

Abstract
This paper describes a detailed investigation of factors controlling the dominance of a directing group in Pd-catalyzed ligand-directed arene acetoxylation. Mechanistic studies, involving reaction kinetics, Hammett analysis, kinetic isotope effect experiments, and the kinetic order in various reagents, have been conducted for a series of different substrates. Initial rates studies of substrates bearing different directing groups showed that these transformations are accelerated by the use of electron withdrawing directing groups. However, in contrast, under conditions where two different directing groups are in competition with one another in the same reaction flask, substrates with electron donating directing groups react preferentially. These results are discussed in the context of the proposed mechanism for Pd-catalyzed arene acetoxylation.
Keywords: Directed, Chelate, Palladium, Catalysis, C–H Activation, Mechanism, Acetoxylation
Introduction
Palladium-catalyzed ligand-directed C–H bond functionalization has emerged as a powerful method for the direct conversion of arenes and alkanes into new products.1,2,3 These reactions allow for the highly site selective transformation of a C–H bond proximal to a coordinating functional group (L in Scheme 1) into a new C–X bond (X = O, Cl, Br, I, F, C or N). Importantly, most natural products, pharmaceuticals, and agrochemicals contain suitable directing groups for this chemistry. As such, these transformations could be valuable for late stage derivatization and analog generation in such important classes of molecules.
Scheme 1.

Palladium-Catalyzed Chelate-Directed C–H Bond Functionalization
The vast majority of Pd-catalyzed directed C–H functionalization reactions in the literature involve simple organic compounds containing a single directing group (Scheme 1).1,2,3 However, many molecules of interest have not just one, but multiple basic functional groups that could bind to a Pd center and direct C–H bond functionalization (for three representative examples, see Figure 1).4 As such, the development of selective, efficient, and high yielding transformations is predicated on a clear understanding of the factors governing product distributions when multiple directing groups are present simultaneously. This report describes a detailed investigation of Pd-catalyzed directed arene acetoxylation as a function of directing group electronics and structure. The implications of these results for both the mechanism and the synthetic application of this chemistry are discussed.
Figure 1.

Examples of Biologically Active Molecules Containing Multiple Potential Directing Groups
Results
Our first goal was to systematically study how the electronic nature of a directing group affects the distribution of products in Pd-catalyzed C–H bond acetoxylation with PhI(OAc)2. As shown in Scheme 2, we designed a series of experiments to compete two directing groups against one another in the same reaction flask (mimicking situations where two potential ligands are present within the same molecule). In these systems, 1 equiv of substrate I and 1 equiv of substrate II were subjected to Pd-catalyzed reaction with 1 equiv of PhI(OAc)2. The ratio of acetoxylated products (I-OAc/II-OAc) was then determined by GC, and this value represents the relative reaction rates of the two directing groups (kIkII) under a given set of conditions.
Scheme 2.

Competition Studies Between Two Different Directing Groups (L and L’)
Our initial studies focused on substituted benzylpyridine derivatives 1a-7a as substrates for these transformations. These substrates were designed with several criteria in mind. First, pyridine derivatives are well-known to serve as highly effective directing groups for Pd-catalyzed C–H bond functionalization.1,2b,f,i,3a,c,i,l,n,s Second, substitution at the meta- and para-positions of the pyridine ring allows for electronic modification of the directing group. Third, these substrates contain a methyl substituent at the meta position of the arene ring to limit competing di-ortho-functionalization, which could complicate product ratio analysis.1g Finally, and most importantly, these substrates contain a methylene spacer between the directing group and the arene, which is expected to limit electronic communication between the pyridine substituent and the C–H bond being functionalized.5 This should allow interpretation of product ratios solely in terms of electronic perturbation of the directing group.6
As summarized in Table 1, all of the substituted pyridine derivatives served as effective directing groups for Pd-catalyzed C–H bond acetoxylation. Under optimized conditions (1 mol % of Pd(OAc)2, 1.02 equiv of PhI(OAc)2 in AcOH/Ac2O at 100 °C), the mono-acetoxylated products 1b-7b were obtained in 70–93% isolated yield. Importantly, these transformations exhibited extremely high (>100 : 1) selectivity for ortho-functionalization of the aromatic ring; furthermore, the less sterically congested ortho-site (para to the methyl substituent) was acetoxylated with > 25 : 1 selectivity in all cases.1g
Table 1.
Acetoxylation of Substituted Benzylpyridine Derivatives
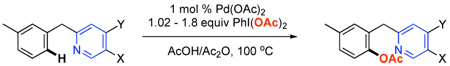 | ||||
|---|---|---|---|---|
| Entry | Substrate | X | Y | Product (Yield) |
| 1 | 1a | H | CH3 | 1b (70%) |
| 2 | 2a | H | OCH3 | 2b (75%) |
| 3 | 3a | H | CF3 | 3b (91%) |
| 4 | 4a | H | Cl | 4b (74%) |
| 5 | 5a | CH3 | H | 5b (74%) |
| 6 | 6a | H | H | 6b (75%) |
| 7 | 7a | F | H | 7b (93%) |
We next carried out competition studies between electronically varied benzylpyridines in AcOH/Ac2O (Scheme 2). In these experiments, a 1 : 1 molar ratio of 2-benzylpyridine 6a and each substituted derivative (1a-5a and 7a) was subjected to 1 equiv of PhI(OAc)2 and 1 mol % of Pd(OAc)2. Upon completion of the reaction, the yields and ratios of acetoxylated products were determined by gas chromatography. In a representative experiment, the reaction of an equimolar quantity of 6a and 2a afforded acetoxylated products 6b and 2b in a ratio of 1 : 0.77 (k6ak2a = 1/0.77) (Scheme 3).
Scheme 3.

Competition Between Benzylpyridines 2a and 6a
The data from these experiments was used to construct a Hammett plot (Figure 2), which showed a non-linear convex relationship between σ and log(kX/kH). Such convex plots can be indicative of a change in rate determining step with electronic variation of the substituents.7 However, in this case, we reasoned that the non-linearity might instead be due to varying degrees of pyridine protonation by the AcOH solvent. The Ka for this acid/base reaction should vary substantially with substitution on the pyridine, thereby changing the concentration of accessible ligand. As such, we hypothesized that correcting for the concentration of unprotonated benzylpyridine in AcOH/Ac2O might provide a linear Hammett plot for these reactions.
Figure 2.

Hammett Plot for Competition Experiments in AcOH/Ac2O
The concentration of each unprotonated benzylpyridine was estimated using standard acid/base equilibria8 based on the approximation that Ka is equal to that of the analogous pyridine derivative (eq 1).9 The experimental ratios of the acetoxylated products (kx/kh) were then corrected based on the calculated concentrations of free benzylpyridine (see supporting information and Table S2 for full details). Gratifyingly, the Hammett plot of this corrected data was linear (R2 = 0.96), and provided a ρ value of −5.46 (Figure 3).10
 |
(1) |
Figure 3.

Hammett Plot for Competition Experiments in AcOH/Ac2O Corrected for Concentration of Unprotonated Benzylpyridine
To further confirm that equilibrium protonation was the source of non-linearity in AcOH/Ac2O, analogous competition studies were conducted in benzene. As anticipated, under optimal conditions (5 mol % of Pd(OAc)2, 1.02 equiv of PhI(OAc)2, 80 °C), a linear Hammett plot (R2 = 0.94) with a ρ value of −2.01 was obtained (Figure 4).10 While the slopes of the Hammett plots in AcOH/Ac2O and benzene differ substantially, the negative ρ values demonstrate that in both solvents substrates bearing more electron rich directing groups react preferentially under competition conditions,.
Figure 4.

Hammett Plot for Competition Experiments in Benzene
We next sought to determine if the reaction rates of 1a-7a in isolation showed a similar trend to the competition studies discussed above. As such, the initial rate of Pd-catalyzed C–H activation/acetoxylation for each benzylpyridine derivative was measured in AcOH/Ac2O (Scheme 4). A Hammett plot was then constructed, and showed a non-linear concave relationship between σ and log(kX/kH) (Figure 5). A concave Hammett plot often reflects a change in mechanism as the electronic nature of the aromatic ring is varied.7 However, based on the results from the competition experiments above, we hypothesized that the non-linearity was more likely due to competitive protonation of the pyridine. Indeed, correction of the benzylpyridine concentrations based on pyridine Ka values9 provided a linear Hammett plot (R2 = 0.96) with a ρ value of +4.74 (Figure 6).
Scheme 4.

Individual Kinetic Studies
Figure 5.

Hammett Plot for Individual Kinetics in AcOH/Ac2O
Figure 6.

Hammett Plot for Individual Kinetics in AcOH/Ac2O Corrected for Concentration of Protonated Benzylpyridine
Again, analogous kinetics experiments were performed in benzene and provided a linear Hammett plot (R2 = 0.96) with a ρ value of +1.40 (Figure 7). The positive ρ value in both solvents shows that electron-withdrawing substituents on the pyridine ring accelerate the rate of acetoxylation. Importantly, this is directly opposite to the results of the competition experiments. As discussed below, these data suggest that two different steps of the catalytic cycle bearing opposite electronic requirements control the relative rates of functionalization in the presence and absence of other directing groups.
Figure 7.

Hammett Plot for Individual Kinetics in Benzene
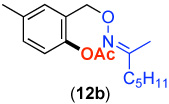
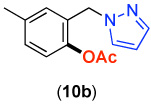
We next investigated whether the electronic effects observed with benzylpyridines 1a-7a were general across a wider range of common directing groups. As shown in Table 2, a series of substrates containing eight different directing groups (L = pyridine, pyrimidine, pyrazine, pyrazole, isoxazoline, methyl oxime ether, benzyloxime ether, and amide) were synthesized.11 Importantly, all contain both a methylene spacer between L and the arene ring in order to attenuate electronic communication between the two halves of the molecule and a meta-methyl substituent to promote mono-acetoxylation.1g As shown in Table 2, each of these substrates underwent clean and high yielding Pd-catalyzed C–H acetoxylation with PhI(OAc)2 in AcOH/AcO2.
Table 2.
Isolated Yields for Substrates Containing Diverse Directing Groups
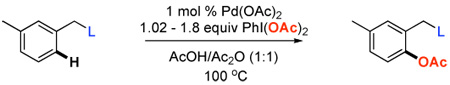 | |||||||
|---|---|---|---|---|---|---|---|
| Entry | Substrate | Product | Yield | Entry | Substrate | Product | Yield |
| 1 | 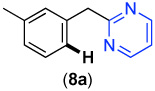 |
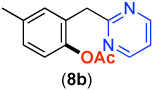 |
90% | 5 |  |
 |
79% |
| 2 |  |
 |
69% | 6 | 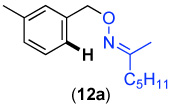 |
 |
70% |
| 3 | 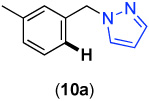 |
 |
88% | 7 |  |
 |
77% |
| 4 |  |
 |
75% | 8 |  |
 |
74% |
Competition studies analogous to those described in Scheme 2 were performed for substrates 6a and 8a-14a in both AcOH/Ac2O and benzene. In a representative experiment, benzylpyridine 6a and benzylpyrazole 10a reacted in benzene to afford a 1 : 0.06 ratio of acetoxylated products 6b and 10b. In AcOH/Ac2O, 6b was still the major product albeit with lower selectivity (1 : 0.4) (Scheme 5). Based on the data compiled from these experiments (Table S2 and Table S3), the relative reactivities of 6a and 8a-14a were ranked (Figure 8). While the trends in the two solvent systems varied slightly, the results were generally consistent with the more basic directing groups dominating the reaction. For example, competitions between the most basic heterocycles (pyridine, pyrimidine, pyrazine, and pyrazole derivatives 6a, 8a, 10a, and 14a) and substrates bearing less basic directing groups such as isoxazoline 9a, oxime ethers 11a and 12a, and amide 13a generally afforded only acetoxylation of the former with >50 : 1 selectivity. These results are consistent with our prior observation1e of selective pyridine-directed acetoxylation in molecules containing both a pyridine and an oxime ether directing group.
Scheme 5.

Competition Between Benzylpyridine 6a and Benzylpyrazole 10a
Figure 8.

Relative Reactivity of Directing Groups from Competition Studies in AcOH/Ac2O and C6H6
The initial rate of Pd-catalyzed C–H bond acetoxylation for each individual substrate was also determined. As summarized in Table 3, the initial rates for acetoxylation of 6a and 8a-14a in AcOH/Ac2O ranged over approximately two orders of magnitude from 0.1 × 10−1 to 6.4 × 10−1 (mol)(L)−1(min)−1. Under these conditions, the three substrates bearing six-membered nitrogen-containing heterocycles – pyridine 6a, pyrimidine 8a, and pyrazine 14a – exhibited very different initial rates, with the pyrimidine reacting 60 times faster than the pyrazine and 3 times faster than the pyridine (Table 3, entries, 1, 4, and 8). Furthermore, in some cases, substrates bearing two very electronically different directing groups, such as methyl oxime ether 11a and pyridine 6a, reacted at nearly identical rates (entries 4 and 5).
Table 3.
Initial Rate Constants for Substrates 6a and 8a-14a in AcOH/Ac2O and Benzene
| Entry | Substrate | kObs(×10−1) | Entry | Substrate | kObs(×10−1) | ||
|---|---|---|---|---|---|---|---|
| AcOH/Ac2O | Benzene | AcOH/Ac2O | Benzene | ||||
| 1 |  |
6.4 | 5.0 | 5 |  |
2.1 | – |
| 2 | 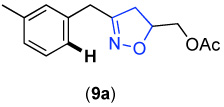 |
2.2 | 1.0 | 6 | 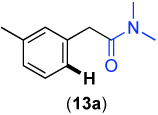 |
1.6 | – |
| 3 | 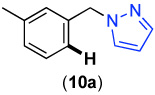 |
2.2 | 0.5 | 7 | 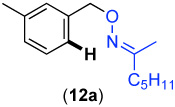 |
1.5 | 0.4 |
| 4 |  |
2.0 | 0.6 | 8 |  |
0.1 | 0.2 |
Solvent also had an effect on both the relative and absolute rates of these transformations. In general, C–H activation/acetoxylation was 2–4 times slower in benzene (with 5 mol % of catalyst) versus AcOH/Ac2O (with 1 mol % of catalyst). Furthermore, while isoxazoline 9a and pyrazole 10a reacted at similar rates in AcOH/Ac2O, 9a reacted 2 times faster than 10a in benzene (Table 3, entries 2 and 3). Additionally, oxime ether 11a and amide 13a showed high reactivity in AcOH/Ac2O; however, these substrates formed only trace amounts of the desired products under standard conditions in benzene.
Having explored the factors affecting the dominant directing group under carefully controlled conditions, our efforts turned to determining whether these insights could be applied to more complex systems. As such, we synthesized substrate 15a, which contains both an amide and an oxime ether directing group. Based on the competition studies discussed above, we predicted that the oxime ether would direct C–H activation/acetoxylation selectively over the amide. We were pleased to find that the reaction of 15a in the presence of 3 mol % of Pd(OAc)2 and 2 equiv of PhI(OAc)2 in AcOH/Ac2O afforded the di-acetoxylated product 15b in 72% yield, and none of the corresponding product of amide-directed C–H acetoxylation was observed. It is important to note that this reaction provided solely product 15b despite the fact that there are not methylene spacers between the directing groups and the aromatic rings being functionalized. Without these spacers, the amide is expected to increase the electron density on the arene and thereby increase its reactivity towards C–H activation,12 while the electron withdrawing oxime ether is expected to have the opposite effect.12 Nonetheless, the trend predicted based on substrates 11a and 13a above held up well in this system.
As shown in Scheme 7, the acetoxylated product 15b could be further elaborated via Pd-catalyzed directed C–H activation reactions. For example, the use of PhI(OAc)2 afforded tri-acetoxylated product 15b-OAc, N-chlorosuccinimide provided chloro product 15b-Cl,1d,e and the iodonium salt [(m-CF3C6H4)2I]BF4 generated the corresponding arylated product 15b-Ar.1h These results demonstrate that the amide is a competent directing group for Pd-catalyzed C–H functionalization reactions, thereby confirming that the high selectivity observed with 15b indeed reflects the relative reactivity of the two directing groups.
Scheme 7.

Amide Directed C–H Functionalization of Product 15b
Discussion
With all these results in hand, we sought to determine which mechanistic steps dictate the relative and absolute reactivity of a directing group in Pd-catalyzed C–H bond acetoxylation. As summarized in Figure 9, the catalytic cycle for these transformations is proposed to involve five steps: (i) ligand coordination to the Pd catalyst to generate complex A, (ii) cyclometalation to form palladacycle B, (iii) oxidation of B by PhI(OAc)2 to afford PdIV intermediate C, (iv) C–O bond forming reductive elimination to generate PdII complex D, and (v) ligand exchange to release the product and coordinate a new substrate to the metal center.
Figure 9.

Proposed Catalytic Cycle for Pd-Catalyzed C–H Bond Acetoxylation
Literature precedent can be used to predict how electronic modification of the directing group (L) will affect each step of the catalytic cycle. Modification of L is expected to have a large influence on the thermodynamics (and therefore Keq) associated with steps i and v, which should be in rapid equilibria under the reaction conditions. Prior work has shown that, with all else being equal, more electron donating ligands form stronger bonds to PdII than their electron deficient analogues.13,14
The cyclopalladation reaction (step ii) is believed to proceed by an electrophilic12b,c,15 mechanism and/or by formation of an agostic intermediate followed by deprotonation.16,17 Both mechanisms involve the Pd acting as an electrophile; as such, the rate of this step is expected to be accelerated by electron withdrawing ancillary ligands (L).12b,d Literature precedent has shown that the oxidation of PdII complexes (step iii of the catalytic cycle) is accelerated with more electron donating ligands, which render the metal center more nucleophilic.18 Finally, the rate of C–O bond-forming reductive elimination from PdIV (step iv) is expected to increase with electron withdrawing ancillary ligands (L).19 We can interpret our experimental data based on this analysis in order to gain insights into the selectivity determining step(s) under individual kinetics and competition conditions.
Individual Rate Studies: Benzylpyridine Derivatives
The individual rate studies with substituted benzylpyridines (Scheme 4, Figure 6 and Figure 7) afforded Hammett ρ values of +1.40 in benzene and + 4.74 in AcOH, indicating that the reaction is accelerated with less basic benzylpyridines. Based on the analysis above, these values suggest that either cyclopalladation (step ii in Figure 9) or C–O bond forming reductive elimination (step iv in Figure 9) is rate determining.20 We propose that cyclopalladation is rate limiting in these systems based on several additional pieces of data. First, literature precedent suggests that reductive elimination will be fast under our reaction conditions (80 °C), since PdIV complexes of general structure C are typically unstable at room temperature.20,21,22 Second, comparison of the initial rates of acetoxylation of substrate 16a versus its deuterated analogue 16a-d5 provided a kH/kD of 3.54 in AcOH/Ac2O and 1.86 in C6H6 (Figure 10). This is consistent with a primary kinetic isotope effect, where C–H(D) bond breaking is involved in the rate-determining step of the reaction. Importantly, similar KIE values (ranging from 1.8 to 4.4) have been observed in related Pd-catalyzed C–H functionalization reactions that proceed by rate-limiting C–H activation.2e,3d,f,h,I,m,t,23 Most relevant, Yu and coworkers observed a KIE of 2.9 in Pd-catalyzed oxazoline-directed C–H acetoxylation reactions that also proceed via 6-membered palladacycles.6,24
Figure 10.

Kinetic Isotope Effect Experiment
Additional support for C–H activation as the rate limiting step came from stoichiometric studies of the reactions of 1a-7a with Pd(OAc)2 (Figure 11). The rates of cyclopalladation were monitored in benzene using UV-vis spectroscopy. As shown in Figure 11, a Hammett plot was constructed and showed a ρ value of +1.77 for stoichiometric C–H activation. This value is similar in both sign and magnitude to that obtained in the catalytic individual rate studies (ρ = +1.4 in benzene), providing further evidence to support turnover-limiting cyclopalladation.
Figure 11.

Hammett Plot for Stoichiometric Cyclopalladation of Benzylpyridines 1a–7a
We note that our catalytic experiments afforded significantly different ρ values in AcOH/Ac2O (+4.74) versus C6H6 (+1.4). Importantly, solvent has also been shown to have a significant effect on the rates of stoichiometric cyclopalladation reactions.25 As such, we propose that the difference in magnitude between the two solvents may be the result of a change in the nature/position of the transition state for C–H activation as a function of reaction medium.
Individual Rate Studies: Other Directing Groups
In contrast to the results with the benzylpyridine derivatives, the individual rate studies with substrates 6a and 8a-14a did not show a strong correlation between kobs and the basicity of the directing group. For example, oxime 12a (pKa ∼ −2.90)26 reacted at a similar rate to pyrazole 10a (pKa ∼ 2.18)27 in benzene, despite a difference of 5 pKa units between the two directing groups. This lack of correlation likely has both steric and electronic origins. First, unlike benzylpyridines 1a-7a, which provide essentially sterically identical coordination environments at the Pd center, compounds 8a-14a differ substantially in terms of both their steric parameters and their conformational flexibility. Literature reports have shown that even relatively small steric changes can have a significant influence on the relative and absolute rates of cyclopalladation.12b,28 In addition, the pKa of a directing group is not an ideal parameter for predicting the subtle electronic influence of these ligands on C–H activation, as it does not take into account the interplay of their σ-donor and π-acceptor/donor abilities.29
Competition Experiments
When multiple potential chelating functionalities are present in solution, substrates containing more electron rich/more basic directing groups react preferentially. This can be concluded based on three key results from the competition studies: (i) the large negative ρ values obtained with substituted benzylpyridines (Figure 3 and Figure 4), (ii) the observation that the most basic directing group (pyridine in substrate 6a) out-competed all of the other directing groups among substrates 8a-14a (Figure 8), and (iii) the fact that heterocyclic ligands with pKa values greater than zero (pyridine, pyrimidine, pyrazine, pyrazole) outcompeted all substrates with pKa values less than zero (oxime ether, amide, and isoxazoline). Based on the considerations discussed above, these results suggest that either ligand binding/exchange (steps i and v in Figure 9) or oxidation (step iii in Figure 9) controls the relative reactivity of the two substrates under these conditions. We were able to rule out the latter based on a study of the order of the reaction in PhI(OAc)2. Under optimal conditions for acetoxylation with substrate 6a, the reaction was found to be zero order in PhI(OAc)2, both in the presence and absence of another substrate (3a) (Figure S6–Figure S7 and Figure S9–Figure S10).
As a result, we propose that selectivity under the competition conditions is controlled by the ligand coordination step. As shown in Scheme 8, there are two possible coordination complexes that can form (A and A’) and that are expected to be in equilibrium under the reaction conditions.30 According to the Curtin Hammett principle, the relative energies of these two complexes (ΔG°) in conjunction with ΔG†† for the turnover limiting C–H activation step from each will determine the product distribution in these transformations. 31
Scheme 8.

Equilibrium for Substrate Binding Under Competition Conditions
As discussed above, literature precedent has shown that coordination of more electron rich ligands to PdII is thermodynamically favored. For example, Hammett ρ values ranging from −0.8 to −1.3 were obtained from Keq measurements of the coordination of substituted pyridines to PdII pincer complexes in CHCl3.13b These values are similar to our results in benzene (ρ = −2.01), which is also a relatively non-polar, non-coordinating solvent. Notably, the literature ρ values for pyridine coordination were found to increase to between −1.7 and −2.1 upon moving to the more polar coordinating solvent DMSO.13a This may provide some explanation for the substantially larger ρ of −5.46 that we observed in the polar protic medium AcOH/Ac2O.
This data suggests that the ligand coordination equilibrium (Scheme 8) dictates the selectivity of acetoxylation reactions in the presence of multiple directing groups.31 However, it is important to note that acidic solvents can significantly perturb this equilibrium by competitively protonating more basic directing groups (hence the convex Hammett plot for benzylpyridine derivatives in Figure 2). Solvent also plays a significant role in the trends observed for substrates 8a-14a (Figure 8). For example, two similar oxime ether derivatives 11a and 12a exhibited very different reactivity when the solvent was changed from AcOH/Ac2O to benzene. Unlike benzyl oxime ether 12a, the methyl oxime ether 11a did not afford any of the acetoxylated product 11b in benzene. Similarly, benzylpyrimidine 8a outcompeted the benzylpyrazole 10a in AcOH/Ac2O; however, a reversal of this selectivity was observed when the solvent was changed to benzene. Additionally, in the competition experiment between benzylpyridine 6a and benzylpyrazole 10a (Scheme 5), selectivity for the benzylpyridine product 6b increased significantly (from 1 : 0.4 to 1 : 0.06) when the solvent was changed from AcOH/Ac2O to benzene.
While the origin of these solvent effects is still under investigation, these results have important implications for future applications of this chemistry. In non-acidic solvents like benzene, the basicity of a directing group appears to serve as a reasonable predictor of its relative reactivity. However, an acidic solvent can be used to attenuate inherent reactivity differences by effectively “protecting” a potential ligand in its protonated form. We anticipate that this and related strategies can be used to obtain, alter, or improve the selectivity of directed C–H functionalization in the context of complex molecules. Future studies will continue to explore how these effects (and the effects of other solvents and additives) translate into predicting and controlling the dominant directing group in more complex systems.
Conclusions
In summary, we have conducted detailed studies to elucidate the electronic requirements of a directing group in Pd-catalyzed directed arene acetoxylation reactions. Under individual kinetics conditions, the reactions are accelerated by electron withdrawing groups and a significant kinetic isotope effect is observed, indicating that cyclopalladation is turnover limiting. However, under competition conditions, substrates with electron donating directing groups react preferentially, suggesting that their relative reactivities are dictated by Keq for substrate coordination under these conditions. Importantly, the current studies have primarily focused on one structural class of substrates where the directing group and the C–H bond are separated by a methylene spacer. As a result, ongoing investigations seek to probe whether the observed effects are generalizable across a broader array of other systems. We anticipate that the mechanistic insights gleaned from this and related work will ultimately prove valuable in future applications of this chemistry.
Experimental Section
General Procedures
NMR spectra were obtained on a Varian Inova 400 (399.96 MHz for 1H; 100.57 MHz for 13C; 376.34 MHz for 19F) unless otherwise noted. 1H NMR chemical shifts are reported in parts per million (ppm) relative to TMS, with the residual solvent peak used as an internal reference.Multiplicities are reported as follows: singlet (s), doublet (d), doublet of doublets (dd), doublet of doublets of doublets (ddd), doublet of triplets (dt), triplet (t), quartet (q), quintet (quin), multiplet (m), and broad resonance (br). IR spectra were obtained on a Perkin-Elmer spectrum BX FT-IR spectrometer. Melting points were determined with a Mel-Temp 3.0, a Laboratory Devices Inc, USA instrument and are uncorrected. HRMS data were obtained on a Micromass AutoSpec Ultima Magnetic Sector mass spectrometer. Gas chromatography was carried out on a Shimadzu 17A using a Restek Rtx®-5 (Crossbond 5% diphenyl – 95 % dimethyl polysiloxane; 15 m, 0.25 mm ID, 0.25 µm df) column.
Materials and Methods
Pd(OAc)2 was obtained from Pressure Chemical and used as received, and PhI(OAc)2 was obtained from Merck Research Laboratories and used as received. Substrates 1a-15a were prepared as described in the Supporting Information. Solvents were obtained from Fisher Chemical and used without further purification unless otherwise noted. Flash chromatography was performed on EM Science silica gel 60 (0.040–0.063 mm particle size, 230–400 mesh) and thin layer chromatography was performed on Merck TLC plates pre–coated with silica gel 60 F254.
General Procedure for Directed C–H Bond Acetoxylation
In a 20 mL scintillation vial, PhI(OAc)2 (0.49–0.86 mmol, 1.02–1.80 equiv) and Pd(OAc)2 (1.08 mg, 0.0048 mmol, 0.01 equiv) were combined in a mixture of AcOH (2 mL) and Ac2O (2 mL). Substrate (0.48 mmol, 1.0 equiv) was added, the vial was sealed with a Teflon-lined cap, and the resulting solution was heated at 100 °C for 3–24 h. The reaction was cooled to room temperature and the solvent was removed under vacuum. The resulting brown oil was purified by chromatography on silica gel. Each substrate was optimized for reaction time and equiv of the oxidant as described in the Supporting Information.
Acetoxylation of Substrate 6a
The reaction was run for 6 h with 1.02 equiv of PhI(OAc)2. The product 6b was obtained as a yellow oil (86.9 mg, 75% yield, Rf = 0.27 in 70% hexanes/30% ethyl acetate). 1H NMR (CDCl3): 5 8.52 (dd, J = 4.8, 1.6 Hz, 1H), 7.54 (dt, J = 7.6, 1.6 Hz, 1H), 7.11–7.02 (multiple peaks, 4H), 6.94 (d, J = 8.0 Hz, 1H), 4.06 (s, 2H), 2.30 (s, 3H), 2.16 (s, 3H). 13C{;1H} NMR (CDCl3): δ 169.37, 159.88, 149.03, 146.79, 136.44, 135.78, 131.72, 130.83, 128.38, 122.85, 122.24, 121.24, 39.21, 20.78, 20.69. IR (thin film): 1759 cm−1. HRMS electrospray (m/z): [M+H]+ calcd for C15H15NO2, 242.1181; found, 242.1177.
General Procedure for Kinetics Experiments
Kinetics experiments were run in two dram vials sealed with Teflon-lined caps. Each data point represents a reaction in an individual vial, with each vial containing an identical concentration of oxidant, catalyst, and substrate. The vials were charged with PhI(OAc)2 (0.0158 g, 0.049 mmol, 1.02 equiv, added as a solid), substrate (0.048 mmol, 1.0 equiv, added as a 0.96 M stock solution in AcOH), and Pd(OAc)2 (0.11 mg, 0.00048 mmol, 0.01 equiv, added as a 0.0096 M stock solution in AcOH), and the resulting mixtures were diluted to a total volume of 400 µL of a 1 : 1 mixture of AcOH and Ac2O. The vials were then heated at 80 °C for various amounts of time. Reactions were quenched by cooling the vial at 0 °C for 5 min, followed by the addition of a 2% solution of pyridine in CH2Cl2 (2 mL). An internal standard (pyrene) was then added, and the reactions were analyzed by gas chromatography. Each reaction was monitored to ∼10% (8.6–11.0%) conversion, and rate constants were calculated using the initial rates method. Each kinetics experiment was run in triplicate, and the data shown in the Hammett plots represent an average of these three runs.
General Procedure for Competition Experiments
A two dram vial was sequentially charged with PhI(OAc)2 (0.0158 g, 0.049 mmol, 1.02 equiv, added as a solid), substrate I (0.048 mmol, 1.0 equiv, added as a 0.96 M stock solution in AcOH), substrate II (0.048 mmol, 1.0 equiv, added as a 0.96 M stock solution in AcOH), and Pd(OAc)2 (0.11 mg, 0.00048 mmol, 0.01 equiv, added as a 0.0096 M stock solution in AcOH), and the resulting mixtures were diluted to a total volume of 400 µL of a 1 : 1 mixture of AcOH and Ac2O. The reaction was heated at 80 °C for 12 h, and then cooled to room temperature. A GC standard (pyrene) was added, and the reaction was analyzed by gas chromatography.
Supplementary Material
Experimental details and spectroscopic and analytical data for new compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
Scheme 6.

Highly Selective Oxime Ether-Directed C–H Acetoxylation of Substrate 15a
Acknowledgements
This work was supported by NIH NIGMS (RO1 GM073836). We also gratefully acknowledge the Arnold and Mabel Beckman Foundation as well as Abbott, Amgen, AstraZeneca, Boehringer-Ingelheim, Bristol Myers Squibb, Eli Lilly, GlaxoSmithKline, Merck Research Laboratories, and Roche for funding. LVD thanks Bristol Myers Squibb for a graduate fellowship. We also acknowledge Dan Dawson (preliminary studies), Nick Deprez (for m-[CF3Ph2I]BF4) and Dipannita Kalyani (editorial assistance).
References
- 1.(a) Hull KL, Sanford MS. J. Am. Chem. Soc. 2007;129:11904. doi: 10.1021/ja074395z. [DOI] [PubMed] [Google Scholar]; (b) Hull KL, Lanni EL, Sanford MS. J. Am. Chem. Soc. 2006;128:14047. doi: 10.1021/ja065718e. [DOI] [PubMed] [Google Scholar]; (c) Hull KL, Anani WQ, Sanford MS. J. Am. Chem. Soc. 2006;128:7134. doi: 10.1021/ja061943k. [DOI] [PubMed] [Google Scholar]; (d) Kalyani D, Dick AR, Anani WQ, Sanford MS. Tetrahedron. 2006;62:11483. [Google Scholar]; (e) Kalyani D, Dick AR, Anani WQ, Sanford MS. Org. Lett. 2006;8:2523. doi: 10.1021/ol060747f. [DOI] [PubMed] [Google Scholar]; (f) Desai LV, Malik HA, Sanford MS. Org. Lett. 2006;8:1141. doi: 10.1021/ol0530272. [DOI] [PubMed] [Google Scholar]; (g) Kalyani D, Sanford MS. Org. Lett. 2005;7:4149. doi: 10.1021/ol051486x. [DOI] [PubMed] [Google Scholar]; (h) Kalyani D, Deprez NR, Desai LV, Sanford MS. J. Am. Chem. Soc. 2005;127:7330. doi: 10.1021/ja051402f. [DOI] [PubMed] [Google Scholar]; (i) Desai LV, Hull KL, Sanford MS. J. Am. Chem. Soc. 2004;126:2300. doi: 10.1021/ja046831c. [DOI] [PubMed] [Google Scholar]; (j) Dick AR, Hull KL, Sanford MS. J. Am. Chem. Soc. 2004;126:2300. doi: 10.1021/ja031543m. [DOI] [PubMed] [Google Scholar]
- 2.For selected examples of palladium-catalyzed directed C–H activation/C–heteroatom bond forming reactions, see: Inamoto K, Saito T, Katsuno M, Sakamoto T, Hiroya K. Org. Lett. 2007;9:2931. doi: 10.1021/ol0711117. Thu HY, Yu WY, Che CM. J. Am. Chem. Soc. 2006;128:9048. doi: 10.1021/ja062856v. Wan X, Ma Z, Li B, Zhang K, Cao S, Zhang S, Shi Z. J. Am. Chem. Soc. 2006;128:7416. doi: 10.1021/ja060232j. Reddy BVS, Reddy LR, Corey E. J. Org. Lett. 2006;8:3391. doi: 10.1021/ol061389j. Wang D, Hao X, Wu D, Yu JQ. Org. Lett. 2006;8:3387. doi: 10.1021/ol061384m. Yu JQ, Giri R, Chen X. Org. Biomol. Chem. 2006;4:4041. doi: 10.1039/b611094k. and references therein. Tsang WCP, Zheng N, Buchwald SL. J. Am. Chem. Soc. 2005;127:14560. doi: 10.1021/ja055353i. Giri R, Liang J, Lei JG, Li JJ, Wang DH, Chen X, Naggar IC, Guo C, Foxman BM, Yu JQ. Angew. Chem., Int. Ed. 2005;44:7420. doi: 10.1002/anie.200502767. Giri R, Chen X, Yu JQ. Angew. Chem., Int. Ed. 2005;44:2112. doi: 10.1002/anie.200462884. Dangel BD, Johnson JA, Sames D. J. Am. Chem. Soc. 2001;123:8149. doi: 10.1021/ja016280f.
- 3.For selected examples of palladium-catalyzed directed C–H activation/C–C bond forming reactions, see: Yu W-Y, Sit WN, Lai K-M, Zhou Z, Chan ASC. J. Am. Chem. Soc. 2008;130:3304. doi: 10.1021/ja710555g. Beccalli EM, Broggini G, Martinelli M, Sottocornola S. Chem. Rev. 2007;107:5318. doi: 10.1021/cr068006f. Alberico D, Scott ME, Lautens M. Chem. Rev. 2007;107:174. doi: 10.1021/cr0509760. Cai G, Fu Y, Li Y, Wan X, Shi Z. J. Am. Chem. Soc. 2007;129:7666. doi: 10.1021/ja070588a. Giri R, Maugel NL, Li JJ, Wang DH, Breazzano SP, Saunders LB, Yu JQ. J. Am. Chem. Soc. 2007;129:3510. doi: 10.1021/ja0701614. Xia J-B, You S-L. Organometallics. 2007;26:4869. Chiong HA, Daugulis O. Org. Lett. 2007;9:1449. doi: 10.1021/ol0702324. Shi ZJ, Li B, Wan X, Cheng J, Fang Z, Cao B, Qin C, Wang Y. Angew. Chem., Int. Ed. 2007;46:5554. doi: 10.1002/anie.200700590. Chen X, Goodhue CE, Yu JQ. J. Am. Chem. Soc. 2006;128:12634. doi: 10.1021/ja0646747. Chen X, Li JJ, Hao XS, Goodhue CE, Yu JQ. J. Am. Chem. Soc. 2006;128:78. doi: 10.1021/ja0570943. Orito K, Miyazawa M, Nakamura T, Horibata A, Ushito H, Nagasaki H, Yuguchi M, Yamashita S, Yamazaki T, Tokuda M. J. Org. Chem. 2006;71:5951. doi: 10.1021/jo060612n. Zaitsev VG, Shabashov D, Daugulis O. J. Am. Chem. Soc. 2005;127:13154. doi: 10.1021/ja054549f. Zaitsev VG, Daugulis O. J. Am. Chem. Soc. 2005;127:4156. doi: 10.1021/ja050366h. Shabashov D, Daugulis O. Org. Lett. 2005;7:3657. doi: 10.1021/ol051255q. Daugulis O, Zaitsev VG. Angew. Chem., Int. Ed. 2005;44:4046. doi: 10.1002/anie.200500589. Orito K, Horibata A, Nakamura T, Ushito H, Nagasaki H, Yuguchi M, Yamashita S, Tokuda M. J. Am. Chem. Soc. 2004;126:14342. doi: 10.1021/ja045342+. Wakui H, Kawasaki S, Satoh T, Miura M, Nomura M. J. Am. Chem. Soc. 2004;126:8658. doi: 10.1021/ja0477874. Hennessy EJ, Buchwald SL. J. Am. Chem. Soc. 2003;125:12084. doi: 10.1021/ja037546g. Ritleng V, Sirlin C, Pfeffer M. Chem. Rev. 2002;102:1731. doi: 10.1021/cr0104330. Boele MDK, van Strijdonck GPF, de Vries AHM, Kamer PCJ, de Vries JG, van Leeuwen PWNM. J. Am. Chem. Soc. 2002;124:1586. doi: 10.1021/ja0176907. Dyker G. Angew. Chem., Int. Ed. Engl. 1999;38:1698. doi: 10.1002/(SICI)1521-3773(19990614)38:12<1698::AID-ANIE1698>3.0.CO;2-6. Tremont SJ, Rahman HU. J. Am. Chem. Soc. 1984;106:5759.
- 4.(a) Ali A, Hunt JA, Kallashi F, Kowalchick JE, Kim D, Smith CJ, Sinclair PJ, Sweis RF, Taylor GE, Thompson CF, Chen L, Quraishi N. PCT Int. Appl. 2007 WO 2007070173. [Google Scholar]; (b) Zhou P, Bernotas RC, Li Y, Nowak PW, Cole DC, Manas ES, Fan Y, Yan Y. PCT Int. Appl. 2007 WO 2007078813. [Google Scholar]; (c) Chen J, Deady LW, Kaye AJ, Finlay GJ, Baguley BC, Denny WA. Bioorg. Med. Chem. 2002;10:2381. doi: 10.1016/s0968-0896(02)00067-6. [DOI] [PubMed] [Google Scholar]
- 5.For previous examples of the stoichiometric cyclopalladation of benzylpyridine, see: Mawo RY, Johnson DM, Wood JL, Smoliakova IP. J. Organomet. Chem. 2008;693:33. and references therein. Hiraki K, Fuchita Y, Takechi K. Inorg. Chem. 1981;20:4316.
- 6.Notably, a similar strategy for electronically isolating an aryl ring from a directing group has been recently reported by Yu and coworkers in mechanistic studies of Pd-catalyzed oxazoline-directed arene oxygenation. Li JJ, Giri R, Yu JQ. Tetrahedron. 2008 in press.
- 7.Anslyn EK, Dougherty DA. Modern Physical Organic Chemistry. Sausalito, CA: University Science Books: University Science Books; 2006. p. 448. [Google Scholar]
- 8.See supporting information for full details of this calculation.
- 9.All pKa values used are in solution phase. (a) research.chem.psu.edu/brpgroup/pKa_compilation.pdf Borowiak-Resterna A, Szymanowski J, Voelkel A. J. Radioanal. Nucl. Chem. Art. 1996;208:75–86.
- 10.The Hammett data looked very similar (ρ =-5.20, R2 = 0.94 in AcOH/Ac2O and ρ =-1.58, R2 = 0.80 in benzene) when these competition experiments were analyzed at low conversion (5–15% combined yield of the two producs) as opposed to at the completion of the reaction.
- 11.Several oxazoline substrates were also examined, as these are widely used as directing groups for Pdcatalyzed C-H functionalization (refs 2, 3, and 6); however, these afforded uncatalyzed acetoxylation of starting substrate. See ref. 24 and the supporting information for full details.
- 12.(a) Ackerman LJ, Sadighi JP, Kurtz DM, Labinger JA, Bercaw JE. Organometallics. 2003;22:3884. [Google Scholar]; (b) Ryabov AD. Chem. Rev. 1990;90:403. and references therein. [Google Scholar]; (c) Ryabov AD, Sakodinskaya IK, Yatsimirsky AK. J. Chem. Soc., Dalton Trans. 1985:2629. [Google Scholar]; (d) Gutierrez MA, Newkome GR, Selbin J. J. Organomet. Chem. 1980;202:341. [Google Scholar]
- 13.(a) Jeon SL, Loveless DM, Yount WC, Craig SL. Inorg. Chem. 2006;45:11060. doi: 10.1021/ic061188b. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Manen H-J, Nakashima K, Shinkai S, Kooijman H, Spek AL, Veggel FCJM, Reinhoudt DN. Eur. J. Inorg. Chem. 2000:2533. [Google Scholar]
- 14.Yagyu T, Iwatsuki S, Aizawa S, Funahashi S. Bull. Chem. Soc. Jpn. 1998;71:1857. [Google Scholar]
- 15.(a) Martin-Matute B, Mateo C, Cardenas DJ, Echavarren AM. Chem. Eur. J. 2001;7:2341. doi: 10.1002/1521-3765(20010601)7:11<2341::aid-chem23410>3.0.co;2-s. [DOI] [PubMed] [Google Scholar]; (b) Marsella A, Agapakis S, Pinna F, Strukul G. Organometallics. 1992;11:3578. [Google Scholar]
- 16.Davies DL, Donald SMA, Macgregor SA. J. Am. Chem. Soc. 2005;127:13754. doi: 10.1021/ja052047w. [DOI] [PubMed] [Google Scholar]
- 17.(a) Lafrance M, Gorelsky SI, Fagnou K. J. Am. Chem. Soc. 2007;129:14570. doi: 10.1021/ja076588s. [DOI] [PubMed] [Google Scholar]; (b) Stuart DR, Fagnou K. Science. 2007;316:1172. doi: 10.1126/science.1141956. [DOI] [PubMed] [Google Scholar]; (c) Lafrance M, Fagnou K. J. Am. Chem. Soc. 2006;128:16496. doi: 10.1021/ja067144j. [DOI] [PubMed] [Google Scholar]; (d) Lafrance M, Rowley CN, Woo TK, Fagnou K. J. Am. Chem. Soc. 2006;128:8754. doi: 10.1021/ja062509l. [DOI] [PubMed] [Google Scholar]; (e) Garcia-Cuadrado D, Braga AAC, Maseras F, Echavarren AM. J. Am. Chem. Soc. 2006;128:1066. doi: 10.1021/ja056165v. [DOI] [PubMed] [Google Scholar]; (f) Campeau LC, Parisien Jean A, Fagnou K. J. Am. Chem. Soc. 2006;128:581. doi: 10.1021/ja055819x. [DOI] [PubMed] [Google Scholar]; (g) Campeau LC, Parisien M, Leblanc M, Fagnou K. J. Am. Chem. Soc. 2004;126:9186. doi: 10.1021/ja049017y. [DOI] [PubMed] [Google Scholar]
- 18.(a) Valk JM, Boersma J, van Koten G. Organometallics. 1996;15:4366. [Google Scholar]; (b) Alsters PL, Teunissen HT, Boersma J, Spek AL, van Koten G. Organometallics. 1993;12:4691. [Google Scholar]
- 19.For examples of analogous electronic effects in reductive elimination from PdII, see: Graham DC, Cavell KJ, Yates BF. Dalton Trans. 2006:1768. doi: 10.1039/b512681a. Nolan SP. In: Palladium in Organic Synthesis. Tsuji J, editor. Vol. 14. Berlin, Germany: Topics in Organometallic Chemistry; Springer-Verlag; 2005. pp. 241–272.
- 20.Stability/reductive elimination from PdIV complexes with one alkyl/aryl ligand and 3 other X-type ligands: Alsters PL, Engel PF, Hogerheide MP, Copijn M, Spek AL, van Koten G. Organometallics. 1993;12:1831.
- 21.Some PdIV complexes containing multiple σ-akyl and/or σ-aryl ligands have been isolated and shown to be stable at room temperature or above. Markies BA, Canty AJ, Boersma J, van Koten G. Organometallics. 1994;13:2053. Byers PK, Canty AJ, Skelton BW, White AH. J. Chem. Commun. 1986:1722. However, the proposed intermediate in the current transformations is a monoaryl PdIV species (B). Limited examples of such complexes have been observed, and all undergo rapid reductive elimination at room temperature (see ref 18).
- 22.We have reported that PdIV complexes containing two σ-aryl ligands can be stable at room temperature Dick AR, Kampf JW, Sanford MS. J. Am. Chem. Soc. 2005;127:12790. doi: 10.1021/ja0541940.
- 23.Chiong HA, Pham Q, Daugulis O. J. Am. Chem. Soc. 2007;129:9879. doi: 10.1021/ja071845e. [DOI] [PubMed] [Google Scholar]
-
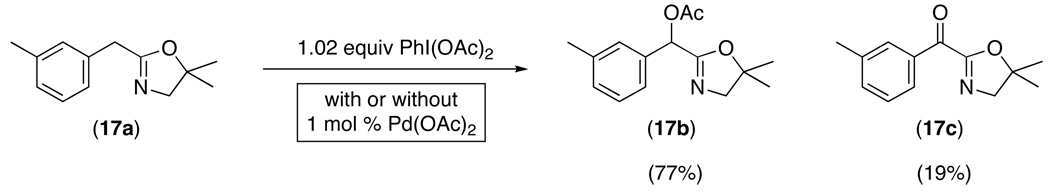
24.The oxazoline directing group from ref. 6 could not be directly compared to the current systems. Under our standard reaction conditions, oxazoline 17a underwent a rapid uncatalyzed reaction with the oxidant to afford 17b (77% NMR yield) and 17c (19% NMR yield). In the presence of Pd(OAc)2, 17b and 17c were still the major products, and <5% of the expected aromatic C–H functionalization product was formed. See supporting information for full details.
- 25.(a) Gomez M, Granell J, Martinez M. Eur. J. Inorg. Chem. 2000:217. [Google Scholar]; (b) Ryabov AD. Inorg. Chem. 1987;26:1252. [Google Scholar]
- 26.Sokolov SD, Tikhomirova GB, Turchin KF. Chem. Heterocycl. Compd. 1985;21:507. [Google Scholar]
- 27.Catalan J, Claramunt RM, Elguero J, Laynez J, Menendez M, Anvia F, Quian JH, Taagepera M, Taft RW. J. Am. Chem. Soc. 1988;110:4105. [Google Scholar]
- 28.(a) Mossi w, Klaus AJ, Rys P. Helv. Chim. Acta. 1992;75:2531. [Google Scholar]; (b) Thummel RP, Jahng Y. J. Org. Chem. 1987;52:73. [Google Scholar]
- 29.(a) Crabtree RH. “The Organometallic Chemistry of the Transition Metals,”. Fourth Edition. Hoboken, NJ: John Wiley and Sons, Inc; 2005. pp. 18–26. [Google Scholar]; (b) Slater JW, Rourke JP. J. Organomet. Chem. 2003;688:112. [Google Scholar]
- 30.A similar ρvalue of –1.7 to –2.1 in DMSO was observed by Craig in studying the substituted pyridine coordination to 1,4-phenylene bridged bimetallic palladium complexes containing tridentate ligands (see ref. 10a).
- 31.Importantly, the proposed effects of the equilibrium for ligand exchange are consistent with the Curtin-Hammett principle as long as the difference in the energy (ΔG) between the two PdII pyridine coordination complexes is greater than the difference in the activation energies (ΔΔG††) for the C–H activation step (the slow step of these transformations). See Supporting Information for further details.

Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Experimental details and spectroscopic and analytical data for new compounds. This material is available free of charge via the Internet at http://pubs.acs.org.