

Abstract
Many genes involved in immunity evolve rapidly. It remains unclear, however, to what extent pattern-recognition receptors (PRRs) of the innate immune system in vertebrates are subject to recurrent positive selection imposed by pathogens, as suggested by studies in Drosophila, or whether they are evolutionarily constrained. Here, we show that Toll-like receptor 5 (TLR5), a member of the Toll-like receptor family of innate immunity genes that responds to bacterial flagellin, has undergone a history of adaptive evolution in primates. We have identified specific residues that have changed multiple times, sometimes in parallel in primates, and are thus likely candidates for selection. Most of these changes map to the extracellular leucine-rich repeats involved in pathogen recognition, and some are likely to have an effect on protein function due to the radical nature of the amino acid substitutions that are involved. These findings suggest that vertebrate PRRs might show similar patterns of evolution to Drosophila PRRs, in spite of the acquisition of the more complex and specific vertebrate adaptive immune system. At shorter timescales, however, we found no evidence of adaptive evolution in either humans or chimpanzees. In fact, we found that one mutation that abolishes TLR5 function is present at high frequencies in many human populations. Patterns of variation indicate that this mutation is not young, and its high frequency suggests some functional redundancy for this PRR in humans.
Keywords: Toll-like receptor 5, TLR5, adaptive evolution, dN/dS, premature stop codon, TLR5392STOP
Introduction
Vertebrate immune systems include acquired and innate components. Pattern-recognition receptors (PRRs) are an essential component of the innate immune system. PRRs recognize and bind pathogen-associated molecular patterns (PAMPs), conserved molecular motifs that are shared by infectious agents but which are absent in the host. The interaction between PRRs and PAMPs illustrates two fundamental aspects of the innate immune system: 1) the ability to discriminate between self and nonself and 2) the targeting of components essential for microbial fitness, which are therefore functionally constrained (Medzhitov and Janeway 1997). Toll-like receptors (TLRs) constitute the best characterized PRRs of the innate immune system of vertebrates, and so far, 10 have been described in humans (Akira and Takeda 2004). After stimulation with their ligands, TLRs form homo- or heterodimers and trigger intracellular signaling cascades that induce the expression of a variety of genes. This in turn leads to the activation of innate immunity effector mechanisms as well as the development of adaptive immunity (Akira and Takeda 2004).
Because TLRs interact with microbial invaders, theory predicts that over evolutionary time they may be engaged in coevolutionary arms races with their microbial ligands. Recent results from the comparison of several Drosophila genomes support this hypothesis, showing that among innate immunity loci, PRRs constitute a functional class that evolves quickly between species (Sackton et al. 2007). It remains unclear whether vertebrates and invertebrates are similar in this respect. On the other hand, given the extremely conserved nature of the molecular patterns targeted by TLRs, they might be evolutionarily inflexible. In fact, they are often cited as an example of evolutionary conservation due to the fundamental constraint imposed by the inability of pathogens to tolerate mutations in molecular motifs that are essential to their fitness (Medzhitov and Janeway 1997).
Here, we attempt to distinguish between these two competing hypotheses using the evolution of Toll-like receptor 5 (TLR5) in primates as an example. So far, the limited evidence about the patterns of molecular evolution of TLRs in primates is inconclusive. Although Ortiz et al. (2008) claimed that all TLRs, except for TLR1, have evolved under purifying selection in primates, Nakajima et al. (2008) using a more extensive phylogenetic sampling suggested that TLR4 has been under positive selection in Old World monkeys.
TLR5 targets monomeric flagellin, the main component of the bacterial flagella and a potent virulence factor (Hayashi et al. 2001; Ramos et al. 2004). Recently, Andersen-Nissen et al. (2005) showed that members of the α and ϵ Proteobacteria that are important human pathogens, such as Campylobacter jejuni and Helicobacter pylori, are able to evade TLR5 recognition by mutating key residues in the TLR5 recognition site. These mutations abolish flagellar motility, but the pathogens acquire compensatory mutations in other parts of the flagellin molecule that restore motility, which is essential for efficient infectivity. These results demonstrate that pathogens can evolve to evade PRR recognition while remaining fully functional and capable of infection. More importantly, these findings suggest opportunities for coevolution between PRRs and their microbial ligands, in spite of some overall functional constraint.
Additional motivation for studying PRRs in primates comes from ideas concerning the relationship between mating systems and disease risk. Based on the finding that the basal number of white blood cells in primates and carnivores is correlated with the degree of sexual promiscuity, but not with other life-history traits expected to influence disease risk, Nunn and colleagues proposed the controversial idea that mating system drives the evolution of the immune system (Nunn et al. 2000, 2003; Nunn 2002). The underlying hypothesis is that in promiscuous species, the increased risk of acquiring sexually transmitted diseases has resulted in the evolution of stronger immune systems. This hypothesis has not been broadly tested at the molecular level. As a secondary goal, we take advantage of the variation in mating system among primate species to test predictions of this hypothesis.
A final motivation for studying TLR5 comes from association studies in humans, which showed that a premature stop codon (TLR5392STOP) was linked to susceptibility to Legionnaire's disease, a type of pneumonia produced by the flagellated bacterium Legionella pneumophila (Hawn et al. 2003) and resistance to two autoimmune disorders: Crohn's disease (Gewirtz et al. 2006) and Systemic Lupus Erythematosus (SLE) (Hawn et al. 2005). TLR5392STOP results in a loss of function, acts in a negative-dominant fashion, and has been reported to segregate in different populations at frequencies between 5% and 10% (Hawn et al. 2003, 2005; Gewirtz et al. 2006). Hawn et al. (2005) suggested that the high population frequency of TLR5392STOP might be due to an evolutionary advantage associated with defective TLR5-mediated signaling, at least in some situations. The “less is more” hypothesis proposed by Olson (1999) and Olson and Varki (2003) suggests that gene loss might be advantageous and an important engine of evolutionary change. This idea has received considerable attention recently in light of several reports of adaptive pseudogenizations in the human lineage (Tournamille et al. 1995; Ali et al. 1998; Wang et al. 2006; Seixas et al. 2007). The idea that TLR5 might be another case of adaptive gene loss in humans is intriguing because of its putative important immunologic function.
Here, we have analyzed the entire TLR5 coding sequence of 22 species of old and new world primates and apes in a phylogenetic framework, and surveyed sequence variation in both coding and noncoding regions in population samples of humans and chimpanzees to answer the following questions: 1) Has TLR5 undergone adaptive evolution in primates? 2) Is there any support for the promiscuity/disease-risk hypothesis in the rates of protein evolution across primates? 3) Are there signatures of positive selection in the patterns of nucleotide variation at TLR5 in humans and chimpanzees? and 4) Has the premature stop codon in humans increased in frequency due to recent positive selection? We found convincing evidence of positive selection at TLR5 throughout the primate phylogeny, involving amino acids that might mediate flagellin recognition, suggesting that innate immunity genes may experience some of the same evolutionary pressures previously described for adaptive immunity genes. Only four of six independent transitions to increased sexual promiscuity were associated with increased rates of protein evolution, arguing against the hypothesis that mating system plays a major role in TLR5 evolution. In humans and chimpanzees, patterns of DNA sequence variation are largely consistent with neutral expectations, suggesting that the relatively high frequency and widespread distribution of the human TLR5392STOP mutation might be a consequence of functional redundancy.
Materials and Methods
Samples
The species used in the phylogenetic analyses are shown in figure 1, and the origins of the samples are given in supplementary table 1, Supplementary Material online. Samples were collected in accordance with Institutional Animal Care and Use Committee guidelines. Additionally, coding sequences of Homo sapiens and Macaca mulatta were retrieved from GenBank (accession numbers NM_003268 and XM_001099501, respectively).
FIG. 1.—

Lineage-specific dN/dS values of TLR5 in primates (A) for the entire gene and (B) for the extracellular domain. Estimated dN/dS values from the branch-based model are shown above branches and the estimated number of nonsynonymous and synonymous changes are shown below branches. Branches with dN/dS values greater than 1 are shown in red. Mating systems categorized as “less promiscuous” (polygyny + monogamy) are indicated with a blue circle, whereas “more promiscuous” (promiscuous + dispersed) mating systems are indicated with a red circle. Arrows show the six unambiguous independent transitions between less and more promiscuous mating systems. For the Old World and New World monkey clades, “circled-pointed arrows” indicate additional transitions between low and high promiscuity according to alternative but equally parsimonious reconstructions.
DNA Samples from 19 Pan troglodytes verus, 3 Pan troglodytes troglodytes and 18 humans (9 Africans, 9 Europeans) from the Y-Chromosome Consortium DNA collection were provided by Dr. Michael Hammer at the University of Arizona. Human sequence data (24 African Americans and 23 European Americans) for two nonoverlapping fragments that together include ∼17 kb were gathered from the Innate Immunity Database (www.innateimmunity.net).
Nine hundred and fifty individuals from the Human Genome Diversity Panel (Cann et al. 1999, 2002) were used to estimate the worldwide frequency and geographic distribution of the TLR5392STOP mutation. This Human Genome Diversity Panel (HGDP) excludes samples previously identified as related individuals or duplicates (Rosenberg 2006).
DNA Amplification and Sequencing
The entire coding region of TLR5 (∼2.5 kb) was polymerase chain reaction (PCR)-amplified and sequenced from the 19 primate species listed above, using primers designed in conserved regions of published primate sequences. Together with the Macaca sequence from GenBank and the human and chimpanzee sequences (see below), the phylogenetic analyses included 22 species.
Two nonoverlapping genomic fragments were PCR amplified and sequenced from 18 humans (∼12 and ∼5 kb) to match similar gene regions available from the Innate Immunity Database (see below). A 5-kb fragment was also PCR amplified and sequenced in 19 P. t. verus and 3 P. t. troglodytes. In humans and chimpanzees, the sequenced regions contain the complete coding region as well as adjacent noncoding sequence.
PCR was performed in 25–50 μl reactions using Platinum Taq High Fidelity DNA Polymerase (Invitrogen, San Diego, CA). A complete list of amplification and sequencing primers for all fragments and the corresponding annealing temperatures and PCR protocols are provided in supplementary tables 2 and 3, Supplementary Material online. PCR products were purified using the Qiagen PCR purification kit (Qiagen, Valencia, CA) and sequenced using an ABI 3700 automated sequencer (Applied Biosystems, Foster City, CA). Sequences have been deposited in GenBank under the following accession numbers: Primates other than humans and chimpanzees: FJ542200–FJ542219; Chimpanzees: FJ546349–FJ546370; and Humans: FJ556974–FJ556991.
Sequence editing and assembly were performed using SEQUENCHER (Gene Codes, Ann Arbor, MI). DNA sequences were aligned using ClustalX (Thompson et al. 1997) with manual alignment of small indels using the amino acid sequence as a reference. Gametic phase was computationally determined using PHASE (Stephens et al. 2001).
Phylogeny-Based Tests of Selection
We tested for positive selection in the primate phylogeny by comparing the number of nonsynonymous substitutions per nonsynonymous site (dN) with the number of synonymous substitutions per synonymous site (dS) in a maximum likelihood (ML) framework. A ratio of dN/dS(ω) greater than one is usually taken as evidence of selection. We used the accepted primate phylogeny (Purvis 1995; Bininda-Emonds et al. 2007) in all analyses. We also used the TLR5 data to estimate phylogenetic relationships using Neighbor-Joining. The resulting tree was similar to the well-accepted primate phylogeny (Bininda-Emonds et al. 2007) with only four branches placed in slightly different positions. Analyses of selection using the TLRs tree yielded very similar results to those obtained using the accepted primate phylogeny, so we report only the latter below.
First, we evaluated selection at individual codons, not allowing variation among lineages. We ran a series of nested models implemented in PAML ver 4 (Yang 1997, 2007), in which the “neutral” models restrict ω to values ≤1, whereas “selection” models include a class of sites with dN/dS > 1. A likelihood ratio test (LRT) was then used to compare nested models (table 1). To check for convergence, all analyses were run twice, using initial ω values of 0.5 and 1.5. Amino acids under selection for model M8 were identified using a Bayes Empirical Bayes approach (BEB) (Yang et al. 2005). Two models of codon frequencies were used: F3x4 and F61.
Table 1.
| Model Categoryb | Models Comparedc | χ2d | dfe | P-Value | psf | ωselg |
| Site models | M1 versus M2 | 22.132 | 2 | <0.0001 | 0.029 | 4.55 |
| M7 versus M8 | 23.002 | 2 | <0.0001 | 0.034 | 4.54 | |
| M8a versus M8 | 22.796 | 1 | <0.0001 | |||
| Branch models | M0 versus Full | 31.115 | 40 | 0.842 |
Analysis using the F3x4 or F61 models of codon frequencies yielded virtually identical results; the results presented here refer to the F3x4 model.
LRTs were performed between nested models that allow variation in dN/dS among codons but not branches (“sites” models) or between models that allow variation among branches but not codons (“branch” models).
In the case of “site models,” we performed three comparisons, each involving a null model (M1, M7, and M8a) and a positive selection model (M2, M8). Specifically, we compared models M1 (two classes of sites with rates, ω0 < 1, ω1=1) versus M2 (three rates ω0 < 1, ω1=1, and ωsel > 1), and M7 (fit to a beta distribution, 10 rates) versus M8 (fit to a beta distribution with an extra rate that allows ωsel > 1) (Nielsen and Yang 1998; Yang et al. 2000). Additionally, the M8a model proposed by Swanson et al. (2003) was compared with M8.
−2lnΔL (where ΔL is the difference in likelihoods between the nested models) is distributed approximately as χ2.
df = Degrees of freedom, equal to the difference in the number of parameters between the models.
Proportion of the sites under selection.
Estimated dN/dS of the sites under selection.
A recent improvement in statistical methods to infer selection in a phylogenetic context is the incorporation of variation in the rate of synonymous substitution (Pond and Muse 2005). Kosakovsky Pond and Frost (2005) proposed a series of models to study selection on a codon basis. They classify previous methods as either “counting methods,” “random effect models,” or “fixed effect models.” Counting methods reconstruct the ancestral sequences to estimate the number of synonymous and nonsynonymous changes at each codon. Random effect models assume a distribution of rates across sites and then infer the rate at which individual sites evolve. Fixed effect models estimate the ratio of nonsynonymous-to-synonymous substitution on a site-by-site basis, without assuming a priori a distribution of rates across sites. Single likelihood ancestor counting (SLAC), random effects likelihood (REL), and fixed effects likelihood (FEL) methods, new versions of the “counting,” “random effect” and “fixed effect” models, respectively, which allow variation in the synonymous substitution rate (Kosakovsky Pond and Frost 2005), were implemented at the DATAMONKEY web server (Pond and Frost 2005).
Finally, to detect variation in ω among lineages, a model with one ω (M0) was compared with a “free-ratio” model that allows each branch to have a separate ω value while keeping variation among sites constant (Nielsen and Yang 1998; Yang 1998). Because a parameter-rich model does not necessarily fit the data better than simpler models, a model selection scheme was performed in DATAMONKEY to find the variable-branch model with the best fit to the data.
Parallel amino acid changes were inferred using maximum parsimony in MacClade (Sinauer Associates, Sunderland, MA).
Population Genetic Analyses and Tests of Selection
Nucleotide heterozygosity, π (Nei and Li 1979), and the proportion of segregating sites, θw (Waterson 1975), were estimated for the entire human and chimpanzee data sets, and also for different functional regions (coding, noncoding), and different human populations separately.
Tajima's D (Tajima 1989) and Fu and Li's D* (Fu and Li 1993) were calculated to assess whether the allele frequency spectrum deviates from neutral expectations. Coalescent simulations, conditioned on the observed number of segregating sites, were used to generate the null distributions of these test statistics. The ratio of nonsynonymous-to-synonymous polymorphisms in humans or chimpanzees was compared with the ratio of nonsynonymous-to-synonymous fixed differences with respect to the orangutan sequence (Mcdonald and Kreitman 1991). These analyses were performed using DnaSP (Rozas et al. 2003) and SITES (Hey and Wakeley 1997). To test for selection at putative regulatory regions as in Andolfatto (2005), we compared the ratio of polymorphism within humans with human–chimpanzee divergence at silent sites in the coding region and at two 1-kb regions directly upstream of two alternative human transcripts.
To study population structure in the chimpanzee data, 50,000 neutral genealogies of 38 chromosomes were simulated under panmixia using the program “ms” (Hudson 2002) using the observed level of variability and the recombination rate estimated from the data. To test for an excess of linkage disequilibrium (LD) due to admixture/population structure in chimpanzees, we computed the number of congruent sites (pb), defined as sites that determine only two haplotypes, and gd, defined as the maximum distance between any two congruent sites, using the script lbcalc (Garrigan et al. 2005). We then compared these values with the simulated distribution to calculate the probability of obtaining values more extreme than the observed ones.
To further evaluate the likelihood of gene flow between the chimpanzee subspecies, we fitted an isolation with migration model (Nielsen and Wakeley 2001; Hey and Nielsen 2004) using a Markov chain Monte Carlo method implemented in the program IMa (Hey and Nielsen 2007). Under this model, two populations split and diverge in isolation, with some level of gene flow. We used the largest nonrecombining region of the combined verus–troglodytes sample, which includes 1,660 bases of noncoding sequence, to run the program with a burn-in period of 2,000,000 steps using 15 chains with a geometric heating scheme. After the burn-in period, we ran the program for 15,399,385 steps, recording the results every 10 steps. We checked for convergence by comparing multiple runs.
Genotyping Assay
The TLR5392STOP mutation was genotyped by restriction analysis with DdeI in the HGDP as in Hawn et al. (2003).
Results
Positive Selection on the Extracellular Domain of Primate TLR5
We obtained the coding sequence of TLR5 for a relatively broad array of primates including New World primates, Old World primates and apes. To address whether specific codons in the protein have been subject repeatedly to positive directional selection in different species, we first investigated models in which the dN/dS ratio is allowed to vary among different classes of sites. LRTs showed that models that incorporate selection fit significantly better than neutral models (table 1). For model 8, the most stringent of the models implemented in PAML, a small proportion of the codons (3.4% or 29 codons) was estimated to be under selection, with a ω value of 4.34, of which 13 were identified by the BEB approach with posterior probabilities above 0.8 (table 2).
Table 2.
TLR5 Amino Acid Sites under Positive Selection Identified Using Different Methods
| AA positiona | Amino Acid Change (No. of Parallel Changes) | Protein Domainb | Maximum Likelihood Method |
Parallel Change | Uf, g | Clade g | |||
| PAML M8c | SLAC | FELd | RELe | ||||||
| 14 | Val-Met (2) | Signal P. | Yes | 0.986 | A, N | ||||
| 29 | Arg-Gln (2) | LRRNT | Yes | 1.045 | A, O | ||||
| 104 | Asp-Ser | LRR3 | 0.908 | 0.158 | 63 | * | A, O, amb. | ||
| Ser-Gly | 1.360 | ||||||||
| Ser-Asn | 2.053 | ||||||||
| 158 | Arg-His (3) | LRR5 | 0.173 | 22 | Yes | 1.317 | A, O, N | ||
| 168 | Lys-Glu (2) | LRR5 | Yes | 0.548 | N, amb | ||||
| [181] | Gln-Lys | LRR6 | 0.192 | 1.466 | O, N | ||||
| Gln-Arg | 1.045 | ||||||||
| [197] | Thr-Met (2) | LRR6 | Yes | 1.007 | O, N | ||||
| [207] | Asn-Ser (2) | LRR7 | Yes | 2.053 | A, N | ||||
| [230] | Thr-Ileu (2) | LRR8 | Yes | 0.750 | A, N | ||||
| [262] | His-Tyr (2) | LRR9 | Yes | 0.665 | O, N | ||||
| [268] | Gly-Ser or Gly-Thr | LRR9 | 0.883 | 1.36 or * | N, amb | ||||
| Ser-Thr | 2.49 | ||||||||
| [280] | Asn-Ser (2) | LRR9 | Yes | 2.053 | A, N | ||||
| [292] | His-Arg (3) | LRR10 | 0.925 | 0.102 | 86 | Yes | 1.317 | A, N, amb | |
| His-Leu or Arg-Leu | 0.56 or 0.414 | ||||||||
| [312] | Gln-Arg | LRR10 | 0.990 | 0.069 | 113 | Yes | 1.045 | A, N, amb | |
| Arg-Gly (2) | 0.534 | ||||||||
| Gln-Lys | 1.466 | ||||||||
| [354] | Ser-Trp or Ser-Leu | LRR12 | 0.972 | 28 | Yes | 0.375 or 0.725 | A, N, amb | ||
| Trp-Leu (2) | 0.793 | ||||||||
| Ser-Ala | 2.38 | ||||||||
| [363] | Ala-Thr (2) | LRR13 | Yes | 1.587 | N, amb | ||||
| [400] | His-Tyr (2) | LRR14 | Yes | 0.665 | O, amb | ||||
| 407 | Asp-Ala (2) | LRR15 | 0.897 | Yes | 0.657 | O, amb | |||
| Asp-Asn (1 or 2) | 1.015 | ||||||||
| 416 | Ala-Val (2) | LRR15 | Yes | 1.017 | N, amb | ||||
| 446 | Arg-Gln or Arg-Glu | LRR16 | 0.187 | 1.045 or * | N, amb | ||||
| Gln-Glu | 1.634 | ||||||||
| 460 | Leu-Phe | LRR17 | 0.185 | 0.732 | A | ||||
| 482 | Glu-Gly (3) | LRR18 | 0.185 | 22 | Yes | 0.553 | O | ||
| 492 | Glu-Gln (3) | LRR18 | 0.951 | Yes | 1.634 | A, O | |||
| Glu-Ala | 0.906 | ||||||||
| 496 | Asp-Asn (2) | LRR18 | Yes | 1.015 | A, O | ||||
| 523 | Ser-Lys (2 or 3) | LRR19 | 0.995 | 0.110 | 166 | Yes | * | O | |
| 530 | Gly-Arg (3) | LRR20 | 0.968 | 0.181 | 0.108 | 73 | Yes | 0.534 | A, O, N |
| Gly-Ala | 1.379 | ||||||||
| 564 | Asp-Asn (2) | LRR21 | Yes | 1.015 | A | ||||
| 567 | Leu-Val (3) | LRR21 | 0.971 | 0.160 | 36 | Yes | 1.329 | A, O, N | |
| Leu-Phe | 0.732 | ||||||||
| 586 | Glu-Ala | LRR22 | 0.172 | 22 | Yes | 0.906 | O | ||
| Ala-Thr (2) | 1.587 | ||||||||
| 592 | Asn-His | LRR22 | 0.906 | 1.382 | A, O | ||||
| Asn-Ser | 2.053 | ||||||||
| Asn-Lys | 1.075 | ||||||||
| His-Arg | 1.317 | ||||||||
| 616 | Leu-Phe (1–2) | LRRCT | Yes | 0.732 | A, amb | ||||
| 628 | Asp-Gly (2) | LRRCT | 0.924 | Yes | 0.548 | A, O, amb | |||
| Asp-Ala or Gly-Ala | 0.657 or 1.379 | ||||||||
| Ala-Val | 1.015 | ||||||||
| 634 | Ileu-Val (2) | LRRCT | Yes | 2.415 | N, amb | ||||
| 644 | Ileu-Val (2) | Transm. | Yes | 2.415 | O, N | ||||
| 650 | Val-Leu | Transm. | 0.153 | 1.329 | O | ||||
| 680 | Lys-Arg (2) | Intracel. | Yes | 1.583 | O | ||||
| 690 | Thr-Met (2) | Intracel. | Yes | 1.007 | O, amb | ||||
| 847 | Ser-Asn (3) | TIR | 0.916 | 0.130 | 74 | Yes | 2.053 | N, amb | |
| Asn-Asp | 1.015 | ||||||||
| 854 | Val-Ileu (2) | TIR | Yes | 2.415 | O, N | ||||
For the codons identified as candidates for positive selection, posterior probabilities, P values or Bayes factors are given (see below). Codons identified by more than one ML method are underlined.
Amino acids between brackets fall within the 228 amino acid region identified by Andersen-Nissen et al. (2007) as important for flagellin recognition.
Signal P = Signal Peptide, LLR = Leucine-rich repeat, NT = N-terminal, CT = C-terminal, Transm. = Transmembrane, Intracel. = Intracellular, and TIR = Toll-IL-1 Receptor Domain.
Posterior probabilities of the BEB analysis.
P values.
Bayes factor.
U is an empirically derived index that measures the likelihood of a nonsynonymous fixation (Tang et al. 2004). * indicates that more than 1-nt change is needed for the amino acid change.
Clade in which amino acid substitution occurred (O = Old World Monkeys, N = New World Monkeys, A = Apes and Humans, amb = more than one equally parsimonious reconstruction).
We then compared these results with those from methods that incorporate synonymous rate variation (table 2). Using significance thresholds of P < 0.2 for SLAC and FEL (consistent with a true Type I error rate of ∼5%, as suggested by Kosakovsky Pond and Frost 2005 and a Bayes factor >20 for REL [corresponding approximately to a P value of 0.05]) SLAC and FEL identified 1 and 14 codons, respectively, and REL identified 11 codons as targets of selection. Eleven codons (104, 158, 292, 312, 354, 482, 523, 530, 567, 586, and 847) were picked by at least two methods.
Although not independent from previous results, we also considered parallel amino acid changes (independent changes at the same codon position, from the same initial state to the same final state) as potential candidates for selection. At TLR5, 24 codon sites show parallel evolution in two lineages and eight sites have evolved in parallel in three lineages (table 2). Most of these do not fall at CpG sites (on either strand) and are thus not likely to be the product of mutational bias and/or increased mutation rate. Ten of the parallel changes (aa 158, 292, 312, 354, 482, 523, 530, 567, 586, and 847) correspond to sites that were identified by more than one ML method as targets of selection. Interestingly, parallel changes have not accumulated on specific branches but instead are relatively scattered across the primate phylogeny. A possible explanation for the high number of parallel changes is functional constraint due to the presence of many leucine-rich repeats in the extracellular domain. Such motifs typically contain a conserved 11 aa motif (LXXLXLXXNXL, where “L” is Leu, Ile, Val, or Phe; “N” is Asn; and X is any amino acid) and a variable region (Matsushima et al. 2007). In this case, all the parallel changes that occurred in the conserved portion of the LRRs involve “X” residues, suggesting that if functional constraint to maintain this motif exists, it does not seem to be responsible for the high number of parallel changes. We thus infer that selection might have played a role in driving these substitutions.
We investigated the radical or conservative nature of amino acid substitutions using U, an empirically derived universal index based on the genetic code that measures amino acid exchangeability during evolution (Tang et al. 2004) (table 2). In principle, more radical changes are more likely to affect function. U varies from 0.241 to 2.490 with lower values representing more radical (less common) changes (Tang et al. 2004). U is weakly correlated with other conventional measures, such as Grantham's distance, that determine amino acid exchangeability based on a combination of physicochemical properties such as volume and polarity (Grantham 1974), but it is a considerably better predictor of the observed pattern of amino acid substitution in a variety of taxa (Tang et al. 2004). Several sites show relatively radical amino acid changes (table 2).
Of the 11 sites that were identified by more than one ML method, amino acids 292, 312, 530, and 567 show the strongest evidence of selection because they were consistently identified by at least three ML methods, they show parallel changes, and they involve relatively radical amino acid changes. Particularly compelling is the evidence for selection on aa 530. This is the only site identified by all four ML methods, and it displays a radical change occurring in three independent lineages. Of the remaining seven sites, two deserve special attention. Site 354 involves a moderately radical change, and together with site 312, falls within the flagellin recognition domain (Andersen-Nissen et al. 2007). Site 847 also shows the same amino acid transition in three independent instances and is located in the very conserved TIR signaling domain.
Disease Risk and Mating System
Having shown that TLR5 evolution in primates is consistent with recurrent positive selection, we were interested in looking for heterogeneity in rates of protein evolution among different lineages and in investigating whether these differences were correlated with reported levels of sexual promiscuity. The best-fit model that allows variation in dN/dS among lineages grouped branches under four different rates: ω = 3.13, ω = 0.51, ω = 0.25, and ω = 0.06. The full model, which assigns a different rate to each branch, had a higher likelihood but not a significantly better fit than a model with a single rate for all branches. Nonetheless, we compared the dN/dS values obtained in this full model with the variation in mating systems among species (fig. 1). We categorized mating systems as less promiscuous (monogamous + polygynous) or more promiscuous (promiscuous + dispersed), based on information compiled by Dixon (1998) and Lindenfors and Tullberg (1998). To avoid the problem of uncertainty in reconstructing mating system along long branches, we focused only on the terminal branches. We observed an increase in the rate of evolution associated with an increase in promiscuity in four of the six independent transitions from less promiscuous to more promiscuous mating systems (fig. 1). This was true when we included all sites or when we included only the extracellular domain where most positively selected sites were located. For the extracellular domain, the average ω for more promiscuous branches (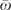 = 0.84; SD = 0.79) was higher than the average ω for less promiscuous branches ( = 0.46; SD = 0.22), but this difference was not significant (t-test, P = 0.093). Thus, there is no compelling evidence for a causal link between mating system and molecular evolution at TLR5 in these data.
= 0.84; SD = 0.79) was higher than the average ω for less promiscuous branches ( = 0.46; SD = 0.22), but this difference was not significant (t-test, P = 0.093). Thus, there is no compelling evidence for a causal link between mating system and molecular evolution at TLR5 in these data.
Human and Chimpanzee Polymorphism at TLR5
Levels of variation at TLR5 in humans are summarized in table 3. In general, both coding and noncoding regions showed polymorphism levels similar to those seen at other genes (Akey et al. 2004). Overall, humans presented an excess of rare variants with negative values of Tajima's D and Fu and Li's D* for both the coding and noncoding regions (table 3). The African samples showed strongly negative values, whereas the European samples showed either less negative values (coding) or slightly positive (noncoding) values. Differences in the level and pattern of variation between the African and European samples in noncoding regions are largely in agreement with well-accepted demographic scenarios for African Americans, and European Americans (Stajich and Hahn 2005). For example, our Tajima's D values are not outliers in the distribution of Tajima's D for a large set of genes sequenced in African Americans and European Americans (Stajich and Hahn 2005), suggesting that demographic effects rather than positive selection best explain the deviations from the null model at noncoding sites.
Table 3.
Levels of Polymorphism and Tests of Neutrality in Humans and Chimpanzees
| na | Coding |
Noncoding |
|||||||||||
| Lb | Sc | π (%) | θ (%) | Tajima's D | Fu and Li's D* | Lb | Sc | π (%) | θ (%) | Tajima's D | Fu and Li's D* | ||
| Humans | |||||||||||||
| Africans | 66 | 2,573 | 13 | 0.048 | 0.106 | −1.560* | −0.124 | 13,724.8 | 112 | 0.123 | 0.171 | −0.984 | −1.803* |
| Europeans | 64 | 2,574 | 12 | 0.057 | 0.099 | −1.203 | −1.387 | 13,571.9 | 56 | 0.092 | 0.087 | 0.176 | 0.698 |
| All | 130 | 2,573 | 19 | 0.054 | 0.136 | −1.684* | −0.722 | 13,647.1 | 122 | 0.109 | 0.164 | −1.087 | −2.110* |
| Chimpanzees | 38 | 2,574 | 4 | 0.036 | 0.037 | −0.0588 | −0.0235 | 1,861 | 5 | 0.082 | 0.064 | 0.703 | 1.1216 |
*Significant at the 0.05 level.
No. of chromosomes.
No. of used sites (including missing data).
No. of polymorphic sites.
For the coding region, both the African and European samples showed a more pronounced excess of low-frequency variants than in the noncoding region (table 3). Tajima's D for nonsynonymous sites was −1.495 and −1.020 for silent sites. This lower value for nonsynonymous polymorphisms is consistent with the idea that some of these mutations may be weakly deleterious. This is also supported by a slightly, but not significantly, higher ratio of polymorphism to divergence for nonsynonymous mutations than for synonymous mutations (table 4) using polymorphism data from both humans and chimpanzees.
Table 4.
Polymorphisms and Fixed Differences for Nonsynonymous and Synonymous Sites
| Comparison | Site | Polymorphismsa | Fixed Differencesb | P Value |
| Human and chimp together | Nonsynonymous | 15 (16) | 23 | 0.08 (0.08) |
| Synonymous | 7 | 28 | ||
| Human alone | Nonsynonymous | 12 (13) | 21 | 0.17 (0.10) |
| Synonymous | 6 | 27 | ||
| Chimp alone | Nonsynonymous | 3 | 21 | 0.61 |
| Synonymous | 1 | 23 |
Thirteen replacement polymorphisms occur in humans. One of them occurs uniquely in the background of the haplotype containing the premature stop. The M–K test was computed including and excluding that replacement change (numbers of polymorphisms and P values in parentheses).
All fixed differences are in comparison with the orangutan sequence.
Of the 13 observed replacement changes observed in humans, three had frequencies above 5% (C1174T [TLR5392STOP], freq = 0.069; A1775G, freq = 0.12; and T1846C, freq = 0.29). The high frequency of these mutations raises the question of whether they represent functional variants maintained at high frequency by selection. Merx et al. (2006) showed that only three of all known nonsynonymous single nucleotide polymorphisms (SNPs) at TLR5 had functional effects when tested on a site-by-site basis in transiently transfected CHO-K1 cells using reporter assays: One was TLR5392STOP and the other two were very rare SNPs not present in our sample. Each of these mutations resulted in a nonresponsive receptor (loss of function) after stimulation with flagellin. The T1846C and A1775G mutations do not have similar effects in this assay, and thus there is no experimental evidence that they affect function. Their high frequency might simply be due to drift.
We used a modified McDonald Kreitman (MK) test to compare the ratio of polymorphism to divergence for silent versus putative regulatory sites as in Andolfatto (2005) and found no deviation from the neutral expectation (table 5).
Table 5.
Polymorphisms and Fixed Differences for Synonymous and Regulatory Sites
| Site | Polymorphisms | Fixed Differencesa | P Value |
| Regulatory 1b | 7 | 18 | 0.49 |
| Synonymous (coding) | 6 | 9 | |
| Regulatory 2c | 9 | 13 | 1.00 |
| Synonymous (coding) | 6 | 9 | |
| Regulatory combined | 16 | 31 | 0.76 |
| Synonymous (coding) | 6 | 9 |
Divergence with respect to the chimpanzee sequence.
1 kb upstream of the transcription start site of transcript ENST00000342210.
1 kb upstream of the transcription start site of transcript ENST00000366881.
Levels of nucleotide variability in western chimpanzees (P. t. verus) are presented in table 3 and are similar to genomewide averages (Yu et al. 2003; Fischer et al. 2006). No significant deviations from neutrality were detected using tests of the allele frequency spectrum (table 3) or the MK test (table 4).
However, examination of the table of polymorphism revealed the presence of two major haplogroups (fig. 2, supplementary table 5, Supplementary Material online). Divergence between these haplogroups was 0.15%, close to the average value between chimpanzee subspecies (Yu et al. 2003; Fischer et al. 2006). To gain more insight into the origin of this variation, we sequenced three individuals of P. t. troglodytes. We found that the least frequent haplotype class (8 of 38) from P. t. verus is present in P. t. troglodytes in five of six chromosomes, whereas another haplotype present only in a single copy in P. t. troglodytes is more closely related to the major haplogroup in P. t. verus (fig. 2).
FIG. 2.—

Haplotype network showing the two divergent haplogroups shared between P.t. verus and P.t. troglodytes. Each circle represents a different haplotype and its size is proportional to its frequency in the sample. Mutations distinguishing haplotypes are shown as marks along the lines, whereas missing haplotypes are shown as black dots.
Three possible explanations for divergent haplotypes shared between subspecies are 1) unsorted ancestral polymorphism, 2) admixture (i.e., gene flow between groups), or 3) old balancing selection. Distinguishing among these is difficult. We note that the estimated divergence time between P. t. verus and P. t. troglodytes of 422,000 years (Won and Hey 2005) is less than the average time required to achieve reciprocal monophyly (E(t) ≈ 4Ne generations = 530,300 years, using the ancestral population size estimated by Won and Hey (2005) of Ne = 5,300 and a generation time of 25 years). Although the variance associated with this estimation is very large (Tajima 1983), this comparison suggests that ancestral variation could still be segregating between these subspecies. However, variation that is ancestral should have relatively little LD, whereas variation that is due to recent admixture should have higher levels of LD, an idea formalized into a test by Wall (2000) to detect ancient admixture in humans. We applied this test to our data. We computed the number of congruent sites (pb) and the maximum distance between any two congruent sites (gd), and compared these values with a simulated distribution generated by sampling neutral genealogies conditioned on the observed level of variation. The probability of obtaining both pb = 6 and gd = 0.285 under panmixia was 0.039 (using the level of recombination estimated from the data), indicating the existence of population structure or historical gene flow. We also fitted an isolation model with gene flow, as in Won and Hey (2005), and found evidence of gene flow between subspecies, although most of this gene flow was from P. t. verus to P. t. troglodytes. In light of the relative excess of LD revealed by the Wall test, some form of admixture or population structure seems to be the most likely explanation for the patterns of variation seen at TLR5 in P. t. verus, although we note that more complex scenarios involving retention of ancestral polymorphism and selection could also contribute to the observed patterns.
Distribution, Frequency, and Haplotype Structure of TLR5392STOP in Humans
Two lines of evidence suggest that TLR5392STOP has functional consequences. First in vitro assays showed that it encodes a defective receptor (Merx et al. 2006). Second, it is associated with disease phenotypes in human populations (Hawn et al. 2003, 2005; Gewirtz et al. 2006). Because of these observations, we were interested in measuring the frequency of TLR5392STOP in different populations and exploring the idea that this mutation might be under recent strong positive selection in humans. We genotyped the mutation in the HGDP and estimated a global frequency of 4.2%. The genotype frequencies were close to Hardy–Weinberg expectations (supplementary table 5, Supplementary Material online). TLR5392STOP is distributed nearly worldwide, with the mutation present in at least one copy in approximately half of the populations sampled (fig. 3). Because the mutation is often relatively rare, it is possible that the mutation is present at low frequencies in more populations than those reported here. Notably, some populations in the Middle East and Southern Asia have considerably higher frequencies, such as Balochi and Baruscho from Pakistan (14.5% and 12.0%, respectively), Miaozu and Naxi from China (10.0% and 11.0%, respectively), Cambodia (16.7%), Papua-New Guinea (14.7%), and Melanesia (22.7%).
FIG. 3.—

Distribution of TLR5392STOP around the world. The frequency of the allele is shown in red.
If TLR5392STOP has increased in frequency due to selection in the recent past, the mutation is expected to be embedded in unusually long haplotypes. For example, selection at G6pd has generated LD over more than 1 Mb (Saunders et al. 2005). However, only two sites (positions 9,946 and 11,185) show significant LD (measured as D') with TLR5392STOP (position 33,309) after Bonferroni correction for multiple testing (table 6). The distances between TLR5392STOP and these sites are 23,963 and 22,724 nt, respectively. Because TLR5392STOP (or any linked marker) is not present in the Hapmap, we were not able to evaluate the extent of LD at longer distances, but the fact that the haplotype containing TLR5392STOP extends less than 25 kb suggests that if selection is responsible for the actual frequencies, it is not recent and strong.
Table 6.
Sites That Show Significant Linkage Disequilibrium with the Premature Stop Mutation in Humans
| Site 1 | TLR5392STOP | Distance (bp) | D′ | Pa | Age of TLR5392STOP (Years)b |
| 9,946 | 33,909 | 23,963 | 0.731 | <0.0001(B) | 27,237 |
| 10,021 | 33,909 | 23,888 | 0.539 | <0.001 | 28,723 |
| 11,185 | 33,909 | 22,724 | 0.731 | <0.0001(B) | 53,893 |
| 11,970 | 33,909 | 21,939 | 0.386 | <0.0001 | 90,382 |
| 13,373 | 33,909 | 20,536 | 0.544 | <0.001 | 61,754 |
B = significant after Bonferroni correction.
The age reported in the text is the average of the five sites.
Discussion
Immunity genes are among the fastest evolving classes of genes in mammalian genomes (Gibbs et al. 2004; Mikkelsen et al. 2005; Nielsen et al. 2005), an observation that is usually interpreted as evidence of positive selection due to their potential engagement in host–pathogen coevolution. Despite this generalization, it has been unclear whether genes of the adaptive and innate branches of immunity show similar patterns of evolution or whether they are characterized by very different levels of functional constraint. By studying both phylogeny-based estimates of evolutionary rates and patterns of nucleotide variation within and between closely related primate species, we sought to provide an integrated understanding of the molecular evolution of an innate immunity receptor at different evolutionary timescales.
Positive Selection at the Extracellular Domain of TLR5 in Primates
The results of several ML approaches provide strong evidence that TLR5 has experienced positive selection in primates. Conservatively, we identified 11 sites that show congruence between different ML methods as the strongest candidates of adaptive evolution. Of these, 10 sites are localized in leucine-rich repeats of the extracelullar domain (table 2), and three are located within a 228 aa region where the putative flagellin recognition site lies (Andersen-Nissen et al. 2007). Although we still do not have a complete picture of the flagellin-TLR5 interaction surface, this observation strengthens the case of adaptive evolution at TLR5. Moreover, based on the modeled 3D structure of the extracellular domain, Andersen-Nissen et al. (2007) hypothesized that amino acids near a conserved concavity within the 228 aa region could mediate species-specific patterns of TLR5 recognition. It is worth noting that site 268 lies adjacent to a residue (267) that was identified by mutagenesis as responsible for differences in specificity between human and mouse TLR5 (Andersen-Nissen et al. 2007). It is possible that residue 268 or some of the other sites identified as candidates for being under selection are also involved in TLR5 species specificity, a matter that functional studies will be able to clarify. “U,” the evolutionary index (Tang et al. 2004), provides additional information about the likelihood that specific mutations affect function and thus may be under selection. Six of the 11 sites under selection (aa292, aa312, aa354, aa482, aa530, and aa567) show relatively radical changes, with U ranging between 0.375 and 0.732.
The identification of several sites under selection within the pathogen interaction domain fits the expectation of coevolutionary models. This is in line with the finding that several flagellated Proteobacteria are able to evade human TLR5 recognition (Andersen-Nissen et al. 2005). However, we note that only a small proportion of sites (11/858 = 1.3%) show strong evidence of positive selection. Thus, most of the protein, including the TIR (signaling) domain, shows strong functional constraint, in agreement with the most generally accepted paradigm of TLR function. This duality of strong positive selection on a few sites against a background of strong purifying selection over most of the TLR5 protein is in sharp contrast with antiretroviral genes such as APOBEC3G, TRIM5α. These genes show a much larger proportion of sites (30% and 18%, respectively) under positive selection (Sawyer et al. 2004, 2005). These differences between TLR5 and APOBEC3G or TRIM5α may reflect general differences between PRRs and genes involved in “intrinsic” immunity (i.e., genes that typically do not participate in the classic innate immunity pathways but nevertheless can confer pre-exposure resistance against certain pathogens). These differences might also reflect differences between genes whose products interact with bacteria versus those whose products interact with viruses. It is possible, for example, that due to their higher mutation rates and faster turnover, viruses impose stronger selection than do bacteria.
Vertebrate immune systems differ from invertebrate immune systems in many ways, but most notably in the presence of an adaptive immune response. The acquisition of adaptive immunity could have fundamentally changed the evolutionary dynamics of vertebrate PRRs. The recent publication of genomewide patterns of evolution of innate immunity genes in Drosophila by Sackton et al. (2007) allows us to start comparing patterns of evolution of PRRs and other classes of innate immunity genes between Drosophila and vertebrates. Using a similar codon-based ML approach as the one used here, Sackton et al. (2007) found that among 245 Drosophila immunity genes, PRRs constitute the class with the highest proportion of positively selected sites (followed by signaling peptides and then antimicrobial peptides) in the D. melanogaster group. In contrast, Schlenke and Begun (2003) reported that adaptive fixations are also common in signaling molecules in Drosophila simulans. In vertebrates, similar genomewide analyses of innate immunity genes are missing, but some evidence points to the possibility that innate immunity genes are also under strong selection. Recent examples include APOBEC3G and TRIM5α (Sawyer et al. 2004, 2005), TRIM22 (Sawyer et al. 2007), TLR4 (Nakajima et al. 2008), RNASEL (Summers and Crespi 2008), and PKR (Elde et al. 2008). Our results demonstrate that some PRRs can also evolve rapidly between species.
Mating System and Molecular Evolution of Immunity Genes
We tested the mating system/disease-risk hypothesis with six phylogenetically independent contrasts between promiscuous and monogamous/polygynous mating systems in the primate phylogeny. We found that dN/dS changed in the predicted direction in four of six cases and that the average dN/dS was not significantly higher in more promiscuous lineages. Thus, rates of molecular evolution at TLR5 do not seem to support this controversial hypothesis and suggest that lineage-specific effects are more important than the effect, if any, of mating system. A more complete test will require analysis of similar data from many immunity genes. An interesting observation is that the increase in ω in the more promiscuous group was accompanied by an increased variance. It is possible that promiscuous mating systems are associated with stronger natural selection on immunity genes only some of the time (or only on a subset of these genes) leading to a higher average ω and also to a greater variance in ω in more promiscuous lineages compared with less promiscuous lineages.
Patterns of Nucleotide Variation in Humans and Chimpanzees
Patterns of nucleotide variation within humans and chimpanzees were largely consistent with neutral expectations. The deviations from neutral predictions in the spectrum of allele frequencies were similar to those seen at other genes, suggesting that demographic effects, rather than selection, are responsible for these patterns. Thus, despite the strong evidence for adaptive evolution at TLR5 over deeper evolutionary timescales in primates (see above), we did not find evidence for adaptive evolution within humans or chimpanzees. This suggests that adaptive evolution at TLR5 may be somewhat episodic, or at least not marked by continual turnover of new adaptive alleles as might be expected under an arms race model of host–pathogen coevolution.
We estimated the rate of adaptive fixations from our phylogenetic comparisons to get a sense of the likelihood of detecting selection within species. Using the 11 sites with the strongest evidence of selection (table 2), we estimated the rate of adaptive fixation by dividing the total number of amino acid substitutions (39) at these sites by the total length of the tree (417.2 My) using divergence times from Bininda-Emonds et al. (2007). This yielded a value of approximately one adaptive fixation every 10 My. This is probably an underestimate of the true rate, because the ML methods used here only have power to detect recurrent positive selection on the same sites. However, even if the true rate were an order of magnitude higher than this estimate, it would not be surprising to fail to find evidence of selection within humans or chimpanzees. Polymorphism-based tests of selection typically have power to detect selection over fairly recent timescales, often on the order of less than N generations (∼250,000 years in humans) (Braverman et al. 1995; Simonsen et al. 1995; Przeworski 2002).
One result worth noting was the observation of low-frequency replacement polymorphisms in humans. These polymorphisms contribute to a ratio of replacement to silent variation that is slightly higher within species than between species (table 4). Along with negative values of Tajima's D for replacement polymorphisms, this suggests that many of these polymorphisms may be weakly deleterious, consistent with the general pattern of functional constraint revealed by the phylogenetic analysis.
Patterns of nucleotide variation within chimpanzees differed from those seen in humans. Levels of variation were lower in chimpanzees, in spite of similar effective population sizes (or slightly higher in P. t. verus) (Yu et al. 2004). The distribution of allele frequencies differed too, with an excess of rare variants in humans and a trend toward an excess of intermediate frequency variants in chimpanzees at noncoding sites. Chimpanzees exhibited two divergent haplogroups in both P. t. verus and in P. t. troglodytes. The presence of these shared haplotypes is probably best explained by gene flow between subspecies at some point in the recent past or by some more complicated form of population structure.
Is the Human TLR5 Redundant?
Recently, several cases of adaptive gene loss in humans have been reported (Tournamille et al. 1995; Ali et al. 1998; Wang et al. 2006; Seixas et al. 2007). This somewhat counterintuitive idea, positive selection favoring gene loss, has been proposed as a potentially important mechanism in human evolution (Olson 1999; Olson and Varki 2003).
TLR5392STOP, a loss-of-function mutation, segregates in humans at a considerable frequency along with the normal variants. This raises the question of whether 1) it is being constantly generated by recurrent mutation, 2) it has increased in frequency due to positive selection, in which case there might be a trade-off between the disadvantage of loosing the function and some other benefit, or 3) it has drifted in the population to its present frequency.
The frequency of TLR5392STOP is clearly not compatible with mutation-selection balance. Assuming a mutation rate, μ, of 2 × 10−8 (or ∼10−7 for a CpG site) (Nachman and Crowell 2000) and an equilibrium frequency, qe, of 0.042 we can calculate the selection coefficient, s, against a dominant mutation as s ≈ μ/qe (Haldane 1932). The estimated s (6.0 × 10−7 or 2.4 × 10−6 for a CpG site) is so small as to be effectively neutral in human populations, where the effective population size is approximately 10,000 (Zhao et al. 2006). If s was 0.01, then the mutation rate would have to be on the order of 10−4 to account for the observed frequencies, and this is clearly unrealistic. Moreover, the fact that the TLR5392STOP always appears on the same haplotype argues against recurrent mutation.
We found no evidence of strong, recent selection on TLR5392STOP either in patterns of LD, which were unremarkable, or in levels of variability, which were average. Moreover, the distribution of allele frequencies at TLR5 fits well with generally accepted demographic models. This leaves drift as the most likely explanation for the present frequency of TLR5392STOP. Given the difficulties of detecting selection from polymorphism data in humans, we cannot rule out the possibility that TLR5392STOP has been under weak positive selection, especially in light of the phenotypes associated with this mutation. For instance, SLE has a relatively high prevalence (up to 160/100,000) and mostly affects women in reproductive age (Danchenko et al. 2006) making the hypothesis of selection for protection against autoimmune diseases at least reasonable. There are marked geographic differences in SLE burden that might reflect underlying genetic variation for resistance/susceptibility or variation in environmental factors. Microbial infections are common triggers of autoimmunity through TLRs (Anders et al. 2005). It would be interesting to correlate worldwide abundance of flagellated pathogens with the prevalence of SLE.
If drift took the mutation to its present frequency, then the mutation must be relatively old. An estimate of the age of an allele based on its frequency, q, is given by E(t) = (−2q)(ln q)/(1 − q), where age is measured in units of 2N generations (Kimura and Ohta 1973). The global frequency of TLR5392STOP is 0.042. Assuming that N = 10,000, the estimated age is 5,560 generations or 139,000 years (assuming a generation of 25 years). Another way to estimate the age of the TLR5392STOP mutation is from the decay of LD as a function of time and recombination rate. The time required to erode linkage to the observed level is given by: (Hedrick 2000), where is the observed LD in the data, is the initial LD (assumed to be complete when the TLR5392STOP mutation arose, D′ = 1), and c is the recombination distance calculated using the average recombination rate for chromosome 1 of 1.2 cM/Mb (Jensen-Seaman et al. 2004). Using five sites that show significant LD (table 6) t was estimated as 2,096 generations or 52,398 years.
The observation that TLR5392STOP, a null variant, is present at frequencies up to 23% in some populations suggests that TLR5 function might be partially compensated by other genes (i.e., functional redundancy for TLR5). A similar case is provided by Verdu et al. (2006) who, based on the patterns of nucleotide variation and absence of extended LD, concluded that MBL2, another innate immune receptor that activates the lectin–complement pathways, is functionally redundant in human innate immune defenses. Redundancy in PAMP recognition might be a common theme in the innate immune response (Miao et al. 2007). The recognition of viral RNA provides a good example in which several TLRs participate in the detection of ssRNA and dsRNA in endosomal compartments, while another suite of genes responds to the same PAMPs in the cytosol. It is possible that this recognition at multiple levels is an important and previously unappreciated feature of the innate immune system. The recent identification of a second flagellin receptor (cytosolic), Ipaf (Franchi et al. 2006), is consistent with this idea. However, the downstream effects of both genes are quite different, and they also respond to different types of bacteria (reviewed in Miao et al. 2007), suggesting that TLR5 and Ipaf might cooperate in recognizing flagellated bacteria rather than being completely functionally redundant.
Supplementary Material
Supplementary tables 1–6 are available at Molecular Biology and Evolution online (http://www.mbe.oxfordjournals.org/).
Acknowledgments
We especially thank the following people/institutions for providing DNA or tissue samples: Drs. M. Hammer, O. Ryder, and B. Beer, The Museum of Vertebrate Zoology, Berkeley, California, The Southwest National Primate Research Center, and the Gladys Porter, Toronto, San Diego, and Los Angeles Zoos. We thank Dr. W. Klimecki for providing access to the raw human sequence data deposited in the Innate Immunity Database, Dr. M. Saunders and Dr. H. Norton for help with analysis, A. Woerner for the use of scripts to handle/analyze polymorphism data, and Donata Vercelli, Armando Geraldes, Matt Dean, Jeff Good Tovah Salcedo, Miguel Carneiro, and Mari Sans-Fuentes for very useful discussions of the data. The findings and conclusions in this report are those of the authors and do not necessarily represent the views of the Centers for Disease Control and Prevention. This work was supported by an NIH grant to M.W.N. (GM074245). The findings and conclusions in this report are those of the authors and do not necessarily represent the views of the Centers for Disease Control and Prevention.
References
- Akey JM, Eberle MA, Rieder MJ, Carlson CS, Shriver MD, Nickerson DA, Kruglyak L. Population history and natural selection shape patterns of genetic variation in 132 genes. Plos Biol. 2004;2:1591–1599. doi: 10.1371/journal.pbio.0020286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akira S, Takeda K. Toll-like receptor signalling. Nat Rev Immunol. 2004;4:499–511. doi: 10.1038/nri1391. [DOI] [PubMed] [Google Scholar]
- Ali M, Rellos P, Cox TM. Hereditary fructose intolerance. J Med Genet. 1998;35:353–365. doi: 10.1136/jmg.35.5.353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anders HJ, Zecher D, Pawar RD, Patole PS. Molecular mechanisms of autoimmunity triggered by microbial infection. Arthritis Res Ther. 2005;7:215–224. doi: 10.1186/ar1818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersen-Nissen E, Smith KD, Bonneau R, Strong RK, Aderem A. A conserved surface on Toll-like receptor 5 recognizes bacterial flagellin. J Exp Med. 2007;204:393–403. doi: 10.1084/jem.20061400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersen-Nissen E, Smith KD, Strobe KL, Barrett SLR, Cookson BT, Logan SM, Aderem A. Evasion of Toll-like receptor 5 by flagellated bacteria. Proc Natl Acad Sci USA. 2005;102:9247–9252. doi: 10.1073/pnas.0502040102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andolfatto P. Adaptive evolution of non-coding DNA in Drosophila. Nature. 2005;437:1149–1152. doi: 10.1038/nature04107. [DOI] [PubMed] [Google Scholar]
- Bininda-Emonds ORP, Cardillo M, Jones KE, MacPhee RDE, Beck RMD, Grenyer R, Price SA, Vos RA, Gittleman JL, Purvis A. The delayed rise of present-day mammals. Nature. 2007;446:507–512. doi: 10.1038/nature05634. [DOI] [PubMed] [Google Scholar]
- Braverman JM, Hudson RR, Kaplan NL, Langley CH, Stephan W. The Hitchhiking effect on the site frequency-spectrum of DNA polymorphisms. Genetics. 1995;140:783–796. doi: 10.1093/genetics/140.2.783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cann HM, de Toma C, Cazes L, et al. (41 co-authors) A human genome diversity cell line panel. Science. 2002;296:261–262. doi: 10.1126/science.296.5566.261b. [DOI] [PubMed] [Google Scholar]
- Cann HM, De Toma C, Marcadet-Troton A, Thomas G, Dausset J, Cavalli-Sforza LL. The HGDP-CEPH human genome diversity panel. Am J Hum Genet. 1999;65:A198. [Google Scholar]
- Danchenko N, Satia JA, Anthony MS. Epidemiology of systemic lupus erythematosus: a comparison of worldwide disease burden. Lupus. 2006;15:308–318. doi: 10.1191/0961203306lu2305xx. [DOI] [PubMed] [Google Scholar]
- Dixon AF. Primate sexuality. New York: Oxford University Press; 1998. [Google Scholar]
- Elde NC, Child SJ, Geballe AP, Malik HS. Protein kinase R reveals an evolutionary model for defeating viral mimicry. Nature. 2009;457:485–489. doi: 10.1038/nature07529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer A, Pollack J, Thalmann O, Nickel B, Paabo S. Demographic history and genetic differentiation in apes. Curr Biol. 2006;16:1133–1138. doi: 10.1016/j.cub.2006.04.033. [DOI] [PubMed] [Google Scholar]
- Franchi L, Amer A, Body-Malapel M, et al. (12 co-authors) Cytosolic flagellin requires Ipaf for activation of caspase-1 and interleukin 1 beta in salmonella-infected macrophages. Nat Immunol. 2006;7:576–582. doi: 10.1038/ni1346. [DOI] [PubMed] [Google Scholar]
- Fu YX, Li WH. Statistical tests of neutrality of mutations. Genetics. 1993;133:693–709. doi: 10.1093/genetics/133.3.693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garrigan D, Mobasher Z, Kingan SB, Wilder JA, Hammer MF. Deep haplotype divergence and long-range linkage disequilibrium at Xp21.1 provide evidence that humans descend from a structured ancestral population. Genetics. 2005;170:1849–1856. doi: 10.1534/genetics.105.041095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gewirtz AT, Vijay-Kumar M, Brant SR, Duerr RH, Nicolae DL, Cho JH. Dominant-negative TLR5 polymorphism reduces adaptive immune response to flagellin and negatively associates with Crohn's disease. Am J Physiol-Gastrointest Liver Physiol. 2006;290:G1157–G1163. doi: 10.1152/ajpgi.00544.2005. [DOI] [PubMed] [Google Scholar]
- Gibbs RA, Weinstock GM, Metzker ML, et al. (230 co-authors) Genome sequence of the Brown Norway rat yields insights into mammalian evolution. Nature. 2004;428:493–521. doi: 10.1038/nature02426. [DOI] [PubMed] [Google Scholar]
- Grantham R. Amino-acid difference formula to help explain protein evolution. Science. 1974;185:862–864. doi: 10.1126/science.185.4154.862. [DOI] [PubMed] [Google Scholar]
- Haldane JBS. The causes of evolution. London: Longmans, Green & Co; 1932. [Google Scholar]
- Hawn TR, Verbon A, Lettinga KD, et al. (13 co-authors) A common dominant TLR5 stop codon polymorphism abolishes flagellin signaling and is associated with susceptibility to legionnaires’ disease. J Exp Med. 2003;198:1563–1572. doi: 10.1084/jem.20031220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawn TR, Wu H, Grossman JM, Hahn BH, Tsao BP, Aderem A. A stop codon polymorphism of Toll-like receptor 5 is associated with resistance to systemic lupus erythematosus. Proc Natl Acad Sci USA. 2005;102:10593–10597. doi: 10.1073/pnas.0501165102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi F, Smith KD, Ozinsky A, Hawn TR, Yi EC, Goodlett DR, Eng JK, Akira S, Underhill DM, Aderem A. The innate immune response to bacterial flagellin is mediated by Toll-like receptor 5. Nature. 2001;410:1099–1103. doi: 10.1038/35074106. [DOI] [PubMed] [Google Scholar]
- Hedrick PW. Genetics of populations. Sudbury: Jones and Bartlett Publishers Inc; 2000. [Google Scholar]
- Hey J, Nielsen R. Multilocus methods for estimating population sizes, migration rates and divergence time, with applications to the divergence of Drosophila pseudoobscura and D-persimilis. Genetics. 2004;167:747–760. doi: 10.1534/genetics.103.024182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hey J, Nielsen R. Integration within the Felsenstein equation for improved Markov chain Monte Carlo methods in population genetics. Proc Natl Acad Sci USA. 2007;104:2785–2790. doi: 10.1073/pnas.0611164104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hey J, Wakeley J. A coalescent estimator of the population recombination rate. Genetics. 1997;145:833–846. doi: 10.1093/genetics/145.3.833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hudson RR. Generating samples under a Wright–Fisher neutral model of genetic variation. Bioinformatics. 2002;18:337–338. doi: 10.1093/bioinformatics/18.2.337. [DOI] [PubMed] [Google Scholar]
- Jensen-Seaman MI, Furey TS, Payseur BA, Lu YT, Roskin KM, Chen CF, Thomas MA, Haussler D, Jacob HJ. Comparative recombination rates in the rat, mouse, and human genomes. Genome Res. 2004;14:528–538. doi: 10.1101/gr.1970304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura M, Ohta T. Age of a neutral mutant persisting in a finite population. Genetics. 1973;75:199–212. doi: 10.1093/genetics/75.1.199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kosakovsky Pond SL, Frost SDW. Not so different after fll: a comparison of methods for detecting amino acid sites under selection. Mol Biol Evol. 2005;22:1208–1222. doi: 10.1093/molbev/msi105. [DOI] [PubMed] [Google Scholar]
- Lindenfors P, Tullberg BS. Phylogenetic analyses of primate size evolution: the consequences of sexual selection. Biol J Linn Soc. 1998;64:413–447. [Google Scholar]
- Matsushima N, Tanaka T, Enkhbayar P, Mikami T, Taga M, Yamada K, Kuroki Y. Comparative sequence analysis of leucine-rich repeats (LRRs) within vertebrate toll-like receptors. BMC Genomics. 2007;8:124. doi: 10.1186/1471-2164-8-124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mcdonald JH, Kreitman M. Adaptive protein evolution at the Adh locus in Drosophila. Nature. 1991;351:652–654. doi: 10.1038/351652a0. [DOI] [PubMed] [Google Scholar]
- Medzhitov R, Janeway CA. Innate immunity: the virtues of a nonclonal system of recognition. Cell. 1997;91:295–298. doi: 10.1016/s0092-8674(00)80412-2. [DOI] [PubMed] [Google Scholar]
- Merx S, Zimmer W, Neumaier M, Ahmad-Nejad P. Characterization and functional investigation of single nucleotide polymorphisms (SNPs) in the human TLR5 gene. Hum Mut. 2006;27:293. doi: 10.1002/humu.9409. [DOI] [PubMed] [Google Scholar]
- Miao EA, Andersen-Nissen E, Warren SE, Aderem A. TLR5 and Ipaf: dual sensors of bacterial flagellin in the innate immune system. Semin Immunopathol. 2007;29:275–288. doi: 10.1007/s00281-007-0078-z. [DOI] [PubMed] [Google Scholar]
- Mikkelsen TS, Hillier LW, Eichler EE, et al. (68 co-authors) Initial sequence of the chimpanzee genome and comparison with the human genome. Nature. 2005;437:69–87. doi: 10.1038/nature04072. [DOI] [PubMed] [Google Scholar]
- Nachman MW, Crowell SL. Estimate of the mutation rate per nucleotide in humans. Genetics. 2000;156:297–304. doi: 10.1093/genetics/156.1.297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakajima T, Ohtani H, Satta Y, Uno Y, Akari H, Ishida T, Kimura A. Natural selection in the TLR-related genes in the course of primate evolution. Immunogenetics. 2008;60:727–735. doi: 10.1007/s00251-008-0332-0. [DOI] [PubMed] [Google Scholar]
- Nei M, Li WH. Mathematical-model for studying genetic-variation in terms of restriction endonucleases. Proc Natl Acad Sci USA. 1979;76:5269–5273. doi: 10.1073/pnas.76.10.5269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen R, Bustamante C, Clark AG, et al. (13 co-authors) A scan for positively selected genes in the genomes of humans and chimpanzees. PLoS Biol. 2005;3:e170. doi: 10.1371/journal.pbio.0030170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen R, Wakeley J. Distinguishing migration from isolation: a Markov chain Monte Carlo approach. Genetics. 2001;158:885–896. doi: 10.1093/genetics/158.2.885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen R, Yang ZH. Likelihood models for detecting positively selected amino acid sites and applications to the HIV-1 envelope gene. Genetics. 1998;148:929–936. doi: 10.1093/genetics/148.3.929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nunn CL. A comparative study of leukocyte counts and disease risk in primates. Evolution. 2002;56:177–190. doi: 10.1111/j.0014-3820.2002.tb00859.x. [DOI] [PubMed] [Google Scholar]
- Nunn CL, Gittleman JL, Antonovics J. A comparative study of white blood cell counts and disease risk in carnivores. Proc Roy Soc Lond B Bio. 2003;270:347–356. doi: 10.1098/rspb.2002.2249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nunn CL, Gittleman JL, Antonovics J. Promiscuity and the primate immune system. Science. 2000;290:1168–1170. doi: 10.1126/science.290.5494.1168. [DOI] [PubMed] [Google Scholar]
- Olson MV. When less is more: gene loss as an engine of evolutionary change. Am J Hum Genet. 1999;64:18–23. doi: 10.1086/302219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olson MV, Varki A. Sequencing the chimpanzee genome: insights into human evolution and disease. Nat Rev Genet. 2003;4:20–28. doi: 10.1038/nrg981. [DOI] [PubMed] [Google Scholar]
- Ortiz M, Kaessmann H, Zhang K, Bashirova A, Carrington M, Quintana-Murci L, Telenti A. The evolutionary history of the CD209 (DC-SIGN) family in humans and non-human primates. Genes Immun. 2008;9:483–492. doi: 10.1038/gene.2008.40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pond SK, Muse SV. Site-to-site variation of synonymous substitution rates. Mol Biol Evol. 2005;22:2375–2385. doi: 10.1093/molbev/msi232. [DOI] [PubMed] [Google Scholar]
- Pond SLK, Frost SDW. Datamonkey: rapid detection of selective pressure on individual sites of codon alignments. Bioinformatics. 2005;21:2531–2533. doi: 10.1093/bioinformatics/bti320. [DOI] [PubMed] [Google Scholar]
- Przeworski M. The signature of positive selection at randomly chosen loci. Genetics. 2002;160:1179–1189. doi: 10.1093/genetics/160.3.1179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purvis A. A composite estimate of primate phylogeny. Philos T Roy Soc B. 1995;348:405–421. doi: 10.1098/rstb.1995.0078. [DOI] [PubMed] [Google Scholar]
- Ramos HC, Rumbo M, Sirard JC. Bacterial flagellins: mediators of pathogenicity and host immune responses in mucosa. Trends Microbiol. 2004;12:509–517. doi: 10.1016/j.tim.2004.09.002. [DOI] [PubMed] [Google Scholar]
- Rosenberg NA. Standardized subsets of the HGDP–CEPH human genome diversity cell line panel, accounting for atypical and duplicated samples and pairs of close relatives. Ann Hum Genet. 2006;70:841–847. doi: 10.1111/j.1469-1809.2006.00285.x. [DOI] [PubMed] [Google Scholar]
- Rozas J, Sanchez-DelBarrio JC, Messeguer X, Rozas R. DnaSP, DNA polymorphism analyses by the coalescent and other methods. Bioinformatics. 2003;19:2496–2497. doi: 10.1093/bioinformatics/btg359. [DOI] [PubMed] [Google Scholar]
- Sackton TB, Lazzaro BP, Schlenke TA, Evans JD, Hultmark D, Clark AG. Dynamic evolution of the innate immune system in Drosophila. Nat Genet. 2007;39:1461–1468. doi: 10.1038/ng.2007.60. [DOI] [PubMed] [Google Scholar]
- Saunders MA, Slatkin M, Garner C, Hammer MR, Nachman MW. The extent of linkage disequilibrium caused by selection on G6PD in humans. Genetics. 2005;171:1219–1229. doi: 10.1534/genetics.105.048140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawyer SL, Emerman M, Malik HS. Ancient adaptive evolution of the primate antiviral DNA-editing enzyme APOBEC3G. Plos Biol. 2004;2:1278–1285. doi: 10.1371/journal.pbio.0020275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawyer SL, Emerman M, Malik HS. Discordant evolution of the adjacent antiretroviral genes TRIM22 and TRIM5 in mammals. Plos Pathog. 2007;3:1918–1929. doi: 10.1371/journal.ppat.0030197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawyer SL, Wu LI, Emerman M, Malik HS. Positive selection of primate TRIM5 alpha identifies a critical species-specific retroviral restriction domain. Proc Natl Acad Sci USA. 2005;102:2832–2837. doi: 10.1073/pnas.0409853102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlenke TA, Begun DJ. Natural selection drives drosophila immune system evolution. Genetics. 2003;164:1471–1480. doi: 10.1093/genetics/164.4.1471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seixas S, Suriano G, Carvalho F, Seruca R, Rocha J, Di Rienzo A. Sequence diversity at the proximal 14q32.1 SERPIN subcluster: evidence for natural selection favoring the pseudogenization of SERPINA2. Mol Biol Evol. 2007;24:587–598. doi: 10.1093/molbev/msl187. [DOI] [PubMed] [Google Scholar]
- Simonsen KL, Churchill GA, Aquadro CF. Properties of statistical tests of neutrality for DNA polymorphism data. Genetics. 1995;141:413–429. doi: 10.1093/genetics/141.1.413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stajich JE, Hahn MW. Disentangling the effects of demography and selection in human history. Mol Biol Evol. 2005;22:63–73. doi: 10.1093/molbev/msh252. [DOI] [PubMed] [Google Scholar]
- Stephens M, Smith NJ, Donnelly P. A new statistical method for haplotype reconstruction from population data. Am J Hum Genet. 2001;68:978–989. doi: 10.1086/319501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Summers K, Crespi B. Molecular evolution of the prostate cancer susceptibility locus RNASEL: evidence for positive selection. Infec Genet Evol. 2008;8:297–301. doi: 10.1016/j.meegid.2008.01.004. [DOI] [PubMed] [Google Scholar]
- Swanson WJ, Nielsen R, Yang QF. Pervasive adaptive evolution in mammalian fertilization proteins. Mol Biol Evol. 2003;20:18–20. doi: 10.1093/oxfordjournals.molbev.a004233. [DOI] [PubMed] [Google Scholar]
- Tajima F. Evolutionary relationship of DNA sequences in finite populations. Genetics. 1983;105:437–460. doi: 10.1093/genetics/105.2.437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tajima F. Statistical-method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics. 1989;123:585–595. doi: 10.1093/genetics/123.3.585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang H, Wyckoff GJ, Lu J, Wu CI. A universal evolutionary index for amino acid changes. Mol Biol Evol. 2004;21:1548–1556. doi: 10.1093/molbev/msh158. [DOI] [PubMed] [Google Scholar]
- Thompson JD, Gibson TJ, Plewniak F, Jeanmougin F, Higgins DG. The CLUSTAL_X windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res. 1997;25:4876–4882. doi: 10.1093/nar/25.24.4876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tournamille C, Colin Y, Cartron JP, Levankim C. Disruption of a Gata motif in the Duffy gene promoter abolishes erythroid gene-expression in Duffy negative individuals. Nat Genet. 1995;10:224–228. doi: 10.1038/ng0695-224. [DOI] [PubMed] [Google Scholar]
- Verdu P, Barreiro LB, Gessain A, et al. (13 co-authors) Evolutionary insights into the high worldwide prevalence of MBL2 deficiency alleles. Hum Mol Genet. 2006;15:2650–2658. doi: 10.1093/hmg/ddl193. [DOI] [PubMed] [Google Scholar]
- Wall JD. Detecting ancient admixture in humans using sequence polymorphism data. Genetics. 2000;154:1271–1279. doi: 10.1093/genetics/154.3.1271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang XX, Grus WE, Zhang JZ. Gene losses during human origins. Plos Biol. 2006;4:366–377. doi: 10.1371/journal.pbio.0040052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waterson GA. On the number of segregating sites in genetical models without recombination. Theor Popul Biol. 1975;7 doi: 10.1016/0040-5809(75)90020-9. [DOI] [PubMed] [Google Scholar]
- Won YJ, Hey J. Divergence population genetics of chimpanzees. Mol Biol Evol. 2005;22:297–307. doi: 10.1093/molbev/msi017. [DOI] [PubMed] [Google Scholar]
- Yang ZH. PAML: a program package for phylogenetic analysis by maximum likelihood. Comput Appl Biosci. 1997;13:555–556. doi: 10.1093/bioinformatics/13.5.555. [DOI] [PubMed] [Google Scholar]
- Yang ZH. Likelihood ratio tests for detecting positive selection and application to primate lysozyme evolution. Mol Biol Evol. 1998;15:568–573. doi: 10.1093/oxfordjournals.molbev.a025957. [DOI] [PubMed] [Google Scholar]
- Yang ZH. PAML 4: phylogenetic analysis by maximum likelihood. Mol Biol Evol. 2007;24:1586–1591. doi: 10.1093/molbev/msm088. [DOI] [PubMed] [Google Scholar]
- Yang ZH, Nielsen R, Goldman N, Pedersen AMK. Codon-substitution models for heterogeneous selection pressure at amino acid sites. Genetics. 2000;155:431–449. doi: 10.1093/genetics/155.1.431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang ZH, Wong WSW, Nielsen R. Bayes empirical Bayes inference of amino acid sites under positive selection. Mol Biol Evol. 2005;22:1107–1118. doi: 10.1093/molbev/msi097. [DOI] [PubMed] [Google Scholar]
- Yu N, Jensen-Seaman MI, Chemnick L, Kidd JR, Deinard AS, Ryder O, Kidd KK, Li WH. Low nucleotide diversity in chimpanzees and bonobos. Genetics. 2003;164:1511–1518. doi: 10.1093/genetics/164.4.1511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu N, Jensen-Seaman MI, Chemnick L, Ryder O, Li WH. Nucleotide diversity in gorillas. Genetics. 2004;166:1375–1383. doi: 10.1534/genetics.166.3.1375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao ZM, Yu N, Fu YX, Li WH. Nucleotide variation and haplotype diversity in a 10-kb noncoding region in three continental human populations. Genetics. 2006;174:399–409. doi: 10.1534/genetics.106.060301. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.