E, Bischofberger J, Oliver D, Knaus HG, Schulte U, Fakler B. BKCa-Cav channel complexes mediate rapid and localized Ca2+-activated K+ signaling. Science. 2006;314:615–620. doi: 10.1126/science.1132915. [DOI] [PubMed] [Google Scholar]
- 25.Bertaso F, Ward RJ, Viard P, Milligan G, Dolphin AC. Mechanism of action of Gq to inhibit G beta gamma modulation of Cav2.2 calcium channels: probed by the use of receptor-G alpha tandems. Mol Pharmacol. 2003;63:832–843. doi: 10.1124/mol.63.4.832. [DOI] [PubMed] [Google Scholar]
- 26.Bichet D, Cornet V, Geib S, Carlier E, Volsen S, Hoshi T, Mori Y, De Waard M. The I-II loop of the Ca2+ channel alpha1 subunit contains an endoplasmic reticulum retention signal antagonized by the beta subunit. Neuron. 2000;25:177–190. doi: 10.1016/s0896-6273(00)80881-8. [DOI] [PubMed] [Google Scholar]
- 27.Bichet D, Haass FA, Jan LY. Merging functional studies with structures of inward-rectifier K+ channels. Nature Rev. 2003;4:957–967. doi: 10.1038/nrn1244. [DOI] [PubMed] [Google Scholar]
- 28.Biddlecome GH, Berstein G, Ross EM. Regulation of phospholipase C-beta1 by Gq and m1 muscarinic cholinergic receptor. Steady-state balance of receptor-mediated activation and GTPase-activating protein-promoted deactivation. J Biol Chem. 1996;271:7999–8007. doi: 10.1074/jbc.271.14.7999. [DOI] [PubMed] [Google Scholar]
- 29.Bielefeldt K, Jackson MB. Intramolecular and intermolecular enzymatic modulation of ion channels in excised membrane patches. Biophys J. 1994;66:1904–1914. doi: 10.1016/S0006-3495(94)80984-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bildl W, Strassmaier T, Thurm H, Andersen J, Eble S, Oliver D, Knipper M, Mann M, Schulte U, Adelman JP, Fakler B. Protein kinase CK2 is coassembled with small conductance Ca2+-activated K+ channels and regulates channel gating. Neuron. 2004;43:847–858. doi: 10.1016/j.neuron.2004.08.033. [DOI] [PubMed] [Google Scholar]
- 31.Bliss TV, Collingridge GL. A synaptic model of memory: long-term potentiation in the hippocampus. Nature. 1993;361:31–39. doi: 10.1038/361031a0. [DOI] [PubMed] [Google Scholar]
- 32.Bloodgood BL, Sabatini BL. Nonlinear regulation of unitary synaptic signals by Cav(2.3) voltage-sensitive calcium channels located in dendritic spines. Neuron. 2007;53:249–260. doi: 10.1016/j.neuron.2006.12.017. [DOI] [PubMed] [Google Scholar]
- 33.Blumenstein Y, Kanevsky N, Sahar G, Barzilai R, Ivanina T, Dascal N. A novel long N-terminal isoform of human L-type Ca2+ channel is up-regulated by protein kinase C. J Biol Chem. 2002;277:3419–3423. doi: 10.1074/jbc.C100642200. [DOI] [PubMed] [Google Scholar]
- 34.Bond CT, Maylie J, Adelman JP. SK channels in excitability, pacemaking and synaptic integration. Curr Opin Neurobiol. 2005;15:305–311. doi: 10.1016/j.conb.2005.05.001. [DOI] [PubMed] [Google Scholar]
- 35.Bourinet E, Soong TW, Sutton K, Slaymaker S, Mathews E, Monteil A, Zamponi GW, Nargeot J, Snutch TP. Splicing of alpha 1A subunit gene generates phenotypic variants of P- and Q-type calcium channels. Nat Neurosci. 1999;2:407–415. doi: 10.1038/8070. [DOI] [PubMed] [Google Scholar]
- 36.Brandon EP, Idzerda RL, McKnight GS. PKA isoforms, neural pathways, and behaviour: making the connection. Curr Opin Neurobiol. 1997;7:397–403. doi: 10.1016/s0959-4388(97)80069-4. [DOI] [PubMed] [Google Scholar]
- 37.Brenner R, Jegla TJ, Wickenden A, Liu Y, Aldrich RW. Cloning and functional characterization of novel large conductance calcium-activated potassium channel beta subunits, hKCNMB3 and hKCNMB4. J Biol Chem. 2000;275:6453–6461. doi: 10.1074/jbc.275.9.6453. [DOI] [PubMed] [Google Scholar]
- 38.Bunemann M, Frank M, Lohse MJ. Gi protein activation in intact cells involves subunit rearrangement rather than dissociation. Proc Natl Acad Sci USA. 2003;100:16077–16082. doi: 10.1073/pnas.2536719100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bunemann M, Gerhardstein BL, Gao T, Hosey MM. Functional regulation of L-type calcium channels via protein kinase A-mediated phosphorylation of the beta(2) subunit. J Biol Chem. 1999;274:33851–33854. doi: 10.1074/jbc.274.48.33851. [DOI] [PubMed] [Google Scholar]
- 40.Caceres A, Binder LI, Payne MR, Bender P, Rebhun L, Steward O. Differential subcellular localization of tubulin and the microtubule-associated protein MAP2B in brain tissue as revealed by immunocytochemistry with monoclonal hybridoma antibodies. J Neurosci. 1984;4:394–410. doi: 10.1523/JNEUROSCI.04-02-00394.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cai C, Coleman SK, Niemi K, Keinanen K. Selective binding of synapse-associated protein 97 to GluR-A α-amino-5-hydroxy-3-methyl-4-isoxazole-4-propionate receptor subunit is determined by a novel sequence motif. J Biol Chem. 2002;277:31484–31490. doi: 10.1074/jbc.M204354200. [DOI] [PubMed] [Google Scholar]
- 42.Cantrell AR, Tibbs VC, Westenbroek RE, Scheuer T, Catterall WA. Dopaminergic modulation of voltage-gated Na+ current in rat hippocampal neurons requires anchoring of cAMP-dependent protein kinase. J Neurosci. 1999;19:RC21. doi: 10.1523/JNEUROSCI.19-17-j0003.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cantrell AR, Tibbs VC, Yu FH, Murphy BJ, Sharp EM, Qu Y, Catterall WA, Scheuer T. Molecular mechanism of convergent regulation of brain Na+ channels by protein kinase C and protein kinase A anchored to AKAP-15. Mol Cell Neurosci. 2002;21:63–80. doi: 10.1006/mcne.2002.1162. [DOI] [PubMed] [Google Scholar]
- 44.Carafoli E. Calcium signaling: a tale for all seasons. Proc Natl Acad Sci USA. 2002;99:1115–1122. doi: 10.1073/pnas.032427999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Carlisle Michel JJ, Dodge KL, Wong W, Mayer NC, Langeberg LK, Scott JD. PKA-phosphorylation of PDE4D3 facilitates recruitment of the mAKAP signalling complex. Biochem J. 2004;381:587–592. doi: 10.1042/BJ20040846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Carlisle Michel JJ, Scott JD. AKAP mediated signal transduction. Annu Rev Pharmacol Toxicol. 2002;42:235–257. doi: 10.1146/annurev.pharmtox.42.083101.135801. [DOI] [PubMed] [Google Scholar]
- 47.Carmody LC, Bauman PA, Bass MA, Mavila N, DePaoli-Roach AA, Colbran RJ. A protein phosphatase-1γ1 isoform selectivity determinant in dendritic spine-associated neurabin. J Biol Chem. 2004;279:21714–21723. doi: 10.1074/jbc.M402261200. [DOI] [PubMed] [Google Scholar]
- 48.Carr DB, Day M, Cantrell AR, Held J, Scheuer T, Catterall WA, Surmeier DJ. Transmitter modulation of slow, activity-dependent alterations in sodium channel availability endows neurons with a novel form of cellular plasticity. Neuron. 2003;39:793–806. doi: 10.1016/s0896-6273(03)00531-2. [DOI] [PubMed] [Google Scholar]
- 49.Castellano A, Wei X, Birnbaumer L, Perez-Reyes E. Cloning and expression of a neuronal calcium channel beta subunit. J Biol Chem. 1993;268:12359–12366. [PubMed] [Google Scholar]
- 50.Castellano A, Wei X, Birnbaumer L, Perez-Reyes E. Cloning and expression of a third calcium channel beta subunit. J Biol Chem. 1993;268:3450–3455. [PubMed] [Google Scholar]
- 51.Catterall WA. From ionic currents to molecular mechanisms: the structure and function of voltage-gated sodium channels. Neuron. 2000;26:13–25. doi: 10.1016/s0896-6273(00)81133-2. [DOI] [PubMed] [Google Scholar]
- 52.Catterall WA. Structure and regulation of voltage-gated Ca2+ channels. Annu Rev Cell Dev Biol. 2000;16:521–555. doi: 10.1146/annurev.cellbio.16.1.521. [DOI] [PubMed] [Google Scholar]
- 53.Catterall WA, Goldin AL, Waxman SG. International Union of Pharmacology. XLVII. Nomenclature and structure-function relationships of voltage-gated sodium channels. Pharmacol Rev. 2005;57:397–409. doi: 10.1124/pr.57.4.4. [DOI] [PubMed] [Google Scholar]
- 54.Catterall WA, Perez-Reyes E, Snutch TP, Striessnig J. International Union of Pharmacology. XLVIII. Nomenclature and structure-function relationships of voltage-gated calcium channels. Pharmacol Rev. 2005;57:411–425. doi: 10.1124/pr.57.4.5. [DOI] [PubMed] [Google Scholar]
- 55.Catterall WA, Striessnig J, Snutch TP, Perez-Reyes E. International Union of Pharmacology. XL. Compendium of voltage-gated ion channels: calcium channels. Pharmacol Rev. 2003;55:579–581. doi: 10.1124/pr.55.4.8. [DOI] [PubMed] [Google Scholar]
- 56.Chen CC, Lamping KG, Nuno DW, Barresi R, Prouty SJ, Lavoie JL, Cribbs LL, England SK, Sigmund CD, Weiss RM, Williamson RA, Hill JA, Campbell KP. Abnormal coronary function in mice deficient in alpha1H T-type Ca2+ channels. Science. 2003;302:1416–1418. doi: 10.1126/science.1089268. [DOI] [PubMed] [Google Scholar]
- 57.Chen L, Chetkovich DM, Petralia RS, Sweeney NT, Kawasaki Y, Wenthold RJ, Bredt DS, Nicoll RA. Stargazing regulates synaptic targeting of AMPA receptors by two distinct mechanisms. Nature. 2000;408:936–943. doi: 10.1038/35050030. [DOI] [PubMed] [Google Scholar]
- 58.Chen L, Kurokawa J, Kass RS. Phosphorylation of the A-kinase-anchoring protein Yotiao contributes to protein kinase A regulation of a heart potassium channel. J Biol Chem. 2005;280:31347–31352. doi: 10.1074/jbc.M505191200. [DOI] [PubMed] [Google Scholar]
- 59.Chen L, Marquardt ML, Tester DJ, Sampson KJ, Ackerman MJ, Kass RS. Mutation of an A-kinase-anchoring protein causes long-QT syndrome. Proc Natl Acad Sci USA. 2007;104:20990–20995. doi: 10.1073/pnas.0710527105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Chen Y, Harry A, Li J, Smit MJ, Bai X, Magnusson R, Pieroni JP, Weng G, Iyengar R. Adenylyl cyclase 6 is selectively regulated by protein kinase A phosphorylation in a region involved in Gαs stimulation. Proc Natl Acad Sci USA. 1997;94:14100–14104. doi: 10.1073/pnas.94.25.14100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Chen Y, Yu FH, Surmeier DJ, Scheuer T, Catterall WA. Neuromodulation of Na+ channel slow inactivation via cAMP-dependent protein kinase and protein kinase C. Neuron. 2006;49:409–420. doi: 10.1016/j.neuron.2006.01.009. [DOI] [PubMed] [Google Scholar]
- 62.Chen YH, Li MH, Zhang Y, He LL, Yamada Y, Fitzmaurice A, Shen Y, Zhang H, Tong L, Yang J. Structural basis of the alpha1-beta subunit interaction of voltage-gated Ca2+ channels. Nature. 2004;429:675–680. doi: 10.1038/nature02641. [DOI] [PubMed] [Google Scholar]
- 63.Chen-Izu Y, Xiao RP, Izu LT, Cheng H, Kuschel M, Spurgeon H, Lakatta EG. Gi-dependent localization of beta(2)-adrenergic receptor signaling to L-type Ca2+ channels. Biophys J. 2000;79:2547–2556. doi: 10.1016/S0006-3495(00)76495-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Chung HJ, Huang YH, Lau LF, Huganir RL. Regulation of the NMDA receptor complex and trafficking by activity-dependent phosphorylation of the NR2B subunit PDZ ligand. J Neurosci. 2004;24:10248–10259. doi: 10.1523/JNEUROSCI.0546-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Chung SK, Reinhart PH, Martin BL, Brautigan D, Levitan IB. Protein kinase activity closely associated with a reconstituted calcium-activated potassium channel. Science. 1991;253:560–562. doi: 10.1126/science.1857986. [DOI] [PubMed] [Google Scholar]
- 66.Clapham DE. Calcium signaling. Cell. 1995;80:259–268. doi: 10.1016/0092-8674(95)90408-5. [DOI] [PubMed] [Google Scholar]
- 67.Clapham DE, Neer EJ. G protein beta gamma subunits. Annu Rev Pharmacol Toxicol. 1997;37:167–203. doi: 10.1146/annurev.pharmtox.37.1.167. [DOI] [PubMed] [Google Scholar]
- 68.Coghlan VM, Perrino BA, Howard M, Langeberg LK, Hicks JB, Gallatin WM, Scott JD. Association of protein kinase A and protein phosphatase 2B with a common anchoring protein. Science. 1995;267:108–111. doi: 10.1126/science.7528941. [DOI] [PubMed] [Google Scholar]
- 69.Cohen RM, Foell JD, Balijepalli RC, Shah V, Hell JW, Kamp TJ. Unique modulation of L-type Ca2+ channels by short auxiliary beta1d subunit present in cardiac muscle. Am J Physiol Heart Circ Physiol. 2005;288:H2363–H2374. doi: 10.1152/ajpheart.00348.2004. [DOI] [PubMed] [Google Scholar]
- 70.Colledge M, Dean RA, Scott GK, Langeberg LK, Huganir RL, Scott JD. Targeting of PKA to glutamate receptors through a MAGUK-AKAP complex. Neuron. 2000;27:107–119. doi: 10.1016/s0896-6273(00)00013-1. [DOI] [PubMed] [Google Scholar]
- 71.Conti M, Richter W, Mehats C, Livera G, Park JY, Jin C. Cyclic AMP-specific PDE4 phosphodiesterases as critical components of cyclic AMP signaling. J Biol Chem. 2003;278:5493–5496. doi: 10.1074/jbc.R200029200. [DOI] [PubMed] [Google Scholar]
- 72.Cooper CB, Arnot MI, Feng ZP, Jarvis SE, Hamid J, Zamponi GW. Cross-talk between G-protein and protein kinase C modulation of N-type calcium channels is dependent on the G-protein beta subunit isoform. J Biol Chem. 2000;275:40777–40781. doi: 10.1074/jbc.C000673200. [DOI] [PubMed] [Google Scholar]
- 73.Csortos C, Zolnierowicz S, Bako E, Durbin S, DePaoli-Roach AA. High complexity in the expression of the B′ subunit of protein phosphatase 2A0. J Biol Chem. 1996;271:2578–2588. doi: 10.1074/jbc.271.5.2578. [DOI] [PubMed] [Google Scholar]
- 74.Curtis BM, Catterall WA. Phosphorylation of the calcium antagonist receptor of the voltage-sensitive calcium channel by cAMP-dependent protein kinase. Proc Natl Acad Sci USA. 1985;82:2528–2532. doi: 10.1073/pnas.82.8.2528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Daaka Y, Luttrell LM, Lefkowitz RJ. Switching of the coupling of the beta2-adrenergic receptor to different G proteins by protein kinase A. Nature. 1997;390:88–91. doi: 10.1038/36362. [DOI] [PubMed] [Google Scholar]
- 76.Dai S, Klugbauer N, Zong X, Seisenberger C, Hofmann F. The role of subunit composition on prepulse facilitation of the cardiac L-type calcium channel. FEBS Lett. 1999;442:70–74. doi: 10.1016/s0014-5793(98)01632-9. [DOI] [PubMed] [Google Scholar]
- 77.Dascal N, Lotan I. Activation of protein kinase C alters voltage dependence of a Na+ channel. Neuron. 1991;6:165–175. doi: 10.1016/0896-6273(91)90131-i. [DOI] [PubMed] [Google Scholar]
- 78.Davare MA, Avdonin V, Hall DD, Peden EM, Burette A, Weinberg RJ, Horne MC, Hoshi T, Hell JW. A beta2 adrenergic receptor signaling complex assembled with the Ca2+ channel Cav1.2. Science. 2001;293:98–101. doi: 10.1126/science.293.5527.98. [DOI] [PubMed] [Google Scholar]
- 79.Davare MA, Dong F, Rubin CS, Hell JW. The A-kinase anchor protein MAP2B and cAMP-dependent protein kinase are associated with class C L-type calcium channels in neurons. J Biol Chem. 1999;274:30280–30287. doi: 10.1074/jbc.274.42.30280. [DOI] [PubMed] [Google Scholar]
- 80.Davare MA, Hell JW. Increased phosphorylation of the neuronal L-type Ca2+ channel Ca(v)1.2 during aging. Proc Natl Acad Sci USA. 2003;100:16018–16023. doi: 10.1073/pnas.2236970100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Davare MA, Horne MC, Hell JW. Protein phosphatase 2A is associated with class C L-type calcium channels (Cav1.2) and antagonizes channel phosphorylation by cAMP-dependent protein kinase. J Biol Chem. 2000;275:39710–39717. doi: 10.1074/jbc.M005462200. [DOI] [PubMed] [Google Scholar]
- 82.David M, Richer M, Mamarbachi AM, Villeneuve LR, Dupre DJ, Hebert TE. Interactions between GABA-B(1) receptors and Kir 3 inwardly rectifying potassium channels. Cell Signal. 2006;18:2172–2181. doi: 10.1016/j.cellsig.2006.05.014. [DOI] [PubMed] [Google Scholar]
- 83.Davies A, Hendrich J, Van Minh AT, Wratten J, Douglas L, Dolphin AC. Functional biology of the alpha(2)delta subunits of voltage-gated calcium channels. Trends Pharmacol Sci. 2007 doi: 10.1016/j.tips.2007.03.005. [DOI] [PubMed] [Google Scholar]
- 84.De Jongh KS, Merrick DK, Catterall WA. Subunits of purified calcium channels: a 212-kDa form of alpha 1 and partial amino acid sequence of a phosphorylation site of an independent beta subunit. Proc Natl Acad Sci USA. 1989;86:8585–8589. doi: 10.1073/pnas.86.21.8585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.De Jongh KS, Murphy BJ, Colvin AA, Hell JW, Takahashi M, Catterall WA. Specific phosphorylation of a site in the full length form of the α1 subunit of the cardiac L-type calcium channel by adenosine 3′,5′-cyclic monophosphate-dependent protein kinase. Biochemistry. 1996;35:10392–10402. doi: 10.1021/bi953023c. [DOI] [PubMed] [Google Scholar]
- 86.De Jongh KS, Warner C, Colvin AA, Catterall WA. Characterization of the two size forms of the alpha 1 subunit of skeletal muscle L-type calcium channels. Proc Natl Acad Sci USA. 1991;88:10778–10782. doi: 10.1073/pnas.88.23.10778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.De Waard M, Hering J, Weiss N, Feltz A. How do G proteins directly control neuronal Ca2+ channel function? Trends Pharmacol Sci. 2005;26:427–436. doi: 10.1016/j.tips.2005.06.008. [DOI] [PubMed] [Google Scholar]
- 88.De Waard M, Liu H, Walker D, Scott VE, Gurnett CA, Campbell KP. Direct binding of G-protein betagamma complex to voltage-dependent calcium channels. Nature. 1997;385:446–450. doi: 10.1038/385446a0. [DOI] [PubMed] [Google Scholar]
- 89.Dell EJ, Connor J, Chen S, Stebbins EG, Skiba NP, Mochly-Rosen D, Hamm HE. The betagamma subunit of heterotrimeric G proteins interacts with RACK1 and two other WD repeat proteins. J Biol Chem. 2002;277:49888–49895. doi: 10.1074/jbc.M202755200. [DOI] [PubMed] [Google Scholar]
- 90.Dell'Acqua ML, Faux MC, Thorburn J, Thorburn A, Scott JD. Membrane-targeting sequences on AKAP79 bind phosphatidylinositol-4, 5-bisphosphate. EMBO J. 1998;17:2246–2260. doi: 10.1093/emboj/17.8.2246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.DeMaria CD, Soong TW, Alseikhan BA, Alvania RS, Yue DT. Calmodulin bifurcates the local Ca2+ signal that modulates P/Q-type Ca2+ channels. Nature. 2001;411:484–489. doi: 10.1038/35078091. [DOI] [PubMed] [Google Scholar]
- 92.Dick IE, Tadross MR, Liang H, Tay LH, Yang W, Yue DT. A modular switch for spatial Ca2+ selectivity in the calmodulin regulation of Cav channels. Nature. 2008;451:830–834. doi: 10.1038/nature06529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Dikic I, Tokiwa G, Lev S, Courtneidge SA, Schlessinger J. A role for Pyk2 and Src in linking G-protein-coupled receptors with MAP kinase activation. Nature. 1996;383:547–549. doi: 10.1038/383547a0. [DOI] [PubMed] [Google Scholar]
- 94.Dilly KW, Kurokawa J, Terrenoire C, Reiken S, Lederer WJ, Marks AR, Kass RS. Overexpression of beta2-adrenergic receptors cAMP-dependent protein kinase phosphorylates and modulates slow delayed rectifier potassium channels expressed in murine heart: evidence for receptor/channel co-localization. J Biol Chem. 2004;279:40778–40787. doi: 10.1074/jbc.M406010200. [DOI] [PubMed] [Google Scholar]
- 95.Dodge KL, Khouangsathiene S, Kapiloff MS, Mouton R, Hill EV, Houslay MD, Langeberg LK, Scott JD. mAKAP assembles a protein kinase A/PDE4 phosphodiesterase cAMP signaling module. EMBO J. 2001;20:1921–1930. doi: 10.1093/emboj/20.8.1921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Dodge-Kafka KL, Soughayer J, Pare GC, Carlisle Michel JJ, Langeberg LK, Kapiloff MS, Scott JD. The protein kinase A anchoring protein mAKAP coordinates two integrated cAMP effector pathways. Nature. 2005;437:574–578. doi: 10.1038/nature03966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Doering CJ, Kisilevsky AE, Feng ZP, Arnot MI, Peloquin J, Hamid J, Barr W, Nirdosh A, Simms B, Winkfein RJ, Zamponi GW. A single Gbeta subunit locus controls cross-talk between protein kinase C and G protein regulation of N-type calcium channels. J Biol Chem. 2004;279:29709–29717. doi: 10.1074/jbc.M308693200. [DOI] [PubMed] [Google Scholar]
- 98.Dolmetsch RE, Pajvani U, Fife K, Spotts JM, Greenberg ME. Signaling to the nucleus by an L-type calcium channel-calmodulin complex through the MAP kinase pathway. Science. 2001;294:333–339. doi: 10.1126/science.1063395. [DOI] [PubMed] [Google Scholar]
- 99.Dolphin AC. Beta subunits of voltage-gated calcium channels. J Bioenerg Biomembr. 2003;35:599–620. doi: 10.1023/b:jobb.0000008026.37790.5a. [DOI] [PubMed] [Google Scholar]
- 100.Dolphin AC. G protein modulation of voltage-gated calcium channels. Pharmacol Rev. 2003;55:607–627. doi: 10.1124/pr.55.4.3. [DOI] [PubMed] [Google Scholar]
- 101.Dorn GW, 2nd, Mochly-Rosen D. Intracellular transport mechanisms of signal transducers. Annu Rev Physiol. 2002;64:407–429. doi: 10.1146/annurev.physiol.64.081501.155903. [DOI] [PubMed] [Google Scholar]
- 102.DuBell WH, Rogers TB. Protein phosphatase 1 and an opposing protein kinase regulate steady-state L-type Ca2+ current in mouse cardiac myocytes. J Physiol. 2004;556:79–93. doi: 10.1113/jphysiol.2003.059329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Dupre DJ, Hebert TE. Biosynthesis and trafficking of seven transmembrane receptor signalling complexes. Cell Signal. 2006;18:1549–1559. doi: 10.1016/j.cellsig.2006.03.009. [DOI] [PubMed] [Google Scholar]
- 104.Dupre DJ, Robitaille M, Ethier N, Villeneuve LR, Mamarbachi AM, Hebert TE. Seven transmembrane receptor core signaling complexes are assembled prior to plasma membrane trafficking. J Biol Chem. 2006;281:34561–34573. doi: 10.1074/jbc.M605012200. [DOI] [PubMed] [Google Scholar]
- 105.Dzhura I, Wu Y, Colbran RJ, Balser JR, Anderson ME. Calmodulin kinase determines calcium-dependent facilitation of L-type calcium channels. Nat Cell Biol. 2000;2:173–177. doi: 10.1038/35004052. [DOI] [PubMed] [Google Scholar]
- 106.Edgerton JR, Reinhart PH. Distinct contributions of small and large conductance Ca2+-activated K+ channels to rat Purkinje neuron function. J Physiol. 2003;548:53–69. doi: 10.1113/jphysiol.2002.027854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Egloff MP, Johnson DF, Moorhead G, Cohen PT, Cohen P, Barford D. Structural basis for the recognition of regulatory subunits by the catalytic subunit of protein phosphatase 1. EMBO J. 1997;16:1876–1887. doi: 10.1093/emboj/16.8.1876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Endoh T. Involvement of Src tyrosine kinase and mitogen-activated protein kinase in the facilitation of calcium channels in rat nucleus of the tractus solitarius by angiotensin II. J Physiol. 2005;568:851–865. doi: 10.1113/jphysiol.2005.095307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Erickson MG, Liang H, Mori MX, Yue DT. FRET two-hybrid mapping reveals function and location of L-type Ca2+ channel CaM preassociation. Neuron. 2003;39:97–107. doi: 10.1016/s0896-6273(03)00395-7. [DOI] [PubMed] [Google Scholar]
- 110.Ertel EA, Campbell KP, Harpold MM, Hofmann F, Mori Y, Perez-Reyes E, Schwartz A, Snutch TP, Tanabe T, Birnbaumer L, Tsien RW, Catterall WA. Nomenclature of voltage-gated calcium channels. Neuron. 2000;25:533–535. doi: 10.1016/s0896-6273(00)81057-0. [DOI] [PubMed] [Google Scholar]
- 111.Erxleben C, Liao Y, Gentile S, Chin D, Gomez-Alegria C, Mori Y, Birnbaumer L, Armstrong DL. Cyclosporin and Timothy syndrome increase mode 2 gating of Cav1.2 calcium channels through aberrant phosphorylation of S6 helices. Proc Natl Acad Sci USA. 2006;103:3932–3937. doi: 10.1073/pnas.0511322103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Esguerra M, Wang J, Foster CD, Adelman JP, North RA, Levitan IB. Cloned Ca2+-dependent K+ channel modulated by a functionally associated protein kinase. Nature. 1994;369:563–565. doi: 10.1038/369563a0. [DOI] [PubMed] [Google Scholar]
- 113.Evans RM, Zamponi GW. Presynaptic Ca2+ channels–integration centers for neuronal signaling pathways. Trends Neurosci. 2006;29:617–624. doi: 10.1016/j.tins.2006.08.006. [DOI] [PubMed] [Google Scholar]
- 114.Faber ES, Delaney AJ, Sah P. SK channels regulate excitatory synaptic transmission and plasticity in the lateral amygdala. Nat Neurosci. 2005;8:635–641. doi: 10.1038/nn1450. [DOI] [PubMed] [Google Scholar]
- 115.Fallon JL, Halling DB, Hamilton SL, Quiocho FA. Structure of calmodulin bound to the hydrophobic IQ domain of the cardiac Ca(v)1.2 calcium channel. Structure. 2005;13:1881–1886. doi: 10.1016/j.str.2005.09.021. [DOI] [PubMed] [Google Scholar]
- 116.Fan G, Shumay E, Wang H, Malbon CC. The scaffold protein gravin (cAMP-dependent protein kinase-anchoring protein 250) binds the beta 2-adrenergic receptor via the receptor cytoplasmic Arg-329 to Leu-413 domain and provides a mobile scaffold during desensitization. J Biol Chem. 2001;276:24005–24014. doi: 10.1074/jbc.M011199200. [DOI] [PubMed] [Google Scholar]
- 117.Ferris CD, Cameron AM, Bredt DS, Huganir RL, Snyder SH. Inositol 1,4,5-trisphosphate receptor is phosphorylated by cyclic AMP-dependent protein kinase at serines 1755 and 1589. Biochem Biophys Res Commun. 1991;175:192–198. doi: 10.1016/s0006-291x(05)81219-7. [DOI] [PubMed] [Google Scholar]
- 118.Few WP, Scheuer T, Catterall WA. Dopamine modulation of neuronal Na+ channels requires binding of A kinase-anchoring protein 15 and PKA by a modified leucine zipper motif. Proc Natl Acad Sci USA. 2007;104:5187–5192. doi: 10.1073/pnas.0611619104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Flockerzi V, Oeken HJ, Hofmann F, Pelzer D, Cavalie A, Trautwein W. Purified dihydropyridine-binding site from skeletal muscle t-tubules is a functional calcium channel. Nature. 1986;323:66–68. doi: 10.1038/323066a0. [DOI] [PubMed] [Google Scholar]
- 120.Foell JD, Balijepalli RC, Delisle BP, Yunker AM, Robia SL, Walker JW, McEnery MW, January CT, Kamp TJ. Molecular heterogeneity of calcium channel beta-subunits in canine and human heart: evidence for differential subcellular localization. Physiol Gen. 2004;17:183–200. doi: 10.1152/physiolgenomics.00207.2003. [DOI] [PubMed] [Google Scholar]
- 121.Forscher P, Oxford GS, Schulz D. Noradrenaline modulates calcium channels in avian dorsal root ganglion cells through tight receptor-channel coupling. J Physiol. 1986;379:131–144. doi: 10.1113/jphysiol.1986.sp016244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Fowler CE, Aryal P, Suen KF, Slesinger PA. Evidence for association of GABAB receptors with Kir3 channels and RGS4 proteins. J Physiol. 2007;580:51–65. doi: 10.1113/jphysiol.2006.123216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Frace AM, Hartzell HC. Opposite effects of phosphatase inhibitors on L-type calcium and delayed rectifier currents in frog cardiac myocytes. J Physiol. 1993;472:305–326. doi: 10.1113/jphysiol.1993.sp019948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Frank M, Thumer L, Lohse MJ, Bunemann M. G Protein activation without subunit dissociation depends on a Gα(i)-specific region. J Biol Chem. 2005;280:24584–24590. doi: 10.1074/jbc.M414630200. [DOI] [PubMed] [Google Scholar]
- 125.Fraser ID, Cong M, Kim J, Rollins EN, Daaka Y, Lefkowitz RJ, Scott JD. Assembly of an A kinase-anchoring protein-beta(2)-adrenergic receptor complex facilitates receptor phosphorylation and signaling. Curr Biol. 2000;10:409–412. doi: 10.1016/s0960-9822(00)00419-x. [DOI] [PubMed] [Google Scholar]
- 126.Fraser IDC, Tavalin SJ, Lester LB, Langeberg LK, Westphal AM, Dean RA, Marrion NV, Scott JD. A novel lipid-anchored A-kinase anchoring protein facilitates cAMP-responsive membrane events. EMBO J. 1998;17:2261–2272. doi: 10.1093/emboj/17.8.2261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Gales C, Van Durm JJ, Schaak S, Pontier S, Percherancier Y, Audet M, Paris H, Bouvier M. Probing the activation-promoted structural rearrangements in preassembled receptor-G protein complexes. Nat Struct Mol Biol. 2006;13:778–786. doi: 10.1038/nsmb1134. [DOI] [PubMed] [Google Scholar]
- 127a.Ganesan AN, Maack C, Johns DC, Sidor A, O'Rourke B. β-Adrenergic stimulation of L-type Ca2+ channels in cardiac myocytes requires the distal carboxy1 terminus of α1C but not serine 1928. Circ Res. 2006;98:e11–18. doi: 10.1161/01.RES.0000202692.23001.e2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Gao T, Cuadra AE, Ma H, Bunemann M, Gerhardstein BL, Cheng T, Eick RT, Hosey MM. C-terminal fragments of the alpha 1C (CaV1.2) subunit associate with and regulate L-type calcium channels containing C-terminal-truncated alpha 1C subunits. J Biol Chem. 2001;276:21089–21097. doi: 10.1074/jbc.M008000200. [DOI] [PubMed] [Google Scholar]
- 129.Gao T, Puri TS, Gerhardstein BL, Chien AJ, Green RD, Hosey MM. Identification and subcellular localization of the subunits of L-type calcium channels and adenylyl cyclase in cardiac myocytes. J Biol Chem. 1997;272:19401–19407. doi: 10.1074/jbc.272.31.19401. [DOI] [PubMed] [Google Scholar]
- 130.Gao T, Yatani A, Dell'Acqua ML, Sako H, Green SA, Dascal N, Scott JD, Hosey MM. cAMP-dependent regulation of cardiac L-type Ca2+ channels requires membrane targeting of PKA and phosphorylation of channel subunits. Neuron. 1997;19:185–196. doi: 10.1016/s0896-6273(00)80358-x. [DOI] [PubMed] [Google Scholar]
- 131.Garcia DE, Li B, Garcia-Ferreiro RE, Hernandez-Ochoa EO, Yan K, Gautam N, Catterall WA, Mackie K, Hille B. G-protein beta-subunit specificity in the fast membrane-delimited inhibition of Ca2+ channels. J Neurosci. 1998;18:9163–9170. doi: 10.1523/JNEUROSCI.18-22-09163.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Gardner LA, Delos Santos NM, Matta SG, Whitt MA, Bahouth SW. Role of the cyclic AMP-dependent protein kinase in homologous resensitization of the beta1-adrenergic receptor. J Biol Chem. 2004;279:21135–21143. doi: 10.1074/jbc.M313652200. [DOI] [PubMed] [Google Scholar]
- 133.Gardner LA, Tavalin SJ, Goehring AS, Scott JD, Bahouth SW. AKAP79-mediated targeting of the cyclic AMP-dependent protein kinase to the beta1-adrenergic receptor promotes recycling and functional resensitization of the receptor. J Biol Chem. 2006;281:33537–33553. doi: 10.1074/jbc.M601809200. [DOI] [PubMed] [Google Scholar]
- 134.Gasparini S, Kasyanov AM, Pietrobon D, Voronin LL, Cherubini E. Presynaptic R-type calcium channels contribute to fast excitatory synaptic transmission in the rat hippocampus. J Neurosci. 2001;21:8715–8721. doi: 10.1523/JNEUROSCI.21-22-08715.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Gerhardstein BL, Gao T, Bunemann M, Puri TS, Adair A, Ma H, Hosey MM. Proteolytic processing of the C terminus of the alpha(1C) subunit of L-type calcium channels and the role of a proline-rich domain in membrane tethering of proteolytic fragments. J Biol Chem. 2000;275:8556–8563. doi: 10.1074/jbc.275.12.8556. [DOI] [PubMed] [Google Scholar]
- 136.Gerhardstein BL, Puri TS, Chien AJ, Hosey MM. Identification of the sites phosphorylated by cyclic AMP-dependent protein kinase on the beta 2 subunit of L-type voltage-dependent calcium channels. Biochemistry. 1999;38:10361–10370. doi: 10.1021/bi990896o. [DOI] [PubMed] [Google Scholar]
- 137.Ghosh A, Greenberg ME. Calcium signaling in neurons: molecular mechanisms and cellular consequences. Science. 1995;268:239–247. doi: 10.1126/science.7716515. [DOI] [PubMed] [Google Scholar]
- 138.Gingrich JR, Pelkey KA, Fam SR, Huang Y, Petralia RS, Wenthold RJ, Salter MW. Unique domain anchoring of Src to synaptic NMDA receptors via the mitochondrial protein NADH dehydrogenase subunit 2. Proc Natl Acad Sci USA. 2004;101:6237–6242. doi: 10.1073/pnas.0401413101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Golding NL, Staff NP, Spruston N. Dendritic spikes as a mechanism for cooperative long-term potentiation. Nature. 2002;418:326–331. doi: 10.1038/nature00854. [DOI] [PubMed] [Google Scholar]
- 140.Gomez AM, Valdivia HH, Cheng H, Lederer MR, Santana LF, Cannell MB, McCune SA, Altschuld RA, Lederer WJ. Defective excitation-contraction coupling in experimental cardiac hypertrophy and heart failure. Science. 1997;276:800–806. doi: 10.1126/science.276.5313.800. [DOI] [PubMed] [Google Scholar]
- 141.Gomez LL, Alam S, Smith KE, Horne E, Dell'Acqua ML. Regulation of A-kinase anchoring protein 79/150-cAMP-dependent protein kinase postsynaptic targeting by NMDA receptor activation of calcineurin and remodeling of dendritic actin. J Neurosci. 2002;22:7027–7044. doi: 10.1523/JNEUROSCI.22-16-07027.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Gorski JA, Gomez LL, Scott JD, Dell'Acqua ML. Association of an A-kinase-anchoring protein signaling scaffold with cadherin adhesion molecules in neurons and epithelial cells. Mol Biol Cell. 2005;16:3574–3590. doi: 10.1091/mbc.E05-02-0134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Graef IA, Mermelstein PG, Stankunas K, Neilson JR, Deisseroth K, Tsien RW, Crabtree GR. L-type calcium channels and GSK-3 regulate the activity of NF-ATc4 in hippocampal neurons. Nature. 1999;401:703–708. doi: 10.1038/44378. [DOI] [PubMed] [Google Scholar]
- 144.Gray PC, Johnson BD, Westenbroek RE, Hays LG, Yates JR, III, Scheuer T, Catterall WA, Murphy BJ. Primary structure and function of an A kinase anchoring protein associated with calcium channels. Neuron. 1998;20:1017–1026. doi: 10.1016/s0896-6273(00)80482-1. [DOI] [PubMed] [Google Scholar]
- 145.Gray PC, Tibbs VC, Catterall WA, Murphy BJ. Identification of a 15-kDa cAMP-dependent protein kinase-anchoring protein associated with skeletal muscle L-type calcium channels. J Biol Chem. 1997;272:6297–6302. doi: 10.1074/jbc.272.10.6297. [DOI] [PubMed] [Google Scholar]
- 146.Groschner K, Schuhmann K, Mieskes G, Baumgartner W, Romanin C. A type 2A phosphatase-sensitive phosphorylation site controls modal gating of L-type Ca2+ channels in human vascular smooth-muscle cells. Biochem J. 1996;318:513–517. doi: 10.1042/bj3180513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Grueter CE, Abiria SA, Dzhura I, Wu Y, Ham AJ, Mohler PJ, Anderson ME, Colbran RJ. L-type Ca2+ channel facilitation mediated by phosphorylation of the beta subunit by CaMKII. Mol Cell. 2006;23:641–650. doi: 10.1016/j.molcel.2006.07.006. [DOI] [PubMed] [Google Scholar]
- 148.Grunnet M, Kaufmann WA. Coassembly of big conductance Ca2+-activated K+ channels and L-type voltage-gated Ca2+ channels in rat brain. J Biol Chem. 2004;279:36445–36453. doi: 10.1074/jbc.M402254200. [DOI] [PubMed] [Google Scholar]
- 149.Gui P, Wu X, Ling S, Stotz SC, Winkfein RJ, Wilson E, Davis GE, Braun AP, Zamponi GW, Davis MJ. Integrin receptor activation triggers converging regulation of Cav1.2 calcium channels by c-Src and protein kinase A pathways. J Biol Chem. 2006;281:14015–14025. doi: 10.1074/jbc.M600433200. [DOI] [PubMed] [Google Scholar]
- 150.Gutman GA, Chandy KG, Grissmer S, Lazdunski M, McKinnon D, Pardo LA, Robertson GA, Rudy B, Sanguinetti MC, Stuhmer W, Wang X. International Union of Pharmacology. LIII. Nomenclature and molecular relationships of voltage-gated potassium channels. Pharmacol Rev. 2005;57:473–508. doi: 10.1124/pr.57.4.10. [DOI] [PubMed] [Google Scholar]
- 151.Haase H, Alvarez J, Petzhold D, Doller A, Behlke J, Erdmann J, Hetzer R, Regitz-Zagrosek V, Vassort G, Morano I. Ahnak is critical for cardiac Ca(V)1.2 calcium channel function and its beta-adrenergic regulation. FASEB J. 2005;19:1969–1977. doi: 10.1096/fj.05-3997com. [DOI] [PubMed] [Google Scholar]
- 152.Haase H, Karczewski P, Beckert R, Krause EG. Phosphorylation of the L-type calcium channel beta subunit is involved in beta-adrenergic signal transduction in canine myocardium. FEBS Lett. 1993;335:217–222. doi: 10.1016/0014-5793(93)80733-b. [DOI] [PubMed] [Google Scholar]
- 153.Hall DD, Davare MA, Shi M, Allen ML, Weisenhaus M, McKnight GS, Hell JW. Critical role of cAMP-dependent protein kinase anchoring to the L-type calcium channel Cav1.2 via A-kinase anchor protein 150 in neurons. Biochemistry. 2007;46:1635–1646. doi: 10.1021/bi062217x. [DOI] [PubMed] [Google Scholar]
- 154.Hall DD, Feekes JA, Arachchige Don AS, Shi M, Hamid J, Chen L, Strack S, Zamponi GW, Horne MC, Hell JW. Binding of protein phosphatase 2A to the L-type calcium channel Cav1.2 next to Ser1928, its main PKA site, is critical for Ser1928 dephosphorylation. Biochemistry. 2006;45:3448–3459. doi: 10.1021/bi051593z. [DOI] [PubMed] [Google Scholar]
- 155.Hall DD, Hell JW. The fourth dimension in cellular signaling. Science. 2001;293:2205. doi: 10.1126/science.293.5538.2204. [DOI] [PubMed] [Google Scholar]
- 156.Halling DB, Aracena-Parks P, Hamilton SL. Regulation of voltage-gated Ca2+ channels by calmodulin. Sci STKE. 2006;2006:er1. doi: 10.1126/stke.3182006er1. [DOI] [PubMed] [Google Scholar]
- 157.Hamid J, Nelson D, Spaetgens R, Dubel SJ, Snutch TP, Zamponi GW. Identification of an integration center for cross-talk between protein kinase C and G protein modulation of N-type calcium channels. J Biol Chem. 1999;274:6195–6202. doi: 10.1074/jbc.274.10.6195. [DOI] [PubMed] [Google Scholar]
- 158.Hartzell HC, Hirayama Y, Petit-Jacques J. Effects of protein phosphatase and kinase inhibitors on the cardiac L-type Ca current suggest two sites are phosphorylated by protein kinase A and another protein kinase. J Gen Physiol. 1995;106:393–414. doi: 10.1085/jgp.106.3.393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Hayes JS, Brunton LL, Mayer SE. Selective activation of particulate cAMP-dependent protein kinase by isoproterenol and prostaglandin E1. J Biol Chem. 1980;255:5113–5119. [PubMed] [Google Scholar]
- 160.He J, Conklin MW, Foell JD, Wolff MR, Haworth RA, Coronado R, Kamp TJ. Reduction in density of transverse tubules and L-type Ca2+ channels in canine tachycardia-induced heart failure. Cardiovasc Res. 2001;49:298–307. doi: 10.1016/s0008-6363(00)00256-x. [DOI] [PubMed] [Google Scholar]
- 161.He JQ, Pi Y, Walker JW, Kamp TJ. Endothelin-1 and photore-leased diacylglycerol increase L-type Ca2+ current by activation of protein kinase C in rat ventricular myocytes. J Physiol. 2000;524:807–820. doi: 10.1111/j.1469-7793.2000.00807.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Hell JW, Appleyard SM, Yokoyama CT, Warner C, Catterall WA. Differential phosphorylation of two size forms of the N-type calcium channel alpha 1 subunit which have different COOH termini. J Biol Chem. 1994;269:7390–7396. [PubMed] [Google Scholar]
- 163.Hell JW, Westenbroek RE, Breeze LJ, Wang KKW, Chavkin C, Catterall WA. N-methyl-d-aspartate receptor-induced proteolytic conversion of postsynaptic class C L-type calcium channels in hippocampal neurons. Proc Natl Acad Sci USA. 1996;93:3362–3367. doi: 10.1073/pnas.93.8.3362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Hell JW, Westenbroek RE, Elliott EM, Catterall WA. Differential phosphorylation, localization, and function of distinct α1 subunits of neuronal calcium channels. Two size forms for class B, C, and D α1 subunits with different COOH-termini. Ann NY Acad Sci. 1994;747:282–293. [PubMed] [Google Scholar]
- 165.Hell JW, Westenbroek RE, Warner C, Ahlijanian MK, Prystay W, Gilbert MM, Snutch TP, Catterall WA. Identification and differential subcellular localization of the neuronal class C and class D L-type calcium channel α1 subunits. J Cell Biol. 1993;123:949–962. doi: 10.1083/jcb.123.4.949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Hell JW, Yokoyama CT, Breeze LJ, Chavkin C, Catterall WA. Phosphorylation of presynaptic and postsynaptic calcium channels by cAMP-dependent protein kinase in hippocampal neurons. EMBO J. 1995;14:3036–3044. doi: 10.1002/j.1460-2075.1995.tb07306.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Hell JW, Yokoyama CT, Wong ST, Warner C, Snutch TP, Catterall WA. Differential phosphorylation of two size forms of the neuronal class C L-type calcium channel α1 subunit. J Biol Chem. 1993;268:19451–19457. [PubMed] [Google Scholar]
- 168.Heriche JK, Lebrin F, Rabilloud T, Leroy D, Chambaz EM, Goldberg Y. Regulation of protein phosphatase 2A by a direct interaction with casein kinase 2α. Science. 1997;276:952–955. doi: 10.1126/science.276.5314.952. [DOI] [PubMed] [Google Scholar]
- 169.Herlitze S, Garcia DE, Mackie K, Hille B, Scheuer T, Catterall WA. Modulation of Ca2+ channels by G-protein beta gamma subunits. Nature. 1996;380:258–262. doi: 10.1038/380258a0. [DOI] [PubMed] [Google Scholar]
- 170.Herzig S, Meier A, Pfeiffer M, Neumann J. Stimulation of protein phosphatases as a mechanism of the muscarinic-receptor-mediated inhibition of cardiac L-type Ca2+ channels. Pflugers Arch. 1995;429:531–538. doi: 10.1007/BF00704158. [DOI] [PubMed] [Google Scholar]
- 171.Herzig S, Neumann J. Effects of serine/threonine protein phosphatases on ion channels in excitable membranes. Physiol Rev. 2000;80:173–210. doi: 10.1152/physrev.2000.80.1.173. [DOI] [PubMed] [Google Scholar]
- 172.Hescheler J, Kameyama M, Trautwein W, Mieskes G, Soling HD. Regulation of the cardiac calcium channel by protein phosphatases. Eur J Biochem. 1987;165:261–266. doi: 10.1111/j.1432-1033.1987.tb11436.x. [DOI] [PubMed] [Google Scholar]
- 173.Hess P, Lansman JB, Tsien RW. Different modes of Ca channel gating behaviour favoured by dihydropyridine Ca agonists and antagonists. Nature. 1984;311:538–544. doi: 10.1038/311538a0. [DOI] [PubMed] [Google Scholar]
- 174.Hibino H, Pironkova R, Onwumere O, Rousset M, Charnet P, Hudspeth AJ, Lesage F. Direct interaction with a nuclear protein and regulation of gene silencing by a variant of the Ca2+-channel beta 4 subunit. Proc Natl Acad Sci USA. 2003;100:307–312. doi: 10.1073/pnas.0136791100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Hille B. Modulation of ion-channel function by G-protein-coupled receptors. Trends Neurosci. 1994;17:531–536. doi: 10.1016/0166-2236(94)90157-0. [DOI] [PubMed] [Google Scholar]
- 176.Hohl CM, Li QA. Compartmentation of cAMP in adult canine ventricular myocytes. Relation to single-cell free Ca2+ transients. Circ Res. 1991;69:1369–1379. doi: 10.1161/01.res.69.5.1369. [DOI] [PubMed] [Google Scholar]
- 177.Holmes TC, Fadool DA, Ren R, Levitan IB. Association of Src tyrosine kinase with a human potassium channel mediated by SH3 domain. Science. 1996;274:2089–2091. doi: 10.1126/science.274.5295.2089. [DOI] [PubMed] [Google Scholar]
- 178.Hoogland TM, Saggau P. Facilitation of L-type Ca2+ channels in dendritic spines by activation of beta2 adrenergic receptors. J Neurosci. 2004;24:8416–8427. doi: 10.1523/JNEUROSCI.1677-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Hoshi N, Langeberg LK, Scott JD. Distinct enzyme combinations in AKAP signalling complexes permit functional diversity. Nat Cell Biol. 2005 doi: 10.1038/ncb1315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Hoshi N, Zhang JS, Omaki M, Takeuchi T, Yokoyama S, Wanaverbecq N, Langeberg LK, Yoneda Y, Scott JD, Brown DA, Higashida H. AKAP150 signaling complex promotes suppression of the M-current by muscarinic agonists. Nat Neurosci. 2003;6:564–571. doi: 10.1038/nn1062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Hruska-Hageman AM, Wemmie JA, Price MP, Welsh MJ. Interaction of the synaptic protein PICK1 (protein interacting with C kinase 1) with the non-voltage gated sodium channels BNC1 (brain Na+ channel 1) and ASIC (acid-sensing ion channel) Biochem J. 2002;361:443–450. doi: 10.1042/0264-6021:3610443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Hu XD, Huang Q, Yang X, Xia H. Differential regulation of AMPA receptor trafficking by neurabin-targeted synaptic protein phosphatase-1 in synaptic transmission and long-term depression in hippocampus. J Neurosci. 2007;27:4674–4686. doi: 10.1523/JNEUROSCI.5365-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Hu XQ, Singh N, Mukhopadhyay D, Akbarali HI. Modulation of voltage-dependent Ca2+ channels in rabbit colonic smooth muscle cells by c-Src and focal adhesion kinase. J Biol Chem. 1998;273:5337–5342. doi: 10.1074/jbc.273.9.5337. [DOI] [PubMed] [Google Scholar]
- 184.Huang CL, Feng S, Hilgemann DW. Direct activation of inward rectifier potassium channels by PIP2 and its stabilization by Gβγ. Nature. 1998;391:803–806. doi: 10.1038/35882. [DOI] [PubMed] [Google Scholar]
- 185.Huang CL, Slesinger PA, Casey PJ, Jan YN, Jan LY. Evidence that direct binding of G beta gamma to the GIRK1 G protein-gated inwardly rectifying K+ channel is important for channel activation. Neuron. 1995;15:1133–1143. doi: 10.1016/0896-6273(95)90101-9. [DOI] [PubMed] [Google Scholar]
- 186.Huang XP, Pi Y, Lokuta AJ, Greaser ML, Walker JW. Arachidonic acid stimulates protein kinase C-epsilon redistribution in heart cells. J Cell Sci. 1997;110:1625–1634. doi: 10.1242/jcs.110.14.1625. [DOI] [PubMed] [Google Scholar]
- 187.Huang Y, Lu W, Ali DW, Pelkey KA, Pitcher GM, Lu YM, Aoto H, Roder JC, Sasaki T, Salter MW, MacDonald JF. CAKbeta/ Pyk2 kinase is a signaling link for induction of long-term potentiation in CA1 hippocampus. Neuron. 2001;29:485–496. doi: 10.1016/s0896-6273(01)00220-3. [DOI] [PubMed] [Google Scholar]
- 188.Hudmon A, Lebel E, Roy H, Sik A, Schulman H, Waxham MN, De Koninck P. A mechanism for Ca2+/calmodulin-dependent protein kinase II clustering at synaptic and nonsynaptic sites based on self-association. J Neurosci. 2005;25:6971–6983. doi: 10.1523/JNEUROSCI.4698-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Hudmon A, Schulman H. Structure-function of the multifunctional Ca2+/calmodulin-dependent protein kinase II. Biochem J. 2002;364:593–611. doi: 10.1042/BJ20020228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Hudmon A, Schulman H, Kim J, Maltez JM, Tsien RW, Pitt GS. CaMKII tethers to L-type Ca2+ channels, establishing a local and dedicated integrator of Ca2+ signals for facilitation. J Cell Biol. 2005;171:537–547. doi: 10.1083/jcb.200505155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Hullin R, Khan IF, Wirtz S, Mohacsi P, Varadi G, Schwartz A, Herzig S. Cardiac L-type calcium channel beta-subunits expressed in human heart have differential effects on single channel characteristics. J Biol Chem. 2003;278:21623–21630. doi: 10.1074/jbc.M211164200. [DOI] [PubMed] [Google Scholar]
- 192.Hulme JT, Ahn M, Hauschka SD, Scheuer T, Catterall WA. A novel leucine zipper targets AKAP15 and cyclic AMP-dependent protein kinase to the C terminus of the skeletal muscle Ca2+ channel and modulates its function. J Biol Chem. 2002;277:4079–4087. doi: 10.1074/jbc.M109814200. [DOI] [PubMed] [Google Scholar]
- 193.Hulme JT, Konoki K, Lin TW, Gritsenko MA, Camp DG, 2nd, Big-elow DJ, Catterall WA. Sites of proteolytic processing and noncovalent association of the distal C-terminal domain of Cav1.1 channels in skeletal muscle. Proc Natl Acad Sci USA. 2005;102:5274–5279. doi: 10.1073/pnas.0409885102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Hulme JT, Lin TW, Westenbroek RE, Scheuer T, Catterall WA. Beta-adrenergic regulation requires direct anchoring of PKA to cardiac Cav1.2 channels via a leucine zipper interaction with A kinase-anchoring protein 15. Proc Natl Acad Sci USA. 2003;100:13093–13098. doi: 10.1073/pnas.2135335100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Hulme JT, Westenbroek RE, Scheuer T, Catterall WA. Phosphorylation of serine-1928 in the distal C-terminal domain of cardiac Cav1.2 channels during beta1-adrenergic regulation. Proc Natl Acad Sci USA. 2006;103:16574–16579. doi: 10.1073/pnas.0607294103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Hulme JT, Yarov-Yarovoy V, Lin TW, Scheuer T, Catterall WA. Autoinhibitory control of the Cav1.2 channel by its proteolytically processed distal C-terminal domain. J Physiol. 2006;576:87–102. doi: 10.1113/jphysiol.2006.111799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Ikeda SR. Voltage-dependent modulation of N-type calcium channels by G-protein beta gamma subunits. Nature. 1996;380:255–258. doi: 10.1038/380255a0. [DOI] [PubMed] [Google Scholar]
- 198.Inanobe A, Yoshimoto Y, Horio Y, Morishige KI, Hibino H, Matsumoto S, Tokunaga Y, Maeda T, Hata Y, Takai Y, Kurachi Y. Characterization of G-protein-gated K+ channels composed of Kir3.2 subunits in dopaminergic neurons of the substantia nigra. J Neurosci. 1999;19:1006–1017. doi: 10.1523/JNEUROSCI.19-03-01006.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Isaacson JS, Murphy GJ. Glutamate-mediated extrasynaptic inhibition: direct coupling of NMDA receptors to Ca2+-activated K+ channels. Neuron. 2001;31:1027–1034. doi: 10.1016/s0896-6273(01)00428-7. [DOI] [PubMed] [Google Scholar]
- 200.Isacson CK, Lu Q, Karas RH, Cox DH. RACK1 is a BKCa channel binding protein. Am J Physiol Cell Physiol. 2007;292:C1459–C1466. doi: 10.1152/ajpcell.00322.2006. [DOI] [PubMed] [Google Scholar]
- 201.Ivanina T, Blumenstein Y, Shistik E, Barzilai R, Dascal N. Modulation of L-type Ca2+ channels by Gbeta gamma and calmodulin via interactions with N and C termini of alpha 1C. J Biol Chem. 2000;275:39846–39854. doi: 10.1074/jbc.M005881200. [DOI] [PubMed] [Google Scholar]
- 202.Ivanina T, Rishal I, Varon D, Mullner C, Frohnwieser-Steinecke B, Schreibmayer W, Dessauer CW, Dascal N. Mapping the Gbeta-gamma-binding sites in GIRK1 and GIRK2 subunits of the G protein-activated K+ channel. J Biol Chem. 2003;278:29174–29183. doi: 10.1074/jbc.M304518200. [DOI] [PubMed] [Google Scholar]
- 203.Ivanina T, Varon D, Peleg S, Rishal I, Porozov Y, Dessauer CW, Keren-Raifman T, Dascal N. G-alpha-i1 and G-alpha-i3 differentially interact with, and regulate, the G protein-activated K+ channel. J Biol Chem. 2004 doi: 10.1074/jbc.M313425200. [DOI] [PubMed] [Google Scholar]
- 204.Iwami G, Kawabe J, Ebina T, Cannon PJ, Homcy CJ, Ishikawa Y. Regulation of adenylyl cyclase by protein kinase A. J Biol Chem. 1995;270:12481–12484. doi: 10.1074/jbc.270.21.12481. [DOI] [PubMed] [Google Scholar]
- 205.Jahn H, Nastainczyk W, Rohrkasten A, Schneider T, Hofmann F. Site-specific phosphorylation of the purified receptor for calcium-channel blockers by cAMP- and cGMP-dependent protein kinases, protein kinase C, calmodulin-dependent protein kinase II and casein kinase II. Eur J Biochem. 1988;178:535–542. doi: 10.1111/j.1432-1033.1988.tb14480.x. [DOI] [PubMed] [Google Scholar]
- 206.Janssens V, Goris J. Protein phosphatase 2A: a highly regulated family of serine/threonine phosphatases implicated in cell growth and signalling. Biochem J. 2001;353:417–439. doi: 10.1042/0264-6021:3530417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Jiang X, Lautermilch NJ, Watari H, Westenbroek RE, Scheuer T, Catterall WA. Modulation of Cav2.1 channels by Ca2+/ calmodulin-dependent protein kinase II bound to the C-terminal domain. Proc Natl Acad Sci USA. 2008;105:341–346. doi: 10.1073/pnas.0710213105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Johnson BD, Brousal JP, Peterson BZ, Gallombardo PA, Hockerman GH, Lai Y, Scheuer T, Catterall WA. Modulation of the cloned skeletal muscle L-type Ca2+ channel by anchored cAMP-dependent protein kinase. J Neurosci. 1997;17:1243–1255. doi: 10.1523/JNEUROSCI.17-04-01243.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Jurevicius J, Skeberdis VA, Fischmeister R. Role of cyclic nucleotide phosphodiesterase isoforms in cAMP compartmentation following beta2-adrenergic stimulation of ICa,L in frog ventricular myocytes. J Physiol. 2003;551:239–252. doi: 10.1113/jphysiol.2003.045211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Kalia LV, Pitcher GM, Pelkey KA, Salter MW. PSD-95 is a negative regulator of the tyrosine kinase Src in the NMDA receptor complex. EMBO J. 2006;25:4971–4982. doi: 10.1038/sj.emboj.7601342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Kamp TJ, Hell JW. Regulation of cardiac L-type calcium channels by PKA and PKC. Circ Res. 2000;87:1095–1102. doi: 10.1161/01.res.87.12.1095. [DOI] [PubMed] [Google Scholar]
- 213.Kang MG, Campbell KP. Gamma subunit of voltage-activated calcium channels. J Biol Chem. 2003;278:21315–21318. doi: 10.1074/jbc.R300004200. [DOI] [PubMed] [Google Scholar]
- 214.Kang MG, Chen CC, Felix R, Letts VA, Frankel WN, Mori Y, Campbell KP. Biochemical and biophysical evidence for gamma 2 subunit association with neuronal voltage-activated Ca2+ channels. J Biol Chem. 2001;276:32917–32924. doi: 10.1074/jbc.M100787200. [DOI] [PubMed] [Google Scholar]
- 215.Kang MG, Chen CC, Wakamori M, Hara Y, Mori Y, Campbell KP. A functional AMPA receptor-calcium channel complex in the postsynaptic membrane. Proc Natl Acad Sci USA. 2006;103:5561–5566. doi: 10.1073/pnas.0601289103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Kapiloff MS, Schillace RV, Westphal AM, Scott JD. mAKAP: an A-kinase anchoring protein targeted to the nuclear membrane of differentiated myocytes. J Cell Sci. 1999;112:2725–2736. doi: 10.1242/jcs.112.16.2725. [DOI] [PubMed] [Google Scholar]
- 217.Kato AS, Zhou W, Milstein AD, Knierman MD, Siuda ER, Dotzlaf JE, Yu H, Hale JE, Nisenbaum ES, Nicoll RA, Bredt DS. New transmembrane AMPA receptor regulatory protein isoform, gamma-7, differentially regulates AMPA receptors. J Neurosci. 2007;27:4969–4977. doi: 10.1523/JNEUROSCI.5561-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Kim J, Ghosh S, Nunziato DA, Pitt GS. Identification of the components controlling inactivation of voltage-gated Ca2+ channels. Neuron. 2004;41:745–754. doi: 10.1016/s0896-6273(04)00081-9. [DOI] [PubMed] [Google Scholar]
- 219.Klauck TM, Faux MC, Labudda K, Langeberg LK, Jaken S, Scott JD. Coordination of three signaling enzymes by AKAP79, a mammalian scaffold protein. Science. 1996;271:1589–1592. doi: 10.1126/science.271.5255.1589. [DOI] [PubMed] [Google Scholar]
- 220.Kleuss C, Scherubl H, Hescheler J, Schultz G, Wittig B. Selectivity in signal transduction determined by gamma subunits of heterotrimeric G proteins. Science. 1993;259:832–834. doi: 10.1126/science.8094261. [DOI] [PubMed] [Google Scholar]
- 221.Koschak A, Obermair GJ, Pivotto F, Sinnegger-Brauns MJ, Striessnig J, Pietrobon D. Molecular nature of anomalous L-type calcium channels in mouse cerebellar granule cells. J Neurosci. 2007;27:3855–3863. doi: 10.1523/JNEUROSCI.4028-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Koyrakh L, Lujan R, Colon J, Karschin C, Kurachi Y, Karschin A, Wickman K. Molecular and cellular diversity of neuronal G-protein-gated potassium channels. J Neurosci. 2005;25:11468–11478. doi: 10.1523/JNEUROSCI.3484-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Kubo Y, Adelman JP, Clapham DE, Jan LY, Karschin A, Kurachi Y, Lazdunski M, Nichols CG, Seino S, Vandenberg CA. International Union of Pharmacology. LIV. Nomenclature and molecular relationships of inwardly rectifying potassium channels. Pharmacol Rev. 2005;57:509–526. doi: 10.1124/pr.57.4.11. [DOI] [PubMed] [Google Scholar]
- 224.Kume H, Takai A, Tokuno H, Tomita T. Regulation of Ca2+-dependent K+-channel activity in tracheal myocytes by phosphorylation. Nature. 1989;341:152–154. doi: 10.1038/341152a0. [DOI] [PubMed] [Google Scholar]
- 225.Kunkel MT, Peralta EG. Identification of domains conferring G protein regulation on inward rectifier potassium channels. Cell. 1995;83:443–449. doi: 10.1016/0092-8674(95)90122-1. [DOI] [PubMed] [Google Scholar]
- 226.Kuroda S, Tokunaga C, Kiyohara Y, Higuchi O, Konishi H, Mizuno K, Gill GN, Kikkawa U. Protein-protein interaction of zinc finger LIM domains with protein kinase C. J Biol Chem. 1996;271:31029–31032. doi: 10.1074/jbc.271.49.31029. [DOI] [PubMed] [Google Scholar]
- 227.Kurokawa J, Motoike HK, Rao J, Kass RS. Regulatory actions of the A-kinase anchoring protein Yotiao on a heart potassium channel downstream of PKA phosphorylation. Proc Natl Acad Sci USA. 2004;101:16374–16378. doi: 10.1073/pnas.0405583101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Kurschner C, Yuzaki M. Neuronal interleukin-16 (NIL-16): a dual function PDZ domain protein. J Neurosci. 1999;19:7770–7780. doi: 10.1523/JNEUROSCI.19-18-07770.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Kwon YG, Huang HB, Desdouits F, Girault JA, Greengard P, Nairn AC. Characterization of the interaction between DARPP-32 and protein phosphatase 1 (PP-1): DARPP-32 peptides antagonize the interaction of PP-1 with binding proteins. Proc Natl Acad Sci USA. 1997;94:3536–3541. doi: 10.1073/pnas.94.8.3536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Lange A, Gebremedhin D, Narayanan J, Harder D. 20-Hydroxyeicosatetraenoic acid-induced vasoconstriction and inhibition of potassium current in cerebral vascular smooth muscle is dependent on activation of protein kinase C. J Biol Chem. 1997;272:27345–27352. doi: 10.1074/jbc.272.43.27345. [DOI] [PubMed] [Google Scholar]
- 231.Lavine N, Ethier N, Oak JN, Pei L, Liu F, Trieu P, Rebois RV, Bouvier M, Hebert TE, Van Tol HH. G protein-coupled receptors form stable complexes with inwardly rectifying potassium channels and adenylyl cyclase. J Biol Chem. 2002;277:46010–46019. doi: 10.1074/jbc.M205035200. [DOI] [PubMed] [Google Scholar]
- 232.Lee A, Scheuer T, Catterall WA. Ca2+/calmodulin-dependent facilitation and inactivation of P/Q-type Ca2+ channels. J Neurosci. 2000;20:6830–6838. doi: 10.1523/JNEUROSCI.20-18-06830.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Lee TS, Karl R, Moosmang S, Lenhardt P, Klugbauer N, Hofmann F, Kleppisch T, Welling A. Calmodulin kinase II is involved in voltage-dependent facilitation of the L-type Cav1.2 calcium channel: identification of the phosphorylation sites. J Biol Chem. 2006;281:25560–25567. doi: 10.1074/jbc.M508661200. [DOI] [PubMed] [Google Scholar]
- 234.Lehnart SE, Wehrens XH, Reiken S, Warrier S, Belevych AE, Harvey RD, Richter W, Jin SL, Conti M, Marks AR. Phosphodiesterase 4D deficiency in the ryanodine-receptor complex promotes heart failure and arrhythmias. Cell. 2005;123:25–35. doi: 10.1016/j.cell.2005.07.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234a.Lemke T, Welling A, Christel CJ, Blaich A, Berhard D, Lenhardt P, Hofmann F, Moosmang S. Unchanged β-adrenergic stimulatin of cardic L-type calcium channels in Cav1.2 phosphorylation site S1928A mutant mice. J Biol Chem. 2008;283:34738–34744. doi: 10.1074/jbc.M804981200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Leonard AS, Bayer KU, Merrill MA, Lim IA, Shea MA, Schulman H, Hell JW. Regulation of calcium/calmodulin-dependent protein kinase II docking to N-methyl-d-aspartate receptors by calcium/calmodulin and α-actinin. J Biol Chem. 2002;277:48441–48448. doi: 10.1074/jbc.M205164200. [DOI] [PubMed] [Google Scholar]
- 236.Leonard AS, Davare MA, Horne MC, Garner CC, Hell JW. SAP97 is associated with the α-amino-3-hydroxy-5-methylisox-azole-4-propionic acid receptor GluR1 subunit. J Biol Chem. 1998;273:19518–19524. doi: 10.1074/jbc.273.31.19518. [DOI] [PubMed] [Google Scholar]
- 237.Leonard AS, Lim IA, Hemsworth DE, Horne MC, Hell JW. Calcium/calmodulin-dependent protein kinase II is associated with the N-methyl-d-aspartate receptor. Proc Natl Acad Sci USA. 1999;96:3239–3244. doi: 10.1073/pnas.96.6.3239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Leroy J, Richards MS, Butcher AJ, Nieto-Rostro M, Pratt WS, Davies A, Dolphin AC. Interaction via a key tryptophan in the I-II linker of N-type calcium channels is required for beta1 but not for palmitoylated beta2, implicating an additional binding site in the regulation of channel voltage-dependent properties. J Neurosci. 2005;25:6984–6996. doi: 10.1523/JNEUROSCI.1137-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Levitzki A. From epinephrine to cyclic AMP. Science. 1988;241:800–806. doi: 10.1126/science.2841758. [DOI] [PubMed] [Google Scholar]
- 240.Li D, Wang F, Lai M, Chen Y, Zhang JF. A protein phosphatase 2c alpha-Ca2+ channel complex for dephosphorylation of neuronal Ca2+ channels phosphorylated by protein kinase C. J Neurosci. 2005;25:1914–1923. doi: 10.1523/JNEUROSCI.4790-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.Li M, West JW, Numann R, Murphy BJ, Scheuer T, Catterall WA. Convergent regulation of sodium channels by protein kinase C and cAMP-dependent protein kinase. Science. 1993;261:1439–1442. doi: 10.1126/science.8396273. [DOI] [PubMed] [Google Scholar]
- 242.Lim IA, Hall DD, Hell JW. Selectivity and promiscuity of the first and second PDZ domains of PSD-95 and synapse-associated protein 102. J Biol Chem. 2002;277:21697–21711. doi: 10.1074/jbc.M112339200. [DOI] [PubMed] [Google Scholar]
- 243.Lim NF, Dascal N, Labarca C, Davidson N, Lester HA. A G protein-gated K channel is activated via beta 2-adrenergic receptors and G beta gamma subunits in Xenopus oocytes. J Gen Physiol. 1995;105:421–439. doi: 10.1085/jgp.105.3.421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244.Lin F, Wang H, Malbon CC. Gravin-mediated formation of signaling complexes in beta 2-adrenergic receptor desensitization and resensitization. J Biol Chem. 2000;275:19025–19034. doi: 10.1074/jbc.275.25.19025. [DOI] [PubMed] [Google Scholar]
- 245.Ling S, Sheng JZ, Braun AP. The calcium-dependent activity of large-conductance, calcium-activated K+ channels is enhanced by Pyk2- and Hck-induced tyrosine phosphorylation. Am J Physiol Cell Physiol. 2004;287:C698–C706. doi: 10.1152/ajpcell.00030.2004. [DOI] [PubMed] [Google Scholar]
- 246.Ling S, Woronuk G, Sy L, Lev S, Braun AP. Enhanced activity of a large conductance, calcium-sensitive K+ channel in the presence of Src tyrosine kinase. J Biol Chem. 2000;275:30683–30689. doi: 10.1074/jbc.M004292200. [DOI] [PubMed] [Google Scholar]
- 247.Lisman J, Schulman H, Cline H. The molecular basis of CaMKII function in synaptic and behavioral memory. Nature Neurosci. 2002;3:175–190. doi: 10.1038/nrn753. [DOI] [PubMed] [Google Scholar]
- 248.Liu G, Dilmac N, Hilliard N, Hockerman GH. Cav3.1 is preferentially coupled to glucose-stimulated insulin secretion in the pancreatic beta-cell line INS-1. J Pharmacol Exp Ther. 2003;305:271–278. doi: 10.1124/jpet.102.046334. [DOI] [PubMed] [Google Scholar]
- 249.Liu G, Shi J, Yang L, Cao L, Park SM, Cui J, Marx SO. Assembly of a Ca2+-dependent BK channel signaling complex by binding to beta2 adrenergic receptor. EMBO J. 2004;23:2196–2205. doi: 10.1038/sj.emboj.7600228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250.Lu L, Zhang Q, Timofeyev V, Zhang Z, Young JN, Shin HS, Knowlton AA, Chiamvimonvat N. Molecular coupling of a Ca2+-activated K+ channel to L-type Ca2+ channels via alpha-actinin2. Circ Res. 2007;100:112–120. doi: 10.1161/01.RES.0000253095.44186.72. [DOI] [PubMed] [Google Scholar]
- 251.Lu WY, Xiong ZG, Lei S, Orser BA, Dudek E, Browning MD, MacDonald JF. G-protein-coupled receptors act via protein kinase C and Src to regulate NMDA receptors. Nat Neurosci. 1999;2:331–338. doi: 10.1038/7243. [DOI] [PubMed] [Google Scholar]
- 252.Luscher C, Jan LY, Stoffel M, Malenka RC, Nicoll RA. G protein-coupled inwardly rectifying K+ channels (GIRKs) mediate postsynaptic but not presynaptic transmitter actions in hippocampal neurons. Neuron. 1997;19:687–695. doi: 10.1016/s0896-6273(00)80381-5. [DOI] [PubMed] [Google Scholar]
- 253.MacKenzie SJ, Baillie GS, McPhee I, Bolger GB, Houslay MD. ERK2 mitogen-activated protein kinase binding, phosphorylation, and regulation of the PDE4D cAMP-specific phosphodiesterases. The involvement of COOH-terminal docking sites and NH2-terminal UCR regions. J Biol Chem. 2000;275:16609–16617. doi: 10.1074/jbc.275.22.16609. [DOI] [PubMed] [Google Scholar]
- 254.Maeno-Hikichi Y, Chang S, Matsumura K, Lai M, Lin H, Na-kagawa N, Kuroda S, Zhang JF. A PKC epsilon-ENH-channel complex specifically modulates N-type Ca2+ channels. Nat Neurosci. 2003;6:468–475. doi: 10.1038/nn1041. [DOI] [PubMed] [Google Scholar]
- 255.Maingret F, Coste B, Hao J, Giamarchi A, Allen D, Crest M, Litchfield DW, Adelman JP, Delmas P. Neurotransmitter modulation of small-conductance Ca2+-activated K+ channels by regulation of Ca2+ gating. Neuron. 2008;59:439–449. doi: 10.1016/j.neuron.2008.05.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 256.Malenka RC, Bear MF. LTP and LTD: an embarrassment of riches. Neuron. 2004;44:5–21. doi: 10.1016/j.neuron.2004.09.012. [DOI] [PubMed] [Google Scholar]
- 257.Malenka RC, Nicoll RA. Long-term potentiation-a decade of progress? Science. 1999;285:1870–1874. doi: 10.1126/science.285.5435.1870. [DOI] [PubMed] [Google Scholar]
- 258.Malinow R. AMPA receptor trafficking and long-term potentiation. Philos Trans R Soc Lond B Biol Sci. 2003;358:707–714. doi: 10.1098/rstb.2002.1233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259.Maltez JM, Nunziato DA, Kim J, Pitt GS. Essential Ca(V)beta modulatory properties are AID-independent. Nat Struct Mol Biol. 2005;12:372–377. doi: 10.1038/nsmb909. [DOI] [PubMed] [Google Scholar]
- 260.Mammen AL, Kameyama K, Roche KW, Huganir RL. Phosphorylation of the α-amino-3-hydroxy-5-methylisoxazole-4-propionic acid receptor GluR1 subunit by calcium/calmodulin-dependent kinase II. J Biol Chem. 1997;272:32528–32533. doi: 10.1074/jbc.272.51.32528. [DOI] [PubMed] [Google Scholar]
- 261.Marbach I, Bar-Sinai A, Minich M, Levitzki A. Beta subunit copurifies with GppNHp-activated adenylyl cyclase. J Biol Chem. 1990;265:9999–10004. [PubMed] [Google Scholar]
- 262.Markram H, Lubke J, Frotscher M, Sakmann B. Regulation of synaptic efficacy by coincidence of postsynaptic APs and EPSPs. Science. 1997;275:213–215. doi: 10.1126/science.275.5297.213. [DOI] [PubMed] [Google Scholar]
- 263.Marks AR. Cardiac intracellular calcium release channels: role in heart failure. Circ Res. 2000;87:8–11. doi: 10.1161/01.res.87.1.8. [DOI] [PubMed] [Google Scholar]
- 264.Marrion NV, Tavalin ST. Selective activation of Ca2+-activated K+ channels by co-localized Ca2+channels in hippocampal neurons. Nature. 1998;395:900–905. doi: 10.1038/27674. [DOI] [PubMed] [Google Scholar]
- 265.Marx SO, Kurokawa J, Reiken S, Motoike H, D'Armiento J, Marks AR, Kass RS. Requirement of a macromolecular signaling complex for beta adrenergic receptor modulation of the KCNQ1-KCNE1 potassium channel. Science. 2002;295:496–499. doi: 10.1126/science.1066843. [DOI] [PubMed] [Google Scholar]
- 266.Marx SO, Reiken S, Hisamatsu Y, Gaburjakova M, Gaburjakova J, Yang YM, Rosemblit N, Marks AR. Phosphorylation-dependent regulation of ryanodine receptors: a novel role for leucine/isoleucine zippers. J Cell Biol. 2001;153:699–708. doi: 10.1083/jcb.153.4.699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 267.Marx SO, Reiken S, Hisamatsu Y, Jayaraman T, Burkhoff D, Rosemblit N, Marks AR. PKA phosphorylation dissociates FKBP12.6 from the calcium release channel (ryanodine receptor): defective regulation in failing hearts. Cell. 2000;101:365–376. doi: 10.1016/s0092-8674(00)80847-8. [DOI] [PubMed] [Google Scholar]
- 268.McCarron JG, McGeown JG, Reardon S, Ikebe M, Fay FS, Walsh JV., Jr Calcium-dependent enhancement of calcium current in smooth muscle by calmodulin-dependent protein kinase II. Nature. 1992;357:74–77. doi: 10.1038/357074a0. [DOI] [PubMed] [Google Scholar]
- 269.McCartney S, Little BM, Langeberg LK, Scott JD. Cloning and characterization of A-kinase anchor protein 100 (AKAP100). A protein that targets A-kinase to the sarcoplasmic reticulum. J Biol Chem. 1995;270:9327–9333. doi: 10.1074/jbc.270.16.9327. [DOI] [PubMed] [Google Scholar]
- 270.McCright B, Rivers AM, Audlin S, Virshup DM. The B56 family of protein phosphatase 2A (PP2A) regulatory subunits encodes differentiation-induced phosphoproteins that target PP2A to both nucleus and cytoplasm. J Biol Chem. 1996;271:22081–22089. doi: 10.1074/jbc.271.36.22081. [DOI] [PubMed] [Google Scholar]
- 271.McEwen DP, Meadows LS, Chen C, Thyagarajan V, Isom LL. Sodium channel beta1 subunit-mediated modulation of Nav1.2 currents and cell surface density is dependent on interactions with contactin and ankyrin. J Biol Chem. 2004;279:16044–16049. doi: 10.1074/jbc.M400856200. [DOI] [PubMed] [Google Scholar]
- 272.McGee AW, Dakoji SR, Olsen O, Bredt DS, Lim WA, Prehoda KE. Structure of the SH3-guanylate kinase module from PSD-95 suggests a mechanism for regulated assembly of MAGUK scaffolding proteins. Mol Cell. 2001;8:1291–1301. doi: 10.1016/s1097-2765(01)00411-7. [DOI] [PubMed] [Google Scholar]
- 273.McGee AW, Nunziato DA, Maltez JM, Prehoda KE, Pitt GS, Bredt DS. Calcium channel function regulated by the SH3-GK module in beta subunits. Neuron. 2004;42:89–99. doi: 10.1016/s0896-6273(04)00149-7. [DOI] [PubMed] [Google Scholar]
- 274.McHugh D, Sharp EM, Scheuer T, Catterall WA. Inhibition of cardiac L-type calcium channels by protein kinase C phosphorylation of two sites in the N-terminal domain. Proc Natl Acad Sci USA. 2000;97:12334–12338. doi: 10.1073/pnas.210384297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 275.Medina I, Krapivinsky G, Arnold S, Kovoor P, Krapivinsky L, Clapham DE. A switch mechanism for G beta gamma activation of I(KACh) J Biol Chem. 2000;275:29709–29716. doi: 10.1074/jbc.M004989200. [DOI] [PubMed] [Google Scholar]
- 276.Meissner G. Ryanodine receptor/Ca2+ release channels and their regulation by endogenous effectors. Annu Rev Physiol. 1994;56:485–508. doi: 10.1146/annurev.ph.56.030194.002413. [DOI] [PubMed] [Google Scholar]
- 277.Merrill MA, Chen Y, Strack S, Hell JW. Activity-driven postsynaptic translocation of CaMKII. Trends Pharm Sci. 2005;26:645–653. doi: 10.1016/j.tips.2005.10.003. [DOI] [PubMed] [Google Scholar]
- 278.Mikala G, Klockner U, Varadi M, Eisfeld J, Schwartz A, Varadi G. cAMP-dependent phosphorylation sites and macroscopic activity of recombinant cardiac L-type calcium channels. Mol Cell Biochem. 1998;185:95–109. doi: 10.1023/a:1006878106672. [DOI] [PubMed] [Google Scholar]
- 279.Mikami A, Imoto K, Tanabe T, Niidome T, Mori Y, Takeshima H, Narumiya S, Numa S. Primary structure and functional expression of the cardiac dihydropyridine-sensitive calcium channel. Nature. 1989;340:230–233. doi: 10.1038/340230a0. [DOI] [PubMed] [Google Scholar]
- 280.Milstein AD, Zhou W, Karimzadegan S, Bredt DS, Nicoll RA. TARP subtypes differentially and dose-dependently control synaptic AMPA receptor gating. Neuron. 2007;55:905–918. doi: 10.1016/j.neuron.2007.08.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 281.Mitterdorfer J, Froschmayr M, Grabner M, Moebius FF, Glossmann H, Striessnig J. Identification of PK-A phosphorylation sites in the carboxyl terminus of L-type calcium channel alpha 1 subunits. Biochemistry. 1996;35:9400–9406. doi: 10.1021/bi960683o. [DOI] [PubMed] [Google Scholar]
- 282.Mochly-Rosen D, Gordon AS. Anchoring proteins for protein kinase C: a means for isozyme selectivity. FASEB J. 1998;12:35–42. [PubMed] [Google Scholar]
- 283.Muller BM, Kistner U, Kindler S, Chung WJ, Kuhlendahl S, Fenster SD, Lau LF, Veh RW, Huganir RL, Gundelfinger ED, Garner CC. SAP102, a novel postsynaptic protein that interacts with NMDA receptor complexes in vivo. Neuron. 1996;17:255–265. doi: 10.1016/s0896-6273(00)80157-9. [DOI] [PubMed] [Google Scholar]
- 284.Mullner C, Vorobiov D, Bera AK, Uezono Y, Yakubovich D, Frohnwieser-Steinecker B, Dascal N, Schreibmayer W. Heterologous facilitation of G protein-activated K+ channels by beta-adrenergic stimulation via cAMP-dependent protein kinase. J Gen Physiol. 2000;115:547–558. doi: 10.1085/jgp.115.5.547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 285.Mullner C, Yakubovich D, Dessauer CW, Platzer D, Schreibmayer W. Single channel analysis of the regulation of GIRK1/GIRK4 channels by protein phosphorylation. Biophys J. 2003;84:1399–1409. doi: 10.1016/S0006-3495(03)74954-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 286.Murakami M, Wissenbach U, Flockerzi V. Gene structure of the murine calcium channel beta3 subunit, cDNA and characterization of alternative splicing and transcription products. Eur J Biochem. 1996;236:138–143. doi: 10.1111/j.1432-1033.1996.t01-1-00138.x. [DOI] [PubMed] [Google Scholar]
- 287.Murphy BJ, Rossie S, De Jongh KS, Catterall WA. Identification of the sites of selective phosphorylation and dephosphorylation of the rat brain Na+ channel alpha subunit by cAMP-dependent protein kinase and phosphoprotein phosphatases. J Biol Chem. 1993;268:27355–27362. [PubMed] [Google Scholar]
- 288.Naguro I, Nagao T, Adachi-Akahane S. Ser(1901) of alpha(1C) subunit is required for the PKA-mediated enhancement of L-type Ca2+ channel currents but not for the negative shift of activation. FEBS Lett. 2001;489:87–91. doi: 10.1016/s0014-5793(01)02079-8. [DOI] [PubMed] [Google Scholar]
- 289.Nakanishi H, Obaishi H, Satoh A, Wada M, Mandai K, Satoh K, Nishioka H, Matsuura Y, Mizoguchi A, Takai Y. Neurabin: a novel neural tissue-specific actin filament-binding protein involved in neurite formation. J Cell Biol. 1997;139:951–961. doi: 10.1083/jcb.139.4.951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 290.Nara M, Dhulipala PD, Wang YX, Kotlikoff MI. Reconstitution of beta-adrenergic modulation of large conductance, calcium-activated potassium (maxi-K) channels in Xenopus oocytes. Identification of the cAMP-dependent protein kinase phosphorylation site. J Biol Chem. 1998;273:14920–14924. doi: 10.1074/jbc.273.24.14920. [DOI] [PubMed] [Google Scholar]
- 291.Navedo MF, Nieves-Cintron M, Amberg GC, Yuan C, Votaw VS, Lederer WJ, McKnight GS, Santana LF. AKAP150 is required for stuttering persistent Ca2+ sparklets and angiotensin II-induced hypertension. Circ Res. 2008;102:e20–e35. doi: 10.1161/CIRCRESAHA.107.167809. [DOI] [PubMed] [Google Scholar]
- 292.Neely A, Wei X, Olcese R, Birnbaumer L, Stefani E. Potentiation by the beta subunit of the ratio of the ionic current to the charge movement in the cardiac calcium channel. Science. 1993;262:575–578. doi: 10.1126/science.8211185. [DOI] [PubMed] [Google Scholar]
- 293.Neher E. Vesicle pools and Ca2+ microdomains: new tools for understanding their roles in neurotransmitter release. Neuron. 1998;20:389–399. doi: 10.1016/s0896-6273(00)80983-6. [DOI] [PubMed] [Google Scholar]
- 294.Neubig RR. Membrane organization in G-protein mechanisms. FASEB J. 1994;8:939–946. doi: 10.1096/fasebj.8.12.8088459. [DOI] [PubMed] [Google Scholar]
- 295.Newcomb R, Szoke B, Palma A, Wang G, Chen X, Hopkins W, Cong R, Miller J, Urge L, Tarczy-Hornoch K, Loo JA, Dooley DJ, Nadasdi L, Tsien RW, Lemos J, Miljanich G. Selective peptide antagonist of the class E calcium channel from the venom of the tarantula Hysterocrates gigas. Biochemistry. 1998;37:15353–15362. doi: 10.1021/bi981255g. [DOI] [PubMed] [Google Scholar]
- 296.Ngo-Anh TJ, Bloodgood BL, Lin M, Sabatini BL, Maylie J, Adelman JP. SK channels and NMDA receptors form a Ca2+-mediated feedback loop in dendritic spines. Nat Neurosci. 2005;8:642–649. doi: 10.1038/nn1449. [DOI] [PubMed] [Google Scholar]
- 297.Nicoll RA, Tomita S, Bredt DS. Auxiliary subunits assist AMPA-type glutamate receptors. Science. 2006;311:1253–1256. doi: 10.1126/science.1123339. [DOI] [PubMed] [Google Scholar]
- 298.Nikolov EN, Ivanova-Nikolova TT. Coordination of membrane excitability through a GIRK1 signaling complex in the atria. J Biol Chem. 2004;279:23630–23636. doi: 10.1074/jbc.M312861200. [DOI] [PubMed] [Google Scholar]
- 299.Nishizuka Y. Protein kinase C and lipid signaling for sustained cellular responses. FASEB J. 1995;9:484–496. [PubMed] [Google Scholar]
- 300.Nobles M, Benians A, Tinker A. Heterotrimeric G proteins pre-couple with G protein-coupled receptors in living cells. Proc Natl Acad Sci USA. 2005;102:18706–18711. doi: 10.1073/pnas.0504778102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 301.Ouchi J, Komukai K, Kusakari Y, Obata T, Hongo K, Sasaki H, Kurihara S. α1-Adrenoceptor stimulation potentiates L-type Ca2+ current through Ca2+/calmodulin-dependent PK II (CaMKII) activation in rat ventricular myocytes. Proc Natl Acad Sci USA. 2005;102:9400–9405. doi: 10.1073/pnas.0503569102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 302.Oancea E, Meyer T. Protein kinase C as a molecular machine for decoding calcium and diacylglycerol signals. Cell. 1998;95:307–318. doi: 10.1016/s0092-8674(00)81763-8. [DOI] [PubMed] [Google Scholar]
- 303.Obermair GJ, Szabo Z, Bourinet E, Flucher BE. Differential targeting of the L-type Ca2+ channel alpha1C (Cav1.2) to synaptic and extrasynaptic compartments in hippocampal neurons. Eur J Neurosci. 2004;19:2109–2122. doi: 10.1111/j.0953-816X.2004.03272.x. [DOI] [PubMed] [Google Scholar]
- 304.Oliveria SF, Dell'acqua ML, Sather WA. AKAP79/150 anchoring of calcineurin controls neuronal L-type Ca2+ channel activity and nuclear signaling. Neuron. 2007;55:261–275. doi: 10.1016/j.neuron.2007.06.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 305.Oliveria SF, Gomez LL, Dell'Acqua ML. Imaging kinase-AKAP79-phosphatase scaffold complexes at the plasma membrane in living cells using FRET microscopy. J Cell Biol. 2003;160:101–112. doi: 10.1083/jcb.200209127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 306.Ono K, Fozzard HA. Phosphorylation restores activity of L-type calcium channels after rundown in inside-out patches from rabbit cardiac cells. J Physiol. 1992;454:673–688. doi: 10.1113/jphysiol.1992.sp019286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 307.Ono K, Fozzard HA. Two phosphatase sites on the Ca2+ channel affecting different kinetic functions. J Physiol. 1993;470:73–84. doi: 10.1113/jphysiol.1993.sp019848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 308.Opatowsky Y, Chen CC, Campbell KP, Hirsch JA. Structural analysis of the voltage-dependent calcium channel beta subunit functional core and its complex with the alpha 1 interaction domain. Neuron. 2004;42:387–399. doi: 10.1016/s0896-6273(04)00250-8. [DOI] [PubMed] [Google Scholar]
- 309.Osterrieder W, Brum G, Hescheler J, Trautwein W, Flockerzi V, Hofmann F. Injection of subunits of cyclic AMP-dependent protein kinase into cardiac myocytes modulates Ca2+ current. Nature. 1982;298:576–578. doi: 10.1038/298576a0. [DOI] [PubMed] [Google Scholar]
- 310.Pawson T, Nash P. Assembly of cell regulatory systems through protein interaction domains. Science. 2003;300:445–452. doi: 10.1126/science.1083653. [DOI] [PubMed] [Google Scholar]
- 311.Peleg S, Varon D, Ivanina T, Dessauer CW, Dascal N. G(alpha)(i) controls the gating of the G protein-activated K+ channel, GIRK. Neuron. 2002;33:87–99. doi: 10.1016/s0896-6273(01)00567-0. [DOI] [PubMed] [Google Scholar]
- 312.Perets T, Blumenstein Y, Shistik E, Lotan I, Dascal N. A potential site of functional modulation by protein kinase A in the cardiac Ca2+ channel alpha 1C subunit. FEBS Lett. 1996;384:189–192. doi: 10.1016/0014-5793(96)00303-1. [DOI] [PubMed] [Google Scholar]
- 313.Perez-Reyes E. Molecular physiology of low-voltage-activated T-type calcium channels. Physiol Rev. 2003;83:117–161. doi: 10.1152/physrev.00018.2002. [DOI] [PubMed] [Google Scholar]
- 314.Perry SJ, Baillie GS, Kohout TA, McPhee I, Magiera MM, Ang KL, Miller WE, McLean AJ, Conti M, Houslay MD, Lefkowitz RJ. Targeting of cyclic AMP degradation to beta 2-adrenergic receptors by beta-arrestins. Science. 2002;298:834–836. doi: 10.1126/science.1074683. [DOI] [PubMed] [Google Scholar]
- 315.Peterson BZ, DeMaria CD, Adelman JP, Yue DT. Calmodulin is the Ca2+ sensor for Ca2+-dependent inactivation of L-type calcium channels. Neuron. 1999;22:549–558. doi: 10.1016/s0896-6273(00)80709-6. [DOI] [PubMed] [Google Scholar]
- 316.Pichler M, Cassidy TN, Reimer D, Haase H, Kraus R, Ostler D, Striessnig J. Beta subunit heterogeneity in neuronal L-type Ca2+ channels. J Biol Chem. 1997;272:13877–13882. doi: 10.1074/jbc.272.21.13877. [DOI] [PubMed] [Google Scholar]
- 317.Piedras-Renteria ES, Tsien RW. Antisense oligonucleotides against alpha1E reduce R-type calcium currents in cerebellar granule cells. Proc Natl Acad Sci USA. 1998;95:7760–7765. doi: 10.1073/pnas.95.13.7760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 318.Pitcher JA, Payne ES, Csortos C, DePaoli-Roach AA, Lefkowitz RJ. The G-protein-coupled receptor phosphatase: a protein phosphatase type 2A with distinct subcellular distribution and substrate specificity. Proc Natl Acad Sci USA. 1995;92:8343–8347. doi: 10.1073/pnas.92.18.8343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 319.Prakriya M, Lingle CJ. BK channel activation by brief depolarizations requires Ca2+ influx through L- and Q-type Ca2+ channels in rat chromaffin cells. J Neurophysiol. 1999;81:2267–2278. doi: 10.1152/jn.1999.81.5.2267. [DOI] [PubMed] [Google Scholar]
- 320.Price NE, Mumby MC. Brain protein serine/threonine phosphatases. Curr Opin Neurobiol. 1999;9:336–342. doi: 10.1016/s0959-4388(99)80049-x. [DOI] [PubMed] [Google Scholar]
- 321.Priel A, Kolleker A, Ayalon G, Gillor M, Osten P, Stern-Bach Y. Stargazin reduces desensitization and slows deactivation of the AMPA-type glutamate receptors. J Neurosci. 2005;25:2682–2686. doi: 10.1523/JNEUROSCI.4834-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 322.Proenza C, O'Brien J, Nakai J, Mukherjee S, Allen PD, Beam KG. Identification of a region of RyR1 that participates in allosteric coupling with the alpha(1S) (Ca(V)1.1) II-III loop. J Biol Chem. 2002;277:6530–6535. doi: 10.1074/jbc.M106471200. [DOI] [PubMed] [Google Scholar]
- 323.Qin N, Platano D, Olcese R, Stefani E, Birnbaumer L. Direct interaction of gbetagamma with a C-terminal gbetagamma-binding domain of the Ca2+ channel alpha1 subunit is responsible for channel inhibition by G protein-coupled receptors. Proc Natl Acad Sci USA. 1997;94:8866–8871. doi: 10.1073/pnas.94.16.8866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 323a.Rankovic V, Landgraf P, Kanyshkova T, Munsch T, Budde T. 2008 Neuroscience Meeting Planner. Washington, DC: Society for Neuroscience; 2008. Calcium-dependent inactivation of L-type Ca2+ channels is modulated via a beta adrenergic signaling cascade in thalamocortical relay neurons (Program no 133.5) [Google Scholar]
- 324.Rebois RV, Hebert TE. Protein complexes involved in heptahelical receptor-mediated signal transduction. Receptors Channels. 2003;9:169–194. [PubMed] [Google Scholar]
- 325.Rebois RV, Robitaille M, Gales C, Dupre DJ, Baragli A, Trieu P, Ethier N, Bouvier M, Hebert TE. Heterotrimeric G proteins form stable complexes with adenylyl cyclase and Kir3.1 channels in living cells. J Cell Sci. 2006;119:2807–2818. doi: 10.1242/jcs.03021. [DOI] [PubMed] [Google Scholar]
- 326.Reinhart PH, Levitan IB. Kinase and phosphatase activities intimately associated with a reconstituted calcium-dependent potassium channel. J Neurosci. 1995;15:4572–4579. doi: 10.1523/JNEUROSCI.15-06-04572.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 327.Reuter H. Calcium channel modulation by neurotransmitters, enzymes and drugs. Nature. 1983;301:569–574. doi: 10.1038/301569a0. [DOI] [PubMed] [Google Scholar]
- 328.Reuveny E, Slesinger PA, Inglese J, Morales JM, Iniguez-Lluhi JA, Lefkowitz RJ, Bourne HR, Jan YN, Jan LY. Activation of the cloned muscarinic potassium channel by G protein beta gamma subunits. Nature. 1994;370:143–146. doi: 10.1038/370143a0. [DOI] [PubMed] [Google Scholar]
- 329.Rich TC, Fagan KA, Tse TE, Schaack J, Cooper DM, Karpen JW. A uniform extracellular stimulus triggers distinct cAMP signals in different compartments of a simple cell. Proc Natl Acad Sci USA. 2001;98:13049–13054. doi: 10.1073/pnas.221381398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 330.Rios E, Pizarro G, Stefani E. Charge movement and the nature of signal transduction in skeletal muscle excitation-contraction coupling. Annu Rev Physiol. 1992;54:109–133. doi: 10.1146/annurev.ph.54.030192.000545. [DOI] [PubMed] [Google Scholar]
- 331.Riven I, Iwanir S, Reuveny E. GIRK channel activation involves a local rearrangement of a preformed G protein channel complex. Neuron. 2006;51:561–573. doi: 10.1016/j.neuron.2006.08.017. [DOI] [PubMed] [Google Scholar]
- 332.Riven I, Kalmanzon E, Segev L, Reuveny E. Conformational rearrangements associated with the gating of the G protein-coupled potassium channel revealed by FRET microscopy. Neuron. 2003;38:225–235. doi: 10.1016/s0896-6273(03)00193-4. [DOI] [PubMed] [Google Scholar]
- 333.Robia SL, Ghanta J, Robu VG, Walker JW. Localization and kinetics of protein kinase C-epsilon anchoring in cardiac myocytes. Biophys J. 2001;80:2140–2151. doi: 10.1016/S0006-3495(01)76187-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 334.Rochais F, Abi-Gerges A, Horner K, Lefebvre F, Cooper DM, Conti M, Fischmeister R, Vandecasteele G. A specific pattern of phosphodiesterases controls the cAMP signals generated by different Gs-coupled receptors in adult rat ventricular myocytes. Circ Res. 2006;98:1081–1088. doi: 10.1161/01.RES.0000218493.09370.8e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 335.Rochais F, Vandecasteele G, Lefebvre F, Lugnier C, Lum H, Mazet JL, Cooper DM, Fischmeister R. Negative feedback exerted by cAMP-dependent protein kinase and cAMP phosphodiesterase on subsarcolemmal cAMP signals in intact cardiac myocytes: an in vivo study using adenovirus-mediated expression of CNG channels. J Biol Chem. 2004;279:52095–52105. doi: 10.1074/jbc.M405697200. [DOI] [PubMed] [Google Scholar]
- 336.Rodriguez P, Bhogal MS, Colyer J. Stoichiometric phosphorylation of cardiac ryanodine receptor on serine 2809 by calmodulin-dependent kinase II and protein kinase A. J Biol Chem. 2003;278:38593–38600. doi: 10.1074/jbc.C301180200. [DOI] [PubMed] [Google Scholar]
- 337.Rohrkasten A, Meyer HE, Nastainczyk W, Sieber M, Hofmann F. cAMP-dependent protein kinase rapidly phosphorylates serine-687 of the skeletal muscle receptor for calcium channel blockers. J Biol Chem. 1988;263:15325–15329. [PubMed] [Google Scholar]
- 338.Ron D, Chen CH, Caldwell J, Jamieson L, Orr E, Mochly-Rosen D. Cloning of an intracellular receptor for protein kinase C: a homolog of the beta subunit of G proteins. Proc Natl Acad Sci USA. 1994;91:839–843. doi: 10.1073/pnas.91.3.839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 339.Rotman EI, Murphy BJ, Catterall WA. Sites of selective cAMP-dependent phosphorylation of the L-type calcium channel alpha 1 subunit from intact rabbit skeletal muscle myotubes. J Biol Chem. 1995;270:16371–16377. doi: 10.1074/jbc.270.27.16371. [DOI] [PubMed] [Google Scholar]
- 340.Rubin CS. A kinase anchor proteins and the intracellular targeting of signals carried by cyclic AMP. Biochim Biophys Acta. 1994;1224:467–479. [PubMed] [Google Scholar]
- 341.Ruiz-Velasco V, Ikeda SR. Multiple G-protein betagamma combinations produce voltage-dependent inhibition of N-type calcium channels in rat superior cervical ganglion neurons. J Neurosci. 2000;20:2183–2191. doi: 10.1523/JNEUROSCI.20-06-02183.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 342.Ruth P, Rohrkasten A, Biel M, Bosse E, Regulla S, Meyer HE, Flockerzi V, Hofmann F. Primary structure of the beta subunit of the DHP-sensitive calcium channel from skeletal muscle. Science. 1989;245:1115–1118. doi: 10.1126/science.2549640. [DOI] [PubMed] [Google Scholar]
- 343.Sadeghi A, Doyle AD, Johnson BD. Regulation of the cardiac L-type Ca2+ channel by the actin-binding proteins alpha-actinin and dystrophin. Am J Physiol Cell Physiol. 2002;282:C1502–C1511. doi: 10.1152/ajpcell.00435.2001. [DOI] [PubMed] [Google Scholar]
- 344.Sakurai T, Hell JW, Woppmann A, Miljanich GP, Catterall WA. Immunochemical identification and differential phosphorylation of alternatively spliced forms of the alpha 1A subunit of brain calcium channels. J Biol Chem. 1995;270:21234–21242. doi: 10.1074/jbc.270.36.21234. [DOI] [PubMed] [Google Scholar]
- 345.Salter MW, Kalia LV. Src kinases: a hub for NMDA receptor regulation. Nature Rev. 2004;5:317–328. doi: 10.1038/nrn1368. [DOI] [PubMed] [Google Scholar]
- 346.Sanguinetti MC, Curran ME, Zou A, Shen J, Spector PS, Atkinson DL, Keating MT. Coassembly of K(V)LQT1 and minK (IsK) proteins to form cardiac I(Ks) potassium channel. Nature. 1996;384:80–83. doi: 10.1038/384080a0. [DOI] [PubMed] [Google Scholar]
- 347.Satoh A, Nakanishi H, Obaishi H, Wada M, Takahashi K, Satoh K, Hirao K, Nishioka H, Hata Y, Mizoguchi A, Takai Y. Neurabin-II/spinophilin. An actin filament-binding protein with one PDZ domain localized at cadherin-based cell-cell adhesion sites. J Biol Chem. 1998;273:3470–3475. doi: 10.1074/jbc.273.6.3470. [DOI] [PubMed] [Google Scholar]
- 348.Saucerman JJ, Zhang J, Martin JC, Peng LX, Stenbit AE, Tsien RY, McCulloch AD. Systems analysis of PKA-mediated phosphorylation gradients in live cardiac myocytes. Proc Natl Acad Sci USA. 2006;103:12923–12928. doi: 10.1073/pnas.0600137103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 349.Schillace RV, Scott JD. Association of the type 1 protein phosphatase PP1 with the A-kinase anchoring protein AKAP220. Curr Biol. 1999;9:321–324. doi: 10.1016/s0960-9822(99)80141-9. [DOI] [PubMed] [Google Scholar]
- 350.Schlegel W, Kempner ES, Rodbell M. Activation of adenylate cyclase in hepatic membranes involves interactions of the catalytic unit with multimeric complexes of regulatory proteins. J Biol Chem. 1979;254:5168–5176. [PubMed] [Google Scholar]
- 351.Schnell E, Sizemore M, Karimzadegan S, Chen L, Bredt DS, Nicoll RA. Direct interactions between PSD-95 and stargazin control synaptic AMPA receptor number. Proc Natl Acad Sci USA. 2002;99:13902–13907. doi: 10.1073/pnas.172511199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 352.Schopperle WM, Holmqvist MH, Zhou Y, Wang J, Wang Z, Griffith LC, Keselman I, Kusinitz F, Dagan D, Levitan IB. Slob, a novel protein that interacts with the Slowpoke calcium-dependent potassium channel. Neuron. 1998;20:565–573. doi: 10.1016/s0896-6273(00)80995-2. [DOI] [PubMed] [Google Scholar]
- 353.Schroder F, Handrock R, Beuckelmann DJ, Hirt S, Hullin R, Priebe L, Schwinger RH, Weil J, Herzig S. Increased availability and open probability of single L-type calcium channels from failing compared with nonfailing human ventricle. Circulation. 1998;98:969–976. doi: 10.1161/01.cir.98.10.969. [DOI] [PubMed] [Google Scholar]
- 354.Schuhmann K, Romanin C, Baumgartner W, Groschner K. Intracellular Ca2+ inhibits smooth muscle L-type Ca2+ channels by activation of protein phosphatase type 2B and by direct interaction with the channel. J Gen Physiol. 1997;110:503–513. doi: 10.1085/jgp.110.5.503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 355.Schumacher MA, Rivard AF, Bachinger HP, Adelman JP. Structure of the gating domain of a Ca2+-activated K+ channel complexed with Ca2+/calmodulin. Nature. 2001;410:1120–1124. doi: 10.1038/35074145. [DOI] [PubMed] [Google Scholar]
- 356.Sculptoreanu A, Rotman E, Takahashi M, Scheuer T, Catterall WA. Voltage-dependent potentiation of the activity of cardiac L-type calcium channel alpha 1 subunits due to phosphorylation by cAMP-dependent protein kinase. Proc Natl Acad Sci USA. 1993;90:10135–10139. doi: 10.1073/pnas.90.21.10135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 357.Seabold GK, Burette A, Lim IA, Weinberg RJ, Hell JW. Interaction of the tyrosine kinase Pyk2 with the N-methyl-d-aspartate receptor complex via the src homology 3 domains of PSD-95 and SAP102. J Biol Chem. 2003;278:15040–15048. doi: 10.1074/jbc.M212825200. [DOI] [PubMed] [Google Scholar]
- 358.Sette C, Conti M. Phosphorylation and activation of a cAMP-specific phosphodiesterase by the cAMP-dependent protein kinase. Involvement of serine 54 in the enzyme activation. J Biol Chem. 1996;271:16526–16534. doi: 10.1074/jbc.271.28.16526. [DOI] [PubMed] [Google Scholar]
- 359.Shenolikar S. Protein serine/threonine phosphatases–new avenues for cell regulation. Annu Rev Cell Biol. 1994;10:55–86. doi: 10.1146/annurev.cb.10.110194.000415. [DOI] [PubMed] [Google Scholar]
- 360.Shih M, Lin F, Scott JD, Wang HY, Malbon CC. Dynamic complexes of beta2-adrenergic receptors with protein kinases and phosphatases and the role of gravin. J Biol Chem. 1999;274:1588–1595. doi: 10.1074/jbc.274.3.1588. [DOI] [PubMed] [Google Scholar]
- 361.Shistik E, Keren-Raifman T, Idelson GH, Blumenstein Y, Dascal N, Ivanina T. The N terminus of the cardiac L-type Ca2+ channel alpha(1C) subunit. The initial segment is ubiquitous and crucial for protein kinase C modulation, but is not directly phosphorylated. J Biol Chem. 1999;274:31145–31149. doi: 10.1074/jbc.274.44.31145. [DOI] [PubMed] [Google Scholar]
- 362.Simen AA, Lee CC, Simen BB, Bindokas VP, Miller RJ. The C terminus of the Ca channel alpha1B subunit mediates selective inhibition by G-protein-coupled receptors. J Neurosci. 2001;21:7587–7597. doi: 10.1523/JNEUROSCI.21-19-07587.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 363.Singh A, Hamedinger D, Hoda JC, Gebhart M, Koschak A, Romanin C, Striessnig J. C-terminal modulator controls Ca2+-dependent gating of Ca(v)1.4 L-type Ca2+ channels. Nat Neurosci. 2006;9:1108–1116. doi: 10.1038/nn1751. [DOI] [PubMed] [Google Scholar]
- 364.Sinnegger-Brauns MJ, Hetzenauer A, Huber IG, Renstrom E, Wietzorrek G, Berjukov S, Cavalli M, Walter D, Koschak A, Waldschutz R, Hering S, Bova S, Rorsman P, Pongs O, Singewald N, Striessnig JJ. Isoform-specific regulation of mood behavior and pancreatic beta cell and cardiovascular function by L-type Ca2+ channels. J Clin Invest. 2004;113:1430–1439. doi: 10.1172/JCI20208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 365.Snutch TP, Tomlinson WJ, Leonard JP, Gilbert MM. Distinct calcium channels are generated by alternative splicing and are differentially expressed in the mammalian CNS. Neuron. 1991;7:45–57. doi: 10.1016/0896-6273(91)90073-9. [DOI] [PubMed] [Google Scholar]
- 366.Souroujon MC, Mochly-Rosen D. Peptide modulators of protein-protein interactions in intracellular signaling. Nat Biotechnol. 1998;16:919–924. doi: 10.1038/nbt1098-919. [DOI] [PubMed] [Google Scholar]
- 367.Staudinger J, Lu J, Olson EN. Specific interaction of the PDZ domain protein PICK1 with the COOH terminus of protein kinase C-alpha. J Biol Chem. 1997;272:32019–32024. doi: 10.1074/jbc.272.51.32019. [DOI] [PubMed] [Google Scholar]
- 368.Stea A, Soong TW, Snutch TP. Determinants of PKC-dependent modulation of a family of neuronal calcium channels. Neuron. 1995;15:929–940. doi: 10.1016/0896-6273(95)90183-3. [DOI] [PubMed] [Google Scholar]
- 369.Stebbins EG, Mochly-Rosen D. Binding specificity for RACK1 resides in the V5 region of beta II protein kinase C. J Biol Chem. 2001;276:29644–29650. doi: 10.1074/jbc.M101044200. [DOI] [PubMed] [Google Scholar]
- 370.Steen RL, Martins SB, Tasken K, Collas P. Recruitment of protein phosphatase 1 to the nuclear envelope by A-kinase anchoring protein AKAP149 is a prerequisite for nuclear lamina assembly. J Cell Biol. 2000;150:1251–1262. doi: 10.1083/jcb.150.6.1251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 371.Steinberg SF, Brunton LL. Compartmentation of G protein-coupled signaling pathways in cardiac myocytes. Annu Rev Pharmacol Toxicol. 2001;41:751–773. doi: 10.1146/annurev.pharmtox.41.1.751. [DOI] [PubMed] [Google Scholar]
- 372.Strack S, Choi S, Lovinger DM, Colbran RJ. Translocation of autophosphorylated calcium/calmodulin-dependent protein kinase II to the postsynaptic density. J Biol Chem. 1997;272:13467–13470. doi: 10.1074/jbc.272.21.13467. [DOI] [PubMed] [Google Scholar]
- 373.Strack S, Colbran RJ. Autophosphorylation-dependent targeting of calcium/calmodulin-dependent protein kinase II by the NR2B subunit of the N-methyl-d-aspartate receptor. J Biol Chem. 1998;273:20689–20692. doi: 10.1074/jbc.273.33.20689. [DOI] [PubMed] [Google Scholar]
- 374.Strack S, McNeill RB, Colbran RJ. Mechanism and regulation of calcium/calmodulin-dependent protein kinase II targeting to the NR2B subunit of the N-methyl-d-aspartate receptor. J Biol Chem. 2000;275:23798–23806. doi: 10.1074/jbc.M001471200. [DOI] [PubMed] [Google Scholar]
- 375.Strauss O, Mergler S, Wiederholt M. Regulation of L-type calcium channels by protein tyrosine kinase and protein kinase C in cultured rat and human retinal pigment epithelial cells. FASEB J. 1997;11:859–867. doi: 10.1096/fasebj.11.11.9285484. [DOI] [PubMed] [Google Scholar]
- 376.Striessnig J, Koschak A, Sinnegger-Brauns MJ, Hetzenauer A, Nguyen NK, Busquet P, Pelster G, Singewald N. Role of voltage-gated L-type Ca2+ channel isoforms for brain function. Biochem Soc Trans. 2006;34:903–909. doi: 10.1042/BST0340903. [DOI] [PubMed] [Google Scholar]
- 377.Suh BC, Inoue T, Meyer T, Hille B. Rapid chemically induced changes of PtdIns(4,5)P2 gate KCNQ ion channels. Science. 2006;314:1454–1457. doi: 10.1126/science.1131163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 378.Suko J, Maurer-Fogy I, Plank B, Bertel O, Wyskovsky W, Hohenegger M, Hellmann G. Phosphorylation of serine 2843 in ryanodine receptor-calcium release channel of skeletal muscle by cAMP-, cGMP- and CaM-dependent protein kinase. Biochim Biophys Acta. 1993;1175:193–206. doi: 10.1016/0167-4889(93)90023-i. [DOI] [PubMed] [Google Scholar]
- 379.Sun X, Gu XQ, Haddad GG. Calcium influx via L- and N-type calcium channels activates a transient large-conductance Ca2+-activated K+ current in mouse neocortical pyramidal neurons. J Neurosci. 2003;23:3639–3648. doi: 10.1523/JNEUROSCI.23-09-03639.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 380.Sun XX, Hodge JJ, Zhou Y, Nguyen M, Griffith LC. The eag potassium channel binds and locally activates calcium/calmodulin-dependent protein kinase II. J Biol Chem. 2004;279:10206–10214. doi: 10.1074/jbc.M310728200. [DOI] [PubMed] [Google Scholar]
- 381.Swartz KJ. Modulation of Ca2+ channels by protein kinase C in rat central and peripheral neurons: disruption of G protein-mediated inhibition. Neuron. 1993;11:305–320. doi: 10.1016/0896-6273(93)90186-u. [DOI] [PubMed] [Google Scholar]
- 382.Swartz KJ, Merritt A, Bean BP, Lovinger DM. Protein kinase C modulates glutamate receptor inhibition of Ca2+ channels and synaptic transmission. Nature. 1993;361:165–168. doi: 10.1038/361165a0. [DOI] [PubMed] [Google Scholar]
- 383.Takahashi T, Momiyama A. Different types of calcium channels mediate central synaptic transmission. Nature. 1993;366:156–158. doi: 10.1038/366156a0. [DOI] [PubMed] [Google Scholar]
- 384.Tao J, Shumay E, McLaughlin S, Wang HY, Malbon CC. Regulation of AKAP-membrane interactions by calcium. J Biol Chem. 2006;281:23932–23944. doi: 10.1074/jbc.M601813200. [DOI] [PubMed] [Google Scholar]
- 385.Tao J, Wang HY, Malbon CC. Protein kinase A regulates AKAP250 (gravin) scaffold binding to the beta2-adrenergic receptor. EMBO J. 2003;22:6419–6429. doi: 10.1093/emboj/cdg628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 386.Tasken KA, Collas P, Kemmner WA, Witczak O, Conti M, Tasken K. Phosphodiesterase 4D and protein kinase a type II constitute a signaling unit in the centrosomal area. J Biol Chem. 2001;276:21999–22002. doi: 10.1074/jbc.C000911200. [DOI] [PubMed] [Google Scholar]
- 387.Tavalin SJ. AKAP79 selectively enhances protein kinase C regulation of GluR1 at a Ca2+-calmodulin-dependent protein kinase II/protein kinase C site. J Biol Chem. 2008;283:11445–11452. doi: 10.1074/jbc.M709253200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 388.Tavalin SJ, Colledge M, Hell JW, Langeberg LK, Huganir RL, Scott JD. Regulation of GluR1 by the A-kinase anchoring protein 79 (AKAP79) signaling complex shares properties with long-term depression. J Neurosci. 2002;22:3044–3051. doi: 10.1523/JNEUROSCI.22-08-03044.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 389.Tavares GA, Panepucci EH, Brunger AT. Structural characterization of the intramolecular interaction between the SH3 and guanylate kinase domains of PSD-95. Mol Cell. 2001;8:1313–1325. doi: 10.1016/s1097-2765(01)00416-6. [DOI] [PubMed] [Google Scholar]
- 390.Terry-Lorenzo RT, Carmody LC, Voltz JW, Connor JH, Li S, Smith FD, Milgram SL, Colbran RJ, Shenolikar S. The neuronal actin-binding proteins, neurabin I and neurabin II, recruit specific isoforms of protein phosphatase-1 catalytic subunits. J Biol Chem. 2002;277:27716–27724. doi: 10.1074/jbc.M203365200. [DOI] [PubMed] [Google Scholar]
- 391.Tian L, Coghill LS, MacDonald SH, Armstrong DL, Shipston MJ. Leucine zipper domain targets cAMP-dependent protein kinase to mammalian BK channels. J Biol Chem. 2003;278:8669–8677. doi: 10.1074/jbc.M211661200. [DOI] [PubMed] [Google Scholar]
- 392.Tian L, Coghill LS, McClafferty H, MacDonald SH, Antoni FA, Ruth P, Knaus HG, Shipston MJ. Distinct stoichiometry of BKCa channel tetramer phosphorylation specifies channel activation and inhibition by cAMP-dependent protein kinase. Proc Natl Acad Sci USA. 2004;101:11897–11902. doi: 10.1073/pnas.0402590101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 393.Tian L, Duncan RR, Hammond MS, Coghill LS, Wen H, Rusinova R, Clark AG, Levitan IB, Shipston MJ. Alternative splicing switches potassium channel sensitivity to protein phosphorylation. J Biol Chem. 2001;276:7717–7720. doi: 10.1074/jbc.C000741200. [DOI] [PubMed] [Google Scholar]
- 394.Tibbs VC, Gray PC, Catterall WA, Murphy BJ. AKAP15 anchors cAMP-dependent protein kinase to brain sodium channels. J Biol Chem. 1998;273:25783–25788. doi: 10.1074/jbc.273.40.25783. [DOI] [PubMed] [Google Scholar]
- 395.Tomaselli GF, Marban E. Electrophysiological remodeling in hypertrophy and heart failure. Cardiovasc Res. 1999;42:270–283. doi: 10.1016/s0008-6363(99)00017-6. [DOI] [PubMed] [Google Scholar]
- 396.Tomita S, Adesnik H, Sekiguchi M, Zhang W, Wada K, Howe JR, Nicoll RA, Bredt DS. Stargazin modulates AMPA receptor gating and trafficking by distinct domains. Nature. 2005;435:1052–1058. doi: 10.1038/nature03624. [DOI] [PubMed] [Google Scholar]
- 397.Tomita S, Chen L, Kawasaki Y, Petralia RS, Wenthold RJ, Nicoll RA, Bredt DS. Functional studies and distribution define a family of transmembrane AMPA receptor regulatory proteins. J Cell Biol. 2003;161:805–816. doi: 10.1083/jcb.200212116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 398.Tsunoda S, Sierralta J, Sun Y, Bodner R, Suzuki E, Becker A, Socolich M, Ziker CS. A multivalent PDZ-domain protein assembles signalling complexes in a G-protein-coupled cascade. Nature. 1997;388:243–249. doi: 10.1038/40805. [DOI] [PubMed] [Google Scholar]
- 399.Tu H, Tang TS, Wang Z, Bezprozvanny I. Association of type 1 inositol 1,4,5-trisphosphate receptor with AKAP9 (Yotiao) and protein kinase A. J Biol Chem. 2004;279:19375–19382. doi: 10.1074/jbc.M313476200. [DOI] [PubMed] [Google Scholar]
- 400.Vallee RB, DiBartolomeis MJ, Theurkauf WE. A protein kinase bound to the projection portion of MAP 2 (microtubule-associated protein 2) J Cell Biol. 1981;90:568–576. doi: 10.1083/jcb.90.3.568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 401.Van Petegem F, Chatelain FC, Minor DL., Jr Insights into voltage-gated calcium channel regulation from the structure of the Cav1.2 IQ domain-Ca2+/calmodulin complex. Nat Struct Mol Biol. 2005;12:1108–1115. doi: 10.1038/nsmb1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 402.Van Petegem F, Clark KA, Chatelain FC, Minor DL., Jr Structure of a complex between a voltage-gated calcium channel beta-subunit and an alpha-subunit domain. Nature. 2004;429:671–675. doi: 10.1038/nature02588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 403.Victor RG, Rusnak F, Sikkink R, Marban E, O'Rourke B. Mechanism of Ca2+-dependent inactivation of L-type Ca2+ channels in GH3 cells: direct evidence against dephosphorylation by calcineurin. J Membr Biol. 1997;156:53–61. doi: 10.1007/s002329900187. [DOI] [PubMed] [Google Scholar]
- 404.Vigil D, Blumenthal DK, Brown S, Taylor SS, Trewhella J. Differential effects of substrate on type I and type II PKA holoenzyme dissociation. Biochemistry. 2004;43:5629–5636. doi: 10.1021/bi0499157. [DOI] [PubMed] [Google Scholar]
- 405.Virshup DM. Protein phosphatase 2A: a panoply of enzymes. Curr Opin Cell Biol. 2000;12:180–185. doi: 10.1016/s0955-0674(99)00074-5. [DOI] [PubMed] [Google Scholar]
- 406.Wahl-Schott C, Baumann L, Cuny H, Eckert C, Griessmeier K, Biel M. Switching off calcium-dependent inactivation in L-type calcium channels by an autoinhibitory domain. Proc Natl Acad Sci USA. 2006;103:15657–15662. doi: 10.1073/pnas.0604621103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 407.Walker D, Bichet D, Geib S, Mori E, Cornet V, Snutch TP, Mori Y, De Waard M. A new beta subtype-specific interaction in alpha1A subunit controls P/Q-type Ca2+ channel activation. J Biol Chem. 1999;274:12383–12390. doi: 10.1074/jbc.274.18.12383. [DOI] [PubMed] [Google Scholar]
- 408.Wang HS, Pan Z, Shi W, Brown BS, Wymore RS, Cohen IS, Dixon JE, McKinnon D. KCNQ2 and KCNQ3 potassium channel subunits: molecular correlates of the M-channel. Science. 1998;282:1890–1893. doi: 10.1126/science.282.5395.1890. [DOI] [PubMed] [Google Scholar]
- 409.Wang HY, Tao J, Shumay E, Malbon CC. G-Protein-coupled receptor-associated A-kinase anchoring proteins: AKAP79 and AKAP250 (gravin) Eur J Cell Biol. 2006;85:643–650. doi: 10.1016/j.ejcb.2005.12.003. [DOI] [PubMed] [Google Scholar]
- 410.Wang J, Zhou Y, Wen H, Levitan IB. Simultaneous binding of two protein kinases to a calcium-dependent potassium channel. J Neurosci. 1999;19:RC4. doi: 10.1523/JNEUROSCI.19-10-j0005.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 411.Wang Y, Townsend C, Rosenberg RL. Regulation of cardiac L-type Ca channels in planar lipid bilayers by G proteins and protein phosphorylation. Am J Physiol Cell Physiol. 1993;264:C1473–C1479. doi: 10.1152/ajpcell.1993.264.6.C1473. [DOI] [PubMed] [Google Scholar]
- 412.Wang Z, Kutschke W, Richardson KE, Karimi M, Hill JA. Electrical remodeling in pressure-overload cardiac hypertrophy: role of calcineurin. Circulation. 2001;104:1657–1663. doi: 10.1161/hc3901.095766. [DOI] [PubMed] [Google Scholar]
- 413.Wang Z, Wilson GF, Griffith LC. Calcium/calmodulin-dependent protein kinase II phosphorylates and regulates the Drosophila eag potassium channel. J Biol Chem. 2002;277:24022–24029. doi: 10.1074/jbc.M201949200. [DOI] [PubMed] [Google Scholar]
- 414.Warmke J, Drysdale R, Ganetzky B. A distinct potassium channel polypeptide encoded by the Drosophila eag locus. Science. 1991;252:1560–1562. doi: 10.1126/science.1840699. [DOI] [PubMed] [Google Scholar]
- 415.Warmke JW, Ganetzky B. A family of potassium channel genes related to eag in Drosophila and mammals. Proc Natl Acad Sci USA. 1994;91:3438–3442. doi: 10.1073/pnas.91.8.3438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 416.Warrier S, Ramamurthy G, Eckert RL, Nikolaev VO, Lohse MJ, Harvey RD. cAMP microdomains and L-type Ca2+ channel regulation in guinea-pig ventricular myocytes. J Physiol. 2007;580:765–776. doi: 10.1113/jphysiol.2006.124891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 417.Wei X, Neely A, Lacerda AE, Olcese R, Stefani E, Perez-Reyes E, Birnbaumer L. Modification of Ca2+ channel activity by deletions at the carboxyl terminus of the cardiac alpha 1 subunit. J Biol Chem. 1994;269:1635–1640. [PubMed] [Google Scholar]
- 418.Wei XY, Perez-Reyes E, Lacerda AE, Schuster G, Brown AM, Birnbaumer L. Heterologous regulation of the cardiac Ca2+ channel alpha 1 subunit by skeletal muscle beta and gamma subunits. Implications for the structure of cardiac L-type Ca2+ channels. J Biol Chem. 1991;266:21943–21947. [PubMed] [Google Scholar]
- 419.Weitmann S, Schultz G, Kleuss C. Adenylyl cyclase type II domains involved in Gbetagamma stimulation. Biochemistry. 2001;40:10853–10858. doi: 10.1021/bi011176w. [DOI] [PubMed] [Google Scholar]
- 420.Wellner-Kienitz MC, Bender K, Pott L. Overexpression of beta 1 and beta 2 adrenergic receptors in rat atrial myocytes. Differential coupling to G protein-gated inward rectifier K+ channels via G(s) and G(i)/o. J Biol Chem. 2001;276:37347–37354. doi: 10.1074/jbc.M106234200. [DOI] [PubMed] [Google Scholar]
- 421.Welsby PJ, Wang H, Wolfe JT, Colbran RJ, Johnson ML, Barrett PQ. A mechanism for the direct regulation of T-type calcium channels by Ca2+/calmodulin-dependent kinase II. J Neurosci. 2003;23:10116–10121. doi: 10.1523/JNEUROSCI.23-31-10116.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 422.Werner P, Hussy N, Buell G, Jones KA, North RA. D2, D3, and D4 dopamine receptors couple to G protein-regulated potassium channels in Xenopus oocytes. Mol Pharmacol. 1996;49:656–661. [PubMed] [Google Scholar]
- 423.West JW, Numann R, Murphy BJ, Scheuer T, Catterall WA. A phosphorylation site in the Na+ channel required for modulation by protein kinase C. Science. 1991;254:866–868. doi: 10.1126/science.1658937. [DOI] [PubMed] [Google Scholar]
- 424.Westphal RS, Anderson KA, Means AR, Wadzinski BE. A signal complex of Ca2+-calmodulin-dependent protein kinase IV and protein phosphatase 2A. Science. 1998;280:1258–1261. doi: 10.1126/science.280.5367.1258. [DOI] [PubMed] [Google Scholar]
- 425.Westphal RS, Coffee RL, Marotta A, Pelech SL, Wadzinski BE. Identification of kinase phosphatase signaling modules composed of p70 S6 kinase-protein phosphatase 2A (PP2A) and p21-activated kinase-PP2A. J Biol Chem. 1999;274:687–692. doi: 10.1074/jbc.274.2.687. [DOI] [PubMed] [Google Scholar]
- 426.Westphal RS, Tavalin SJ, Lin JW, Alto NM, Fraser IDC, Langeberg LK, Sheng M, Scott JD. Regulation of NMDA receptors by an associated phosphatase-kinase signaling complex. Science. 1999;285:93–96. doi: 10.1126/science.285.5424.93. [DOI] [PubMed] [Google Scholar]
- 427.Wickman KD, Iniguez-Lluhl JA, Davenport PA, Taussig R, Krapivinsky GB, Linder ME, Gilman AG, Clapham DE. Recombinant G-protein beta gamma-subunits activate the muscarinic-gated atrial potassium channel. Nature. 1994;368:255–257. doi: 10.1038/368255a0. [DOI] [PubMed] [Google Scholar]
- 428.Wiechen K, Yue DT, Herzig S. Two distinct functional effects of protein phosphatase inhibitors on guinea-pig cardiac L-type Ca2+ channels. J Physiol. 1995;484:583–592. doi: 10.1113/jphysiol.1995.sp020688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 429.Willoughby D, Wong W, Schaack J, Scott JD, Cooper DM. An anchored PKA and PDE4 complex regulates subplasmalemmal cAMP dynamics. EMBO J. 2006;25:2051–2061. doi: 10.1038/sj.emboj.7601113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 430.Wilson SM, Toth PT, Oh SB, Gillard SE, Volsen S, Ren D, Philipson LH, Lee EC, Fletcher CF, Tessarollo L, Copeland NG, Jenkins NA, Miller RJ. The status of voltage-dependent calcium channels in alpha 1E knock-out mice. J Neurosci. 2000;20:8566–8571. doi: 10.1523/JNEUROSCI.20-23-08566.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 431.Wolfart J, Roeper J. Selective coupling of T-type calcium channels to SK potassium channels prevents intrinsic bursting in dopaminergic midbrain neurons. J Neurosci. 2002;22:3404–3413. doi: 10.1523/JNEUROSCI.22-09-03404.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 432.Wolfe JT, Wang H, Perez-Reyes E, Barrett PQ. Stimulation of recombinant Ca(v)3.2, T-type, Ca2+ channel currents by CaMKIIgamma(C) J Physiol. 2002;538:343–355. doi: 10.1113/jphysiol.2001.012839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 433.Wong W, Scott JD. AKAP signalling complexes: focal points in space and time. Nat Rev Mol Cell Biol. 2004;5:959–970. doi: 10.1038/nrm1527. [DOI] [PubMed] [Google Scholar]
- 434.Wu X, Davis GE, Meininger GA, Wilson E, Davis MJ. Regulation of the L-type calcium channel by alpha 5beta 1 integrin requires signaling between focal adhesion proteins. J Biol Chem. 2001;276:30285–30292. doi: 10.1074/jbc.M102436200. [DOI] [PubMed] [Google Scholar]
- 435.Wu Y, MacMillan LB, McNeill RB, Colbran RJ, Anderson ME. CaM kinase augments cardiac L-type Ca2+ current: a cellular mechanism for long Q-T arrhythmias. Am J Physiol Heart Circ Physiol. 1999;276:H2168–H2178. doi: 10.1152/ajpheart.1999.276.6.H2168. [DOI] [PubMed] [Google Scholar]
- 436.Xia J, Zhang X, Staudinger J, Huganir RL. Clustering of AMPA receptors by the synaptic PDZ domain-containing protein PICK1. Neuron. 1999;22:179–187. doi: 10.1016/s0896-6273(00)80689-3. [DOI] [PubMed] [Google Scholar]
- 437.Xiang Y, Naro F, Zoudilova M, Jin SL, Conti M, Kobilka B. Phosphodiesterase 4D is required for beta2 adrenoceptor subtype-specific signaling in cardiac myocytes. Proc Natl Acad Sci USA. 2005;102:909–914. doi: 10.1073/pnas.0405263102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 438.Xiao J, Tian X, Jones PP, Bolstad J, Kong H, Wang R, Zhang L, Duff HJ, Gillis AM, Fleischer S, Kotlikoff M, Copello JA, Chen SR. Removal of FKBP126 does not alter the conductance and activation of the cardiac ryanodine receptor or the susceptibility to stress-induced ventricular arrhythmias. J Biol Chem. 2007;282:34828–34838. doi: 10.1074/jbc.M707423200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 439.Xiao RP, Cheng H, Lederer WJ, Suzuki T, Lakatta EG. Dual regulation of Ca2+/calmodulin-dependent kinase II activity by membrane voltage and by calcium influx. Proc Natl Acad Sci USA. 1994;91:9659–9663. doi: 10.1073/pnas.91.20.9659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 440.Xiao RP, Avdonin P, Zhou YY, Cheng H, Akhter SA, Eschenhagen T, Lefkowitz RJ, Koch WJ, Lakatta EG. Coupling of beta2-adrenoceptor to Gi proteins and its physiological relevance in murine cardiac myocytes. Circ Res. 1999;84:43–52. doi: 10.1161/01.res.84.1.43. [DOI] [PubMed] [Google Scholar]
- 441.Xiao RP, Cheng H, Zhou YY, Kuschel M, Lakatta EG. Recent advances in cardiac beta(2)-adrenergic signal transduction. Circ Res. 1999;85:1092–1100. doi: 10.1161/01.res.85.11.1092. [DOI] [PubMed] [Google Scholar]
- 442.Yan Z, Hsieh-Wilson L, Feng J, Tomizawa K, Allen PB, Fienberg AA, Nairn AC, Greengard P. Protein phosphatase 1 modulation of neostriatal AMPA channels: regulation by DARPP-32 and spinophilin. Nat Neurosci. 1999;2:13–17. doi: 10.1038/4516. [DOI] [PubMed] [Google Scholar]
- 443.Yang J, Drazba JA, Ferguson DG, Bond M. A-kinase anchoring protein 100 (AKAP100) is localized in multiple subcellular compartments in the adult rat heart. J Cell Biol. 1998;142:511–522. doi: 10.1083/jcb.142.2.511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 444.Yang J, Tsien RW. Enhancement of N- and L-type calcium channel currents by protein kinase C in frog sympathetic neurons. Neuron. 1993;10:127–136. doi: 10.1016/0896-6273(93)90305-b. [DOI] [PubMed] [Google Scholar]
- 445.Yang L, Liu G, Zakharov SI, Morrow JP, Rybin VO, Steinberg SF, Marx SO. Ser1928 is a common site for Cav1.2 phosphorylation by protein kinase C isoforms. J Biol Chem. 2005;280:207–214. doi: 10.1074/jbc.M410509200. [DOI] [PubMed] [Google Scholar]
- 446.Yang S, Fletcher WH, Johnson DA. Regulation of cAMP-dependent protein kinase: enzyme activation without dissociation. Biochemistry. 1995:6267–6271. doi: 10.1021/bi00019a002. [DOI] [PubMed] [Google Scholar]
- 447.Yang SN, Larsson O, Branstrom R, Bertorello AM, Leibiger B, Leibiger IB, Moede T, Kohler M, Meister B, Berggren PO. Syntaxin 1 interacts with the L(D) subtype of voltage-gated Ca2+ channels in pancreatic beta cells. Proc Natl Acad Sci USA. 1999;96:10164–10169. doi: 10.1073/pnas.96.18.10164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 448.Yao J, Davies LA, Howard JD, Adney SK, Welsby PJ, Howell N, Carey RM, Colbran RJ, Barrett PQ. Molecular basis for the modulation of native T-type Ca channels in vivo by Ca/calmodulin-dependent protein kinase II. J Clin Invest. 2006;116:2403–2412. doi: 10.1172/JCI27918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 449.Yasuda R, Sabatini BL, Svoboda K. Plasticity of calcium channels in dendritic spines. Nat Neurosci. 2003;6:948–955. doi: 10.1038/nn1112. [DOI] [PubMed] [Google Scholar]
- 450.Yokoyama CT, Myers SJ, Fu J, Mockus SM, Scheuer T, Catterall WA. Mechanism of SNARE protein binding and regulation of Cav2 channels by phosphorylation of the synaptic protein interaction site. Mol Cell Neurosci. 2005;28:1–17. doi: 10.1016/j.mcn.2004.08.019. [DOI] [PubMed] [Google Scholar]
- 451.Yokoyama CT, Sheng ZH, Catterall WA. Phosphorylation of the synaptic protein interaction site on N-type calcium channels inhibits interactions with SNARE proteins. J Neurosci. 1997;17:6929–6938. doi: 10.1523/JNEUROSCI.17-18-06929.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 452.Yokoyama CT, Westenbroek RE, Hell JW, Soong TW, Snutch TP, Catterall WA. Biochemical properties and subcellular distribution of the neuronal class E calcium channel alpha 1 subunit. J Neurosci. 1995;15:6419–6432. doi: 10.1523/JNEUROSCI.15-10-06419.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 453.Yu FH, Catterall WA. The VGL-chanome: a protein superfamily specialized for electrical signaling and ionic homeostasis. Sci STKE. 2004;2004:re15. doi: 10.1126/stke.2532004re15. [DOI] [PubMed] [Google Scholar]
- 454.Yu XM, Askalan R, Keil GJ, II, Salter MW. NMDA channel regulation by channel-associated protein tyrosine kinase Src. Science. 1997;275:674–678. doi: 10.1126/science.275.5300.674. [DOI] [PubMed] [Google Scholar]
- 455.Zaccolo M, Pozzan T. Discrete microdomains with high concentration of cAMP in stimulated rat neonatal cardiac myocytes. Science. 2002;295:1711–1715. doi: 10.1126/science.1069982. [DOI] [PubMed] [Google Scholar]
- 456.Zamah AM, Delahunty M, Luttrell LM, Lefkowitz RJ. Protein kinase A-mediated phosphorylation of the beta 2-adrenergic receptor regulates its coupling to Gs and Gi. Demonstration in a reconstituted system. J Biol Chem. 2002;277:31249–31256. doi: 10.1074/jbc.M202753200. [DOI] [PubMed] [Google Scholar]
- 457.Zamponi GW, Bourinet E, Nelson D, Nargeot J, Snutch TP. Crosstalk between G proteins and protein kinase C mediated by the calcium channel alpha1 subunit. Nature. 1997;385:442–446. doi: 10.1038/385442a0. [DOI] [PubMed] [Google Scholar]
- 458.Zhang R, Dzhura I, Grueter CE, Thiel W, Colbran RJ, Anderson ME. A dynamic alpha-beta inter-subunit agonist signaling complex is a novel feedback mechanism for regulating L-type Ca2+ channel opening. FASEB J. 2005;19:1573–1575. doi: 10.1096/fj.04-3283fje. [DOI] [PubMed] [Google Scholar]
- 459.Zhang ZH, Johnson JA, Chen L, El-Sherif N, Mochly-Rosen D, Boutjdir M. C2 region-derived peptides of beta-protein kinase C regulate cardiac Ca2+ channels. Circ Res. 1997;80:720–729. doi: 10.1161/01.res.80.5.720. [DOI] [PubMed] [Google Scholar]
- 460.Zhou Y, Schopperle WM, Murrey H, Jaramillo A, Dagan D, Griffith LC, Levitan IB. A dynamically regulated 14-3-3, Slob, and Slowpoke potassium channel complex in Drosophila presynaptic nerve terminals. Neuron. 1999;22:809–818. doi: 10.1016/s0896-6273(00)80739-4. [DOI] [PubMed] [Google Scholar]
- 461.Zhou Y, Wang J, Wen H, Kucherovsky O, Levitan IB. Modulation of Drosophila slowpoke calcium-dependent potassium channel activity by bound protein kinase a catalytic subunit. J Neurosci. 2002;22:3855–3863. doi: 10.1523/JNEUROSCI.22-10-03855.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 462.Zhou YY, Cheng H, Bogdanov KY, Hohl C, Altschuld R, Lakatta EG, Xiao RP. Localized cAMP-dependent signaling mediates beta 2-adrenergic modulation of cardiac excitation-contraction coupling. Am J Physiol Heart Circ Physiol. 1997;273:H1611–H1618. doi: 10.1152/ajpheart.1997.273.3.H1611. [DOI] [PubMed] [Google Scholar]
- 463.Zolnierowicz S, Van Hoof C, Andjelkovic N, Cron P, Stevens I, Merlevede W, Goris J, Hemmings BA. The variable subunit associated with protein phosphatase 2A0 defines a novel multimember family of regulatory subunits. Biochem J. 1996;317:187–194. doi: 10.1042/bj3170187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 464.Zong X, Schreieck J, Mehrke G, Welling A, Schuster A, Bosse E, Flockerzi V, Hofmann F. On the regulation of the expressed L-type calcium channel by cAMP-dependent phosphorylation. Pflügers Arch. 1995;430:340–347. doi: 10.1007/BF00373908. [DOI] [PubMed] [Google Scholar]
- 465.Zuhlke RD, Pitt GS, Deisseroth K, Tsien RW, Reuter H. Calmodulin supports both inactivation and facilitation of L-type calcium channels. Nature. 1999;399:159–162. doi: 10.1038/20200. [DOI] [PubMed] [Google Scholar]
- 466.Zuhlke RD, Pitt GS, Tsien RW, Reuter H. Ca2+-sensitive inactivation and facilitation of L-type Ca2+ channels both depend on specific amino acid residues in a consensus calmodulin-binding motif in the α1C subunit. J Biol Chem. 2000;275:21121–21129. doi: 10.1074/jbc.M002986200. [DOI] [PubMed] [Google Scholar]