

Abstract
We analyze patterns of genetic variation in extant human polymorphism data from the National Institute of Environmental Health Sciences single nucleotide polymorphism project to estimate human demographic parameters. We update our previous work by considering a larger data set (more genes and more populations) and by explicitly estimating the amount of putative admixture between modern humans and archaic human groups (e.g., Neandertals, Homo erectus, and Homo floresiensis). We find evidence for this ancient admixture in European, East Asian, and West African samples, suggesting that admixture between diverged hominin groups may be a general feature of recent human evolution.
Keywords: admixture, population structure, human evolution
Introduction
One of the enduring questions in human evolutionary genetics surrounds the relationship between modern humans and their “archaic” human contemporaries (e.g., Neandertals or Asian Homo erectus). Did the archaic groups die out in the recent past or did they interbreed with modern humans and leave their genes in present-day individuals? Despite a tremendous amount of research on this question, there is still no consensus on what contribution, if any, archaic humans made to the contemporary gene pool (reviewed in Wall and Hammer 2006).
Two main methods that utilize DNA sequence data are available for determining the evolutionary relationship between archaic and modern humans. The first method involves a direct comparison between homologous archaic and contemporary human DNA sequences. This started with a comparison of mitochondrial DNA between modern humans and Neandertals (Krings et al. 1997; Serre et al. 2004; Green et al. 2008) and lately has expanded to comparisons of nuclear DNA between modern humans and Neandertals (Green et al. 2006; Noonan et al. 2006; Krause et al. 2007; Lalueza-Fox et al. 2007). Although some of these studies may be flawed due to contamination with modern human DNA (Wall and Kim 2007; Coop et al. 2008), there seems to be a rough consensus that the contribution of Neandertals to the modern European gene pool was either small or negligible—estimates of admixture rates consistent with the data are generally from 0% to 20% (Serre et al. 2004; Noonan et al. 2006).
A separate method utilizes only sequence polymorphism data from extant humans. This approach analyzes patterns of linkage disequilibrium (LD) to detect the presence of ancient admixture (Wall 2000; Garrigan et al. 2005; Plagnol and Wall 2006). It has the advantage of not relying on scarce or damaged ancient DNA samples, making it more generally applicable (e.g., for putative admixture with archaic human groups without ancient DNA samples), but has the disadvantage of making simplifying assumptions about human demographic history that are generally not testable. Application of this indirect approach to European and African sequence data (Environmental Genome Project, cf. Livingston et al. 2004) found strong evidence of ancient admixture (Plagnol and Wall 2006). This was interpreted as evidence for a substantial contribution of Neandertal DNA to the modern European gene pool, though the method did not allow specific estimates of the admixture proportion.
In this paper, we expand upon the analysis of Plagnol and Wall (2006) by 1) considering a larger data set of 222 genes from the Environmental Genome Project; 2) separately analyzing data from East Asian samples to test for potential admixture with H. erectus or Homo floresiensis; and 3) obtaining point estimates and confidence intervals (CIs) for all demographic parameters, including an estimate of the ancient admixture proportion. Our analysis also corrects a misuse of the popular coalescent simulator ms (Hudson 2002) in our previous study (Plagnol and Wall 2006). This error changes the qualitative results found earlier, and an updated version of ms is available from R. Hudson's web site. See supplemental note S1, Supplementary Material online, for a description of the problem, as well as a comparison between the results of this study and those of Plagnol and Wall (2006).
Methods
We downloaded data from the Environmental Genome Project web site (http://egp.gs.washington.edu) in September 2007 and limited ourselves to autosomal genes where individuals of known ethnicity were sequenced. This left data from 222 genes, comprising a total of 5.3 MB of sequence and over 26,000 total single nucleotide polymorphisms (SNPs). We concentrated on individuals of European, Asian, and African ancestry and analyzed sequence polymorphism data from 22 European-Americans, 12 West Africans (Yoruba), and 24 East Asians (Han Chinese).
We used the polymorphism data to estimate demographic parameters for a two-population model consisting of one African and one non-African population (see fig. 1). The European and Asian data were analyzed separately. The model incorporates several features thought to be relevant to recent human history, including recent population growth (Rogers and Harpending 1992; Pluzhnikov et al. 2002; Adams and Hudson 2004) and a bottleneck in the non-African population (Tishkoff et al. 1996; Reich et al. 2001; Schaffner et al. 2005; Voight et al. 2005). In total, there are six parameters to be estimated: time of onset of population growth in population 1 (g1) and population 2 (g2), the time since the end of the bottleneck in population 2 (tb), the strength of the bottleneck (b), the time when the two populations split from each other (T), and the migration rate between the two populations (m). Our model employs a fixed bottleneck duration of 1,000 years. Previous work has shown that the effect of a bottleneck on patterns of genetic variation depends primarily on the ratio of the bottleneck duration and the bottleneck strength (e.g., Reich et al. 2001). So, coestimating both accurately is extremely difficult, and we chose to fix the bottleneck duration for computational convenience.
FIG. 1.—

Schematic of the population model used. A panmictic ancestral population (Ne = 104) splits at time T into two descendant populations 1 and 2 (both panmictic with Ne = 104). The migration rate (per individual per generation) between the descendant populations is m. Population 2 experiences a population bottleneck (a b-fold reduction in population size) 1,000 years in duration, and ending at time tb. 100-fold population growth commences at time g1 in population 1 and g2 in population 2.
We follow the same parameter estimation process as in Plagnol and Wall (2006). Specifically, we summarize the data using several summary statistics, then calculate the (composite)-likelihood of the summarized data on a grid of parameter values. We assume a mean generation time of 25 years and run 5 × 105 simulations for each grid point. The grid uses increments of 5,000 years for population growth and population bottleneck times, 20 for bottleneck strength (the factor by which the population size is reduced), 20,000 years for divergence time, and 1 × 10−5/generation for the migration rate. Simulations assume that recombination rates are constant within loci but vary across loci. (If f = ρ/θ, then the mean and variance of f across loci are 0.36 and 0.50, respectively).
Summary statistics are divided into two categories. The first category includes Tajima's D (Tajima 1989) from each population, Fu and Li's D* (Fu and Li 1993) in the non-African population, and FST (Wright 1921) between the two populations. Both D and D* are measures of the frequency spectrum, whereas FST measures the level of divergence between populations. For each parameter combination, we estimate the joint likelihood of these statistics by fitting the data to a multivariate Normal distribution. Coalescent simulations are used to estimate the vector of means and the covariance matrix.
The second category of summary statistics divides the SNPs at a locus into four categories: private SNPs in the Yoruba, private SNPs in the non-African population, shared SNPs with minor allele frequency (MAF) in the total sample ≤ 0.1, and shared SNPs with MAF > 0.1. We label these summaries s1, s2, s3, s4, respectively. For each branch of the ancestral recombination graph, all mutations on this branch will belong to a single category, so we can estimate probabilities f1, f2, f3, and f4 that a particular SNP will fall into one of the four categories defined above. Conditional on the graph and the total number of SNPs, the distribution of (s1, s2, s3, s4) is multinomial and can be estimated explicitly by averaging over the computed probabilities for each simulated ancestral recombination graph.
These two sets of summaries are correlated, but we cannot estimate their joint distribution. For computational simplicity, we calculate a composite likelihood by assuming they are independent of each other. We do this separately for each locus, then multiply them together to obtain the overall (composite) likelihood of the data. Given this assumption, it is not clear whether CIs (obtained from standard asymptotic likelihood assumptions) are conservative or not (see also the last paragraph of Methods).
We also calculate a summary of LD, S*, specifically designed to be sensitive to ancient admixture (cf. Plagnol and Wall 2006). S* looks for population-specific SNPs that are in strong LD with each other (e.g., r2 ≈ 1). If there were ancient admixture, then “archaic” SNPs on introgressed sequences would be in strong LD with each other. Simulations suggest that both the number of such SNPs and the total distance spanned by such SNPs are elevated when ancient admixture occurs (Wall 2000). S* tries to exploit both of these observations and also tries to account for the effects of intragenic recombination (see Plagnol and Wall 2006 for more details). We determine the significance of S* values from the actual data by running simulations using the previously estimated demographic parameters to obtain a distribution of S* values under the null hypothesis of no (ancient) admixture. Significantly high S* values are interpreted as departures from the null model in the direction of some unknown ancient population structure, though it is not possible to definitively tie these results to specific archaic groups such as Neandertals.
We also employ a likelihood-based method to directly estimate the amount of ancient admixture with putative archaic human groups. To do this, we modify the model shown in figure 1 to include an archaic population that is completely separated from modern humans for a fixed period of time (from 50 to 400 kya [thousand years ago] for putative Neandertal admixture and from 50 to 1,600 kya for putative H. erectus admixture). Then, more recently, this archaic population instantly mixes with the non-African population (see dashed lines in fig. 1). We then proceed as before, using the same summaries of the joint frequency spectrum to estimate demographic parameters. This time we coestimate one additional parameter, a, measuring the genetic contribution (ranging from 0% to 100%) of the archaic population to the non-African population's gene pool. We obtain both a point estimate for a (and the other parameters) as well as approximate 95% CIs by estimating profile likelihood curves. For greater precision, the profile likelihood curves were based on 10 times more simulation replicates (5 × 106). We emphasize that the estimated values of a are dependent in part on model assumptions, including the recombination model, generation time, and time when the archaic population is isolated.
To test the accuracy of our estimated CIs, we generated simulated data with known parameter values, then used our composite-likelihood machinery to estimate the seven demographic parameters in our model. We tabulated how often the estimated 95% CIs contained the true value of the parameter. Each simulation replicate consisted of 100 loci, each 25 kb in length. We ran five replicates with a = 0, g1 = 25 kya, g2 = 0 kya, tb = 30 kya, b = 160, T = 120 kya, and m = 7 × 10−5 and five replicates with a = 0.1, g1 = 35 kya, g2 = 0 kya, tb = 35 kya, b = 200, T = 60 kya, and m = 7 × 10−5. The simulated archaic population branched off from modern humans 400 kya and instantaneously admixed 50 kya, comparable with parameters appropriate for putative Neandertal admixture. Each simulation replicate took approximately 3 weeks to run on a quad-core Intel 3.16-GHz processor.
Results
First, we used the sequence polymorphism data from the Environmental Genome Project to estimate parameters of a demographic null model (fig. 1) without any ancient admixture. This null model includes one African and one non-African population, so the European and East Asian data were analyzed separately. Parameter estimates, along with approximate CIs, are given in table 1. In general, these parameter estimates are broadly consistent with estimates obtained from different data sets and with other methods (e.g., Reich et al. 2001; Pluzhnikov et al. 2002; Schaffner et al. 2005; Voight et al. 2005; Keinan et al. 2007). We find support for recent population growth (∼25–35 kya) in the Yoruba but not in the non-African populations. We also find strong evidence for a recent population bottleneck in both non-African populations, though its estimated timing (30 kya) is more recent than predicted by some versions of the Recent African Origin model, but is compatible with estimates obtained using other methods (Keinan et al. 2007; Lohmueller et al. 2009). The fact that the point estimates for tb and b are the same in both non-African populations is probably a coincidence, because we analyzed the two populations separately (see also Discussion).
Table 1.
Point Estimates of Demographic Parameters with 95% CIs in Parentheses
| African–European Data | African–East Asian Data | |
| g1 (kya) | 25 (19–33) | 35 (26–42) |
| g2 (kya) | 0 (0–0) | 0 (0–1) |
| tb (kya) | 30 (24–35) | 30 (25–33) |
| b | 160-fold reduction (123–177) | 160-fold reduction (140–185) |
| T (kya) | 120 (86–148) | 80 (60–93) |
| m (× 10−5) | 7 (5.6–8) | 5 (4–6.1) |
See text or figure 1 for parameter definitions.
Next, we compared a range of summary statistics produced by the best-fit model with the corresponding values of the summaries from the actual data (supplemental table S2, Supplementary Material online). In 21 of 22 cases, the summaries from the actual data fit within the middle 95% of the distribution of simulated values (based on 100 replicates of 222 simulated loci), suggesting that the best-fit model provides a reasonable fit to the true data. Although this is not surprising for summaries used in the composite-likelihood calculations (e.g., Tajima's D), it is also true for a summary of LD independent of the parameter estimation process (, cf. Hudson 2001). In particular, this suggests that simulations of the best-fit model have overall levels of LD that are comparable with the levels of LD found in the actual data.
We then considered the best-fit model as a null model in a hypothesis testing framework and calculated a statistic (S*) that is especially sensitive to the particular long-range LD caused by ancient admixture (Plagnol and Wall 2006). We used coalescent simulations to estimate a P value for S* at each locus and for the whole data set. The results were significant for both the European (P = 4.8 × 10−3) and the East Asian (P = 4.2 × 10−3) data. Interestingly, we also find evidence for ancient admixture in the Yoruba (P < 10−7). A list of all loci that were significant (at the 5% level) is provided in supplemental table S3, Supplementary Material online.
To test the idea of putative ancient admixture further, we modified the model shown in figure 1 to include an additional parameter, a, denoting the proportion of European (or East Asian) ancestry derived from an archaic human population (see Methods). We then calculated the profile likelihood curve for a in both non-African populations (fig. 2). We estimate admixture proportions of 14% (95% CI: 8–20%) in the European-American sample and 1.5% (95% CI: 0.5–2.5%) in the East Asian sample. In both cases, the relative log-likelihood for a = 0 (i.e., no ancient admixture) is significantly lower than the maximum likelihood (likelihood-ratio test, P < 10−3), which provides additional evidence (along with the S* results in the previous paragraph) that ancient admixture occurred. The estimates of admixture rates in Europeans are consistent with estimates of Neandertal admixture obtained from analyses of Neandertal DNA (Serre et al. 2004; Noonan et al. 2006), though with some caveats discussed below. More information on the parameter estimates obtained as a function of a is provided in supplemental table S4, Supplementary Material online. Sample ms command line parameters are given in supplemental note S5, Supplementary Material online.
FIG. 2.—
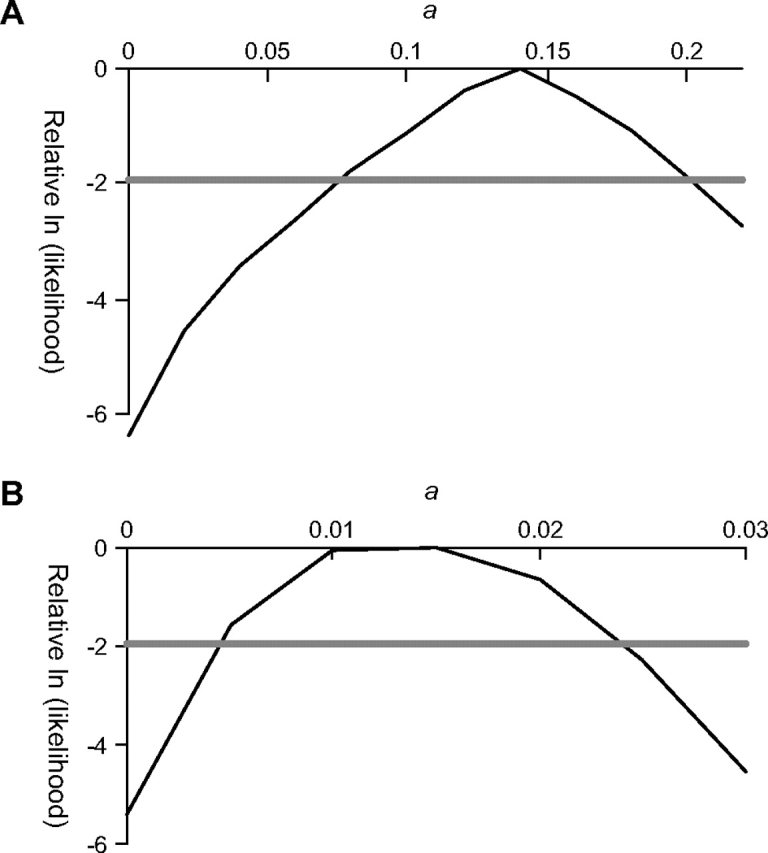
Profile likelihood curves for a (the amount of ancient admixture) in the European (a) and the East Asian (b) samples. See Methods for more details.
Discussion
Understanding demographic history has long been of interest to evolutionary biologists, but there are only a few studies that have used multi-locus genetic variation data sets to infer demographic parameters. Despite the variety of data sets used and the range of models and methods employed, different studies have come to relatively similar results. This study, along with previous ones, has found no signal of recent population growth in non-African populations but a weak signal of recent population growth in West African populations (Pluzhnikov et al. 2002; Adams and Hudson 2004). Also, as with Keinan et al. (2007), we find evidence for recent population bottlenecks in both European and Asian population samples. In both studies and both populations, these bottlenecks are estimated to have happened within the last 35,000 years, which substantially postdates the initial migration of modern humans out of Africa and into both Europe and East Asia. Although it is possible that these population bottlenecks were not related to either the migration out of Africa or the colonization of new continents, perhaps a more likely explanation is that there were actually multiple population bottlenecks and that constraining the model to have a single bottleneck leads to an “average” time over which multiple events actually took place. When Keinan et al. (2007) incorporate a two-bottleneck model, the estimated timing of the two is compatible with an initial bottleneck associated with the migration of modern humans out of Africa (80–140 kya) as well as a more recent bottleneck (16–18 kya) associated with the last glacial maximum. Given our computationally intensive parameter estimation scheme, it would be difficult for us to obtain comparable parameter estimates.
Unlike previous studies, we incorporated admixture between archaic and modern humans as an additional demographic parameter to be coestimated. Interestingly, we could exclude no admixture (i.e., exclude a = 0) in both of the non-African populations studied. To explore how reliable our composite-likelihood method is in estimating demographic parameters, we also analyzed several simulated data sets with known parameter values. In general, the estimated 95% CIs included the true parameter value roughly 95% of the time (67 out of 70 times). We note, however, that computational constraints limited us to just a few replicates (see Methods).
The question of whether Neandertals contributed to the modern European gene pool has been extensively studied, and our estimate of a in European–Americans, although on the high side, is consistent with previous estimates of Neandertal admixture rates. We caution though that these estimates are not directly comparable because the model in figure 1 does not specify the identity of the archaic human group. So, our results could potentially reflect ancient admixture with any archaic group, not just Neandertals. In addition, our and previous estimates of ancient admixture rates (Serre et al. 2004; Noonan et al. 2006) are sensitive to assumptions about the admixture time, Neandertal split time, and other basic parameter values. For example, more recent admixture times and/or older Neandertal split times will lead to lower estimated admixture rates (results not shown).
To our knowledge, the question of ancient admixture in other parts of the world has been relatively neglected by the evolutionary genetics community (but see Garrigan et al. 2005). We estimate low levels of ancient admixture in East Asia, perhaps with either Asian H. erectus or H. floresiensis. Both archaic groups are thought to have been contemporaneous with modern humans in Southeast Asia within the last 50,000 years (Roberts et al. 1994; Swisher et al. 1996; Morwood et al. 2004). Homo erectus left Africa more than 1.5 Ma (Swisher et al. 1994; Gabunia and Vekua 1995; Huang et al. 1995) and may have been completely isolated until recent contact with modern humans. This length of isolation is comparable with the length estimated between sister species of chimpanzees and baboons that can potentially hybridize in nature (Vervaecke and van Elsacker 1992; Jolly 2001). For simulations under our best-fit model, approximately 6% of loci have at least some archaic ancestry (i.e., have at least one sequence that inherited DNA from the archaic Asian population).
Our signal of ancient admixture (as measured by S*) is strongest in the West African samples, though the spotty fossil record in sub-Saharan Africa makes it difficult to speculate about potential source populations or the times and locations of admixture. This said, there was thought to be a substantial amount of hominin taxonomic diversity within Africa during the Pleistocene. We note that our simple two-population model does not allow for any population structure within continental groups, and there may be a substantial amount of unsampled population structure within Africa (e.g., between West Africans and pygmies or San, cf. Wall et al. 2008) that serves as a confounding factor. The observation that all (three) populations studied seem to have evidence for ancient admixture suggests that ancient population structure may be a common feature of all contemporary human populations, and this ancient structure may predate the initial expansion of modern humans out of Africa. Future work that estimates the lengths of the putative chunks of sequence inherited from archaic populations may help to estimate the timing of ancient admixture events.
Supplementary Material
Supplemental notes S1 and S5 and supplemental Tables S2–S4 are available at Molecular Biology and Evolution online (http://www.mbe.oxfordjournals.org/).
Acknowledgments
J.D.W. was supported in part by grants BCS-0423123 from the National Science Foundation and HG004049 from the National Institutes of Health. V.P. is a JDRF funded advanced postdoctoral fellow.
References
- Adams AM, Hudson RR. Maximum-likelihood estimation of demographic parameters using the frequency spectrum of unlinked single-nucleotide polymorphisms. Genetics. 2004;168:1699–1712. doi: 10.1534/genetics.104.030171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coop G, Bullaughey K, Luca F, Przeworski M. The timing of selection at the human FOXP2 gene. Mol Biol Evol. 2008;25:1257–1259. doi: 10.1093/molbev/msn091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu YX, Li WH. Statistical tests of neutrality of mutations. Genetics. 1993;133:693–709. doi: 10.1093/genetics/133.3.693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabunia L, Vekua A. A Plio-Pleistocene hominid from Dmanisi, East Georgia, Caucasus. Nature. 1995;373:509–512. doi: 10.1038/373509a0. [DOI] [PubMed] [Google Scholar]
- Garrigan D, Mobasher Z, Severson T, Wilder JA, Hammer MF. Evidence for archaic Asian ancestry on the human X chromosome. Mol Biol Evol. 2005;22:189–192. doi: 10.1093/molbev/msi013. [DOI] [PubMed] [Google Scholar]
- Green RE, Krause J, Ptak SE, et al. Analysis of one million base pairs of Neanderthal DNA. Nature. 2006;444:330–336. doi: 10.1038/nature05336. [DOI] [PubMed] [Google Scholar]
- Green RE, Malaspinas AS, Krause J, et al. A complete Neandertal mitochondrial genome sequence determined by high-throughput sequencing. Cell. 2008;134:416–426. doi: 10.1016/j.cell.2008.06.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang W, Ciochon R, Gu Y, Larick R, Qiren F, Schwarcz H, Yonge C, de Vos J, Rink W. Early Homo and associated artefacts from Asia. Nature. 1995;378:275–278. doi: 10.1038/378275a0. [DOI] [PubMed] [Google Scholar]
- Hudson RR. Two-locus sampling distributions and their application. Genetics. 2001;159:1805–1817. doi: 10.1093/genetics/159.4.1805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hudson RR. Generating samples under a Wright–Fisher neutral model. Bioinformatics. 2002;18:337–338. doi: 10.1093/bioinformatics/18.2.337. [DOI] [PubMed] [Google Scholar]
- Jolly CJ. A proper study for mankind: analogies from the Papionin monkeys and their implications for human evolution. Am J Phys Anthropol. 2001;33:177–204. doi: 10.1002/ajpa.10021. [DOI] [PubMed] [Google Scholar]
- Keinan A, Mullikin JC, Patterson N, Reich D. Measurement of the human allele frequency spectrum demonstrates greater genetic drift in East Asians than in Europeans. Nat Genet. 2007;39:1251–1255. doi: 10.1038/ng2116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krause J, Lalueza-Fox C, Orlando L, et al. The derived FOXP2 variant of modern humans was shared with Neandertals. Curr Biol. 2007;17:1908–1912. doi: 10.1016/j.cub.2007.10.008. [DOI] [PubMed] [Google Scholar]
- Krings M, Stone A, Schmitz RW, Krainitzki H, Stoneking M, Pääbo S. Neandertal DNA sequences and the origin of modern humans. Cell. 1997;90:19–30. doi: 10.1016/s0092-8674(00)80310-4. [DOI] [PubMed] [Google Scholar]
- Lalueza-Fox C, Rompler H, Caramelli D, et al. A melanocortin 1 receptor allele suggests varying pigmentation among Neanderthals. Science. 2007;318:1453–1455. doi: 10.1126/science.1147417. [DOI] [PubMed] [Google Scholar]
- Livingston RJ, von Niederhausern A, Jegga AG, Crawford DC, Carlson CS, Rieder MJ, Gowrisankar S, Aronow BJ, Weiss RB, Nickerson DA. Pattern of sequence variation across 213 environmental response genes. Genome Res. 2004;14:1821–1831. doi: 10.1101/gr.2730004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lohmueller KE, Bustamante CD, Clark AG. Methods for human demographic inference using haplotype patterns from genome-wide SNP data. Genetics. 2009;182:217–231. doi: 10.1534/genetics.108.099275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morwood MJ, Soejono RP, Roberts RG, et al. Archaeology and age of a new hominin from Flores in eastern Indonesia. Nature. 2004;431:1087–1091. doi: 10.1038/nature02956. [DOI] [PubMed] [Google Scholar]
- Noonan JP, Coop G, Kudaravalli S, et al. Sequencing and analysis of Neanderthal genomic DNA. Science. 2006;314:1113–1118. doi: 10.1126/science.1131412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plagnol V, Wall JD. Possible ancestral structure in human populations. PLoS Genet. 2006;2:e105. doi: 10.1371/journal.pgen.0020105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pluzhnikov A, Di Rienzo A, Hudson RR. Inferences about human demography based on multilocus analyses of noncoding sequences. Genetics. 2002;161:1209–1218. doi: 10.1093/genetics/161.3.1209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reich DE, Cargill M, Bolk S, et al. Linkage disequilibrium in the human genome. Nature. 2001;411:199–204. doi: 10.1038/35075590. [DOI] [PubMed] [Google Scholar]
- Roberts RG, Jones R, Spooner NA, Head MJ, Murray AS, Smith MA. The human colonization of Australia: optical dates of 53,000 and 60,000 years bracket human arrival at Deaf Adder Gorge, Northern Territory. Q Sci Rev. 1994;13:575–583. [Google Scholar]
- Rogers AR, Harpending H. Population growth makes waves in the distribution of pairwise genetic differences. Mol Biol Evol. 1992;9:552–569. doi: 10.1093/oxfordjournals.molbev.a040727. [DOI] [PubMed] [Google Scholar]
- Schaffner SF, Foo C, Gabriel S, Reich D, Daly MJ, Altshuler D. Calibrating a coalescent simulation of human genome sequence variation. Genome Res. 2005;15:1576–1583. doi: 10.1101/gr.3709305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serre D, Langaney A, Chech M, Teschler-Nicola M, Paunovic M, Mennecier P, Hofreiter M, Possnert G, Pääbo S. No evidence of Neandertal mtDNA contribution to early modern humans. PLoS Biol. 2004;2:e57. doi: 10.1371/journal.pbio.0020057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swisher CC, Curtis GH, Jacob T, Getty AG, Suprijo A, Widiasmoro Age of the earliest known hominids in Java, Indonesia. Science. 1994;263:1118–1121. doi: 10.1126/science.8108729. [DOI] [PubMed] [Google Scholar]
- Swisher CC, Rink WJ, Anton SC, Schwarcz HP, Curtis GH, Suprijo A, Widiasmoro Latest Homo erectus of Java: potential contemporaneity with Homo sapiens in Southeast Asia. Science. 1996;274:1870–1874. doi: 10.1126/science.274.5294.1870. [DOI] [PubMed] [Google Scholar]
- Tajima F. Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics. 1989;123:585–595. doi: 10.1093/genetics/123.3.585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tishkoff SA, Dietzsch E, Speed W, Pakstis AJ, Kidd JR, Cheung K, Bonne-Tamir B, Santachiara-Benerecetti AS, Moral P, Krings M. Global patterns of linkage disequilibrium at the CD4 locus and modern human origins. Science. 1996;271:1380–1387. doi: 10.1126/science.271.5254.1380. [DOI] [PubMed] [Google Scholar]
- Vervaecke H, van Elsacker L. Hybrids between common chimpanzees (Pan troglodytes) and pygmy chimpanzees (Pan paniscus) in captivity. Mammalia. 1992;56:667–669. [Google Scholar]
- Voight BF, Adams AM, Frisse LA, Qian Y, Hudson RR, Di Rienzo A. Interrogating multiple aspects of variation in a full resequencing data set to infer human population size changes. Proc Natl Acad Sci USA. 2005;102:18508–18513. doi: 10.1073/pnas.0507325102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wall JD. Detecting ancient admixture in humans using sequence polymorphism data. Genetics. 2000;154:1271–1279. doi: 10.1093/genetics/154.3.1271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wall JD, Cox MP, Mendez FL, Woerner A, Severson T, Hammer MF. A novel DNA sequence database for analyzing human demographic history. Genome Res. 2008;18:1354–1361. doi: 10.1101/gr.075630.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wall JD, Hammer MF. Archaic admixture in the human genome. Curr Opin Genet Dev. 2006;16:606–610. doi: 10.1016/j.gde.2006.09.006. [DOI] [PubMed] [Google Scholar]
- Wall JD, Kim SK. Inconsistencies in Neanderthal genomic DNA sequences. PLoS Genet. 2007;3:1862–1866. doi: 10.1371/journal.pgen.0030175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright S. Systems of mating. II. The effects of inbreeding on the genetic composition of a population. Genetics. 1921;6:124–143. doi: 10.1093/genetics/6.2.124. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.