

Abstract
Spinal muscular atrophy (SMA) is the leading genetic cause of infant mortality. Most SMA cases are associated with the low levels of SMN owing to deletion of Survival Motor Neuron 1 (SMN1). SMN2, a nearly identical copy of SMN1, fails to compensate for the loss of SMN1 due to predominant skipping of exon 7. Hence, correction of aberrant splicing of SMN2 exon 7 holds the potential for cure of SMA. Here we report an 8-mer antisense oligonucleotide (ASO) to have a profound stimulatory response on correction of aberrant splicing of SMN2 exon 7 by binding to a unique GC-rich sequence located within intron 7 of SMN2. We confirm that the splicing-switching ability of this short ASO comes with a high degree of specificity and reduced off-target effect compared to larger ASOs targeting the same sequence. We further demonstrate that a single low nanomolar dose of this 8-mer ASO substantially increases the levels of SMN and a host of factors including Gemin 2, Gemin 8, ZPR1, hnRNP Q and Tra2-β1 known to be down-regulated in SMA. Our findings underscore the advantages and unmatched potential of very short ASOs in splicing modulation in vivo.
Keywords: survival motor neuron (SMN), SMN1, SMN2, alternative splicing, intron 7, exon 7, ISS-N1, GC-rich sequence, antisense oligonucleotide (ASO), 8-mer ASO, SMA
Introduction
Alternative splicing is an essential process in the generation of protein diversity and has been a major contributory force to genome evolution.1 Splicing is catalyzed by a spliceosome, a complex macromolecular machine.2,3 However, control of alternative splicing rests on non-spliceosomal factors that bind to pre-mRNA sequences called exonic or intronic splicing enhancers (ESEs or ISEs) and silencers (ESSs or ISSs).4-6 Enhancer and silencer motifs promote or suppress splice-site (ss) selection, respectively. Methods to decipher critical splicing motifs are continuing to evolve.7-10 An additional regulatory role is provided by RNA structures that enforce accessibility to splicing elements, as well as bring two distantly located cis-elements into close proximity.11-14 Alternative splicing has been implicated in a growing number of human diseases.15 In this regard antisense oligonucleotides (ASOs) targeting regulatory elements have emerged as the powerful tools to modulate alternative splicing in pathological conditions.15-19
Humans have two nearly identical copies of the Survival Motor Neuron (SMN) gene: SMN1 and SMN2.20 The two SMN genes code for identical proteins, however, SMN2 predominantly generates a short transcript due to skipping of exon 7, producing a truncated SMN that is highly unstable.21 Other non-productive SMN2 mRNAs lacking exons 5 and 3 have also been reported.22 The inability of SMN2 to compensate for the loss of SMN1 results in spinal muscular atrophy (SMA), a debilitating disease of children and infants.15,16,23 Further, low levels of SMN are also associated with certain forms of amyotrophic lateral sclerosis (ALS), a progressive neurological disease that attacks the nerve cells responsible for controlling voluntary muscles.24,25 SMN is a housekeeping protein, with its most important function being the assembly of U snRNPs, the essential components of the spliceosomal machinery.26 Consistently, depletion of SMN causes tissue-specific perturbations in the repertoire of snRNAs and widespread defects in splicing.27 Other important functions associated with SMN include transcriptional regulation, telomerase regeneration and cellular trafficking.28 Consistently, higher organisms lacking SMN are not viable.28
SMN1 and SMN2 differ by a critical C to T substitution at position 6 (C6U transition) of exon 7 in SMN2.29 The significance of an additional difference within intron 7 of SMN1 and SMN2 has been reported.30 Both SMN1 and SMN2 share the same branch site for the lariat formation during the removal of intron 6.31 Yet C6U weakens the 3'-splice site (3' ss) of exon 7 in SMN2.32 Published reports suggest C6U abrogates an enhancer and/or creates a silencer.33-36 In vivo selection of the entire exon revealed three major cis-elements within exon 7.37 Two of them, “Exinct” and “3'-Cluster,” are negative regulators of splicing. They flank “Conserved Tract”, the positive element located in the middle of exon 7.29 The identification of “Conserved Tract” is in agreement with the role of the previously reported Tra2-β1-associated enhancer.38 Recently, an antisense micro-walk within exon 7 independently supported the nature of these three cis-elements.39 Several intronic cis-elements have been implicated in regulation of SMN2 exon 7 splicing as well.30,40-43
Since SMN2 is almost universally present in SMA patients, correction of SMN2 exon 7 splicing using target-specific ASOs holds the promise for cure. A number of studies have focused on use of large ASOs with bifunctional properties to restore SMN2 exon 7 inclusion.44-47 Other studies have focused on targeting large regulatory motifs or a combination of regulatory motifs to induce SMN2 exon 7 inclusion.39,42,43,48,49 However, it is not known if sequestering of a short splicing motif by a short ASO could prevent exon skipping and restore SMN2 exon 7 inclusion. Here we report the splicing-switching ability of an 8-mer ASO that binds to a unique regulatory sequence and fully restores SMN2 exon 7 inclusion. We also demonstrate that this 8-mer ASO offers a high degree of specificity and reduced off-target effect compared to larger ASOs targeting the same region. Considering short ASOs offer several advantages including low cost of synthesis, ease of chemical modifications, reduced chances of immune response, and higher probability of crossing biological barriers, our findings serve as a major step forward for splicing modulation in a genetic disease.
Results
An ultra-refined antisense microwalk revealed shortest motif for splicing correction
Recent reports have confirmed the presence of a negative context located downstream of the 5' splice site (5' ss) of SMN2 exon 7.42,43 This negative context is defined by a 15-nucleiotide cis-element, ISS-N1 that harbors two putative hnRNP A1/A2 motifs (Fig. 1A).42,43 ISS-N1 partially overlaps with an octamer sequence CUGCCAGC, which is the only GC-rich sequence in the first half of the intron 7 of human SMN. This sequence is predicted to reside in a single-stranded region sandwiched between two stem-loop structures (Fig. 1A).28 Combined with an easy accessibility and the high GC-rich content (75%), this octamer sequence has a potential to provide an ideal ASO target. However, it is not known if a short RNA:RNA duplex formed between CUGCCAGC and an ASO could displace an interacting protein and/or drastically change the negative context to reverse the splicing pattern. To explore such possibility, we performed an ultra-refined antisense microwalk downstream of the 5' ss of SMN2 exon 7. All ASOs used in our study incorporated 2'-O-methyl modification and phosphorothioate backbone (abbreviated as “2OMePS”), a widely used RNA modification with proven stability in vivo.16,18,19 We performed our experiments in commercially available SMA type I patient cells (GM03813), which serves as an ideal system for testing of splicing-correcting compounds in the context of the disease caused by the lack of SMN1.39,42,43
Figure 1.

Untra-refined antisense microwalk to identify the shortest stimulatory ASO. (A) Diagrammatic representation of ASOs targeting sequences upstream of ISS-N1. Exon 7 is boxed and the first 24 residues of human SMN intron 7 are shown. Numbering starts from the first position of intron 7. The 5' ss of exon 7 is indicated by a vertical arrow. ASOs blocking different regions are shown as horizontal bars. Sequences of these ASOs are given in Table 1. Boundary of ISS-N1 is demarcated.42 hnRNP A1 motifs are indicated.43 Green bars represent ASOs that promote SMN2 exon 7 inclusion. Intensity of green color reflects the strength of stimulatory effect. Tan bars represent ASOs that have no effect on SMN2 exon 7 inclusion. Area highlighted in pink represents the only GC-rich sequence in the first half of human intron 7. Area highlighted in light blue represents the core sequence of the antisense target. Right panel shows the relative positioning of ISS-N1, GC-rich sequence in the context of predicted RNA structure. Green bar represents 3UP8, the shortest ASO to stimulate SMN2 exon 7 inclusion. (B) Splicing pattern of endogenous SMN2 in SMA type I patient fibroblasts (GM03813) treated with different ASOs. Cells were transfected with 20 nM of 2OMePS ASOs and the total RNA for splicing assay was isolated 24 h post transfection. Results were analyzed as described earlier.35 3UP8 was the shortest ASO to show stimulatory response (highlighted by green box).
Our ultra-refined antisense microwalk used four groups of ASOs of varying sizes. The ASOs from each group sequestered 0, 1, 2 or 3 residues upstream of ISS-N1. Accordingly, ASOs were named as F, 1UP, 2UP and 3UP, followed by a number representing the size of the ASO (Table 1). To discriminate between the most and the least efficient ASOs, the antisense microwalk was performed at four concentrations: 1 nM, 10 nM, 50 nM and 100 nM (Table 1). Figure 1 shows the splicing pattern of representative ASOs performed at 20 nM. We observed a decrease in the antisense effect on exon 7 inclusion with a decrease in the size of F, 1UP and 2UP ASOs (Table 1, Fig. 1B). However, the results were drastically different with 3UP ASOs: shortening of ASOs from 14 nucleotides to 8 nucleotides produced no significant changes in their stimulatory effects on exon 7 splicing (Fig. 1B). However, the stimulatory effect drastically decreased when ASO size was further reduced from 8 nucleotides to 7 nucleotides. Hence, we conclude that the shortest ASO to effectively restore SMN2 exon 7 inclusion was 3UP8, an 8-mer ASO that sequestered the entire octamer sequence, CUGCCAGC, discussed above. Remarkably, 3UP8 was able to fully restore SMN2 exon 7 inclusion at a relatively low concentration of 50 nM (Table 1).
Table 1.
Effect of ASOs on skipping of SMN2 exon 7 in SMA type I fibroblasts (GM03183)
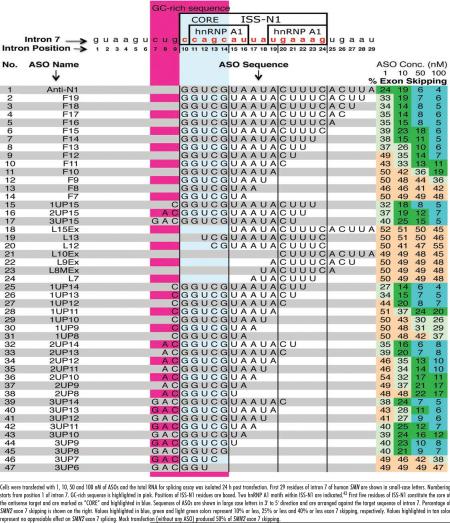 |
The finding that 3UP8 restores SMN2 exon 7 inclusion marks the discovery of the shortest ASO among ∼200 ASOs tested thus far in SMA patient cells (Table 1).39,42-49 In addition to revealing the shortest stimulatory ASO, the ultra-refined microwalk was able to accurately define the first five residues (CCAGC) of ISS-N1 as the core sequence of the antisense target. Our finding of core sequence brings a parallel with seed sequence required for miRNA and siRNA response.50 Similar to seed sequence, sequestering of the core sequence was essential but not sufficient for the antisense response. In addition to sequestration of core sequence, sequestration of additional three residues (CUG) upstream of ISS-N1 was found to be essential to obtain the shortest stimulatory ASO. In the absence of the sequestration of CUG residues, an 11-nucleotide or longer target was required for realizing the stimulatory response (Table 1). Underscoring the overlapping nature of splicing cis-elements and their hard-to-predict accessibility during the dynamic process of splicing, we found no direct correlation between the size of ASOs and their stimulatory response. Similarly, we found no direct correlation between sequestration of any of the individual hnRNP A1 motifs and the level of stimulatory response. For instance, F10 and L13 fully sequestered the 1st and the 2nd hnRNP A1 motifs, respectively, and yet did not produce any significant stimulatory response even at higher concentration of 100 nM (Table 1). On the other hand, 3UP8 restored SMN2 exon 7 by sequestering an 8-mer motif that only partially overlaps the first hnRNP A1 motif.
Antisense effect is specific to base paring with the target
Having discovered that a short intronic motif could be targeted for splicing modulation of endogenous pre-mRNA, we next examined the efficacy and specificity of short ASO in SMN2 minigene system. Here HeLa cells were co-transfected with the minigene (0.1 μg) and an ASO of interest (50 nM) and the effect on splicing was accessed by RT-PCR. As shown in Figure 2B, the ASO effect on splicing of minigene-derived exon 7 was consistent with the results for the endogenous SMN2 with 3UP8 being the shortest ASO to fully restore exon 7 inclusion. To compare the target specificity between long and short ASOs, we generated a mutant minigene, SMN2/64A. This minigene has a single C to A substitution at the first position of ISS-N1, hence has capability to weaken the RNA:RNA duplex between the antisense and the target (Fig. 2A). Indeed, our shortest ASO (3UP8) lost all its stimulatory response in SMN2/64A. As expected, a mutant 8-mer ASO (3UP8/64A) that reinstated the base pairing with the mutated target fully restored exon 7 inclusion in SMN2/64A minigene; at the same time 3UP8/64A had no stimulatory effect on splicing of SMN2 minigene (Fig. 2B). Note that the stimulatory impact of 3UP8/64A in SMN2/64A minigene was realized despite the fact that the C-to-A mutation reduced the GC content of the target from 75% to 62.5%. Unlike 8-mer ASOs, all four 15-mer ASOs we used effectively restored exon 7 inclusion in SMN2/64A. These results clearly suggest that an increase in ASO size could have drastic (negative) consequences on the specificity of the antisense response since longer ASO appear to be more “tolerant” to single-nucleotide mismatches.
Figure 2.

Antisense effect is specific to its target sequence. (A) Diagrammatic representation of intron 7 of SMN2 minigene and its mutant, SMN2/64A. Numbering starts from the first position of human SMN intron 7. ISS-N1 sequence is highlighted in gray. Mutated residue is highlighted in black. (B) Effect of ASOs on splicing of SMN2 minigene and its mutant, SMN2/64A. HeLa cells were transfected with 50 nM of a given ASO and 0.1 μg of minigene in a 24-well plate. Splicing was determined 24 h after transfection. Results were analyzed as described earlier.35
The shortest stimulatory ASO has no off-target effect on other SMN2 exons
To examine the possible off-target effect of ASOs that promote SMN2 exon 7 inclusion in GM03813 cells, we focused on splicing of SMN2 exons 3 and 5. These exons are known to undergo alternative splicing;22 and therefore, have a potential to regulate the levels of full-length SMN. We have earlier reported that a 5 nM concentration of Anti-N1 had no detectable effect on splicing pattern of SMN2 exons 3 and 5.42 Here we increased the ASO concentration to 20 and 100 nM. We chose to use Anti-N1, F14 and 3UP8 to represent the longest, the intermediate and the shortest stimulatory ASO, respectively. F8 served as a negative control.
We started with the amplification of endogenous SMN2-spliced products using a pair of primers located within exons 4 and 8. This primer combination provided an added advantage of simultaneous detection of skipping of exons 5 and 7. To compare the amount of the spliced products in broad size range, we used an end-labeled primer. As shown in Figure 3A, all three functional ASOs: Anti-N1, F14 and 3UP8, were highly efficient in promoting exon 7 inclusion at 20 nM concentration, while F8 produced no effect. None of the ASOs appeared to have a pronounced effect on splicing of exon 5 at both ASO concentrations. At the same time, Anti-N1, F14 and 3UP8 caused a decrease in the amount of the co-exclusion product (mRNAs lacking both exons 5 and 7), especially at 100 nM (Fig. 3A). Skipping of exon 5 was separately measured with another primer pair that annealed to exons 4 and 6. We found no significant difference on effect on exon 5 splicing in cells treated with any of the tested ASOs (Fig. 3B). To monitor skipping of SMN2 exon 3, we used a forward primer that annealed to exon 1 and the reverse primer that annealed to exon 4. As shown in Figure 3C, F14 and 3UP8 produced no detectable change in the level of exon 3 skipping as compared to the mock-transfected sample. Contrary to this, Anti-N1 produced a substantial increase in SMN2 exon 3 skipping at 100 nM (Fig. 3C). Mechanism by which Anti-N1 elicits this off-target effect is not understood, although it clearly underscores the disadvantage of long ASOs as the therapeutic molecules.
Figure 3.

Effect of ASOs on alternative splicing of different exons of endogenous SMN2. SMA type I patient fibroblasts (GM03813) were transfected with 20 or 100 nM of selected ASOs in 6-well plates. The total RNA for splicing assay was isolated 24 h post transfection. Spliced products were amplified by RT-PCR with one of the primers being end-labeled. Annealing positions of primers are shown by bars. (A) Left panel depicts the diagrammatic representation of expected spliced products. Right panel shows the results of RT-PCR. Exon 7 skipped, exon 5 skipped and co-excluded products are marked. (B) Left panel depicts the diagrammatic representation of expected spliced products due to skipping of exon 5. Right panel shows the results of RT-PCR. Exon 5 included and exon 5 skipped products are marked. (C) Left panel depicts the diagrammatic representation of expected spliced products due to skipping of exon 3. Right panel shows the results of RT-PCR. Exon 3 included and exon 3 skipped products are marked.
Restoration of SMN levels by shortest ASO in SMA patient cells
The next goal of our study was to determine whether the correction of SMN2 exon 7 splicing by 3UP8 resulted in SMN protein increase in patient cells. In particular, we wanted to compare the stimulatory effect of 3UP8 with a longer ASO, Anti-N1. F8 served as the negative control. The experiments were performed with 40 nM of a given ASO and protein levels were determined 48 hours after transfection. Simultaneously, we monitored the levels of SMN2 exon 7 inclusion. As shown in Figure 4A, mock-treatment (mock) or treatment with F8 did not produce any change in SMN levels (left) as well as in levels of SMN2 exon 7 inclusion (right). In contrast, treatment with 3UP8 resulted in a substantial upregulation of SMN levels (Fig. 4A, left) and SMN2 exon 7 inclusion (Fig. 4A, right). Significantly, the effect of 3UP8 on SMN levels was comparable to the effect produced by Anti-N1 treatment. To determine whether increase in SMN levels in ASO-treated patient cells was accompanied by a change in cellular metabolism, we performed western blot for a number of proteins that are generally downregulated in SMA. As shown in Figure 4A (left), treatment of patient cells with 3UP8 was accompanied by a marked increase in the levels of Gemin 2 and Gemin 8. These factors are associated with SMN complex, a macromolecule essential for the housekeeping role of snRNP biogenesis.26,27 We also observed an increase in the levels of ZPR1 (Fig. 4A), another SMN-interacting protein, reduced expression of which is associated with the progressive loss of motor neurons.51,52 Interestingly, the correction of splicing by 3UP8 resulted in increase of levels of splicing factors Tra2-β1 and hnRNP Q. Tra2-β1 and hnRNP Q1 isoform have been shown to promote SMN2 exon 7 inclusion and are generally downregulated in SMA.38,53-55 Thus, our findings suggest that SMN may be a part of a positive feedback loop that provides signals to increase the levels of different splicing factors.
Figure 4.

Effect of the shortest stimulatory ASO (3UP8) on levels of cellular proteins in SMA patient cells. (A) Western blot showing the effect of different ASOs. SMA type I patient cells (GM03813) were transfected with 40 nM of selected ASOs and cells were harvested 48 h after transfection. Left panel represents the results of western blot of different proteins, whereas the right panel represents the results of RT-PCR. (B) Time course of 3UP8 effect on the levels of SMN and other factors. SMA type I patient cells (GM03813) were transfected with a single dose of 40 nM of 3UP8 and harvested after every 24 h for six days. Left panel represents the results of western blot of different proteins, whereas the right panel represents the results of RT-PCR.
Cell division and degradation of ASOs are bound to attenuate the stimulatory effect of ASOs with respect to time. To determine the sustainability of a single 3UP8 treatment, we performed a time course analysis in which levels of SMN and other factors were examined at 24 hr intervals for six days. Simultaneously, we also monitored ASO effect on SMN2 exon 7 splicing. A single dose of 40 nM of 3UP8 was sufficient to sustain the increased levels of SMN for five days (Fig. 4B, left). Effect on other proteins varied with respect to time. For example, levels of Gemin 2, Gemin 8, ZPR1 and hnRNP Q peaked at day three but started decreasing after that, whereas the levels of Tra2-β1 reached maximum on day three and remained high till day five (Fig. 4B, left). As for the effect on exon 7 splicing, levels of exon inclusion remained high for two days followed by a graduate decrease (Fig. 4B, right). It is possible that increase in Tra2-β1 and hnRNP Q levels contributed to exon 7 inclusion.
SMA patient cells are usually deficient in SMN-containing sub-nuclear bodies or gems. To test whether increase in SMN levels can induce its nuclear accumulation in gems, we performed immunofluorescence analysis of 3UP8 treated GM03813 cells. Here F8 was used as a negative control. As shown in Figure 5, transfection of cells with 3UP8 was accompanied by a profound increase in the number of gems containing SMN. We also observed that 3UP8 but not F8 resulted in increase and redistribution to gems of SMN-interacting protein, ZPR1 (Fig. 5). It is known that ZPR1 is required for accumulation of SMN in these sub-nuclear structures.56,57 Our finding that 3UP8 is able to increase the number of gems confirms a proper assembly of SMN in the nucleus. This also marks the first evidence of a stimulatory response by a very short ASO leading to the massive macromolecular reorganization in the nucleus of a patient cell.
Figure 5.

Confocal images confirming that treatment with short ASO (3UP8) promotes nuclear accumulation of SMN in SMA patient cells. The fibroblasts from SMA type I patient (GM03813) were cultured on coverslips and transfected with 40 nM of F8 (control) or 3UP8 CY3-labeled ASOs. Cells were fixed 48 h after transfection and stained with anti-SMN (Green) and anti-ZPR1 (Red) antibodies. Cells transfected with ASOs were detected by Cy3 fluorescence and presented in pseudo-color (Cyan). DNA was stained with DAPI (blue). The scale bar is 10 mm.
Discussion
SMA is the second most common genetic disorder of children and infants caused by insufficient levels of SMN protein due to the loss of the SMN1 gene. Presence of a defective gene, SMN2, makes SMA a unique genetic disease that could be avoided and possibly cured by redirecting SMN2 exon 7 splicing. Among several approaches to correct aberrant splicing, an ASO-based approach provides a superior alternative due to the anticipated target specificity. Size of an ASO is an important determinant in success of an ASO-based strategy. Despite the expected advantages, it is not known if very short ASOs could anneal to the target and bring desired changes in a sequence-specific manner, particularly at the low nanomolar concentrations.
Here we report an 8-mer ASO (3UP8) as the shortest ASO to correct the aberrant splicing of SMN2 exon 7 in SMA patient cells. To the best of our knowledge, this is the first report in which an 8-mer ASO is able to effectively correct aberrant splicing in a patient cell line. Identification of this ASO was achieved through a systematic approach of ultra-refined antisense microwalk in an intronic region adjacent to the 5' ss of exon 7. The 8-mer ASO exerts its stimulatory effect through binding to a GC-rich sequence (CUGCCAGC) spanning from the 7th to 14th position of intron 7 (Fig. 1). Underscoring an evolutionary significance, this intronic region is not conserved between human and mice.42 CUGCCAGC target sequence seems to be highly accessible since low nanomolar concentrations of 3UP8 fully restores SMN2 exon 7 inclusion (Table 1). Consistently, the predicted secondary structure puts this target sequence in an internal loop flanked by terminal stem-loop structures (Fig. 1A).28
Our ultra-refined antisense microwalk with about 50 ASOs captured relative strength of multiple antisense targets that differed by a single nucleotide. As a consequence, it also revealed positions of high significance, wherein sequestering of the last five residues (CCAGC) of the GC-rich target was found to be absolutely required for the stimulatory response on SMN2 exon 7 inclusion (Table 1). Hence CCAGC residues could be considered as the core motif, analogous to the seed sequence of the micro-RNA target.50 However, unlike microRNAs that require assembly of a RNA-induced silencing complex (RISCs) on an 18-mer or longer sequence, our antisense response is solely based on the short RNA:RNA duplex. Based on the published reports, it is highly unlikely that protein factors could form a stable complex with a short RNA:RNA duplex. However, we cannot rule out the possibility of secondary contacts that might have been affected.
The GC-rich target described here does not resemble any known binding motif of a splicing factor, although, it overlaps with the first five residues of ISS-N1, an intronic element that harbors two putative hnRNP A1 binding sites.42,43 The C residue at the first position (1C) of ISS-N1 is not the part of hnRNP A1 motif, yet sequestering of this position was found to be absolutely necessary for the antisense response. Further, several ASOs that did not sequester 1C produced an inhibitory effect even though they fully sequestered both hnRNP A1 motifs (data not shown). These results suggest that the stimulatory response of ASOs is a combination of effects not necessarily caused by blocking of hnRNP A1 motifs.
Various mechanisms may account for the stimulatory response exerted by 3UP8. The most obvious among them is the strong target affinity of 3UP8 compared to an inhibitory factor that may transiently interact with the same target during the dynamic process of splicing. It is also possible that the RNA:RNA duplex formed between 3UP8 and the GC-rich target helps bring a subtle change in the RNA structure in the vicinity of the 5' ss. Such a structural change may help improve U1 snRNP recruitment and/or the 5' ss recognition. We have previously shown that recruitment of U1 snRNP at the 5' ss of exon 7 is a limiting step for SMN2 exon 7 inclusion.13,29 Our results also suggest that the catalytic core of splicing is not affected by a RNA:RNA duplex formed between an ASO and its target immediately downstream of the U1 snRNA binding site. However, dissociation of ASO from the target sequence through a helicase reaction during the catalytic core formation could not be ruled out. In this scenario, the same antisense will be recycled several times on different SMN2 pre-mRNAs. This is an obvious advantage of short ASOs in an ASO-based therapy because frequency of drug (ASO) administration could be minimized.
Our work underscores the high target specificity of very short ASOs during RNA:RNA interactions. For instance, a single mismatch in the middle of the target caused a drastic decrease in the stimulatory response by 3UP8. On the contrary, longer ASOs tolerated this mismatch mutation due to a large duplex formed between an ASO and the target. Tolerance of mismatched mutations provides an inherent drawback and therapeutic risk associated with longer ASOs. Consistently, high concentrations of a 20-mer ASO (Anti-N1) targeting intron 7 produced an off-target effect on SMN2 exon 3 splicing, whereas identical concentrations of 3UP8 had no effect (Fig. 3C).
Owing to the high target specificity and an efficient antisense response by a short ASO, 3UP8 increased levels of SMN in SMA patient cells. It also restored levels of several key proteins that are generally downregulated in SMA (Fig. 4B). These include factors involved in snRNP biogenesis (Gemin 2 and Gemin 8) and RNA splicing (Tra2-β1 and hnRNP Q).38,55 hnRNP Q proteins have been also implicated in other aspect of RNA metabolism, such as RNA transcription, translation, stability and trafficking.53-55 Increase in ZPR1 in 3UP8-treated cells suggests that a short ASO is capable of restoring SMN-interacting factors, reduced expressions of which are associated with the progressive loss of motor neurons.56,57 Despite a gradual decrease in the levels of SMN2 exon 7 inclusion after two days, high SMN levels were maintained for five days after single treatment with 40 nM 3UP8. These findings suggest a substantially longer half-life of SMN owing to the stabilization of SMN through association with itself and/or with other factors. Consistent with the restoration of the SMN-interacting partners, 3UP8-treated cells showed increased numbers of sub-nuclear bodies (gems) in the nucleus (Fig. 5).
Currently SMA has no cure, although several small compounds capable of increasing levels of SMN in SMA have been identified.23 Mechanisms of actions and side effects of these compounds remain unknown. Earlier ASO-based strategies promised high target specificity and focused on large ASOs in the anticipation that small motifs could not be targeted by small ASOs.42-49 In general, literature is replete with studies using 15-mer or longer ASOs for modulation of alternative splicing. Our work provides the first evidence of high target specificity for a very short ASO and sets a unique precedence of pathogenic splicing modulation by RNA molecules less than half the size of the most reported ASOs. Compared to large ASOs that carry the inherent risk of partial sequestration of different kinds of small motifs and tolerate mismatch mutations, we show that the stimulatory activity of a small ASO is exclusively dependent upon the perfect match with a single motif that is uniquely located within an accessible region of a negative context. Short ASOs offer additional advantages including low cost of synthesis, ease of chemical modifications, reduced chances of immune response, and higher probability of crossing biological barriers.58 When promotion of exon inclusion is the goal, a short intronic target brings the desired benefits of noninterference with nuclear export and translation. Hence, our findings represent further advancement towards an ASO-based therapy of SMA and bring a unique perspective to our understanding of splicing regulation of a defective gene associated with a major genetic disease of children and infants.
Materials and Methods
Plasmids, cells and ASOs
Construction of SMN2 minigene is described earlier.35 Construct SMN2/64A contains a C-to-A mutation in SMN2 minigene and was generated by PCR using Phusion High-Fidelity DNA polymerase (New England Biolabs). HeLa cells were obtained from the American Type Culture Collection and were cultured in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum (FBS). Primary fibroblast cell line from SMA type I patient (Repository number GM03813) and a healthy control (Repository number AG06814) were obtained from Coriell Cell Repositories. These cells were maintained in MEM supplemented with 2 mM GlutaMAX-I and 15% FBS. All tissue culture media and supplements were purchased from Invitrogen. RNA ASOs used in our study were synthesized by Dharmacon Inc., These ASOs incorporated 2'-O-methyl modification and phosphorothioate backbone (2OMePS) as described earlier.42
Transfections and in vivo splicing assays
Transient transfections of cells with plasmid DNA and/or with ASOs were performed using Lipofectamine 2000 (Invitrogen) following the manufacturer's recommendations. Briefly, cells were plated 24 h prior to transfection so that their density on the day of transfection was ∼80%. Oligonucleotide concentration ranged from 1 to 100 nM. In a given experiment, the total amount of oligonucleotide was maintained constant by adding the control oligonucleotide (5'TGA CAT CCA CTT TGC CTT TCT CTC3'). Total RNA was isolated at the indicated time points using Trizol reagent (Invitrogen). To generate cDNA reverse-transcription was carried out using SuperScript III Reverse Transcriptase (Invitrogen) and Oligo (dT) primer (Invitrogen). 1 μg and 3 μg of total RNA were used per 20 μl of reaction for amplification of minigene-specific and endogenous spliced products, respectively. Minigene-specific spliced products were identified using Taq polymerase (Invitrogen) and the pair of primers P1 and P2 for SMN2 minigenes.35 For PCR amplification of endogenous exons the following primer combinations were used: N-24 and P2 for SMN exon 7;42 5'Ex4 hSMN-RP (5'GGC CAA GAC TGG GAC CAG G3') and 3'SPL8 (5'TGG TGT CAT TTA GTG CTG CT3') or 5'Ex4last1192 (5'AGG GCC AAG ACT GGG ACC AGG AAA GG3') and 3'Exon6SMN (5'CAT ATA ATA GCC AGT ATG ATA GCC3') for SMN exon 5; 5'Exon1SMN (5'CTG TTC CGG CGC GGC ACA GGC CAG3') and 3'Exon4SMN (5'TCA CTT TCA TCT GTT GAA ACT TGG3') for SMN exon 3. PCR reactions were performed either in the presence of a trace amount of [α-32P] dATP (3,000 Ci/mmole) or with one of the primers being 5'end-labeled. Primers were end-labeled using [γ-32P]ATP (3,000 Ci/mmole) and T4 polynucleotide kinase (New England Biolabs), followed by phenol:chloroform extraction and spinning through a Micro Bio-spin 30 Chromatography Column (Bio-Rad) to get rid of unincorporated [γ-32P] ATP. Analysis and quantifications of spliced products were performed using a FPL-5000 Image Reader and Multi Gauge software (Fuji Photo Film Inc.,). Results were confirmed by at least three independent experiments.
Western blot analysis
Whole-cell extracts were prepared using ice-cold RIPA buffer (Boston BioProducts) supplemented with protease inhibitor cocktail (Roche Applied Science). Protein concentrations were determined using BSA Protein Assay Kit (Thermo Scientific). Cell extracts were resolved on a 10% (w/v) SDS-PAGE gel and transferred onto polyvinylidene fluoride (BioTrace PVDF) membrane (Pall Life Sciences). The following primary and secondary antobodies were used: mouse monoclonal anti-SMN (BD Transduction Laboratories), mouse monoclonal anti-hnRNP Q (Sigma), rabbit polyclonal anti-Tra2 (Abcam), rabbit polyclonal anti-β-actin (Sigma), horseradish-peroxidase-conjugated secondary antibodies against mouse (Sigma) and rabbit (Jackson Immuno Research). Mouse monoclonal anti-Gemin 2 and anti-Gemin 8 antibodies were kindly provided by Dr. Gideon Dreyfuss. Mouse monoclonal anti-ZPR was the same as described earlier.51,57 In most cases, the membranes were stripped (15 min at room temperature) using Restore western Blot Stripping Buffer (Thermo Scientific) and re-probed. The membranes were scanned using UVP BioSpectrum AC Imaging System (UVP). Signal intensities were quantified using Vision works LS Image Acquisition and Analysis software (UVP). Results were confirmed by at least three independent experiments.
Immunofluorescence analysis
Patient fibroblasts (GM03813) were cultured on coverslips and transfected with 40 nM of F8 (control) and 3UP8 CY3-labeled ASOs using Lipofectamine 2000 as described above. Cell were harvested 48 hr post-transfection, washed, fixed and processed for immunofluorescence.51,57 Double labeling (ZPR1/SMN) was carried out by sequential incubations with anti-SMN (clone 8, BD Transduction laboratories), Alexa 633-conjugated anti-mouse IgG secondary antibody (Molecular Probes) and then with FITC-conjugated LG1 (anti-ZPR1).51,57 The cover slips were mounted on slides using Vectashield with DAPI (Vector Laboratories) and examined by indirect immunoflourescence using LSM510 confocal microscope (Carl Ziess) equipped with 405 nm diode laser.
Acknowledgements
This work was supported by grants from United States National Institutes of Health (R01NS055925 and R21NS055149) and Muscular Dystrophy Association, USA (MDA USA) to R.N.S. L.G. was supported by a grant from MDA, USA. R.N.S. acknowledges support of Salsbury Endowment at the Iowa State University. Authors would like to thank Ms. Mary Ann deVries for providing editorial assistance and valuable comments on the manuscript. Authors acknowledge their gratitude to Dr. Gideon Dreyfuss for providing antibodies against Gemin 2 and Gemin 8.
Abbreviations
- SMA
spinal muscular atrophy
- SMN
survival motor neuron
- ISS-N1
intronic splicing silencer N1
- C6U
a C-to-T mutation at the 6th position of SMN2 exon 7
- ASO
antisense oligonucleotide
- Anti-N1
a 20-mer stimulatory ASO
- 3UP8
an 8-mer stimulatory ASO
References
- 1.Xing Y, Lee C. Relating alternative splicing to proteome complexity and genome evolution. Adv Exp Med Biol. 2007:623:42–9. doi: 10.1007/978-0-387-77374-2_3. [DOI] [PubMed] [Google Scholar]
- 2.Nilsen TW. The spliceosome: the most complex macromolecular machine in the cell. Bioessays. 2003;25:1147–9. doi: 10.1002/bies.10394. [DOI] [PubMed] [Google Scholar]
- 3.Matlin AJ, Moore MJ. Spliceosome assembly and composition. Adv Exp Med Biol. 2007;623:14–35. doi: 10.1007/978-0-387-77374-2_2. [DOI] [PubMed] [Google Scholar]
- 4.Hertel KJ. Combinatorial control of exon recognition. J Biol Chem. 2008;283:1211–5. doi: 10.1074/jbc.R700035200. [DOI] [PubMed] [Google Scholar]
- 5.Lin S, Fu XD. SR proteins and related factors in alternative splicing. Adv Exp Med Biol. 2007;623:107–22. doi: 10.1007/978-0-387-77374-2_7. [DOI] [PubMed] [Google Scholar]
- 6.Martinez-Contreras R, Cloutier P, Shkreta L, Fisette JF, Revil T, Chabot B. hnRNP proteins and splicing control. Adv Exp Med Biol. 2007;623:123–47. doi: 10.1007/978-0-387-77374-2_8. [DOI] [PubMed] [Google Scholar]
- 7.Singh RN. Unfolding the mystery of alternative splicing through a unique method of in vivo selection. Front Biosci. 2007;12:3263–72. doi: 10.2741/2310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chasin LA. Searching for splicing motifs. Adv Exp Med Biol. 2007;623:85–106. doi: 10.1007/978-0-387-77374-2_6. [DOI] [PubMed] [Google Scholar]
- 9.David CJ, Manley JL. The search for alternative splicing regulators: new approaches offer a path to a splicing code. Genes Dev. 2008;22:279–85. doi: 10.1101/gad.1643108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang Z, Burge CB. Splicing regulation: from a parts list of regulatory elements to an integrated splicing code. RNA. 2008;14:802–13. doi: 10.1261/rna.876308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Buratti E, Baralle FE. Influence of RNA secondary structure on the pre-mRNA splicing process. Mol Cell Biol. 2004;24:10505–14. doi: 10.1128/MCB.24.24.10505-10514.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Graveley BR. Mutually exclusive splicing of the insect Dscam pre-mRNA directed by competing intronic RNA secondary structures. Cell. 2005;123:65–73. doi: 10.1016/j.cell.2005.07.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Singh NN, Singh RN, Androphy EJ. Modulating role of RNA structure in alternative splicing of a critical exon in the spinal muscular atrophy genes. Nucleic Acids Res. 2007;35:371–89. doi: 10.1093/nar/gkl1050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hiller M, Zhang Z, Backofen R, Stamm S. Pre-mRNA secondary structures influence exon recognition. PLoS Genet. 2007;3:204. doi: 10.1371/journal.pgen.0030204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cooper TA, Wan L, Dreyfuss G. RNA and disease. Cell. 2009;136:777–93. doi: 10.1016/j.cell.2009.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Garcia-Blanco MA. Alternative splicing: therapeutic target and tool. Prog Mol Subcell Biol. 2006;44:47–64. doi: 10.1007/978-3-540-34449-0_3. [DOI] [PubMed] [Google Scholar]
- 17.Madsen EC, Morcos PA, Mendelsohn BA, Gitlin JD. In vivo correction of a Menkes disease model using antisense oligonucleotides. Proc Natl Acad Sci USA. 2008;105:3909–14. doi: 10.1073/pnas.0710865105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.van Deutekom JC, Janson AA, Ginjaar IB, Frankhuizen WS, Aartsma-Rus A, Bremmer-Bout M, et al. Local dystrophin restoration with antisense oligonucleotide PRO051. N Engl J Med. 2008;357:2677–86. doi: 10.1056/NEJMoa073108. [DOI] [PubMed] [Google Scholar]
- 19.Bauman J, Jearawiriyapaisarn N, Kole R. Therapeutic potential of splice switching oligo-nucleotides. Oligonucleotides. 2009;19:1–14. doi: 10.1089/oli.2008.0161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lefebvre S, Burglen L, Reboullet S, Clermont O, Burlet P, Viollet L, et al. Identification and characterization of a spinal muscular atrophy-determining gene. Cell. 1995;80:1–5. doi: 10.1016/0092-8674(95)90460-3. [DOI] [PubMed] [Google Scholar]
- 21.Vitte J, Fassier C, Tiziano FD, Dalard C, Soave S, Roblot N, et al. Refined characterization of the expression and stability of the SMN gene products. Am J Pathol. 2007;171:1269–80. doi: 10.2353/ajpath.2007.070399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hsieh-Li HM, Chang JG, Jong YJ, Wu MH, Wang NM, Tsai CH, Li H. A mouse model for spinal muscular atrophy. Nat Genet. 2002;24:66–70. doi: 10.1038/71709. [DOI] [PubMed] [Google Scholar]
- 23.Wirth B, Brichta L, Hahnen E. Spinal muscular atrophy and therapeutic prospects. Prog Mol Subcell Biol. 2006;44:109–32. doi: 10.1007/978-3-540-34449-0_6. [DOI] [PubMed] [Google Scholar]
- 24.Corcia P, Camu W, Praline J, Gordon PH, Vourch P, Andres C. The importance of the SMN genes in the genetics of sporadic ALS. Amyotroph Lateral Scler. 2009;6:1–5. doi: 10.3109/17482960902759162. [DOI] [PubMed] [Google Scholar]
- 25.Turner BJ, Parkinson NJ, Davies KE, Talbot K. Survival motor neuron deficiency enhances progression in an amyotrophic lateral sclerosis mouse model. Neurobiol Dis. 2009 doi: 10.1016/j.nbd.2009.03.005. [Epub ahead of print] PMID: 19332122. [DOI] [PubMed] [Google Scholar]
- 26.Gabanella F, Butchbach ME, Saieva L, Carissimi C, Burghes AH, Pellizzoni L. Ribonucleoprotein assembly defects correlate with spinal muscular atrophy severity and preferentially affect a subset of spliceosomal snRNPs. PLoS ONE. 2007;2:921. doi: 10.1371/journal.pone.0000921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhang Z, Lotti F, Dittmar K, Younis I, Wan L, Kasim M, Dreyfuss G. SMN deficiency causes tissue-specific perturbations in the repertoire of snRNAs and widespread defects in splicing. Cell. 2008;133:585–600. doi: 10.1016/j.cell.2008.03.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Singh NN, Androphy EJ, Singh RN. The regulation and regulatory activities of alternative splicing of the SMN gene. Crit Rev Eukaryote Gene Expr. 2004;14:271–85. doi: 10.1615/critreveukaryotgeneexpr.v14.i4.30. [DOI] [PubMed] [Google Scholar]
- 29.Singh RN. Evolving concepts on human SMN pre-mRNA splicing. RNA Biol. 2007;4:7–10. doi: 10.4161/rna.4.1.4535. [DOI] [PubMed] [Google Scholar]
- 30.Kashima T, Rao N, Manley JL. An intronic element contributes to splicing repression in spinal muscular atrophy. Proc Natl Acad Sci USA. 2007;104:3426–31. doi: 10.1073/pnas.0700343104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Scholl R, Marquis J, Meyer K, Schümperli D. Spinal muscular atrophy: position and functional importance of the branch site preceding SMN exon 7. RNA Biol. 2007;4:34–7. doi: 10.4161/rna.4.1.4534. [DOI] [PubMed] [Google Scholar]
- 32.Martins de Araújo M, Bonnal S, Hastings ML, Krainer AR, Valcárcel J. Differential 3' splice site recognition of SMN1 and SMN2 transcripts by U2AF and U2 snRNP. RNA. 2009;15:515–23. doi: 10.1261/rna.1273209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cartegni L, Krainer AR. Disruption of an SF2/ASF-dependent exonic splicing enhancer in SMN2 causes spinal muscular atrophy in the absence of SMN1. Nat Genet. 2002;30:377–84. doi: 10.1038/ng854. [DOI] [PubMed] [Google Scholar]
- 34.Kashima T, Manley JL. A negative element in SMN2 exon 7 inhibits splicing in spinal muscular atrophy. Nat Genet. 2003;34:460–3. doi: 10.1038/ng1207. [DOI] [PubMed] [Google Scholar]
- 35.Singh NN, Androphy EJ, Singh RN. An extended inhibitory context causes skipping of exon 7 of SMN2 in spinal muscular atrophy. Biochem Biophys Res Commun. 2004;315:381–8. doi: 10.1016/j.bbrc.2004.01.067. [DOI] [PubMed] [Google Scholar]
- 36.Cartegni L, Hastings ML, Calarco JA, de Stanchina E, Krainer AR. Determinants of exon 7 splicing in the spinal muscular atrophy genes, SMN1 and SMN2. Am J Hum Genet. 2006;78:63–77. doi: 10.1086/498853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Singh NN, Androphy EJ, Singh RN. In vivo selection reveals combinatorial controls that define a critical exon in the spinal muscular atrophy genes. RNA. 2004;10:1291–305. doi: 10.1261/rna.7580704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hofmann Y, Lorson CL, Stamm S, Androphy EJ, Wirth B. Htra2-beta1 stimulates an exonic splicing enhancer and can restore full-length SMN expression to survival motor neuron 2 (SMN2). Proc Natl Acad Sci USA. 2000;97:9618–23. doi: 10.1073/pnas.160181697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hua Y, Vickers TA, Baker BF, Bennett CF, Krainer AR. Enhancement of SMN2 exon 7 inclusion by antisense oligonucleotides targeting the exon. PLoS Biol. 2007;5:73. doi: 10.1371/journal.pbio.0050073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Miyajima H, Miyaso H, Okumura M, Kurisu J, Imaizumi K. Identification of a cis-acting element for the regulation of SMN exon 7 splicing. J Biol Chem. 2002;277:23271–7. doi: 10.1074/jbc.M200851200. [DOI] [PubMed] [Google Scholar]
- 41.Miyaso H, Okumura M, Kondo S, Higashide S, Miyajima H, Imaizumi K. An intronic splicing enhancer element in Survival Motor Neuron (SMN) pre-mRNA. J Biol Chem. 2003;278:15825–31. doi: 10.1074/jbc.M209271200. [DOI] [PubMed] [Google Scholar]
- 42.Singh NK, Singh NN, Androphy EJ, Singh RN. Splicing of a critical exon of human Survival Motor Neuron is regulated by a unique silencer element located in the last intron. Mol Cell Biol. 2006;26:1333–46. doi: 10.1128/MCB.26.4.1333-1346.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hua Y, Vickers TA, Okunola HL, Bennett CF, Krainer AR. Antisense masking of an hnRNP A1/A2 intronic splicing silencer corrects SMN2 splicing in transgenic mice. Am J Hum Genet. 2008;82:834–48. doi: 10.1016/j.ajhg.2008.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cartegni L, Krainer AR. Correction of disease-associated exon skipping by synthetic exon-specific activators. Nat Struct Biol. 2003;10:120–5. doi: 10.1038/nsb887. [DOI] [PubMed] [Google Scholar]
- 45.Skordis LA, Dunckley MG, Yue B, Eperon IC, Muntoni F. Bifunctional antisense oligonucleotides provide a trans-acting splicing enhancer that stimulates SMN2 gene expression in patient fibroblasts. Proc Natl Acad Sci USA. 2003;100:4114–9. doi: 10.1073/pnas.0633863100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Marquis J, Meyer K, Angehrn L, Kämpfer SS, Rothen-Rutishauser B, Schümperli D. Spinal muscular atrophy: SMN2 pre-mRNA splicing corrected by a U7 snRNA derivative carrying a splicing enhancer sequence. Mol Ther. 2007;15:1479–86. doi: 10.1038/sj.mt.6300200. [DOI] [PubMed] [Google Scholar]
- 47.Coady TH, Baughan TD, Shababi M, Passini MA, Lorson CL. Development of a single vector system that enhances trans-splicing of SMN2 transcripts. PLoS ONE. 2008;3:3468. doi: 10.1371/journal.pone.0003468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lim SR, Hertel KJ. Modulation of survival motor neuron pre-mRNA splicing by inhibition of alternative 3' splice site pairing. J Biol Chem. 2001;276:45476–83. doi: 10.1074/jbc.M107632200. [DOI] [PubMed] [Google Scholar]
- 49.Madocsai C, Lim SR, Geib T, Lam BJ, Hertel KJ. Correction of SMN2 Pre-mRNA splicing by antisense U7 small nuclear RNAs. Mol Ther. 2005;12:1013–22. doi: 10.1016/j.ymthe.2005.08.022. [DOI] [PubMed] [Google Scholar]
- 50.Rana TM. Illuminating the silence: understanding the structure and function of small RNAs. Nat Rev Mol Cell Biol. 2007;8:23–36. doi: 10.1038/nrm2085. [DOI] [PubMed] [Google Scholar]
- 51.Gangwani L, Mikrut M, Theroux S, Sharma M, Davis RJ. Spinal muscular atrophy disrupts the interaction of ZPR1 with the SMN protein. Nat Cell Biol. 2001;3:376–83. doi: 10.1038/35070059. [DOI] [PubMed] [Google Scholar]
- 52.Doran B, Gherbesi N, Hendricks G, Flavell RA, Davis RJ, Gangwani L. Deficiency of the zinc finger protein ZPR1 causes neurodegeneration. Proc Natl Acad Sci USA. 2006;103:7471–5. doi: 10.1073/pnas.0602057103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Helmken C, Hofmann Y, Schoenen F, Oprea G, Raschke H, Rudnik-Schöneborn S, et al. Evidence for a modifying pathway in SMA discordant families: reduced SMN level decreases the amount of its interacting partners and Htra2-beta1. Hum Genet. 2003;114:11–21. doi: 10.1007/s00439-003-1025-2. [DOI] [PubMed] [Google Scholar]
- 54.Rossoll W, Kröning AK, Ohndorf UM, Steegborn C, Jablonka S, Sendtner M. Specific interaction of Smn, the spinal muscular atrophy determining gene product, with hnRNPR and gry-rbp/hnRNP-Q: a role for Smn in RNA processing in motor axons. Hum Mol Genet. 2002;11:93–105. doi: 10.1093/hmg/11.1.93. [DOI] [PubMed] [Google Scholar]
- 55.Chen HH, Chang JG, Lu RM, Peng TY, Tarn WY. The RNA binding protein hnRNP Q modulates the utilization of exon 7 in the survival motor neuron 2 (SMN2) gene. Mol Cell Biol. 2008;28:6929–38. doi: 10.1128/MCB.01332-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lefebvre S, Burlet P, Liu Q, Bertrandy S, Clermont O, Munnich A, et al. Correlation between severity and SMN protein level in spinal muscular atrophy. Nat Genet. 1997;16:265–9. doi: 10.1038/ng0797-265. [DOI] [PubMed] [Google Scholar]
- 57.Gangwani L, Flavell RA, Davis RJ. ZPR1 is essential for survival and is required for localization of the survival motor neurons (SMN) protein to Cajal bodies. Mol Cell Biol. 2005;25:2744–56. doi: 10.1128/MCB.25.7.2744-2756.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Jaeger LB, Banks WA. Transport of antisense across the Blood-brain barrier. In: Phillips MI, editor. Methods in Molecular Medicine: Antisense Therapeutics. Vol. 106. Humana Press; Totowa, NJ: 2005. pp. 237–51. [DOI] [PubMed] [Google Scholar]