

Abstract
Pseudoautosomal regions (PARs) shared by avian Z and W sex chromosomes are typically small homologous regions within which recombination still occurs and are hypothesized to share the properties of autosomes. We capitalized on the unusual structure of the sex chromosomes of emus, Dromaius novaehollandiae, which consist almost entirely of PAR shared by both sex chromosomes, to test this hypothesis. We compared recombination, linkage disequilibrium (LD), GC content, and nucleotide diversity between pseudoautosomal and autosomal loci derived from 11 emu bacterial artificial chromosome (BAC) clones that were mapped to chromosomes by fluorescent in situ hybridization. Nucleotide diversity (π = 4Neμ) was not significantly lower in pseudoautosomal loci (14 loci, 1.9 ± 2.4 × 10−3) than autosomal loci (8 loci, 4.2 ± 6.1 × 10−3). By contrast, recombination per site within BAC-end sequences (ρ = 4Nc) (pseudoautosomal, 3.9 ± 6.9 × 10−2; autosomal, 2.3 ± 3.7 × 10−2) was higher and average LD (D′) (pseudoautosomal, 4.2 ± 0.2 × 10−1; autosomal, 4.7 ± 0.5 × 10−1) slightly lower in pseudoautosomal sequences. We also report evidence of deviation from a simple neutral model in the PAR and in autosomal loci, possibly caused by departures from demographic equilibrium, such as population growth. This study provides a snapshot of the population genetics of avian sex chromosomes at an early stage of differentiation.
Keywords: bacterial artificial chromosomes, fluorescent in situ hybridization, nucleotide diversity, pseudoautosomal region, recombination, sex chromosomes
Sex chromosomes are thought to have evolved from pairs of ancestral autosomes that gained a sex-specific gene and function (Charlesworth 1996; Rice 1996), which in turn caused cessation of recombination, leading to degeneration of one of the pair of sex chromosomes. For example, in humans, the male-specific Y sex chromosome appears to be a degenerated homolog of the X sex chromosome. The degeneration is thought to be a consequence of higher frequency of mutation on the Y, ineffective selection in the absence of X–Y recombination and the lower effective population size of Y chromosomes compared with X chromosomes. As a result of this degradation, some loci are found on only one sex chromosome because their gametologs have decayed on the other sex chromosome (Garcia-Moreno and Mindell 2000). For example, male birds carry 2 Z chromosomes and females carry 1 Z chromosome and 1 W chromosome that appears to be a degenerated homolog of the Z.
Avian sex chromosomes comprise Z- or W-specific regions and one or more shared pseudoautosomal regions (PARs) that recombine between the Z and W. As recombination ceases and one of a pair of sex chromosomes begins to degenerate, PARs become proportionally smaller, presumably because deletions are not selected against and there is no opportunity for repair from the other chromosome. Because pseudoautosomal loci recombine at meiosis, they are expected to be more similar in diversity and dynamics to autosomal loci than they are to Z- or W-specific loci (Ellis et al. 1990; Toder and Graves 1998).
The gene content of the Z chromosome is well conserved even between distantly related carinate and ratite birds, although reorganization of Z-linked genes appears to have occurred between chicken, Gallus gallus, and zebra finch, Taeniopygia guttata (Backstrom, Brandstrom, et al. 2006; Itoh et al. 2006). However, comparative painting indicates complete homology at a broad scale of the Z between chicken and zebra finch (Shetty et al. 1999). By contrast, bird species vary considerably in the length of the W chromosome, and as a consequence, the proportion of the sex chromosomes that is pseudoautosomal varies among taxa. For example, in chicken, the Z chromosome is more than twice as large as the W chromosome, whereas the sex chromosomes of emu, Dromaius novaehollandiae, are nearly identical in size (Figure 1A), and chicken Z-specific chromosome paint hybridizes to all but a small region of the emu sex chromosomes (Shetty et al. 1999). Only one gene marker, DMRT1, is known to be Z specific in emu (Shetty et al. 2002), and no W-specific markers have yet been identified in this species. Why the PAR appears to have degenerated more slowly in ratites like emus than in other paleognathes like tinamou (Backstrom, Brandstrom, et al. 2006; Mank and Ellegren 2007; Tsuda et al. 2007) and neognathes like chicken is unknown.
Figure 1.
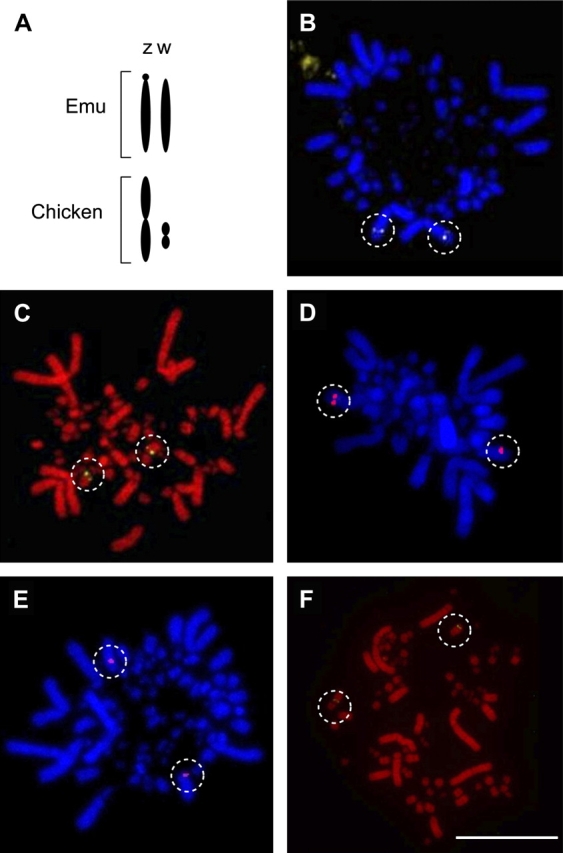
(A) Sex chromosomes from emu and chicken. The PAR covers a larger proportion of the sex chromosomes in emu than in chicken. (B–F) Fluorescently labeled BAC clones mapped to chromosomes of a female emu. Randomly selected BAC clones (B: AL3 and C: AL5) were mapped to autosomes. BAC #AL3 mapped to a macrochromosome, and BAC #AL5 mapped to a microchromosome. BAC clones mapped to the PAR of sex chromosomes were selected for the presence of target loci (D: PAR2; IREBP, E: PAR4; CHD, and F: PAR6; ZOV3). White dotted circles indicate the hybridized probes and scale bar represents 10 μm.
Nucleotide diversity typically differs between sex chromosomes and autosomes in a broad range of animals (Charlesworth et al. 1987; McAllister and Charlesworth 1999; Bachtrog and Charlesworth 2000; Ellegren 2003; Axelsson et al. 2004; Berlin and Ellegren 2004, 2006; Sundstrom et al. 2004; Handley et al. 2006), usually being lower on the Z (X) and lower still on the W (Y). A 3-fold reduction in π was reported on the sex chromosomes of domestic strains of chickens, with Z-linked noncoding π being 2.0 ± 0.1 × 10−3 and autosomal noncoding π being 6.5 ± 0.3 × 10−3 (Sundstrom et al. 2004). Similarly, Saetre et al. (2003) found lower diversity in Z-linked loci than in autosomal loci of Ficedula flycatchers. In principle, this reduction could be partially explained by differences in effective population size between Z- or W-specific loci and autosomal loci (Nisbet and Hatch 1999). However, diversity on chicken and flycatcher sex chromosomes was substantially less than would be predicted by effective population size, even if sex ratios were strongly skewed toward males. For this reason, sex-linked reduction in diversity in chicken and flycatcher has been interpreted as evidence of selective sweeps within nonrecombining regions (Berlin and Ellegren 2004; Borge et al. 2005).
In this study, we examine a range of factors modulating levels of nucleotide diversity in autosomal and pseudoautosomal genes in the emu, a large, flightless Australian bird belonging to the monophyletic group of living ratites (van Tuinen et al. 1998). Emus are thought to have diverged from their sister genus, represented by the cassowary, Casuarius casuarius, approximately 41 million years ago (Pereira and Baker 2006). Emus are ideal for studying genomic variation at an early stage of sex chromosome differentiation because their sex chromosomes are only slightly diverged (Pigozzi and Solari 1999; Shetty et al. 2002). In bird or mammal populations, the ratio of autosomal loci to pseudoautosomal loci is 1:1 regardless of sex ratio (Graves et al. 1998). Effective population size, a direct determinant of neutral nucleotide diversity, does not differ between autosomal and pseudoautosomal loci by definition. As is always the case for PAR loci, we expect the number of population copies of pseudoautosomal and autosomal loci to be equal so that they can be directly compared for differences in diversity without the confounding effects of population size.
We consider influences of population growth and structure, mutation, recombination, and linkage disequilibrium (LD) on levels of diversity between pseudoautosomal and autosomal loci (Aquadro et al. 1994; Wall 2001). Also, by using end sequences from bacterial artificial chromosome (BAC) clones, we were able to explore the relationship between LD and physical distance at the sequence level, which has rarely been attempted in natural populations of birds though it has been investigated in other taxa (Awadalla and Charlesworth 1999; Awadalla et al. 1999; Meunier and Eyre-Walker 2001; Nachman 2002; Reich et al. 2002; Myers et al. 2005). LD typically decreases with distance though positive selection has been shown to increase LD in the genomic neighborhood of a selected locus (Meiklejohn et al. 2004). Previous studies of LD in birds present inconsistent results. In red-winged blackbirds, Agelaius phoeniceus, no LD was detected among loci within a 40-kb autosomal region of the major histocompatibility complex (Edwards and Dillon 2004), whereas most of 23 analyzed Z-linked genes (each about 500 bp and spanning many megabases) were reported to be in perfect LD in collared flycatchers, Ficedula albicollis (Backstrom, Ovarnstrom, et al. 2006). Also, high LD was reported across spans of up to 5 cM in commercial, likely inbred chicken populations (Heifetz et al. 2005). LD is an important force modulating genetic diversity (Wakeley and Lessard 2003), hence our interest in comparing this parameter in the PAR and autosomes of emu.
A long-standing hypothesis predicts that the PAR and autosomal loci will have equivalent population genetic dynamics (Ellis et al. 1990). However, it has long been known, for example, that there are greatly increased male recombination rates (20-fold higher) of the human PAR1 compared with autosomes (Lien et al. 2000), and the proximity of the PAR to the chromosomally diverging sex-specific region could alter the population genetics of the PAR. Thus, a major question is whether and to what extent the PAR is influenced by being attached to a differentiating region, especially a degenerating W (Y) region. Z- and W-specific loci were likely, at one time, pseudoautosomal. Differences between the PAR and autosomal loci could indicate an initial stage of chromosomal degeneration leading to contraction of the PAR. Differences in diversity between autosomal and pseudoautosomal loci would suggest that the PAR is influenced by linkage to Z- or W-specific loci, or for some reason experiences selection intrinsic to genes in the region. Equivalent diversity between autosomal and pseudoautosomal loci would suggest that evidence of selective sweeps that have reduced sex chromosome diversity in other birds is predominantly limited to Z- or W-specific, not pseudoautosomal, loci.
Methods and Materials
Tissues and DNAs
Tissue samples from wild-caught emus, stored in 95% ethanol, were gathered from Western Australia (n = 10), Queensland (n = 2), Southern Australia (n = 4), and New South Wales (n = 8). Tissue samples from cassowary, C. casuarius, were used as outgroups to measure levels of divergence. Genomic DNA was extracted from tissue samples using the DNeasy Blood and Tissue Kit (Qiagen, Valencia, CA) following manufacturer's instructions.
Selection and Amplification of Target Loci
Five avian candidate sex-linked loci were identified from GenBank (records AB002056, AB006694, AY095498, AB006695, and AF288505; Table 1). These loci consisted of a presumptive pseudogene (EE0.6), an iron-responsive element-binding protein gene (IREBP), the protein-encoding spindlin gene (SPIN), a member of the Ig superfamily (ZOV3), and a chromo-helicase DNA-binding protein gene (CHD1). EE0.6, IREBP, SPIN, ZOV3, and CHD1 sequences were available for emu (Ogawa et al. 1998; Garcia-Moreno and Mindell 2000; de Kloet RS and de Kloet SR 2003). Sequences were amplified from emus, purified, and cloned into XL-10 Gold ultracompetent cells (Shedlock et al. 2008) to be used as probes.
Table 1.
Autosomal and pseudoautosomal loci in emu surveyed for polymorphism
| Locus name | Well location | BAC insert size (bp) | Chromosome | GenBank record | Reference |
| AL1a | 194e24 (SubA) | 163 400 | Autosome | EU200931 | This study |
| AL1b | 194e24 (SubB) | 163 400 | Autosome | EU200931 | This study |
| AL2a | 149N1A | 137 200 | Autosome | ET041500 | This study |
| AL2b | 149N1B | 137 200 | Autosome | ET041501 | This study |
| AL3a | 146N2A | 115 400 | Autosome | ET041502 | This study |
| AL4a | 306L20B | 90 700 | Autosome | ET041515 | This study |
| AL5a | 19C13A | 112 800 | Autosome | ET041512 | This study |
| AL5b | 19C13B | 112 800 | Autosome | ET041513 | This study |
| EE0.6 | PAR | AB002056 | Ogawa et al. (1998) | ||
| IREBP | PAR | AB006694 | Ogawa et al. (1998) | ||
| SPIN | PAR | AY095498 | de Kloet RS and de Kloet SR (2003) | ||
| ZOV3 | PAR | AB006695 | Ogawa et al. (1998) | ||
| PAR1a | 146L2B (Irebp) | 103 600 | PAR | ET041507 | This study |
| PAR2a | 19E14A (Irebp) | 129 200 | PAR | ET041520 | This study |
| PAR2b | 19E14B (Irebp) | 129 200 | PAR | ET041521 | This study |
| PAR3a | 280L21A (Chd) | 130 900 | PAR | ET041516 | This study |
| PAR3b | 280L21B (Chd) | 130 900 | PAR | ET041517 | This study |
| PAR4a | 149M2A (Chd) | 104 800 | PAR | ET041508 | This study |
| PAR4b | 149M2B (Chd) | 104 800 | PAR | ET041509 | This study |
| PAR5a | 280L24A (Chd) | 134 900 | PAR | ET041510 | This study |
| PAR6a | 146L1A (Zov3) | 119 100 | PAR | ET041518 | This study |
| PAR6b | 146L1B (Zov3) | 119 100 | PAR | ET041519 | This study |
Novel loci were selected from ends and subclones of BACs screened from the emu library using 5 previously published loci as probes.
Retrieval of BAC Clones
A BAC library for emu was available through the US Department of Energy's Joint Genome Institute (http://www.jgi.doe.gov/; now from SymBio Corporation, www.sym-bio.com), and high-density filters were screened as previously described with radiolabeled cloned fragments (Shedlock et al. 2008). Six BAC clones containing CHD1 (n = 3), IREBP (n = 2), and ZOV3 (n = 1) were retrieved from the library. Presence of target sequences on clones was confirmed by amplification of loci using PCR and sequencing using original primers. We also selected 5 random BAC clones from the library to find autosomal loci for comparison to previously reported and novel pseudoautosomal loci. Aside from the 4 previously published loci, loci in this study were named either AL as an autosomal locus or PAR as found in the pseudoautosomal region. The number assigned to each locus distinguishes BAC inserts from which they were sequenced. The letters a and b distinguish ends of the same BAC insert.
Fluorescent In Situ Hybridization (FISH)
To confirm the chromosomal location of loci and BACs used in the study, we conducted FISH, even when the locus had been previously localized by other workers. Metaphase chromosome spreads were harvested from fibroblast cell lines of emu, and slides were prepared as described in Ezaz et al. (2005).
BAC clones were grown overnight in 15 ml of Difco LB broth at 37 °C with shaking. DNAs were purified as previously described (Shedlock et al. 2008). In addition to DNAs from 5 randomly selected BAC clones, DNAs from 6 BAC clones containing pseudoautosomal targets were labeled with Biotin-16-deoxyuridine triphosphate by nick translation and hybridized to emu metaphase chromosome spreads as described by Janes et al. (2008). Images were captured using a Zeiss Axioplan epifluorescence microscope equipped with a charge-coupled device camera (RT-Spot, Diagnostic Instrument, Sterling Heights, MI) using filters 02, 10, or 15 from the Zeiss fluorescence filter set or the Pinkel filter set (filter set 8300, Chroma Technologies, Rockingham, VT). The camera was controlled by an Apple Macintosh computer. IPLab scientific imaging software (V.3.9, Scanalytics Inc., Rockville, MD) was used to capture and superimpose gray scale images to produce color images.
End Sequencing, Subcloning, and Fingerprinting of Bacterial Artificial Chromosomes
To generate additional noncoding loci for analysis, we end sequenced the first 500–1000 bp on either end of BAC inserts (GenBank accession numbers ETO41500–ETO41522). Standard alkaline lysis miniprep techniques were used to purify BAC DNA from clones grown overnight in 1 ml chloramphenicol-supplemented 2YT medium. To sequence BAC ends, sequencing reactions were performed with BigDye Terminator chemistry version 3.1 (Applied Biosystems, Foster City, CA) and M13 forward (5′CAGGAAACAGCTATGACC3′) and M13 reverse (5′TGTAAAACGACGGCCAGT3′) universal primers. Samples were analyzed on a 3730xl DNA analyzer (Applied Biosystems), and vector sequences were removed manually. BAC insert sizes were measured by fingerprinting to consider distances between paired BAC ends when measuring recombination and LD. Clones were cleaved with HindIII restriction enzymes and analyzed by agarose gel electrophoresis (1% Tris-acetate electrophoresis buffer [TAE]). Band sizes were measured and summed to calculate total size of BAC inserts (Liu et al. 2006). Two additional autosomal loci were produced from subclones of BAC insert #AL1. These 2 loci are not BAC-end sequences but rather were sequenced from subcloned regions within the insert (Shedlock et al. 2008).
Amplification of Target Loci
Primers were designed for a total of 8 autosomal and 14 pseudoautosomal loci with the Oligo Primer Analysis Software (Molecular Biology Insights, Cascade, CO). Sixteen of the 22 target loci are ends of BAC clones collected from library screening. The other 6 target loci consist of 2 subclone sequences and 4 loci from the literature (Table 1). Amplification reactions were performed using an initial denaturation step at 94 °C for 2 min, followed by 35 cycles of 94 °C for 40 s, varying annealing temperatures depending on target (Table 2) for 1 min, and 72 °C for 1 min, followed by a final step of 72 °C for 5 min. Amplified fragments were purified using Multiscreen PCRμ96 Filter Plates (Millipore, Billerica, MA) and sequenced directly using BigDye Terminator Cycle Sequencing chemistry with original primers (Applied Biosystems, Foster City, CA). Sequences were recorded with an ABI3100 automated sequencing instrument (Applied Biosystems). Homologous loci were also amplified from the cassowary, C. casuarius, to compare emu loci and correct nucleotide diversity estimates for divergence between the 2 sister species.
Table 2.
Primers and annealing temperatures for amplification of autosomal and pseudoautosomal loci in emu
| Locus name | Forward | Reverse | Annealing temperature (°C) |
| AL1a | TTCTTTAGGGCATAGCATAGG | AGCACTTTGCCGGTAA | 58.5 |
| AL1b | ATTTGTTGGCACCGTGTCAGC | CCGTCACCCAGGATTCGAGA | 57.6 |
| AL2a | ACATTGTGTCAGGATCCCTA | CAAAAGCAAGACAGGGTATC | 53.7 |
| AL2b | CCTTGGCTTCCATACTCA | CTGTGGCAGAGTCAGTAACA | 53.1 |
| AL3a | CATTCAGCCCATAGTCACA | TTTAATGTTGGGTGTAGGGA | 53.1 |
| AL4a | ACTACCTAATCCAGCGTTAC | TGCCCAGTAGACTTGC | 56.3 |
| AL5a | ACCTTTGCCTTGCACCTAA | ATCAGAAGGATGCGTGGTAA | 59.8 |
| AL5b | TCAGGCCAGTCTCGGAGAG | ATGCTGTACCATCGCTTTGC | 55.2 |
| EE0.6 | TTGATTCAAGGAGGGTACCTAC | GGAATCAAGCCAATGCTC | 55 |
| IREBP | GCTTAGCCCTGCTTGCACCTA | GTGCTTAACTCCGCCCAATGC | 54.3 |
| SPIN | AAACTTTAGACTACGCATTG | CGCCTTAGTGTAACATGA | 56.3 |
| ZOV3 | TCCCAAATTAATTGACTAAA | GAATCATTAATGGCAATCT | 52.7 |
| PAR1a | AACACTGGGCAATGGT | ACTGGGCTATGATTCTACAA | 53.1 |
| PAR2a | GGTTGGGAAGGATTAAG | AACTGTTGCCATGTATCTAT | 54.3 |
| PAR2b | ACTTAACTCCACCAGTTGTT | CCAGACCTCGCTGATA | 59.8 |
| PAR3a | CCTGGAAAACAACTTGAGC | AATGCAGAAAAACGTGGATA | 52.1 |
| PAR3b | GCAAAAGCTGGAAAGTCATA | TGTTGTGCTAGACCCAAGA | 59.8 |
| PAR4a | ATAATTCAAGCATGGCAATA | GACTTTGACGCTTTAGGAG | 58.9 |
| PAR4b | GGGACCGCCAGTAATG | TATCAATCCGCTTGCTAATG | 52.7 |
| PAR5a | CTTGGACTTGGCTAACAATC | TAAGTTGCCTCCGAGTTCTA | 52.1 |
| PAR6a | TTCAAATCTTGTGCCAATAG | CACTTGCCTCTTTACGC | 54.3 |
| PAR6b | GTGATGAGAGGGAAGTTGTT | AGGAGCAGGTGGTAAGC | 59.8 |
Sequence Analysis and Statistics
Forward and reverse sequences from each amplicon were reconciled with each other using Sequencher v. 4.5 (Gene Codes, Ann Arbor, MI). Sequences were aligned among the 24 individuals, and haplotypes were estimated by PHASE (Stephens et al. 2001). PHASE outfiles were converted to Nexus files with python script (Chapus C, personal communication) and analyzed by DNAsp (Rozas et al. 2003) for nucleotide diversity, Tajima's D, and Fu and Li's D (Tajima 1989; Fu and Li 1993). Sequence divergences were estimated between each emu locus and its homolog from cassowary by alignment in ClustalX (Thompson et al. 1997; van Tuinen et al. 1998). Nucleotide diversity for each locus was corrected for mutation rate using percentage of divergence from cassowary and weighted by length (base pair) and number of samples. Corrections were intended to control for influences of mutation rate and sample size differences. Weighted diversity from autosomal and pseudoautosomal loci were compared by student's t-test using JMP software (version 6, Cary, NC, 2006). The most likely substitution model and the GC content of each locus were measured by MODELTEST software (Posada and Crandall 1998). Twelve of the study's 22 loci were comprised of 6 pairs of BAC ends from the same clones. Two pairs of BAC ends (AL2a,b and AL5a,b; Table 1) carried autosomal sequences, and 4 pairs of BAC ends (PAR2a,b; PAR3a,b; PAR4a,b; and PAR6a,b; Table 1) carried pseudoautosomal sequences. To detect selection using data from interspecific divergence, the Hudson-Kreitman-Aguade (HKA) test (Hudson et al. 1987) was applied to sequence data from both species using J. Hey's HKA software (http://lifesci.rutgers.edu/∼heylab/HeylabSoftware.htm#HKA). Autosomal loci and pseudoautosomal loci were analyzed with HKA separately and together. The data set was characterized for population genetic structure by Bayesian Analysis of Population Structure (BAPS) software, which employs a Bayesian Markov chain Monte Carlo (MCMC) method of inference for possible population structure (Corander and Marttinen 2006). Population structure was also measured with 25 000 000 replicates of the model-based cluster method implemented in Structure software (Pritchard et al. 2000).
Recombination rate per site (ρ = 4Nc) within and between loci was estimated in PHASE. Edwards and Dillon (2004) estimated ρ for a major histocompatibility complex (MHC)-linked locus (0.0767–0.0512) and multiple noncoding loci (0.0488–0.0586) in red-winged blackbirds, A. phoeniceus, the only wild bird for which ρ per site has been reported. Values for noncoding loci from that study were averaged to generate the prior value (0.0588) for analysis in emu. The data set was also run with a standard human prior of 0.0004 (Stephens et al. 2004). LD between sites within and between pairs of BAC ends was measured by Haploview software (Barrett et al. 2005). Distances between paired BAC ends were calculated from fingerprint results and incorporated into LD matrices. Differences in recombination across BAC ends on autosomal and pseudoautosomal loci were evaluated by Wilcoxon/Kruskal-Wallis test for rank sums using JMP. To obtain multilocus estimates of genetic diversity and recombination rate, θ = 4Nμ was estimated for a single population using MIGRATE (Beerli 2006), and the scaled recombination rate (r = c/μ, where c equals the per site recombination rate that was estimated by the Markov Chain Monte Carlo method implemented in the program LAMARC (Kuhner et al. 2000; Kuhner 2006; Kuhner and Smith 2007) using default settings except for the bird prior (0.0588) estimated from Edwards and Dillon (2004). Also, evidence of population growth (G = g/μ, where g equals the population growth rate) was estimated by LAMARC for autosomal and pseudoautosomal loci. To evaluate the signal for significant LD within these locus pairs, LdHat (McVean et al. 2002) compared the regression of observed LD and distance to regressions from 5000 shufflings of sites across a pair of BAC ends.
Results
FISH
FISH results from emu showed that the pseudoautosomal markers CHD1, IREBP, and ZOV3 all mapped to comparable positions on the Z and W chromosomes. This matched expectations from previously published accounts of the chromosomal location of these genes in emu (Ogawa et al. 1998; Fridolfsson and Ellegren 2000). Metaphase chromosomes derived from a female as well as a male emu exhibited 2 copies of FISH probes containing pseudoautosomal target loci, and the 5 randomly selected BAC clones mapped to autosomes (Figure 1B–F; Table 1).
Population Genetics
A total of 4.82 kb of sequence for each of the 24 emus was analyzed in this study: 3.48 kb from the PAR of the sex chromosomes and 1.34 kb from noncoding autosomal markers. Fourteen pseudoautosomal loci comprised EE0.6, IREBP, SPIN, and ZOV3 and 10 BAC-end sequences. All 8 autosomal loci were BAC-end sequences. Five BACS were mapped to emu autosomes but only 8 of the 10 possible BAC-end sequences could be amplified from our sample of emus. Corrected for divergence from cassowary and weighted for length (bp) and number of samples, diversity did not differ significantly between pseudoautosomal sequences (mean π = 1.9 ± 2.4 × 10−3) and autosomal sequences (mean π = 4.2 ± 6.1 × 10−3; t = 0.63788; P = 0.55; Figure 2A–B). One autosomal locus (AL3a) and 2 pseudoautosomal loci (IREBP and PAR3a) had surprisingly high divergences between cassowary and emu, suggesting an inability to amplify orthologous cassowary sequences. For this reason, these 3 loci were excluded from analysis of corrected diversity and Figure 2. Removal of correction factors did not change results. Diversity uncorrected for divergence from cassowary also did not differ significantly between pseudoautosomal and autosomal loci. All but 2 of the studied loci had negative Tajima's D values (Table 3), although only 3 of these were significantly different from zero. Nine of the 22 loci had negative Fu and Li's D values, but only 2 loci in each class had a significantly extreme Fu and Li's D (Table 3). MODELTEST detected no noticeable differences in substitution models between autosomal and pseudoautosomal loci (Supplementary Table 1). GC content was lower in the PAR (mean GCPAR = 0.389, GCAUTO = 0.453; t = 2.17; P = 0.05), a result that does not match expectation between regions of higher and lower recombination rate (see below; Eyre-walker 1993; Webster et al. 2006).
Figure 2.

Diversities and divergences of autosomal and pseudoautosomal loci in emu. (A) Diversities (π) of 8 autosomal and 14 pseudoautosomal loci vary widely within categories of loci (autosomal or pseudoautosomal). (B) Sequence divergence between each emu locus and its ortholog from cassowary. See Results for explanation of unrepresented loci.
Table 3.
Diversities, divergences, sample sizes, and summary statistics for autosomal and pseudoautosomal loci in emu. Divergences were measured by comparing homologous sequences between emu and the sister species cassowary
| Locus name (bp) | Polymorphic Sites | Nucleotide Diversity (×10−3) | Divergence (%) | N (alleles) | Tajima's D | Fu and Li's D | ||
| AL1a (292) | 14 | 2.500 | 2.7 | 34 | −2.03143 | P < 0.05 | -3.23469 | P < 0.02 |
| AL1b (170) | 9 | 12.85 | 6 | 30 | −1.65602 | −0.96003 | ||
| AL2a (137) | 4 | 4.470 | 12.7 | 18 | −1.43164 | −1.6133 | ||
| AL2b (140) | 7 | 17.57 | 7.3 | 24 | 0.06806 | 1.37323 | P < 0.05 | |
| AL3a (186) | 6 | 7.290 | Not determined | 10 | −1.49289 | −1.51001 | ||
| AL4a (279) | 32 | 19.21 | 1.1 | 38 | −1.0516 | 0.64737 | ||
| AL5a (152) | 13 | 13.71 | 2.7 | 32 | −1.15414 | −0.75026 | ||
| AL5b (171) | 11 | 6.370 | 6.2 | 46 | −1.66988 | 0.31441 | ||
| EE0.6 (353) | 2 | 0.760 | 10.8 | 42 | −1.32551 | −0.84256 | ||
| IREBP (110) | 4 | 14.09 | Not determined | 16 | 0.89417 | 1.14136 | ||
| SPIN (511) | 5 | 1.720 | 0.6 | 36 | −0.70594 | −0.6902 | ||
| ZOV3 (230) | 7 | 5.610 | 2.2 | 44 | −0.53901 | 1.24988 | ||
| PAR1a (232) | 3 | 3.510 | 5.7 | 14 | −0.42241 | −1.03687 | ||
| PAR2a (222) | 3 | 1.710 | 5 | 46 | −0.93188 | −1.68049 | ||
| PAR2b (416) | 14 | 6.580 | 5.3 | 42 | −0.49614 | 0.61452 | ||
| PAR3a (386) | 10 | 4.240 | Not determined | 36 | −1.18677 | −0.14337 | ||
| PAR3b (227) | 5 | 1.690 | 2.7 | 50 | −1.63511 | 0.13389 | ||
| PAR4a (70) | 3 | 1.860 | 8.7 | 50 | −1.8623 | P < 0.05 | −3.40466 | P < 0.02 |
| PAR4b (228) | 11 | 9.400 | 4 | 36 | −0.5954 | 1.44052 | P < 0.05 | |
| PAR5a (229) | 5 | 1.750 | 5.7 | 44 | −1.62267 | 0.16599 | ||
| PAR6a (400) | 25 | 12.69 | 5.1 | 44 | −0.89516 | 0.52649 | ||
| PAR6b (329) | 19 | 15.00 | 1.7 | 36 | 0.25922 | 0.59384 |
BAPS and Structure software reported conflicting results on population structure. Despite the range of individuals’ origins, the Bayesian analysis detected no clustering among the 24 individuals (log (marginal likelihood) of optimal partition = −3143.0662). However, Structure reported significantly higher likelihood (−1758.04; P < 0.001) for a 3-population model than for a single-population model, but individuals were assigned to populations that did not match their geographic origins, usually with probability lower than 50%. We consider BAPS and Structure results as evidence of little or no effect of geography on diversity.
The 6 pairs of BAC ends in this sample are separated by 104–138 kb (Table 1). Recombination rates estimated in PHASE did not differ significantly between ends of BAC inserts carrying either autosomal or pseudoautosomal loci (Figure 3). Recombination at sites among pairs of BAC ends was not significantly lower in the PAR than on autosomes (P = 0.47; Figure 4A). Also, no correlation was found between recombination rates of pairs of ends from the same BAC insert and the distance between those pairs. This could mean that recombination rates vary on larger scales than are provided by a BAC clone. The results for recombination were not changed by use of a human prior in PHASE. However, when considering only sites within individual BAC-end loci and loci retrieved from other subclones (rather than between locus pairs), recombination rates (ρ) were higher in the pseudoautosomal loci than autosomal loci (pseudoautosomal, 3.9 ± 6.9 × 10−2; autosomal, 2.3 ± 3.7 × 10−2; P < 0.05; Figure 4B).
Figure 3.
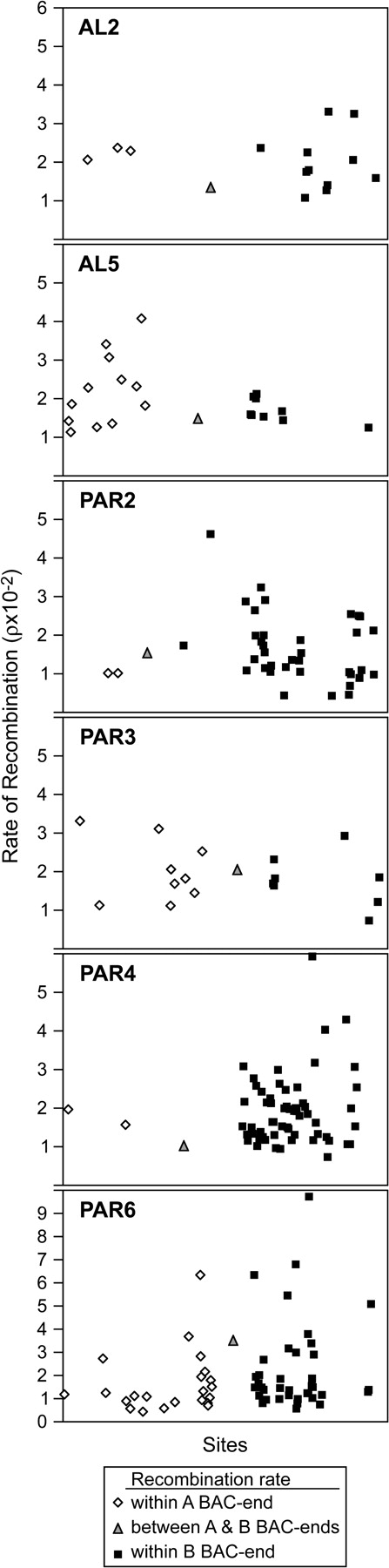
Recombination estimates between variable sites in ends of emu BAC inserts. Each graph (AL2; AL5; PAR2; PAR3; PAR4; and PAR6) shows the median posterior probability of ρ calculated by PHASE software for each variable site in both ends of each of 6 BAC inserts. The first 2 graphs present data from autosomal sequences. The following 4 graphs present data from pseudoautosomal sequences. In each graph, diamonds represent estimates of ρ between variable sites within one BAC end and squares represent estimates of ρ between variable sites from the other BAC end. Triangles represent estimated ρ between the 2 most proximal sites for loci from the 2 ends of the BAC clone.
Figure 4.
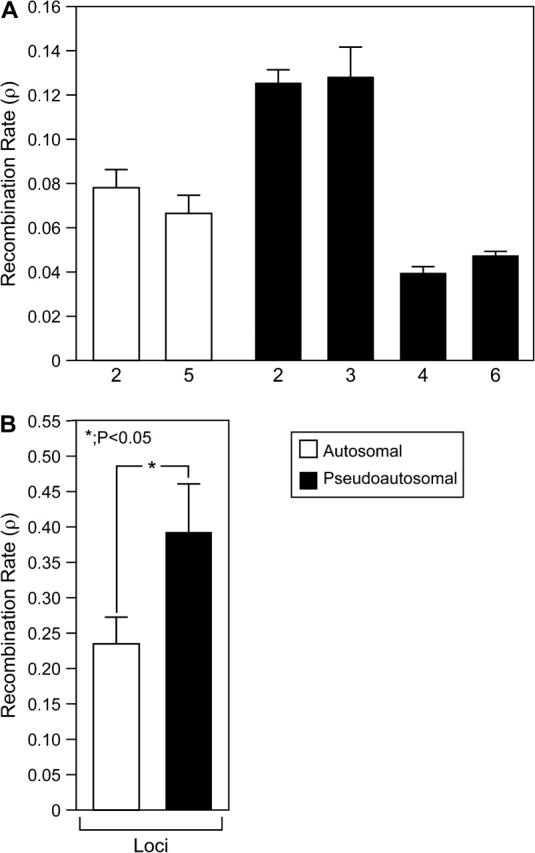
Recombination between (A) and within (B) ends of BAC inserts carrying either autosomal or pseudoautosomal loci from emu. Autosomal loci are represented by dark bars, and pseudoautosomal loci are represented by light bars.
Analysis of scaled recombination rate (r) of loci within BAC ends and subclones using LAMARC also suggested higher recombination rates within pseudoautosomal than autosomal loci (pseudoautosomal, 3.58 × 10−1 [95% CI, 0.237–0.508]; autosomal, 2.25 × 10−1 [95% CI, 0.12–0.378]). LAMARC tests of population growth did not yield consistent patterns across autosomal and pseudoautosomal loci. Tested independently, both autosomal and pseudoautosomal groups of loci yielded evidence of population growth (G = 19.38 ± 4.21 and 57.15 ± 28.78, respectively). However, when combined, all loci yield a negative G (−17.94 ± 0.84).
LD matrices (Figure 5) reveal slightly lower LD in pseudoautosomal loci (4.2 ± 0.2 × 10−1) than autosomal loci (4.7 ± 0.5 × 10−1) and greater LD between sites within loci than between sites on pairs of loci linked on a single BAC. In BAC inserts carrying either autosomal or pseudoautosomal loci, R2 significantly or nearly significantly declined as physical distance between sites increased (autosomes-AL2: P < 0.001 and AL5: P < 0.001; PAR–PAR2: P = 0.06, PAR3: P < 0.001, PAR4: P < 0.01, and PAR6: P < 0.001; Figure 6).
Figure 5.
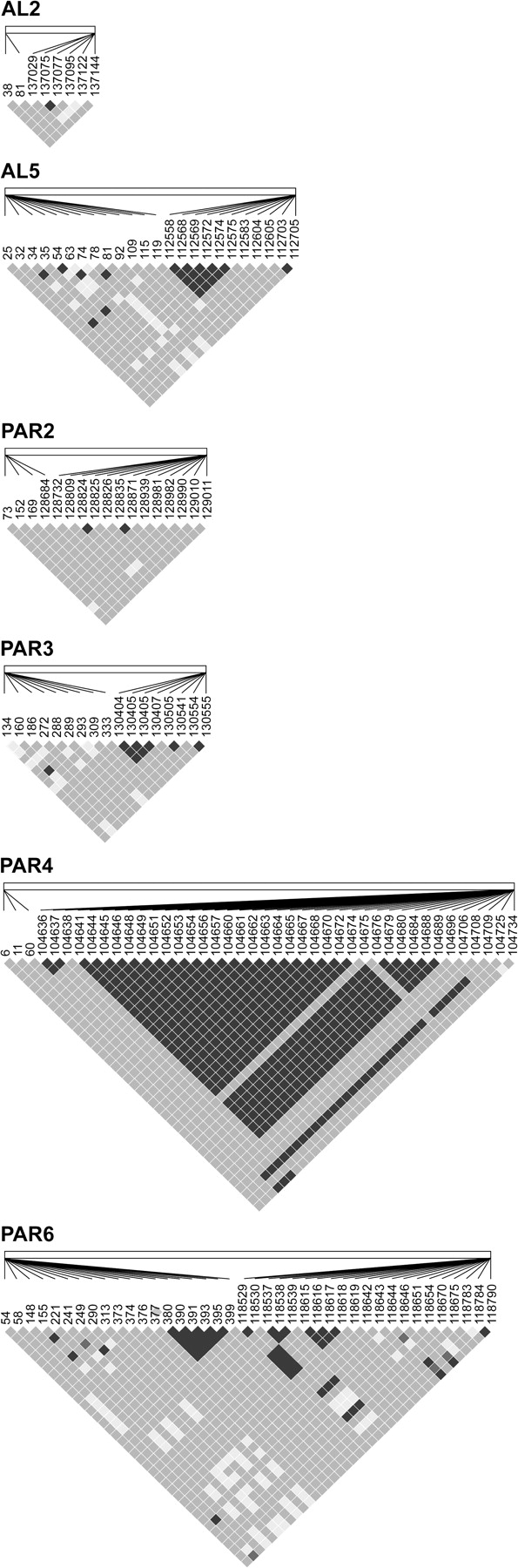
LD matrices across 2 emu BAC inserts containing autosomal sequence (AL2 and AL5) and 4 inserts containing pseudoautosomal sequences (PAR2, PAR3, PAR4, and PAR6). Color of squares represents paired site comparison details (Black: D′ = 1, log of odds (LOD) > 2; dark gray: D′ = 1, LOD < 2; and light gray: D′ < 1, LOD < 2). Linkage disequilibrium is measured between 2 sites either within or between ends of BAC inserts. Lines connect the sites to their position within the insert.
Figure 6.

LD (R2) across emu BAC inserts. On BAC inserts containing either autosomal or pseudoautosomal loci, linkage between paired sites significantly decreases as distance between sites increases. Data points below the left bracket on the x axis refer to paired sites on the same BAC end. Below the right bracket, data points refer to paired sites on loci at different ends of BAC clones. Dotted trendline indicates autosomal trend. Solid trendline indicates pseudoautosomal trend.
Lastly, the multilocus HKA test on all loci combined yielded a sum of deviations of 42.43 (P < 0.0001), indicating that, when measured together, diversity in both pseudoautosomal and autosomal loci deviates from neutral expectations and is not explained solely by mutation rate variation at these loci. When analyzed separately, both pseudoautosomal and autosomal sums of deviations rejected the neutral model (24.89; P < 0.001 and 12.74; P < 0.015, respectively).
Discussion
Our results suggest that levels of diversity in the PAR of the sex chromosomes and autosomes in the emu are indistinguishable. Across 8 autosomal and 14 pseudoautosomal loci, average nucleotide diversity (π) was not significantly lower in the PAR, consistent with the hypothesis that autosomal and pseudoautosomal population genetics are similar. Additionally, other measured demographic and mutational variables that could differentially affect diversity in sex-specific regions did not differ between autosomal and pseudoautosomal loci in this study. We conclude that, if any difference in population genetics distinguishes the PAR from autosomes, it is that the PAR is characterized by slightly higher rates of recombination and slightly lower LD than autosomal loci and that both genomic compartments appear to depart from a standard neutral model of a single population, as measured by the multilocus HKA test.
Although the overall size (4.82 kb) of our data set is small in terms of total sequence length compared with other population genetics studies of avian sex chromosomes (e.g., Berlin and Ellegren 2004, 2006) and might be construed as limited when describing whole chromosomal regions, the number of loci (22) in the data set is relatively large. The sampling strategy of our study followed Felsenstein (2006) and Carling and Brumfield (2007) by sampling many smaller loci as opposed to the more common practice of sampling a few, large loci. For measuring diversity, for example, Felsenstein (2006) suggests sampling more smaller loci rather than a few large loci, given a constraint on the total number of bases sequenced per individual. We sought to test this approach by additional analysis of our data set. Using MIGRATE (Beerli and Felsenstein 2001), we analyzed subsets of our data to see whether total amount of sequence or the number of independently segregating loci produced more confident estimates of diversity (θ). We found that the number of independent loci was a more precise estimator of θ, with a smaller variance in the estimate than the total sequence length. For example, variance in estimates of θ decreased as the number of analyzed autosomal loci increased. A similar inverse relationship between variance in estimates of θ and number of analyzed loci was also seen in pseudoautosomal loci. Increasing length of analyzed sequences, however, did not decrease variance (Supplementary Figure 1). Thus, aside from sampling longer sequences for each of our 22 loci, our strategy of sampling many short loci appears superior to sampling a smaller number of longer loci, at least when describing overall diversity.
Reduced recombination can facilitate reductions in diversity, as seen on the neo-Y chromosome of Drosophila americana (McAllister and Charlesworth 1999), by enhancing the effect of selective sweeps, either due to directional or background selection. The high LD reported for Z-linked genes using widely spaced microsatellites in collared flycatchers is consistent with absence of recombination in the Z-specific region. A problem in comparing recombination within sex chromosomes between different species is that demography has been shown to influence estimates of recombination rates and LD in humans (Wall 2001). Population structure and bottlenecks cause underestimates of recombination, whereas population growth causes overestimates. In this case, population growth does not seem to have had a significant effect on recombination because individual loci exhibit sufficient variation in growth (G) to produce varying results, depending on whether autosomal and pseudoautosomal loci are tested separately or together. Our analysis of population structure suggests that demography has had a weak or inconsistent influence on the population genetics of the emu. This suggestion is not surprising given the widespread accounts of emus throughout Australia. Emus are typically inland, semiarid birds with many widely distributed breeding populations (Barrett et al. 2003).
The loci compared in this study do not appear to be telomeric or centromeric as judged by our FISH results (Figure 1). This is relevant because nucleotide diversity may also be influenced by chromosomal position. Andolfatto and Wall (2003) reported significantly less LD near the telomere of the X chromosome in Drosophila melanogaster, a pattern also found in mammals (Mikkelsen et al. 2007). Also, recombination rates tend to decrease near centromeres of metacentric chromosomes (Nachman 2002).
However, demography is not expected to influence the PAR and autosomal loci differently because of their equivalent population sizes (Bachtrog and Andolfatto 2006). In this study, recombination rates were not significantly different over long distances (>100 kb) between autosomal and pseudoautosomal loci (Figure 4), although recombination rates were significantly higher in the PAR when measured within loci rather than between them. Such a result would not be surprising because recombination rate in human PAR1 is ∼20-fold greater than the human genomic average rate (Lien et al. 2000; Chen et al. 2006). The emu PAR is proportionally much larger than the human PAR1, so a less significant difference between pseudoautosomal and average genomic recombination is expected.
Similarly, we found that LD in both autosomal and PARs diminished detectably and at similar rates over spans of sequence greater than 138 kb long (Figure 6). This is the first report of a relationship between LD and physical distance on the autosomes of a wild bird, adding to a report of such a relationship over much longer physical distances on the Z chromosomes of flycatchers (Backstrom, Ovarnstrom, et al. 2006). Evidence of high LD was reported from a study of microsatellites in chickens but domestication has probably altered the signatures of selection and naturally occurring recombination in this species (Heifetz et al. 2005). The similarity in rates of decay of LD with distance is another way in which the population dynamics of emu PAR and autosomes are indistinguishable.
Another finding was the significant result from the HKA test for evidence of a departure from a standard neutral model in both autosomal and pseudoautosomal loci. This could indicate that selection influences nucleotide diversity in the emu PAR, but the precise form of selection (directional or balancing) is unclear without a panel of unambiguously neutral loci to compare with the PAR. Similar evidence of selection in autosomal loci suggests a systematic influence on loci that may be demographic. Our results suggest no significant differences in demography between autosomal and pseudoautosomal loci, and demography may be driving the signal of departure from demographic equilibrium we report in both groups of loci.
By examining nucleotide diversity in PAR of sex chromosomes at an early stage of divergence, we have confirmed conclusions from other work that Z- or W-specific, but not pseudoautosomal, loci may be under systematically different selective forces than those experienced by autosomes. Our results establish a system within which experiments can be designed to test influences of chromosomal location and function on diversity, recombination, and LD within and between emu chromosomes. For example, if function rather than chromosomal location primarily influences diversity in loci that are hemizygous in some species but autosomal or pseudoautosomal in others, then a similar reduction in diversity should be seen in homologs from species that lack sex chromosomes, a hypothesis that can be tested using species representing the diversity of Z–W differences found in birds and reptiles.
Supplementary Material
Supplementary Figure 1 and Table 1 can be found at http://www.jhered.oxfordjournals.org/.
Funding
National Institutes of Health National Research Service Award to D.E.J.; National Science Foundation (DEB-0315806) to S.V.E.
Supplementary Material
Acknowledgments
This manuscript greatly benefited from critical suggestions from Hans Ellegren and an anonymous reviewer. Tissue samples were provided by the American Museum of Natural History, the Australian Museum, the Commonwealth Scientific and Industrial Research Organization, the National Museum of Natural History, the South Australian Museum, the Australian National Wildlife Collection, and Louisiana State University. Samples were shipped to D.E.J. under United States Department of Agriculture's Animal and Plant Health Inspection Service Permit #54119. Bacterial artificial chromosomes were fingerprinted at Washington University by Wesley Warren and William Courtney. All work was approved under Animal Welfare Assurance #A3593-01 provided by the university's Standing Committee on the Use of Animals in Research and Teaching.
References
- Andolfatto P, Wall JD. Linkage disequilibrium patterns across a recombination gradient in African Drosophila melanogaster. Genetics. 2003;165:1289–1305. doi: 10.1093/genetics/165.3.1289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aquadro CF, Begun DJ, Kindahl EC. Selection, recombination and DNA polymorphism in Drosophila. In: Golding B, editor. Non-neutral evolution: theories and molecular data. New York: Chapman and Hall; 1994. pp. 46–56. [Google Scholar]
- Awadalla P, Charlesworth D. Recombination and selection at Brassica self-incompatibility loci. Genetics. 1999;152:413–425. doi: 10.1093/genetics/152.1.413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Awadalla P, Eyre-Walker A, Smith JM. Linkage disequilibrium and recombination in hominid mitochondrial DNA. Science. 1999;286:2524–2525. doi: 10.1126/science.286.5449.2524. [DOI] [PubMed] [Google Scholar]
- Axelsson E, Smith NGC, Sundstrom H, Berlin S, Ellegren H. Male-biased mutation rate and divergence in autosomal, Z-linked and W-linked introns of chicken and turkey. Mol Biol Evol. 2004;21:1538–1547. doi: 10.1093/molbev/msh157. [DOI] [PubMed] [Google Scholar]
- Bachtrog D, Andolfatto P. Selection, recombination and demographic history in Drosophila miranda. Genetics. 2006;174:2045–2059. doi: 10.1534/genetics.106.062760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bachtrog D, Charlesworth B. Reduced levels of microsatellite variability on the neo-Y chromosome of Drosophila miranda. Curr Biol. 2000;10:1025–1031. doi: 10.1016/s0960-9822(00)00656-4. [DOI] [PubMed] [Google Scholar]
- Backstrom N, Brandstrom M, Gustafsson L, Qvarnstrom A, Cheng H, Ellegren H. Genetic mapping in a natural population of collared flycatchers (Ficedula albicollis): conserved synteny but gene order rearrangements on the avian Z chromosome. Genetics. 2006;174:377–386. doi: 10.1534/genetics.106.058917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Backstrom N, Ovarnstrom A, Gustafsson L, Ellegren H. Levels of linkage disequilibrium in a wild bird population. Biol Lett. 2006;2:435–438. doi: 10.1098/rsbl.2006.0507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrett G, Silcocks A, Barry S, Cunningham R, Poulter R. The new atlas of Australian birds. Victoria (Australia): Royal Australian Ornithologists Union; 2003. [Google Scholar]
- Barrett JC, Fry B, Maller J, Daly MJ. Haploview: analysis and visualization of LD and haplotype maps. Bioinformatics. 2005;21:263–265. doi: 10.1093/bioinformatics/bth457. [DOI] [PubMed] [Google Scholar]
- Beerli P. Comparison of Bayesian and maximum likelihood inference of population genetic parameters. Bioinformatics. 2006;22:341–345. doi: 10.1093/bioinformatics/bti803. [DOI] [PubMed] [Google Scholar]
- Beerli P, Felsenstein J. Maximum likelihood estimation of a migration matrix and effective population sizes in n subpopulations by using a coalescent approach. Proc Natl Acad Sci USA. 2001;98:4563–4568. doi: 10.1073/pnas.081068098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berlin S, Ellegren H. Chicken W: a genetically uniform chromosome in a highly variable genome. Proc Natl Acad Sci USA. 2004;101:15967–15969. doi: 10.1073/pnas.0405126101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berlin S, Ellegren H. Fast accumulation of nonsynonymous mutations on the female-specific W chromosome in birds. J Mol Evol. 2006;62:66–72. doi: 10.1007/s00239-005-0067-6. [DOI] [PubMed] [Google Scholar]
- Borge T, Webster MT, Andersson G, Saetre GP. Contrasting patterns of polymorphism and divergence on the Z chromosome and autosomes in two Ficedula flycatcher species. Genetics. 2005;171:1861–1873. doi: 10.1534/genetics.105.045120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carling MD, Brumfield RT. Gene sampling strategies for multi-locus population estimates of genetic diversity. PLoS One. 2007;2:e160. doi: 10.1371/journal.pone.0000160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charlesworth B. The evolution of chromosomal sex determination and dosage compensation. Curr Biol. 1996;6:149–162. doi: 10.1016/s0960-9822(02)00448-7. [DOI] [PubMed] [Google Scholar]
- Charlesworth B, Coyne JA, Barton NH. The relative rates of evolution of sex-chromosomes and autosomes. Am Nat. 1987;130:113–146. [Google Scholar]
- Chen JF, Lu F, Chen SS, Tao SH. Significant positive correlation between the recombination rate and GC content in the human pseudoautosomal region. Genome. 2006;49:413–419. doi: 10.1139/g05-124. [DOI] [PubMed] [Google Scholar]
- Corander J, Marttinen P. Bayesian identification of admixture events using multilocus molecular markers. Mol Ecol. 2006;15:2833–2843. doi: 10.1111/j.1365-294X.2006.02994.x. [DOI] [PubMed] [Google Scholar]
- de Kloet RS, de Kloet SR. Evolution of the spindlin gene in birds: independent cessation of the recombination of sex chromosomes at the spindlin locus in neognathous birds and tinamous, a paleognathous avian family. Genetica. 2003;119:333–342. doi: 10.1023/b:gene.0000003842.72339.df. [DOI] [PubMed] [Google Scholar]
- Edwards SV, Dillon M. Hitchhiking and recombination in birds: evidence from Mhc-linked and unlinked loci in Red-winged Blackbirds (Agelaius phoeniceus) Genet Res. 2004;84:175–192. doi: 10.1017/s0016672304007189. [DOI] [PubMed] [Google Scholar]
- Ellegren H. Levels of polymorphism on the sex-limited chromosome: a clue to Y from W? Bioessays. 2003;25:163–167. doi: 10.1002/bies.10228. [DOI] [PubMed] [Google Scholar]
- Ellis N, Taylor A, Bengtsson BO, Kidd J, Rogers J, Goodfellow P. Population-structure of the human pseudoautosomal boundary. Nature. 1990;344:663–665. doi: 10.1038/344663a0. [DOI] [PubMed] [Google Scholar]
- Eyre-walker A. Recombination and mammalian genome evolution. Proc R Soc Lond B Biol Sci. 1993;252:237–243. doi: 10.1098/rspb.1993.0071. [DOI] [PubMed] [Google Scholar]
- Ezaz T, Quinn AE, Miura I, Sarre SD, Georges A, Graves JAM. The dragon lizard Pogona vitticeps has ZZ/ZW micro-sex chromosomes. Chromosome Res. 2005;13:763–776. doi: 10.1007/s10577-005-1010-9. [DOI] [PubMed] [Google Scholar]
- Felsenstein J. Accuracy of coalescent likelihood estimates: do we need more sites, more sequences, or more loci? Mol Biol Evol. 2006;23:691–700. doi: 10.1093/molbev/msj079. [DOI] [PubMed] [Google Scholar]
- Fridolfsson AK, Ellegren H. Molecular evolution of the avian CHD1 genes on the Z and W sex chromosomes. Genetics. 2000;155:1903–1912. doi: 10.1093/genetics/155.4.1903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu YX, Li WH. Statistical tests of neutrality of mutations. Genetics. 1993;133:693–709. doi: 10.1093/genetics/133.3.693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Moreno J, Mindell DP. Rooting a phylogeny with homologous genes on opposite sex chromosomes (gametologs): a case study using avian CHD. Mol Biol Evol. 2000;17:1826–1832. doi: 10.1093/oxfordjournals.molbev.a026283. [DOI] [PubMed] [Google Scholar]
- Graves JAM, Wakefield MJ, Toder R. The origin and evolution of the pseudoautosomal regions of human sex chromosomes. Hum Mol Genet. 1998;7:1991–1996. doi: 10.1093/hmg/7.13.1991. [DOI] [PubMed] [Google Scholar]
- Handley LJL, Berset-Brandli L, Perrin N. Disentangling reasons for low Y chromosome variation in the greater white-toothed shrew (Crocidura russula) Genetics. 2006;173:935–942. doi: 10.1534/genetics.105.050203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heifetz EM, Fulton JE, O'Sullivan N, Zhao H, Dekkers JCM, Soller M. Extent and consistency across generations of linkage disequilibrium in commercial layer chicken breeding populations. Genetics. 2005;171:1173–1181. doi: 10.1534/genetics.105.040782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hudson RR, Kreitman M, Aguade M. A test of neutral molecular evolution based on nucleotide data. Genetics. 1987;116:153–159. doi: 10.1093/genetics/116.1.153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itoh Y, Kampf K, Arnold AP. Comparison of the chicken and zebra finch Z chromosomes shows evolutionary rearrangements. Chromosome Res. 2006;14:805–815. doi: 10.1007/s10577-006-1082-1. [DOI] [PubMed] [Google Scholar]
- Janes DE, Ezaz T, Graves JAM, Edwards SV Forthcoming 2008. Characterization, chromosomal location, and genomic neighborhood of a ratite ortholog of a gene with gonadal expression in mammals. Integr Comp Biol. doi: 10.1093/icb/icn024. doi:10.1093/icb/icn024. [DOI] [PubMed] [Google Scholar]
- Kuhner MK. LAMARC 2.0: maximum likelihood and Bayesian estimation of population parameters. Bioinformatics. 2006;22:768–770. doi: 10.1093/bioinformatics/btk051. [DOI] [PubMed] [Google Scholar]
- Kuhner MK, Smith LP. Comparing likelihood and Bayesian coalescent estimation of population parameters. Genetics. 2007;175:155–165. doi: 10.1534/genetics.106.056457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhner MK, Yamato J, Felsenstein J. Maximum likelihood estimation of recombination rates from population data. Genetics. 2000;156:1393–1401. doi: 10.1093/genetics/156.3.1393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lien S, Szyda J, Schechinger B, Rappold G, Arnheim N. Evidence for heterogeneity in recombination in the human pseudoautosomal region: high resolution analysis by sperm typing and radiation-hybrid mapping. Am J Hum Genet. 2000;66:557–566. doi: 10.1086/302754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu HB, Liu K, Wang JF, Ma RLZ. A BAC clone-based physical map of ovine major histocompatibility complex. Genomics. 2006;88:88–95. doi: 10.1016/j.ygeno.2006.02.006. [DOI] [PubMed] [Google Scholar]
- Mank JE, Ellegren H. Parallel divergence and degradation of the avian W sex chromosome. Trends Ecol Evol. 2007;22:389–391. doi: 10.1016/j.tree.2007.05.003. [DOI] [PubMed] [Google Scholar]
- McAllister BF, Charlesworth B. Reduced sequence variability on the neo-Y chromosome of Drosophila americana americana. Genetics. 1999;153:221–233. doi: 10.1093/genetics/153.1.221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McVean G, Awadalla P, Fearnhead P. A coalescent-based method for detecting and estimating recombination from gene sequences. Genetics. 2002;160:1231–1241. doi: 10.1093/genetics/160.3.1231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meiklejohn CD, Kim Y, Hartl DL, Parsch J. Identification of a locus under complex positive selection in Drosophila simulans by haplotype mapping and composite-likelihood estimation. Genetics. 2004;168:265–279. doi: 10.1534/genetics.103.025494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meunier J, Eyre-Walker A. The correlation between linkage disequilibrium and distance: implications for recombination in hominid mitochondria. Mol Biol Evol. 2001;18:2132–2135. doi: 10.1093/oxfordjournals.molbev.a003756. [DOI] [PubMed] [Google Scholar]
- Mikkelsen TS, Wakefield MJ, Aken B, et al. Genome of the marsupial Monodelphis domestica reveals innovation in non-coding sequences. Nature. 2007;447:167–178. doi: 10.1038/nature05805. [DOI] [PubMed] [Google Scholar]
- Myers S, Bottolo L, Freeman C, McVean G, Donnelly P. A fine-scale map of recombination rates and hotspots across the human genome. Science. 2005;310:321–324. doi: 10.1126/science.1117196. [DOI] [PubMed] [Google Scholar]
- Nachman MW. Variation in recombination rate across the genome: evidence and implications. Curr Opin Genet Dev. 2002;12:657–663. doi: 10.1016/s0959-437x(02)00358-1. [DOI] [PubMed] [Google Scholar]
- Nisbet ICT, Hatch JJ. Consequences of a female-biased sex-ratio in a socially monogamous bird: female-female pairs in the Roseate Tern Sterna dougallii. Ibis. 1999;141:307–320. [Google Scholar]
- Ogawa A, Murata K, Mizuno S. The location of Z- and W-linked marker genes and sequence on the homomorphic sex chromosomes of the Ostrich and the Emu. Proc Natl Acad Sci USA. 1998;95:4415–4418. doi: 10.1073/pnas.95.8.4415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pereira SL, Baker AJ. A mitogenomic timescale for birds detects variable phylogenetic rates of molecular evolution and refutes the standard molecular clock. Mol Biol Evol. 2006;23:1731–1740. doi: 10.1093/molbev/msl038. [DOI] [PubMed] [Google Scholar]
- Pigozzi MI, Solari AJ. The ZW pairs of two paleognath birds from two orders show transitional stages of sex chromosome differentiation. Chromosome Res. 1999;7:541–551. doi: 10.1023/a:1009241528994. [DOI] [PubMed] [Google Scholar]
- Posada D, Crandall KA. MODELTEST: testing the model of DNA substitution. Bioinformatics. 1998;14:817–818. doi: 10.1093/bioinformatics/14.9.817. [DOI] [PubMed] [Google Scholar]
- Pritchard JK, Stephens M, Donnelly P. Inference of population structure using multilocus genotype data. Genetics. 2000;155:945–959. doi: 10.1093/genetics/155.2.945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reich DE, Schaffner SF, Daly MJ, et al. Human genome sequence variation and the influence of gene history, mutation and recombination. Nat Genet. 2002;32:135–142. doi: 10.1038/ng947. [DOI] [PubMed] [Google Scholar]
- Rice WR. Evolution of the Y sex chromosome in animals. Bioscience. 1996;46:331–343. [Google Scholar]
- Rozas J, Sanchez-DelBarrio JC, Messeguer X, Rozas R. DnaSP, DNA polymorphism analyses by the coalescent and other methods. Bioinformatics. 2003;19:2496–2497. doi: 10.1093/bioinformatics/btg359. [DOI] [PubMed] [Google Scholar]
- Saetre GP, Borge T, Lindroos K, et al. Sex chromosome evolution and speciation in Ficedula flycatchers. Proc R Soc Lond B Biol Sci. 2003;270:53–59. doi: 10.1098/rspb.2002.2204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shedlock AM, Janes DE, Edwards SV. Amniote phylogenomics: testing evolutionary hypotheses with BAC library scanning and targeted clone analysis of large-scale DNA sequences from reptiles. In: Murphy W, editor. Phylogenomics. Totowa, NJ: Humana Press; 2008. [DOI] [PubMed] [Google Scholar]
- Shetty S, Griffin DK, Graves JAM. Comparative painting reveals strong chromosome homology over 80 million years of bird evolution. Chromosome Res. 1999;7:289–295. doi: 10.1023/a:1009278914829. [DOI] [PubMed] [Google Scholar]
- Shetty S, Kirby P, Zarkower D, Graves JAM. DMRT1 in a ratite bird: evidence for a role in sex determination and discovery of a putative regulatory element. Cytogenet Genome Res. 2002;99:245–251. doi: 10.1159/000071600. [DOI] [PubMed] [Google Scholar]
- Stephens M, Smith NJ, Donnelly P. A new statistical method for haplotype reconstruction from population data. Am J Hum Genet. 2001;68:978–989. doi: 10.1086/319501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephens M, Smith NJ, Donnelly P. Documentation for PHASE version 2.1. Seattle (WA): University of Washington; 2004. [Google Scholar]
- Sundstrom H, Webster MT, Ellegren H. Reduced variation on the Chicken Z chromosome. Genetics. 2004;167:377–385. doi: 10.1534/genetics.167.1.377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tajima F. Statistical-method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics. 1989;123:585–595. doi: 10.1093/genetics/123.3.585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson JD, Gibson TJ, Plewniak F, Jeanmougin F, Higgins DG. The CLUSTAL_X windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res. 1997;25:4876–4882. doi: 10.1093/nar/25.24.4876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toder R, Graves JAM. CSF2RA, ANT3, and STS are autosomal in marsupials: implications for the origin of the pseudoautosomal region of mammalian sex chromosomes. Mamm Genome. 1998;9:373–376. doi: 10.1007/s003359900772. [DOI] [PubMed] [Google Scholar]
- Tsuda Y, Nishida-Umehara C, Ishijima J, Yamada K, Matsuda Y. Comparison of the Z and W sex chromosomal architectures in elegant crested tinamou (Eudromia elegans) and ostrich (Struthio camelus) and the process of sex chromosome differentiation in palaeognathous birds. Chromosoma. 2007;116:159–173. doi: 10.1007/s00412-006-0088-y. [DOI] [PubMed] [Google Scholar]
- van Tuinen M, Sibley CG, Hedges SB. Phylogeny and biogeography of ratite birds inferred from DNA sequences of the mitochondrial ribosomal genes. Mol Biol Evol. 1998;15:370–376. doi: 10.1093/oxfordjournals.molbev.a025933. [DOI] [PubMed] [Google Scholar]
- Wakeley J, Lessard S. Theory of the effects of population structure and sampling on patterns of linkage disequilibrium applied to genomic data from humans. Genetics. 2003;164:1043–1053. doi: 10.1093/genetics/164.3.1043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wall JD. Insights from linked single nucleotide polymorphisms: what we can learn from linkage disequilibrium. Curr Opin Genet Dev. 2001;11:647–651. doi: 10.1016/s0959-437x(00)00248-3. [DOI] [PubMed] [Google Scholar]
- Webster MT, Axelsson E, Ellegren H. Strong regional biases in nucleotide substitution in the chicken genome. Mol Biol Evol. 2006;23:1203–1216. doi: 10.1093/molbev/msk008. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.