

Abstract
Pulmonary clearance of the encapsulated yeast Cryptococcus neoformans requires the development of T1-type immunity. CCR2-deficient mice infected with C. neoformans develop a non-protective T2 immune response and persistent infection. The mechanisms responsible for this aberrant response are unknown. The objective of this study was to define the number, phenotype, and micro-anatomic location of dendritic cells (DC) residing within the lung of CCR2+/+ or CCR2−/− mice throughout a time course following infection with C. neoformans. Results demonstrate the CCR2-mediated recruitment of conventional DC expressing modest amounts of co-stimulatory molecules. DC recruitment was preceded by the up-regulation in the lung of the CCR2 ligands CCL2 and CCL7. Co-localization of numerous DC and CD4+ T cells within bronchovascular infiltrates coincided with increased expression of IL-12 and IFN-γ. By contrast, in the absence of CCR2, DC recruitment was markedly impaired, bronchovascular infiltrates were diminished, and mice developed features of T2 responses, including bronchovascular collagen deposition and IL-4 production. Our results demonstrate that CCR2 is required for the recruitment of large numbers of conventional DC to bronchovascular infiltrates in mice mounting a T1 immune response against a fungal pathogen. These findings shed new insight into the mechanism(s) by which DC recruitment alters T cell polarization in response to an infectious challenge within the lung.
Keywords: Dendritic cells; Cell Trafficking; Lung; Chemokines; Rodent, Cryptococcus
Introduction
Cryptococcus neoformans, an encapsulated yeast acquired via the respiratory tract, causes significant morbidity and mortality in patients with AIDS, lymphoid or hematological malignancies, or those receiving immunosuppressive therapy secondary to autoimmune disease or organ transplantation (1–3). Antibiotic therapy is often inadequate (4), underscoring the need to better understand host defense against this organism. In normal hosts, the development of a T1 cell-mediated immune response is crucial to eradicate this organism (5–14). However, the mechanisms responsible for T1 polarization in response to C. neoformans infection remain incompletely understood.
Disruption of the chemokine and chemokine receptor axis alters T cell polarization and adversely affects microbial clearance in murine models of C. neoformans pulmonary infections (15–21). CCR2-expressing (CCR2+/+) mice develop a T1 immune response and clear C. neoformans infection, whereas CCR2-deficient (CCR2−/−) mice develop T2 responses as evidenced by: (a) reduced levels of IFN-γ; (b) increased production of IL-4 and IL-13 by lung leukocytes; (c) prominent eosinophilic infiltrates; and (d) impaired clearance of C. neoformans (19). The mechanism responsible for impaired T1 polarization in CCR2-deficient mice has not been identified. We have previously correlated early reductions in TNF-α and IFN-γ elaboration in the lungs with decreased IL-12 production, T2 responses, and impaired clearance of C. neoformans infection (9, 10, 14, 22). Our previous studies demonstrating CCR2-mediated lung DC recruitment in another model system, the response to particulate, non-infectious antigen (23), imply that impaired DC recruitment to the lung could account for the differential T cell polarization observed between CCR2+/+ and CCR2−/− mice in cryptococcal infection.
The role of DC in host defense against C. neoformans is not well understood. Dendritic cells (DC) ingest whole C. neoformans or associated mannoproteins, and stimulate a C. neoformans-specific T cell line when assessed in vitro (24–26). DC migrate to regional nodes when exposed to the organism in vivo (11). As part of the initial response to infection, sensitized CD4+ and CD8+ T cells proliferate within draining lymph nodes; however, lymphocytes do not express an activated phenotype or produce IFN-γ in an Ag-specific manner until they have migrated to the lung (27). Whether T cell activation in the lung requires additional interactions with professional APC (possibly DC) has not been established definitively. We hypothesized that newly recruited lymphocytes require additional interactions with DC in the lung to achieve activated and polarized phenotype. The objective of this study was to investigate this hypothesis by defining the number, phenotype, and micro-anatomic location of DC residing within the lung of CCR2+/+ or CCR2−/− mice throughout a time course following infection with C. neoformans.
Materials and Methods
Mice
Specific pathogen-free inbred female BALB/c (designated CCR2+/+) mice purchased from Charles River Laboratory Inc. (Wilmington, MA) were used, except as specified. CCR2−/− mice (C57BL/6 × J129 (C57/J129) back-crossed eight times onto a BALB/c background) were provided by W. Kuziel (Molecular Genetics and Microbiology, University of Texas, Austin, TX) (28) and were bred on-site. Mice were housed at the University of Michigan Unit for Laboratory Animal Medicine Facilities (Ann Arbor, MI), which is fully accredited by the American Association for Accreditation of Laboratory Animal Care. Mice were kept under specific pathogen-free conditions in enclosed filter top cages, and provided standard animal chow and chlorinated tap water ad libitum. Mice were 8–12 wk of age at the time of infection and there were no age-related differences in the responses of these mice to C. neoformans infection. Experiments were approved by the Animal Care and Use Committee at the University of Michigan.
C. neoformans
C. neoformans strain 52D was obtained from the American Type Culture Collection (24067; Manassas, VA). For infection, yeast were grown to stationary phase (48–72 h) at 37°C in Sabouraud dextrose broth (1% neopeptone and 2% dextrose: DIFCO, Detroit, MI) on a shaker. Cultured C. neoformans was then washed in non-pyrogenic saline, counted using Trypan Blue on a hemocytometer, and diluted to 3.3 × 105 CFU/ml in sterile non-pyrogenic saline.
Surgical intratracheal inoculation
Mice were anesthetized by intraperitoneal injection of ketamine (100 mg/kg; Fort Dodge Laboratories, Fort Dodge, IA) and xylazine (6.8 mg/kg; Lloyd Laboratories, Shenandoah, IA) and restrained on a small surgical board. A small incision was made through the skin over the trachea, and the underlying tissue was separated. A 30-gauge needle was attached to a 1-ml tuberculin syringe filled with diluted C. neoformans culture. The needle was inserted into the trachea, and 30 μl of inoculums (104 CFU) was dispensed into the lungs. The needle was removed, and the skin was closed with cyanoacrylate adhesive. The mice recovered with minimal visible trauma.
Monoclonal Abs
The following mAbs purchased from BD Biosciences PharMingen (San Diego, CA) were used: M1/70 (anti-murine CD11b, rat IgG2b); HL3 (anti-murine CD11c, hamster IgG1); 2.4G2 (“Fc block”) (anti-murine CD16/CD32, rat IgG2b); 3/23 (anti-murine CD40, rat IgG2a); 30-F11 (anti-murine CD45, rat IgG2b); 16-10A1 (anti-murine CD80, hamster IgG2); GL1 (anti-murine CD86, rat IgG2a); AMS-32.1 (“MHC Class II”) (anti-murine I-Ad, mouse IgG2b); 145-2C11 (anti-murine CD3e, hamster IgG1, k), and ID3 (anti-murine CD19, rat IgG2a). The mAb, BM8 (anti-murine F4/80, rat IgG2b), was purchased from Caltag Laboratories (Burlingame CA). Monoclonal Abs were primarily conjugated with FITC, PE; PerCP-Cy5.5, or allophycocyanin. Isotype-matched irrelevant control mAbs (PharMingen) were tested simultaneously in all experiments.
Antibody staining and flow cytometric analysis
Staining, including blockade of Fc receptors, and analysis by flow cytometry were performed as described previously (29, 30). Data was collected on a FACS VANTAGE flow cytometer using Cell Quest software (both from Becton Dickinson Immunocytometry Systems, Mountain View, CA) and analyzed using FlowJo software (Tree Star Inc., San Carlos, CA). 10,000 to 100,000 cells were analyzed per sample.
Experimental Design
In all experiments, primary cryptococcal infection was induced in the lungs of CCR2+/+ (Wild type, BALB/c) and syngeneic CCR2−/− mice by the intratracheal administration of 104 CFU of C. neoformans in 30 μL of normal saline. Uninfected (untreated) mice are referred to as “day 0” mice and served as controls. Following euthanasia, all mice underwent perfusion via the right heart with 5 ml of PBS containing 0.5 mM EDTA to reduce intravascular leukocytes. Thereafter, five types of experiments were performed at various time points following infection. First, DC were physically isolated (using CD11c magnetic beads) from lung leukocytes which had been depleted of lung macrophages (by bronchoalveolar lavage and an adherence step). The phenotype of recovered DC was then assessed using flow cytometric analysis (FACS). Second, to identify DC from bulk lung leukocyte populations (without lavage or the use of CD11c beads) three- and four-color FACS was performed on single-cell suspensions generated from enzymatically-digested lung tissue. A gating strategy was developed that identified conventional DC, and distinguished DC from lung macrophages and monocytes (23, 31, 32). Gates were kept consistent in all experiments. This approach allowed for quantitative, comparative analysis of the total numbers of DC present in the lung of CCR2+/+ or CCR2−/− mice throughout a kinetic time course following C. neoformans infection. Third, the differential recruitment of DC to the alveolar and interstitial compartments was assessed by comparing the number and phenotype of DC within bronchoalveolar lavage fluid versus those retained in the lung mince. Fourth, the expression of CCR2 ligands and cytokines within the lungs of infected mice was evaluated with real-time quantitative PCR using RNA samples generated from lung homogenates. Fifth, the micro-anatomic location of DC and T cells within the lungs of infected mice was determined using routine histology on paraffin-embedded tissues and two-color immunohistochemistry.
Physical isolation and characterization of lung DC using CD11c magnetic beads
Following euthanasia, the trachea was cannulated with PE50 tubing (Clay-Adams, Parsippany, NJ) and lungs were lavaged twice with 0.8 ml of PBS containing 5 mM EDTA to remove macrophages (which express CD11c). Lavage fluid was discarded. Lungs were excised, minced, and enzymatically digested for 30 min at 37°C in 15 ml of digestion buffer (RPMI 1640, 5% fetal calf serum, 100 U/ml penicillin, 100 μg/ml streptomycin sulfate, 1 mg/ml collagenase, 30 μg/ml DNase). The cell suspension and undigested fragments were further dispersed by being drawn up-and-down 20 times through the bore of a 10-ml syringe. After erythrocyte lysis using NH4Cl buffer (0.83% NH4Cl, 0.1% KHCO3, 0.037% Na2 EDTA, pH 7.4), cells were washed, filtered over 70 μm mesh, resuspended in complete medium (RPMI 1640, 5% fetal calf serum, 100 U/ml penicillin, 100 μg/ml streptomycin sulfate) and centrifuged for 30 min at 2,000 × g in the presence of 20% Percoll (Sigma-Aldrich) to separate leukocytes from cell debris and epithelial cells. The isolated leukocytes were resuspended in complete medium. To further remove contaminating macrophages, leukocytes were next cultured in 100 × 20 mm tissue culture dishes containing complete medium (RPMI 1640, 5% fetal calf serum, 100 U/ml penicillin, and 100 μg/ml streptomycin sulfate) at a concentration of 2 × 106 cells/ml for one-hour at 37°C. Non-adherent cells (including DC) were removed by gentle pipetting. Adherent cells (primarily macrophages) were discarded. Non-adherent cells were treated with Fc block and DC isolation was performed using CD11c magnetic beads (MACS system, Miltenyi Biotech, Auburn, CA) per the manufacturer's guidelines. The resultant population was assessed by 3-color FACS and DC were identified as cells expressing MHC Class II (I-Ad) (FITC; FL-1), CD11c (allophycocyanin; FL-4), CD11b (PE; FL-2) and low amounts of F4/80 (PE; FL-2). Note that due to the purity of this population, it was not necessary to employ the gating strategy outlined below (for identifying DC from bulk leukocyte populations). Cytospins of this purified population were performed using Hematoxylin-eosin stains (VWR. Westchester, PA) to verify DC morphology.
Total lung leukocyte isolation
To obtain a single population of total lung leukocytes, individual lungs were first excised (this time without lavage), minced, and enzymatically digested as described above. The cell suspension and undigested fragments were further dispersed, filtered, and separated using a Percoll gradient (also as described above). The resultant cell population included the total lung leukocytes (all cells within the alveolar and interstitial compartments). These cells were resuspended in complete medium and total numbers of viable lung leukocytes were assessed in the presence of trypan blue using a hemocytometer. This population was subjected to FACS analysis using the gating strategy outlined below to enumerate the total number of DC per lung.
Differential lung leukocyte isolation from the alveolar versus interstitial compartments
To obtain differential populations of lung leukocytes from the alveolar and interstitial compartments, mice first underwent bronchoalveolar lavage (BAL). Briefly, the trachea was cannulated with PE50 tubing (Clay-Adams, Parsippany, NJ) and a total of 10 lavages with. 8ml of PBS containing 5 mM EDTA were performed. Viable leukocytes recovered from the lavage fluid were enumerated in the presence of trypan blue using a hemocytometer and placed in culture medium for subsequent analysis (as the “BAL” population). Lungs were then excised, minced, enzymatically digested and processed thereafter as described above to generate a single cell suspension. This population of cells was termed “Lung Mince” and consists of cells retained within the interstitial compartment of the lung. Both populations (BAL and Lung Mince) were later assessed by FACS using the gating strategy outlined below to identify DC within each population.
Gating strategy using FACS to identify lung DC within bulk leukocyte populations
Cell populations assessed included 1) Total lung leukocytes, 2) BAL leukocytes, and 3) Lung Mince leukocytes (as defined above). Initial gates were set based on light scatter characteristics to eliminate debris, red cells, and cell clusters. Samples were stained with anti-MHC Class II (I-Ad) (FITC; FL-1), anti-CD11c (allophycocyanin; FL-4), anti-CD3 and anti-CD19 (PerCP Cy5.5; FL-3), and a panel of additional antibodies of interest (PE; FL-2). An initial FSC vs. FL-3 scatter plot was used to exclude T cells (CD3+), B cells (CD19+), and macrophages (large autofluorescent cells), as described previously (33). DC were then identified among the remaining non-T cell, non-B cell, non-autofluorescent population (Gate 1 in Figure 2A) as MHC Class II (I-Ad)+, CD11c+ cells. A panel of antibodies (CD11b, F4/80, CD40, CD80, and CD86 PE; FL-2) were used to further characterize the DC population. Substitution of PE-conjugated anti-CD11c (FL-2) antibody (for anti-CD11c allophycocyanin) permitted DC identification in three-color experiments. To maintain complete consistency, cytometer parameters and gate position were held constant during analysis of all samples. The percentage of DC obtained from flow cytometry was used to calculate the total number of DC from each tissue by multiplying the frequency of DC by the total number of leukocytes (the percent of CD45+ cells multiplied by the original hemocytometer count of total cells) identified within that sample.
Figure 2.

A flow cytometric analysis strategy identifies cDC amongst total lung leukocytes in the lungs of mice infected with C. neoformans. (A–E) A strategy was devised to identify cDC from bulk mononuclear cell populations obtained from CCR2+/+ mice infected with C. neoformans. Lungs were removed (without lavage) 14 days post-infection and a single cell suspension of all mononuclear cells from enzyme-digested lung mince was obtained. All cells were stained with mAbs to CD3 and CD19 (PerCpCy5.5) to generate an initial plot of FL-3 vs. FSC (A) which identified three cell populations: Lymphocytes (L), small cells which stained for either CD3 or CD19 (PerCpCy5.5); Macrophages (M), large auto-fluorescent cells; and mononuclear cells within Gate 1 (which contained a variety of cell types including cDC). Cells within Gate 1 (B–E) were assessed with the following Ab combinations: (B) isotype controls, (C) MHC Class II (I-Ad, FITC) and CD11c (allophycocyanin); (D) CD11b (PE) and CD11c (allophycocyanin), or (E) F4/80 (PE) and CD11c (allophycocyanin). cDC were identified (C–E) as CD11c+ cells co-expressing MHC Class II (I-Ad), CD11b, and low amounts of F4/80 (representative scatter plots, circular gates labeled cDC).
RT-PCR
For PCR, lungs were homogenized in Trizol and immediately stored in RNAlater (Ambion, Austin, TX). Total RNA was prepared using a RiboPure RNA Isolation kit (Ambion). Contaminating DNA was removed with DNA-free (Ambion). Samples were reverse-transcribed using a RETROscript kit (Ambion). Quantitative real-time RT-PCR for chemokines CCL2 (MCP-1), CCL7 (MCP-3), CCL12 (MCP-5), CCL11 (eotaxin-1), cytokines IL-4, IL-12p35, IL-12p40, IFN-γ, and the housekeeping gene GAPDH was performed using Taqman Assay kits obtained from Applied Biosystems (Foster City, CA). cDNA conversion, amplification, and data analysis were performed as previously described (23). Data are presented as fold-increase in specific mRNA transcripts relative to a pooled RNA sample obtained from the lungs of five, uninfected wild type (BALB/c) mice not used elsewhere in this study.
Histology
The trachea was cannulated with PE50 tubing (Clay-Adams, Parsippany, NJ) and inflated with 1 ml of 10% neutral buffered formalin. The fixed lung specimens were stored in 10% neutral-buffered formalin until dehydrated in 70% ethanol and then paraffin embedded. Sections (5 μm) were cut, deparaffinized, stained with hematoxylin and eosin (± PAS or Masson's trichrome where indicated), and viewed by light microscopy.
Immunohistochemistry
Lung sections were prepared and stained for immunohistochemical analysis as previously described in detail (23). Briefly, lungs were inflated first with 1 ml air followed by 1 ml of an 80:20 mixture of RPMI (InVitrogen Life Sciences) to OCT (Tissue Tek, Sakura Finetek, Torrance, CA). Lungs were snap frozen in liquid N2 and stored overnight at −20°C. Lung sections (5-μm-thickness) were blocked with hamster serum, and Ags were visualized using consecutive peroxidase (biotinylated anti-CD11c, anti-CD4, or isotype control mAb; red reaction product) and alkaline phosphatase (biotinylated anti-MHC Class II (I-Ad), anti-CD11b, or isotype control mAb; black reaction product) reactions. Slides were counterstained in hematoxylin and coverslipped using aqueous mounting media (Biomeda).
Statistical analysis
All data were expressed as mean ± SEM. Continuous ratio scale data were evaluated by unpaired Student t test (for comparison between two samples) or by ANOVA (for multiple comparisons) with post hoc analysis by two-tailed Dunnett test, which compares treatment groups to a specific control group (34). Statistical calculations were performed on a Dell 270 computer using GraphPad Prism version 3.00 for Windows, GraphPad Software, San Diego California USA. Statistical difference was accepted at p<0.05.
Results
Conventional DC are reduced in the lungs of CCR2-deficient mice in response to infection with C. neoformans
To characterize lung DC during experimental cryptococcal infection, we first devised a strategy to purify lung DC. Using CD11c magnetic beads, we isolated DC from macrophage-depleted lung leukocytes obtained from CCR2+/+ mice two weeks post-infection with C. neoformans. DC were identified (Fig. 1) within a purified, ungated population as MHC Class II (I-Ad)+, CD11c+ cells (Fig. 1B, circular gate labeled cDC) also expressing the cell surface markers CD11b (Fig. 1C) and low amounts of F4/80 (Fig. 1D). DC displayed a lobulated nucleus with multiple fine cytoplasmic extensions, consistent with classic DC morphology (Fig. 1E). This phenotype is most consistent with that described for conventional DC (cDC); their accumulation within an inflamed peripheral tissue (the lung) further suggests they represent an inflammatory subset (35, 36). Note that the term, myeloid DC (mDC), is often still used in the literature interchangeably with cDC.
Figure 1.

DC isolation identifies the presence of cDC within the lungs of mice infected with C. neoformans. Lungs from CCR2+/+ mice (14 days post-infection) were lavaged (to remove alveolar macrophages) and enzymatically digested. The resultant single cell suspension of mononuclear cells was cultured (1 hr) on tissue culture plates to further deplete macrophages (by adherence). The non-adherent cells were incubated with CD11c-coated magnetic beads to isolate CD11c positive cells. This population (A–E) was stained with mAbs to either isotype controls (A) or (B) MHC Class II (I-Ad, FITC) and CD11c (allophycocyanin) (representative scatter plots without prior gating). cDC (circular gate labeled cDC) were identified as MHC Class II (I-Ad)+ and CD11c+cells which expressed (C) CD11b and (D) low amounts of F4/80 (representative histograms, open histograms represents Ab specific staining, shaded histograms represents isotype control.). (E) Hematoxylin-eosin staining of these cells confirms a DC morphology with a lobulated nucleus and multiple, fine, cytoplasmic extensions.
The use of CD11c magnetic beads to isolate cDC was not conducive to a thorough study of lung DC recruitment; the procedure is labor intensive, costly, and DC are lost in the steps designed to eliminate contaminating macrophages (i.e. the lavage or brief in vitro culture). Therefore, using this defined phenotype (Fig. 1) for lung cDC as a reference, we next used multi-parameter FACS to identify cDC from bulk lung leukocyte preparations derived from individual infected CCR2+/+ mice (Fig. 2). A specific gating strategy proven to identify lung DC accurately in allergen-exposed mice (32, 33) served as the basis of our approach. Briefly, care was taken to first exclude auto-fluorescent alveolar macrophages (which express CD11c but not CD11b) and T or B lymphocytes (Fig. 2A) (32, 33). cDC were identified within a gated population (Fig. 2A, gate 1) as MHC Class II (I-Ad)+, CD11c+ cells (Fig. 2C, circular gate). Four-color experiments confirmed the MHC Class II (I-Ad)+, CD11c+ population co-expressed the cell surface marker CD11b, and low amounts of F4/80 (similar to Fig. 1C and 1D respectively; data not shown). Others have defined lung cDC (or mDC) as non-autofluorescent cells which are CD11b+, CD11c+, or CD11c+, F4/80lo (23, 37–41). After incorporating these two alternative combinations into our gating strategy (Fig. 2D, E), the percentage of cDC identified amongst lung leukocytes was similar (7–11%) to that of the MHC Class II (I-Ad)+, CD11c+ population. Thus, cDC could accurately be identified amongst recruited lung leukocyte populations using FACS alone.
This gating strategy was then utilized to assess the frequency and total number of cDC within the lungs of individual CCR2+/+ or CCR2−/− mice at baseline and at multiple time points following infection with C. neoformans (Fig. 3). Infection of CCR2+/+ mice with C. neoformans resulted in massive leukocyte recruitment to the lungs (Fig. 3A), peaking at day 14 (1.2 ± 0.1 × 108 total leukocytes/mouse, n = 4–7 mice per strain in 2–3 separate time courses). Total leukocyte recruitment was initially delayed in CCR2−/− mice. This finding is not attributable to differences in fungal burden, as our previous studies demonstrated no differences in C. neoformans numbers in the lungs of CCR2+/+ versus CCR2−/− mice until 21 days post-infection (19). Total leukocyte numbers in CCR2−/− mice eventually surpassed those of CCR2+/+ mice. This difference is largely attributable to a significant eosinophil influx (data not shown) and to persistent infection in CCR2−/− mice, consistent with previous findings in this model (19).
Figure 3.

Kinetics of total lung cDC accumulation in CCR2+/+ and CCR2−/− mice following infection with C. neoformans. CCR2+/+ and CCR2−/− mice were infected with C. neoformans; total lung digests (without BAL) were performed on mice at days 0 (uninfected) and 3, 7, 10, 14, 21, and 28 post-infection. (A) Kinetics of total lung leukocyte (CD45+) accumulation. (B) cDC (elliptical gates) were identified using the gating strategy outlined in the Methods section and in the legend to Figure 2. Representative scatter plots (x-axis, I-Ad; y-axis CD11c) of cDC in the lungs of either CCR2+/+ mice (left panels) or CCR2−/− mice (right panels) at day 0 (D0; top panels) or 10 (D10; bottom panels) post-IT infection with C. neoformans. (C) Frequency of lung cDC (% of CD45+ cells) throughout the observed time course. (D) Numbers of total lung cDC (Total leukocytes × frequency of cDC) throughout the observed time course. CCR2+/+, black line or bar; CCR2−/− mice, dashed gray line or bar. Data represents a mean ± SEM of 4–7 mice assayed individually per time-point in 2–3 experiments; * p<0.05 ANOVA with Dunnet's post-hoc analysis vs. Day 0 (uninfected) of mice of the same CCR2 expression profile, ‡p<0.05 by unpaired student t-test when values from CCR2+/+ mice or CCR2−/− mice were compared against each other at the designated time point.
In the uninfected state, cDC comprised 3.1 ± 0.1% (mean ± SEM) of the total lung leukocyte population in CCR2+/+ mice and 2.2 ± 0.1% in CCR2−/− mice (Fig. 3B representative scatter plot D0, 3C cumulative data). Absolute numbers of cDC per lung at baseline were similar in the two strains (CCR2+/+, 2.0 ± 0.2 × 105 total mDC/mouse vs. CCR2−/−, 2.1 ±. 0.1 × 105; n=5–7 mice per strain in 2–3 experiments per time-point) (Fig. 3D). Post-infection, the percentage (Fig. 3C) and absolute numbers (Fig. 3D) of lung cDC increased substantially in CCR2+/+ mice. Numbers of cDC increased maximally (77-fold increase from uninfected mice) by day 14 post-infection in CCR2+/+ mice (12.4 ± 1.2% of total leukocytes, 1.5 ± 0.2 × 107 cDC/mouse). By contrast, the accumulation of lung cDC in CCR2−/− mice following infection with C. neoformans was markedly reduced relative to CCR2+/+ mice. The difference was greatest at day 10 post-infection (Figs. 3B representative scatter plots D10; and Figs. 3C, 3D cumulative data), representing a 96% decrease in CCR2−/− mice (1.1 ± 0.1 × 107 lung cDC/CCR2+/+ mouse vs. 6.5 ± 0.4 × 105 lung cDC/CCR2−/− mouse). Despite greater numbers of total lung leukocytes in CCR2−/− mice by day 21 post-infection, this disparity in cDC numbers persisted.
cDC localized primarily to the lung interstitium of CCR2+/+ mice and were phenotypically immature
These results indicate that CCR2 mediates the recruitment of cDC to the lung (in response to C. neoformans infection) but do not identify the microanatomic compartment within the lung where these cells accumulated. Therefore, we used the FACS gating strategy described above (Fig. 2) to identify cDC within leukocytes recovered from the lung interstitium (Lung Mince) or the alveolar space (BAL). CCR2+/+ mice and CCR2−/− mice were evaluated at day 14 post-infection (the time of maximal DC recruitment; Fig. 3D). In CCR2+/+ mice, the percentage of cDC (relative to other leukocytes) in the lung interstitium was significantly increased relative to those recovered from the alveolar space (Fig. 4A) representing an 82-fold enrichment (8.2 ± 3.5 × 106 cDC/Lung Mice vs 1.0 ± 0.03 × 105 cDC/BAL). In the absence of CCR2, the frequency of cDC amongst lung leukocytes within both the lung interstitium and the alveolar space was reduced relative to CCR2+/+ mice (Fig. 4A). Similar results were obtained using either alternative definition of cDC (CD11c+, CD11b+; Fig. 2D or CD11c+, F4/80lo; Fig. 2E) (data not shown). This data supports our conclusion that total lung cDC recruitment is impaired in CCR2-deficient mice (Fig. 2C, D). The frequency (Fig. 4A) and total numbers of cDC in the lung interstitium of CCR2−/− mice were significantly increased when compared to those within the alveolar space (1.3 ± 0.06 × 106 cDC/Lung Mice vs 4.3 ±0.6 × 104 cDC/BAL). Collectively, these findings strongly suggest that the vast majority of cDC in the lungs of C. neoformans-infected mice (of either strain) localize within the interstitium.
Figure 4.

Comparison of cDC percentage and phenotype amongst cells isolated from the lung interstitial and alveolar compartments. CCR2+/+ and CCR2−/− mice were infected with C. neoformans. At day 14 post-infection single cell populations were obtained from two sources: lung mince (enzyme digested lung post-lavage) or bronchoalveolar lavage (BAL). cDC were identified as MHC Class II (I-Ad)+/CD11c+ cells using the gating strategy outlined in Methods (and per Figure 2). (A) Frequency of lung cDC (% of CD45+ cells) in lung mince and BAL (CCR2+/+, black bar; CCR2−/− mice, gray bar). Data represents a mean ± SEM of 5 mice assayed individually per time-point; p values indicate unpaired student t-test between comparative groups. (B) Representative scatter plots (I-Ad vs CD11c) and histograms (cDC, circular gates) depict staining for the indicated receptors within representative cDC populations identified from lung mince (top panels) and BAL (bottom panels). Open histograms represent specific staining; solid shading show isotype-control staining. Note that the paucity of cDC recovered from the BAL of CCR2−/− mice (bottom-most panels) made analysis of co-stimulatory molecule expression difficult.
Results of the many studies on the maturational state of lung DC in response to allergic, inflammatory, and infectious stimuli have varied widely, with both mature DC (42–44) and immature DC (23, 45, 46) described. To assess co-stimulatory molecule expression on lung cDC in this model system, the four-color FACS and gating strategy (described in methods and Fig. 2) was applied to these leukocyte populations obtained from both the interstitial (Lung Mince) and alveolar compartments (BAL). We observed that cDC within both compartments (and strains) expressed low to moderate levels of the co-stimulatory molecules CD40, CD80, and CD86 (Fig. 4B), consistent with an immature phenotype. CD80 expression appeared slightly increased on cDC from CCR2+/+ mice (compared with CCR2−/− mice); the significance of this small difference is uncertain. Experiments performed on total lung leukocyte populations at day 10 and 17 post-infection revealed a similar phenotypic profile for cDC in both strains. Appreciable differences in MHC Class II expression (over time or between strains) were also not observed (data not shown). Thus, cDC located within the lungs of CCR2+/+ mice and CCR2−/− mice appeared phenotypically immature and qualitatively similar as assessed at multiple time-points following infection with C. neoformans.
The CCR2 ligands CCL2 and CCL7, but not CCL12, were up-regulated early in response to infection with C. neoformans
CCR2 ligands are rapidly up-regulated in response to a variety of infectious and inflammatory stimuli in the lung (15, 23, 47, 48), whereby they mediate the early recruitment of CCR2-bearing cells. The ligand(s) mediating DC recruitment in this model has not been fully defined. To assess the expression of the three known murine CCR2 ligands (CCL2 /monocyte chemotactic protein, MCP-1; CCL7/MCP-3; and CCL12/MCP-5) real time RT-PCR was performed on total lung mRNA obtained from CCR2+/+ and CCR2−/− mice, both at baseline and in response to infection with C. neoformans (Fig. 5). In CCR2+/+ mice, CCL2 mRNA levels were markedly upregulated (36-fold) by day 7 post-infection (Fig. 5A). CCL7 mRNA increased with similar magnitude and kinetics (Fig. 5B). In contrast, lung levels of CCL12 mRNA in CCR2+/+ mice increased only modestly (7-fold) and at a much later time point (day 28) (Fig. 5C).
Figure 5.
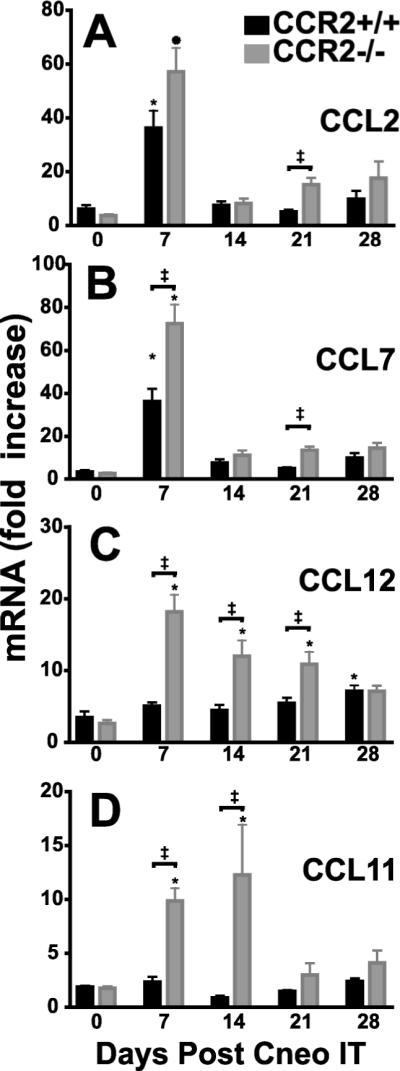
CCR2 ligands are upregulated in the lung in response to pulmonary infection with C. neoformans. RNA was prepared from lung homogenates obtained from CCR2+/+ mice and CCR2−/− mice at baseline and at days 7, 14, 21, and 28 post-IT infection with C. neoformans. A separate RNA preparation from lung homogenates of uninfected CCR2+/+ mice (n=5) was used as a reference standard. RT-PCR was performed to evaluate the expression of the three known murine ligands for CCR2 (A–C): (A) CCL2 (MCP-1), (B) CCL 5 (MCP-3), (C) CCL12 (MCP-5) and (D) CCL11 (eotaxin-1). CCR2+/+, black bar; CCR2−/− mice, gray bar. Note difference in scales between individual panels. Data represent a mean ± SEM of 4–5 mice assayed individually per time-point in 2 experiments; * p<0.05 ANOVA with Dunnet's post-hoc analysis vs. Day 0 (uninfected) of mice of the CCR2 expression profile, ‡p<0.05 by unpaired student t-test when values from CCR2+/+ mice or CCR2−/− mice were compared at the designated time point.
In lungs of C. neoformans-infected CCR2−/− mice, mRNA levels of CCL2, CCL7, and CCL12 also significantly increased from baseline (Fig. 5A–C). These levels exceeded those of CCR2+/+ mice at some time-points. mRNA levels of CCL11 (Eotaxin-1) peaked at day 14 post-infection in CCR2−/− mice (Fig. 5D) accounting for the observed influx of eosinophils that occurs at this time (19) (and data not shown). Thus, CCR2 ligands (CCL2 and CCL7) are expressed in the lung with appropriate kinetics to mediate the CCR2-dependent DC recruitment observed in this model. Moreover, production of these ligands does not depend on recruitment of CCR2+ cells.
Early bronchovascular infiltrates are diminished in CCR2-deficient mice infected with C. neoformans
We next evaluated the lungs of both strains of mice morphologically for evidence of differing patterns of inflammation at day 10, the period of active DC recruitment in CCR2+/+ mice. Prominent inflammatory cell infiltrates developed in CCR2+/+ mice by 10 days post-infection in two micro-anatomic locations: 1) surrounding bronchovascular bundles, and 2) within alveolar spaces (Fig. 6A, C, E). Bronchovascular infiltrates consisted of densely packed mononuclear cells surrounding small arterioles and venules. Many of the cells were lymphocytes, as evident by their compact nuclei and minimal cytoplasm. The vessels appear thickened, and often contained flattened, adherent leukocytes, suggestive of an activated endothelium (Fig. 6E, high power). Less well-developed infiltrates were seen in the submucosal regions underlying small airways. Infiltrates filling alveolar spaces consisted of loose granulomatous inflammation containing C. neoformans, granulocytes, macrophages, and other mononuclear cells. In contrast, bronchovascular infiltrates identified in infected CCR2−/− mice appeared smaller and less well-developed (Fig. 6B, D, F). Infiltrates contained fewer leukocytes; however, perivascular, extracellular matrix was more apparent (Fig. 6F, high power). Alveolar infiltrates contained higher numbers of eosinophils as expected.
Figure 6.

Early bronchovascular infiltrates are diminished in CCR2−/− mice infected with C. neoformans. CCR2+/+ (A, C, E) and CCR2−/− (B, D, F) mice were inoculated IT with C. neoformans and lungs were harvested at day 10 post-infection. Samples were processed for histological evaluation using H & E stain as described in Methods. (A, B) Photomicrographs (×100 magnification) depicting two spatially distinct regions of lung inflammation: bronchovascular infiltrates (BVI) and alveolar infiltrates (AI). Note the BVI adjacent to the airway (AW). BVI are more prominent in CCR2+/+ mice (A) whereas they appear less developed in CCR2−/− mice (B) despite the presence of substantial inflammation. At higher power, (C, D ×200) and (E, F ×400), note that BVI in CCR2+/+ mice (C, E) contain predominantly mononuclear cells and lymphocytes that tightly pack regions surrounding small blood vessels. In contrast, BVI in CCR2−/− mice (D, F) appear reduced both in cell number and density, whereas extracellular matrix in the perivascular space is increased. Individual C. neoformans and infected macrophages (arrow) are identified confirming the presence of infection in these regions.
Patterns of inflammation in CCR2+/+ and CCR2−/− mice continued to diverge at a later time-point (day 21) post-infection (Fig. 7). Bronchovascular infiltrates were diminished in CCR2−/− mice (Fig. 7B) relative to CCR2+/+ mice (Fig. 7A), although differences between strains were less apparent than at the earlier time-point. Trichrome staining (Fig. 7C, D) revealed increased bronchovascular collagen deposition in CCR2−/− mice (Fig. 7D). Increased airway mucus production, smooth muscle hypertrophy, and tissue eosinophilia was also observed (in CCR2−/− mice, Fig. 7D, and data not shown). Collectively, these data demonstrate prominent bronchovascular infiltrate formation in CCR2+/+ mice correlates with the known development of T1 immunity and clearance of C. neoformans in these mice (20). In contrast, diminished formation of bronchovascular infiltrates in CCR2-deficient mice correlates with the characteristic morphological features of pulmonary T2 responses by three weeks post-infection with C. neoformans.
Figure 7.

Increased bronchovascular collagen deposition is evident during later stages of C. neoformans infection in CCR2−/− mice. CCR2+/+ (A, C) and CCR2−/− (B, D) mice were inoculated IT with C. neoformans and lungs were harvested at day 21 post-infection. Samples were processed for histological evaluation as described in Methods. (A), Photomicrographs (H & E staining, ×400 magnification) in CCR2+/+ (A) and CCR2−/− (B) mice depict a persistent reduction in bronchovascular infiltrates (BVI) adjacent to airways (AW) in CCR2−/− mice. Trichrome staining in CCR2+/+ (C) and CCR2−/− (D) mice (×400) reveals increased bronchovascular fibrosis (blue) in CCR2−/− mice. Note that airway mucus production (B, arrows) and smooth muscle (D, block arrowheads) are more apparent in CCR2−/− mice.
DC localized to bronchovascular infiltrates in CCR2+/+ mice infected with C. neoformans
In contrast to the abundant literature on the micro-anatomic location of lung DC in diseases of larger airways such as asthma (39, 49), it is unknown where lung DC localize during fungal pulmonary infections, in which inflammation is restricted to terminal bronchioles and alveolar spaces. Our FACS analysis suggests that the majority of cDC accumulate within the interstitial compartment of the lung in mice infected with C. neoformans (described above and Fig. 3A). We further investigated the specific microanatomic location of lung DC in the two strains of mice using immunohistochemistry (Fig. 8). DC were identified as cells that expressed both MHC Class II and CD11c and displayed multiple cytoplasmic extensions. They were readily distinguished from lung macrophages which expressed only CD11c and had distinct morphologic features including vacuolated cytoplasm and on occasion, intracellular C. neoformans (Fig. 8B). In CCR2+/+ mice, DC formed a near-continuous network of tightly intertwined cells specifically surrounding bronchovascular structures (Fig. 8C, D). In contrast, lung DC were notably reduced in CCR2−/− mice (Fig. 8E), consistent with our quantitative analysis. Instead, larger numbers of large, vacuolated cells lacking DC morphology were seen; these cells expressed only CD11c and are presumably macrophages (Fig. 8F).
Figure 8.

Conventional DC form cellular networks within early bronchovascular infiltrates in the lungs of CCR2+/+ mice infected with C. neoformans. CCR2+/+ (A–D, G, H) and CCR2−/− (E, F) mice were inoculated IT with C. neoformans and lungs were harvested at day 10 post-infection. Samples were snap-frozen and later stained with either isotype contols (A), anti-CD11c (red peroxidase reaction product) and anti-MHC Class II (I-Ad; black alkaline phosphatase reaction product) (B–F), or anti-CD11c (red) and anti-CD11b (black) (G–H). (A) isotype control staining (×100 magnification). (B) Dendritic cell (DC) expressing both MHC Class II and CD11c (note reddish-gray color), and displaying branching cytoplasmic extensions is identified adjacent to a visible C. neoformans (CN) (×1000). Also depicted is a CD11c-expressing macrophage (M) (note strong red staining and vacuolated appearance) which has ingested a C. neoformans. (C) Photomicrograph (×200) taken from a CCR2+/+ mouse demonstrating a scaffold-like network of DC within a bronchovascular infiltrate (BVI) adjacent an airway (AW). (D) Higher power (×400) image further identifying DC (block arrowheads). (E) Photomicrograph (×200) from a CCR2−/− mouse confirms the paucity of DC and demonstrates that the majority of CD11c positive (red) cells within these infiltrates are macrophages (arrows). (F) Higher power (×400) image confirms macrophage morphology of these CD11c positive cells (arrows), many of which have ingested C. neoformans. (G) Photomicrograph (×200) from a CCR2+/+ mouse co-stained with CD11c (red) and CD11b (black) depicting a dense bronchovascular infiltrate. (H) Higher power (×400) image reveals that numerous cells with branching cytoplasmic extensions express both CD11c and CD11b (arrows, note reddish-gray color) confirming their cDC phenotype.
To further confirm that bronchovascular infiltrates were enriched for cDC, the combination of anti-CD11b and anti-CD11c antibodies were applied to lung sections from CCR2+/+ mice infected with C. neoformans. CD11b is absent on lung macrophages (23, 40, 41) but is expressed on cDC (and a broad population of other lung leukocytes including neutrophils, monocytes, and eosinophils). Results revealed that numerous cells within bronchovascular infiltrates expressed both markers (CD11b and CD11c) and displayed classic DC morphology thereby confirming their cDC phenotype (Fig. 8G, H). Macrophages were readily identified as large, vacuolated CD11c+ cells lacking expression of CD11b (data not shown but similar to the macrophages depicted in Fig. 8B) and appeared more numerous in CCR2−/− mice (similar to that shown in Fig. 8E, F).
Co-localization of DC and CD4+ T within bronchovascular infiltrates is associated with increased expression of IL-12 and IFN-γ mRNA in the lungs of CCR2+/+ mice infected with C. neoformans
T cell polarization against fungal pathogens is completed once T cells arrive within the lung (27, 50). We hypothesize that T cell polarization requires interaction(s) with APC. We therefore investigated whether DC specifically co-localized with CD4+ T cells. In this experiment, DC were identified by expression of MHC Class II and characteristic morphology. T cells were smaller, more compact, and expressed CD4. In the lungs of infected CCR2+/+ mice, we observed DC within bronchovascular infiltrates interdigitating with numerous CD4+ T cells (Fig. 9A). Individual DC formed contacts with multiple CD4+ lymphocytes simultaneously (Fig. 9A-inset). Few DC/CD4+ interactions were observed in CCR2−/− mice due to both the reduction in bronchovascular infiltrates, and the paucity of lung DC (data not shown). Thus, the co-localization of DC and CD4+ T cells within bronchovascular infiltrates is associated with CCR2 expression in mice infected with C. neoformans.
Figure 9.

Co-localization of DC and CD4+ lymphocytes within bronchovascular infiltrates coincides with increased mRNA expression of IL-12 and IFN-γ in CCR2+/+ mice. (A) CCR2+/+ mice were inoculated IT with C. neoformans and lungs removed at day 10 post-infection. Samples were snap-frozen and later stained for anti-MHC Class II (I-Ad; black alkaline phosphatase reaction) to identify DC with characteristic morphology and anti-CD4 (red peroxidase reaction) to identify CD4+ lymphocytes. Photomicrograph (×200 magnification) depicting a bronchovascular infiltrate adjacent to an airway (AW). Note the extensive co-localization of DC with CD4+ lymphocytes. At higher magnification (see inset, ×1000), individual DC are identified interacting with multiple CD4+ lymphocytes. (B–D) RNA was prepared from lung homogenates obtained from CCR2+/+ mice and CCR2−/− mice at baseline and day 14 post-IT infection with C. neoformans. A separate RNA preparation from uninfected CCR2+/+ mice (n=5) was used as a reference standard. RT-PCR was performed to evaluate the expression of (B) IL-12p35, (C) IFN-γ and (D) IL-4. CCR2+/+mice, black bar; CCR2−/− mice, gray bar. Data represents a mean ± SEM of 5 mice assayed individually per time-point in 2 experiments; * p<0.05 by unpaired student t-test vs. Day 0 (uninfected) mice of the same CCR2 expression profile, ‡p<0.05 by unpaired student t-test when values from CCR2+/+ mice vs. CCR2−/− mice were compared at the designated time point.
DC production of IL-12 is a key determinant of T1 immune responses (39, 51). IL-12 and IFN-γ are strongly associated with the development of T1 immune responses and clearance of C. neoformans infection (10, 13, 22, 52). In this study, we found that expression of both IL-12 (p35 subunit, Fig. 9B; p40 subunit, data not shown) and IFN-γ (Fig. 9C) was significantly increased (relative to baseline) in CCR2+/+ mice two weeks post-infection with C. neoformans, whereas the increase from baseline in CCR2−/− mice was less marked. In contrast, expression of IL-4 remained unchanged in CCR2+/+ mice, but increased in CCR2−/− mice at day 14 (Fig. 9C). Collectively, these data demonstrate that DC recruitment and T cell co-localization observed in CCR2+/+ mice correlates with IL-12 and T1 cytokine expression in response to C. neoformans infection.
Discussion
This study defines for the first time the effect of CCR2 deficiency on the numbers, phenotype, and micro-anatomical location of lung DC throughout an extended time course of a fungal pulmonary infection. We report that in mice infected intratracheally with C. neoformans: (1) large numbers of immature cDC accumulated predominantly in the lung interstitium of mice expressing CCR2; (2) lung DC recruitment was temporally associated with the early upregulation of two ligands for murine CCR2, CCL2 and CCL7; (3) in CCR2-expressing mice, the formation of bronchovascular infiltrates correlated temporally and spatially with recruitment of cDC to these structures; (4) co-localization of DC and CD4+ T cells within bronchovascular infiltrates coincides with increased expression of the T1-associated cytokines IL-12 and IFN-γ; and (5) bronchovascular infiltrates were markedly reduced in CCR2−/− mice, which, as we have previously shown, develop a T2 response ineffectual for fungal clearance. Collectively, our findings demonstrate that CCR2 mediates lung DC recruitment and the formation of bronchovascular infiltrates, a form of lymphoneogenesis, in response to pulmonary infection with C. neoformans. The specific identification of DC/CD4+ T cell interactions within bronchovascular infiltrates provides additional evidence that DC are critical determinants of T1 polarization and effective pulmonary host defense against a fungal pathogen.
Several aspects of the experimental design are worthy of mention. First, this murine model system has defined the importance of T1 immunity for clearance of C. neoformans (5–14), an important opportunistic pathogen in immunocompromised hosts. This knowledge is crucial for the development of novel therapeutics or vaccines. Second, because lack of CCR2 in the gene-targeted mice is specific, absolute, and irreversible, the role of CCR2 can be evaluated without concern for residual receptor function or need to block multiple CCR2 ligands simultaneously. Third, our flow cytometric method, which carefully distinguishes DC from monocytes and macrophages, permits more informative assessment of the recruited mononuclear cell population than previously achieved by visual identification alone. Fourth, the thorough temporal and spatial analysis of recruited lung DC and chemokines enhances data interpretation and applicability to other studies of fungal infection.
The observed striking accumulation of lung cDC, accounting for 10–12% of total recruited leukocytes early in the infection, demonstrates the potent inflammation induced by C. neoformans (8, 15, 19, 53, 54). An early feature of this inflammation is marked up-regulation of CCL2 and CCL7. The kinetics of mRNA levels for these transcriptionally-regulated chemokines, increasing immediately before the onset of DC accumulation, implicates these CCR2 ligands as DC chemoattractants. They may also function by facilitating DC (or DC precursor) release from the bone marrow (55, 56). Reduced numbers of DC in the lungs of infected CCR2−/− mice cannot be explained by lack of chemoattractant signals, as levels of CCL2 and CCL7 were equivalent to wild-type mice. Nor, based on previous data, can the disparity in DC numbers by attributed to a difference in microbial burden (19).
The early appearance of DC within bronchovascular infiltrates in CCR2+/+ mice but not in CCR2−/− mice illustrates the critical role of chemokines, and their receptors, as mediators of DC transit between distinct tissue compartments (23, 56, 57). In the absence of CCR2, DC lack access to the bronchovascular compartment, thereby diminishing inflammatory infiltrates in this region. Lymphatics originating in these neolymphoid organs serve as a portal of entry for microbial antigens draining from the alveolar environment and DC egress into lung-associated lymph nodes (58). Thus, the novel finding of diminished bronchovascular infiltrates in the absence of CCR2 probably also account for the reduced numbers of DC arriving in the lung- associated lymph nodes of CCR2−/− mice infected with C. neoformans (20).
By showing for the first time that T cells recruited to the lungs early in the infection encounter a dense network of DC within bronchovascular infiltrates, this study enhances understanding of CCR2-dependent differences in T cell polarization and clearance of fungal infection. T cell priming to fungal pathogens such as C. neoformans and A. fumigatus is initiated in regional lymph nodes, but polarization is not completed until T cells arrive within the lung (27, 50). Although T cell priming in the node is undoubtedly mediated by DC, the mechanism(s) responsible for T cell polarization within the lung have remained poorly understood. Bronchovascular bundles are important sites of cellular recruitment from blood to lung via high endothelial venules (59). Lung DC residing in this region are thereby poised to provide “up to the minute” information regarding the etiology and intensity of the infection to Ag-specific T cells upon their arrival in the lung. The temporal-spatial interactions we show here correlate directly with the kinetics with which T cell polarization develops in these models (19, 20, 27, 50). These interactions in the lungs of CCR2-expressing mice may insure that recruited CD4+ T cells are exposed to highly focused concentrations of IL-12, an essential determinant of T1 polarization against many microbial pathogens, including C. neoformans (9, 10, 22). In addition, DC interactions with C. neoformans-containing macrophages might facilitate DC cross-priming of CD8+ T cells, which play an important role in defense against fungal infections (60, 61).
Diminished lung DC recruitment in the absence of CCR2 suggests that subsequent DC-T cell interactions within bronchovascular infiltrates are reduced. This finding represents a potential common mechanistic link between our studies and other investigations previously demonstrating impaired T1 immune responses and/or microbial clearance in CCR2-deficient mice (19, 62–65). However, our results contrast with a recent report by Winters et al., investigating the effect of lung DC numbers on microbial clearance (40). Although that study defined cDC in much the same way as we did, subcutaneous injections of FMS-like tyrosine kinase 3 (Flt-3) ligand increased total numbers of lung DC prior to infection with Steptococcus pneumoniae but did not enhance microbial clearance. We believe this disparity can be accounted by at least two features which differ in our respective studies. First, C. neoformans is a eukaryotic and predominantly intracellular fungal pathogen that differs significantly from the prokaryotic bacteria used in this study. Clearance of C. neoformans is highly dependent upon the successful initiation of a T1 adaptive immune response (occurring over weeks) whereas elimination of bacterial pathogens (in non-immune animals) relies primarily upon innate immunity (occurring over days). Secondly, these authors enhanced lung DC numbers prior to the onset of infection. This approach, although interesting, differs significantly from our approach in which lung DC numbers are equivalent between CCR2+/+ and CCR2−/− mice prior to infection.
Our analysis of lung DC co-stimulatory molecule expression raises three interesting points. First, the absence of significant in-situ DC maturation in either strain of mice may seem surprising. Bone-marrow derived DC from both CCR2+/+ and CCR2−/− mice will mature in response to LPS (data not shown). However, we have previously shown weak co-stimulatory molecule expression on bone-marrow derived DC pulsed with mannoproteins derived from C. neoformans (14). This result suggests that cryptococcal products may interfere with DC maturation. Second, the data imply that the differential T cell polarization observed in this model does not result from substantial differences in the expression of CD40, CD80, or CD86 by lung DC in either the lung interstitium or alveolar space, as their pattern of expression was similar in CCR2+/+ and CCR2−/− mice. It remains possible that differential expression of other co-stimulatory molecules (ICOSL, OX40L) or Notch ligands (Delta, Jagged) by DC might also influence T cell polarization in this model system. Third, the finding that DC in CCR2+/+ mice expressed low levels of CD40, CD80, and CD86 indicates that effective T1 pulmonary immune responses can develop without high expression in the lungs of traditional co-stimulatory molecules. The current findings are not simply the result of the primary nature of this response, as we found a similar lung DC phenotype during the secondary response to a non-infectious particulate antigen (23). Because Ag-sensitized T cells require less co-stimulation for activation than do naïve T cells (66), low expression of these co-stimulatory molecules by lung DC might ensure that only Ag-sensitized T cells arriving from the node {or a memory population (67)} will be activated. Theoretically, diminished co-stimulatory molecule expression by lung DC might protect against deleterious non-specific inflammation within the pulmonary parenchyma in an effort to preserve gas exchange. We believe this model is ideally suited for further studies investigating the role of lung DC co-stimulatory expression on T cell responses directed against a fungal pathogen.
In summary, large numbers of immature cDC accumulate in the lung interstitium following infection with C. neoformans. DC recruitment is CCR2-dependent, and temporally related to the expression of the CCR2 ligands CCL2 and CCL7. CCR2 expression was required for the formation of prominent bronchovascular infiltrates containing DC and CD4+ lymphocytes and the production of T1-promoting cytokines. These novel findings shed significant insight into T cell polarization in response to this clinically significant fungal pathogen. Further definition of the temporal and spatial relationships between DC and T cells within the lung is needed to advance understanding of host defense and aberrant immune responses within the lung.
Acknowledgements
none
Supported by Merit Review Awards (G.B.T. and J.L.C.) and a Career Development Award-2 (J.J.O) from the Biomedical Laboratory Research & Development Service, Department of Veterans Affairs and by National Institutes of Health Grants RO1-HL065912 (to G.B.H.), RO1-AI059201 (to G.B.H.), RO1-HL051082 (to G.B.T.), and T32-AI07413 (to J.J.O.).
The abbreviations used are
- DC
dendritic cells
- cDC
conventional dendritic cells
- mDC
myeloid dendritic cells
- CCR
chemokine receptor
- CCL
chemokine ligand
- CCR2−/−
chemokine receptor 2 gene knockout
- i.t.
intratracheal
- HEV
high endothelial venule
- IL
interleukin
- IFN
interferon
- Ag
antigen
- FACS
flow cytometric analysis
Footnotes
Publisher's Disclaimer: “This is an author-produced version of a manuscript accepted for publication in The Journal of Immunology (The JI). The American Association of Immunologists, Inc. (AAI), publisher of The JI, holds the copyright to this manuscript. This version of the manuscript has not yet been copyedited or subjected to editorial proofreading by The JI; hence, it may differ from the final version published in The JI (online and in print). AAI (The JI) is not liable for errors or omissions in this author-produced version of the manuscript or in any version derived from it by the U.S. National Institutes of Health or any other third party. The final, citable version of record can be found at www.jimmunol.org.”
Disclosures None
Portions of this work have been presented previously at the International Conference of the American Thoracic Society, San Diego, CA, May 22, 2005 and May 22, 2006 and have been published in abstract form (Proc. Am. Thorac. Soc. (2);A364, May 2005 and Proc. Am. Thorac. Soc. (3):A346, 2006) and as an oral presentation at the Keystone Symposium: Determinants of Host Resistance, Susceptibility or Immunopathology to Pathogens: Integrating Knowledge from Experimental Models to Human Disease”, Keystone, CO, January 2006
References
- 1.Kwon-Chung KJ, Sorrell TC, Dromer F, Fung E, Levitz SM. Cryptococcosis: clinical and biological aspects. Med. Mycol. 2000;38(Suppl 1):205–213. [PubMed] [Google Scholar]
- 2.Levitz SM. Activation of human peripheral blood mononuclear cells by interleukin-2 and granulocyte-macrophage colony-stimulating factor to inhibit Cryptococcus neoformans. Infect. Immun. 1991;59:3393–3397. doi: 10.1128/iai.59.10.3393-3397.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Shoham S, Levitz SM. The immune response to fungal infections. Br. J. Haematol. 2005;129:569–582. doi: 10.1111/j.1365-2141.2005.05397.x. [DOI] [PubMed] [Google Scholar]
- 4.Rapp RP. Changing strategies for the management of invasive fungal infections. Pharmacotherapy. 2004;24:4S–28S. quiz 29S–32S. [PubMed] [Google Scholar]
- 5.Huffnagle GB, Yates JL, Lipscomb MF. T cell-mediated immunity in the lung: a Cryptococcus neoformans pulmonary infection model using SCID and athymic nude mice. Infect. Immun. 1991;59:1423–1433. doi: 10.1128/iai.59.4.1423-1433.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Huffnagle GB, Lipscomb MF, Lovchik JA, Hoag KA, Street NE. The role of CD4+ and CD8+ T cells in the protective inflammatory response to a pulmonary cryptococcal infection. J. Leukoc. Biol. 1994;55:35–42. doi: 10.1002/jlb.55.1.35. [DOI] [PubMed] [Google Scholar]
- 7.Huffnagle GB, Lipscomb MF. Cells and cytokines in pulmonary cryptococcosis. Res. Immunol. 1998;149 doi: 10.1016/s0923-2494(98)80762-1. [DOI] [PubMed] [Google Scholar]
- 8.Huffnagle GB, Toews GB, Burdick MD, Boyd MB, McAllister KS, McDonald RA, Kunkel SL, Strieter RM. Afferent phase production of TNF-alpha is required for the development of protective T cell immunity to Cryptococcus neoformans. J. Immunol. 1996;157:4529–4536. [PubMed] [Google Scholar]
- 9.Kawakami K, Tohyama M, Xie Q, Saito A. IL-12 protects mice against pulmonary and disseminated infection caused by Cryptococcus neoformans. Clin. Exp. Immunol. 1996;104:208–214. doi: 10.1046/j.1365-2249.1996.14723.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Herring AC, Lee J, McDonald RA, Toews GB, Huffnagle GB. Induction of interleukin-12 and gamma interferon requires tumor necrosis factor alpha for protective T1-cell-mediated immunity to pulmonary Cryptococcus neoformans infection. Infect. Immun. 2002;70:2959–2964. doi: 10.1128/IAI.70.6.2959-2964.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bauman SK, Huffnagle GB, Murphy JW. Effects of tumor necrosis factor alpha on dendritic cell accumulation in lymph nodes draining the immunization site and the impact on the anticryptococcal cell-mediated immune response. Infect. Immun. 2003;71:68–74. doi: 10.1128/IAI.71.1.68-74.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Arora S, Hernandez Y, Erb-Downward JR, McDonald RA, Toews GB, Huffnagle GB. Role of IFN-gamma in regulating T2 immunity and the development of alternatively activated macrophages during allergic bronchopulmonary mycosis. J. Immunol. 2005;174:6346–6356. doi: 10.4049/jimmunol.174.10.6346. [DOI] [PubMed] [Google Scholar]
- 13.Chen GH, McDonald RA, Wells JC, Huffnagle GB, Lukacs NW, Toews GB. The gamma interferon receptor is required for the protective pulmonary inflammatory response to Cryptococcus neoformans. Infect. Immun. 2005;73:1788–1796. doi: 10.1128/IAI.73.3.1788-1796.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Herring AC, Falkowski NR, Chen GH, McDonald RA, Toews GB, Huffnagle GB. Transient neutralization of tumor necrosis factor alpha can produce a chronic fungal infection in an immunocompetent host: potential role of immature dendritic cells. Infect. Immun. 2005;73:39–49. doi: 10.1128/IAI.73.1.39-49.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Huffnagle GB, Strieter RM, Standiford TJ, McDonald RA, Burdick MD, Kunkel SL, Toews GB. The role of monocyte chemotactic protein-1 (MCP-1) in the recruitment of monocytes and CD4+ T cells during a pulmonary Cryptococcus neoformans infection. J. Immunol. 1995;155:4790–4797. [PubMed] [Google Scholar]
- 16.Huffnagle GB, Strieter RM, McNeil LK, McDonald RA, Burdick MD, Kunkel SL, Toews GB. Macrophage inflammatory protein-1alpha (MIP-1alpha) is required for the efferent phase of pulmonary cell-mediated immunity to a Cryptococcus neoformans infection. J. Immunol. 1997;159:318–327. [PubMed] [Google Scholar]
- 17.Olszewski MA, Huffnagle GB, Traynor TR, McDonald RA, Cook DN, Toews GB. Regulatory effects of macrophage inflammatory protein 1alpha/CCL3 on the development of immunity to Cryptococcus neoformans depend on expression of early inflammatory cytokines. Infect. Immun. 2001;69:6256–6263. doi: 10.1128/IAI.69.10.6256-6263.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Olszewski MA, Huffnagle GB, McDonald RA, Lindell DM, Moore BB, Cook DN, Toews GB. The role of macrophage inflammatory protein-1 alpha/CCL3 in regulation of T cell-mediated immunity to Cryptococcus neoformans infection. J. Immunol. 2000;165:6429–6436. doi: 10.4049/jimmunol.165.11.6429. [DOI] [PubMed] [Google Scholar]
- 19.Traynor TR, Kuziel WA, Toews GB, Huffnagle GB. CCR2 expression determines T1 versus T2 polarization during pulmonary Cryptococcus neoformans infection. J. Immunol. 2000;164:2021–2027. doi: 10.4049/jimmunol.164.4.2021. [DOI] [PubMed] [Google Scholar]
- 20.Traynor TR, Herring AC, Dorf ME, Kuziel WA, Toews GB, Huffnagle GB. Differential roles of CC chemokine ligand 2/monocyte chemotactic protein-1 and CCR2 in the development of T1 immunity. J. Immunol. 2002;168:4659–4666. doi: 10.4049/jimmunol.168.9.4659. [DOI] [PubMed] [Google Scholar]
- 21.Huffnagle GB, McNeil LK, McDonald RA, Murphy JW, Toews GB, Maeda N, Kuziel WA. Cutting edge: Role of C-C chemokine receptor 5 in organ-specific and innate immunity to Cryptococcus neoformans. J. Immunol. 1999;163:4642–4646. [PubMed] [Google Scholar]
- 22.Hoag KA, Lipscomb MF, Izzo AA, Street NE. IL-12 and IFN-gamma are required for initiating the protective Th1 response to pulmonary cryptococcosis in resistant C.B-17 mice. Am. J. Respir. Cell. Mol. Biol. 1997;17:733–739. doi: 10.1165/ajrcmb.17.6.2879. [DOI] [PubMed] [Google Scholar]
- 23.Osterholzer JJ, Ames T, Polak T, Sonstein J, Moore BB, Chensue SW, Toews GB, Curtis JL. CCR2 and CCR6, but not endothelial selectins, mediate the accumulation of immature dendritic cells within the lungs of mice in response to particulate antigen. J. Immunol. 2005;175:874–883. doi: 10.4049/jimmunol.175.2.874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wozniak KL, Vyas JM, Levitz SM. In vivo role of dendritic cells in a murine model of pulmonary cryptococcosis. Infect. Immun. 2006;74:3817–3824. doi: 10.1128/IAI.00317-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Syme RM, Spurrell JC, Amankwah EK, Green FH, Mody CH. Primary dendritic cells phagocytose Cryptococcus neoformans via mannose receptors and Fcgamma receptor II for presentation to T lymphocytes. Infect. Immun. 2002;70:5972–5981. doi: 10.1128/IAI.70.11.5972-5981.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kelly RM, Chen J, Yauch LE, Levitz SM. Opsonic requirements for dendritic cell-mediated responses to Cryptococcus neoformans. Infect. Immun. 2005;73:592–598. doi: 10.1128/IAI.73.1.592-598.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lindell DM, Moore TA, McDonald RA, Toews GB, Huffnagle GB. Distinct compartmentalization of CD4+ T-cell effector function versus proliferative capacity during pulmonary cryptococcosis. Am. J. Pathol. 2006;168:847–855. doi: 10.2353/ajpath.2006.050522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kuziel WA, Morgan SJ, Dawson TC, Griffin S, Smithies O, Ley K, Maeda N. Severe reduction in leukocyte adhesion and monocyte extravasation in mice deficient in CC chemokine receptor 2. Proc. Natl. Acad. Sci. U S A. 1997;94:12053–12058. doi: 10.1073/pnas.94.22.12053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Curtis JL, Kim S, Scott PJ, Buechner-Maxwell VA. Adhesion receptor phenotypes of murine lung CD4+ T cells during a pulmonary immune response to sheep erythrocytes. Am. J. Respir. Cell. Mol. Biol. 1995;12:520–530. doi: 10.1165/ajrcmb.12.5.7537969. [DOI] [PubMed] [Google Scholar]
- 30.Hu B, Sonstein J, Christensen PJ, Punturieri A, Curtis JL. Deficient in vitro and in vivo phagocytosis of apoptotic T cells by resident murine alveolar macrophages. J. Immunol. 2000;165:2124–2133. doi: 10.4049/jimmunol.165.4.2124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.van Rijt LS, Jung S, Kleinjan A, Vos N, Willart M, Duez C, Hoogsteden HC, Lambrecht BN. In vivo depletion of lung CD11c+ dendritic cells during allergen challenge abrogates the characteristic features of asthma. J. Exp. Med. 2005;201:981–991. doi: 10.1084/jem.20042311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.van Rijt LS, Kuipers H, Vos N, Hijdra D, Hoogsteden HC, Lambrecht BN. A rapid flow cytometric method for determining the cellular composition of bronchoalveolar lavage fluid cells in mouse models of asthma. J. Immunol. Methods. 2004;288:111–121. doi: 10.1016/j.jim.2004.03.004. [DOI] [PubMed] [Google Scholar]
- 33.van Rijt LS, Prins JB, Leenen PJ, Thielemans K, de Vries VC, Hoogsteden HC, Lambrecht BN. Allergen-induced accumulation of airway dendritic cells is supported by an increase in CD31(hi)Ly-6C(neg) bone marrow precursors in a mouse model of asthma. Blood. 2002;100:3663–3671. doi: 10.1182/blood-2002-03-0673. [DOI] [PubMed] [Google Scholar]
- 34.Zar JH. Biostatistical analysis. Englewood Cliffs; Prentice-Hall: 1974. [Google Scholar]
- 35.Shortman K, Naik SH. Steady-state and inflammatory dendritic-cell development. Nat. Rev. Immunol. 2007;7:19–30. doi: 10.1038/nri1996. [DOI] [PubMed] [Google Scholar]
- 36.Wu L, Liu YJ. Development of dendritic-cell lineages. Immunity. 2007;26:741–750. doi: 10.1016/j.immuni.2007.06.006. [DOI] [PubMed] [Google Scholar]
- 37.Constant SL, Brogdon JL, Piggott DA, Herrick CA, Visintin I, Ruddle NH, Bottomly K. Resident lung antigen-presenting cells have the capacity to promote Th2 T cell differentiation in situ. J. Clin. Invest. 2002;110:1441–1448. doi: 10.1172/JCI16109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Peters W, Cyster JG, Mack M, Schlondorff D, Wolf AJ, Ernst JD, Charo IF. CCR2-dependent trafficking of F4/80dim macrophages and CD11cdim/intermediate dendritic cells is crucial for T cell recruitment to lungs infected with Mycobacterium tuberculosis. J. Immunol. 2004;172:7647–7653. doi: 10.4049/jimmunol.172.12.7647. [DOI] [PubMed] [Google Scholar]
- 39.Vermaelen K, Pauwels R. Pulmonary dendritic cells. Am. J. Respir. Crit. Care Med. 2005;172:530–551. doi: 10.1164/rccm.200410-1384SO. [DOI] [PubMed] [Google Scholar]
- 40.Winter C, Taut K, Langer F, Mack M, Briles DE, Paton JC, Maus R, Srivastava M, Welte T, Maus UA. FMS-like tyrosine kinase 3 ligand aggravates the lung inflammatory response to Streptococcus pneumoniae infection in mice: role of dendritic cells. J. Immunol. 2007;179:3099–3108. doi: 10.4049/jimmunol.179.5.3099. [DOI] [PubMed] [Google Scholar]
- 41.Winter C, Taut K, Srivastava M, Langer F, Mack M, Briles DE, Paton JC, Maus R, Welte T, Gunn MD, Maus UA. Lung-specific overexpression of CC chemokine ligand (CCL) 2 enhances the host defense to Streptococcus pneumoniae infection in mice: role of the CCL2-CCR2 axis. J. Immunol. 2007;178:5828–5838. doi: 10.4049/jimmunol.178.9.5828. [DOI] [PubMed] [Google Scholar]
- 42.Vermaelen KY, Pauwels RA. Accelerated airway dendritic cell maturation, trafficking and elimination in a mouse model of asthma. Am. J. Respir. Cell. Mol. Biol. 2003;29:405–409. doi: 10.1165/rcmb.2003-0008OC. [DOI] [PubMed] [Google Scholar]
- 43.Vermaelen KY, Carro-Muino I, Lambrecht BN, Pauwels RA. Specific migratory dendritic cells rapidly transport antigen from the airways to the thoracic lymph nodes. J. Exp. Med. 2001;193:51–60. doi: 10.1084/jem.193.1.51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Julia V, Hessel EM, Malherbe L, Glaichenhaus N, O'Garra A, Coffman RL. A restricted subset of dendritic cells captures airborne antigens and remains able to activate specific T cells long after antigen exposure. Immunity. 2002;16:271–283. doi: 10.1016/s1074-7613(02)00276-5. [DOI] [PubMed] [Google Scholar]
- 45.Cochand L, Isler P, Songeon F, Nicod LP. Human lung dendritic cells have an immature phenotype with efficient mannose receptors. Am. J. Respir. Cell. Mol. Biol. 1999;21:547–554. doi: 10.1165/ajrcmb.21.5.3785. [DOI] [PubMed] [Google Scholar]
- 46.Gonzalez-Juarrero M, Orme IM. Characterization of murine lung dendritic cells infected with Mycobacterium tuberculosis. Infect. Immun. 2001;69:1127–1133. doi: 10.1128/IAI.69.2.1127-1133.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Maus U, von Grote K, Kuziel WA, Mack M, Miller EJ, Cihak J, Stangassinger M, Maus R, Schlondorff D, Seeger W, Lohmeyer J. The role of CC chemokine receptor 2 in alveolar monocyte and neutrophil immigration in intact mice. Am. J. Respir. Crit. Care Med. 2002;166:268–273. doi: 10.1164/rccm.2112012. [DOI] [PubMed] [Google Scholar]
- 48.Haeberle HA, Kuziel WA, Dieterich HJ, Casola A, Gatalica Z, Garofalo RP. Inducible expression of inflammatory chemokines in respiratory syncytial virus-infected mice: role of MIP-1alpha in lung pathology. J. Virol. 2001;75:878–890. doi: 10.1128/JVI.75.2.878-890.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lambrecht BN. Dendritic cells and the regulation of the allergic immune response. Allergy. 2005;60:271–282. doi: 10.1111/j.1398-9995.2005.00708.x. [DOI] [PubMed] [Google Scholar]
- 50.Rivera A, Ro G, Van Epps HL, Simpson T, Leiner I, Sant'Angelo DB, Pamer EG. Innate immune activation and CD4+ T cell priming during respiratory fungal infection. Immunity. 2006;25:665–675. doi: 10.1016/j.immuni.2006.08.016. [DOI] [PubMed] [Google Scholar]
- 51.Banchereau J, Briere F, Caux C, Davoust J, Lebecque S, Liu YJ, Pulendran B, Palucka K. Immunobiology of dendritic cells. Annu. Rev. Immunol. 2000;18:767–811. doi: 10.1146/annurev.immunol.18.1.767. [DOI] [PubMed] [Google Scholar]
- 52.Kawakami K, Qureshi MH, Zhang T, Koguchi Y, Shibuya K, Naoe S, Saito A. Interferon-gamma (IFN-gamma)-dependent protection and synthesis of chemoattractants for mononuclear leucocytes caused by IL-12 in the lungs of mice infected with Cryptococcus neoformans. Clin. Exp. Immunol. 1999;117:113–122. doi: 10.1046/j.1365-2249.1999.00955.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Huffnagle GB, Lipscomb MF. Pulmonary cryptococcosis. Am. J. Pathol. 1992;141:1517–1520. [PMC free article] [PubMed] [Google Scholar]
- 54.Huffnagle GB, Yates JL, Lipscomb MF. Immunity to a pulmonary Cryptococcus neoformans infection requires both CD4+ and CD8+ T cells. J. Exp. Med. 1991;173:793–800. doi: 10.1084/jem.173.4.793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Robays LJ, Maes T, Lebecque S, Lira SA, Kuziel WA, Brusselle GG, Joos GF, Vermaelen KV. Chemokine receptor CCR2 but not CCR5 or CCR6 mediates the increase in pulmonary dendritic cells during allergic airway inflammation. J. Immunol. 2007;178:5305–5311. doi: 10.4049/jimmunol.178.8.5305. [DOI] [PubMed] [Google Scholar]
- 56.Serbina NV, Pamer EG. Monocyte emigration from bone marrow during bacterial infection requires signals mediated by chemokine receptor CCR2. Nat. Immunol. 2006;7:311–317. doi: 10.1038/ni1309. [DOI] [PubMed] [Google Scholar]
- 57.Vanbervliet B, Homey B, Durand I, Massacrier C, Ait-Yahia S, de Bouteiller O, Vicari A, Caux C. Sequential involvement of CCR2 and CCR6 ligands for immature dendritic cell recruitment: possible role at inflamed epithelial surfaces. Eur. J. Immunol. 2002;32:231–242. doi: 10.1002/1521-4141(200201)32:1<231::AID-IMMU231>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]
- 58.Albertine KHW, Hyde Mary C., Dallas M. Anatomy and Development of the Respiratory Tract. In: Mason RJ, Broaddus, Murray VC, Nadel John F., Jay A, editors. Textbook of Respiratory Medicine. 4th ed. Eksevier Saunders; Philadelphia: 2005. pp. 3–29. [Google Scholar]
- 59.Xu B, Wagner N, Pham LN, Magno V, Shan Z, Butcher EC, Michie SA. Lymphocyte homing to bronchus-associated lymphoid tissue (BALT) is mediated by L-selectin/PNAd, alpha4beta1 integrin/VCAM-1, and LFA-1 adhesion pathways. J. Exp. Med. 2003;197:1255–1267. doi: 10.1084/jem.20010685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lindell DM, Moore TA, McDonald RA, Toews GB, Huffnagle GB. Generation of antifungal effector CD8+ T cells in the absence of CD4+ T cells during Cryptococcus neoformans infection. J. Immunol. 2005;174:7920–7928. doi: 10.4049/jimmunol.174.12.7920. [DOI] [PubMed] [Google Scholar]
- 61.Lin JS, Yang CW, Wang DW, Wu-Hsieh BA. Dendritic cells cross-present exogenous fungal antigens to stimulate a protective CD8 T cell response in infection by Histoplasma capsulatum. J. Immunol. 2005;174:6282–6291. doi: 10.4049/jimmunol.174.10.6282. [DOI] [PubMed] [Google Scholar]
- 62.Sato N, Ahuja SK, Quinones M, Kostecki V, Reddick RL, Melby PC, Kuziel WA, Ahuja SS. CC chemokine receptor (CCR)2 is required for langerhans cell migration and localization of T helper cell type 1 (Th1)-inducing dendritic cells. Absence of CCR2 shifts the Leishmania major-resistant phenotype to a susceptible state dominated by Th2 cytokines, b cell outgrowth, and sustained neutrophilic inflammation. J. Exp. Med. 2000;192:205–218. doi: 10.1084/jem.192.2.205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Held KS, Chen BP, Kuziel WA, Rollins BJ, Lane TE. Differential roles of CCL2 and CCR2 in host defense to coronavirus infection. Virology. 2004;329:251–260. doi: 10.1016/j.virol.2004.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Peters W, Scott HM, Chambers HF, Flynn JL, Charo IF, Ernst JD. Chemokine receptor 2 serves an early and essential role in resistance to Mycobacterium tuberculosis. Proc. Natl. Acad. Sci. U S A. 2001;98:7958–7963. doi: 10.1073/pnas.131207398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Robben PM, LaRegina M, Kuziel WA, Sibley LD. Recruitment of Gr-1+ monocytes is essential for control of acute toxoplasmosis. J. Exp. Med. 2005;201:1761–1769. doi: 10.1084/jem.20050054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Dubey C, Croft M, Swain SL. Naive and effector CD4 T cells differ in their requirements for T cell receptor versus costimulatory signals. J. Immunol. 1996;157:3280–3289. [PubMed] [Google Scholar]
- 67.Lindell DM, Ballinger MN, McDonald RA, Toews GB, Huffnagle GB. Immunologic homeostasis during infection: coexistence of strong pulmonary cell-mediated immunity to secondary Cryptococcus neoformans infection while the primary infection still persists at low levels in the lungs. J. Immunol. 2006;177:4652–4661. doi: 10.4049/jimmunol.177.7.4652. [DOI] [PubMed] [Google Scholar]