

Abstract
The putative role of (TG/CA)n repeats in the regulation of transcription has recently been reported for several cancer- and disease-related genes, including the genes encoding the epidermal growth factor receptor (EGFR), hydroxysteroid (11-beta) dehydrogenase 2 (HSD11B2) and interferon-gamma (IFNG). These studies indicated a correlation between gene expression levels and the presence or length of (TG/CA)n repeats in their intron 1 regions. A genome-wide search for genes with similar features may provide evidence of whether these dinucleotide repeats represent a class of universal regulators of gene expression, which has recently begun to be investigated as a quantitative complex phenotype. Using a public database of simple repeats, we identified 330 genes containing potentially polymorphic long (TG/CA)n repeats (n ≥ 12) in their intron 1 regions. One known physiological pathway, the calcium signalling pathway, was found to be enriched among the genes containing long repeats. In addition, certain biological processes, such as cation transport, signal transduction and ion transport, were found to be enriched in these genes. Genotyping of the long repeats showed that the majority of these dinucleotide repeats were polymorphic in the HapMap CEU (Caucasians from Utah, USA) samples of northern and western European ancestry. Evidence for a significant association between these repeats and gene expression was not observed in the genes selected based on their expression profiles in the HapMap CEU samples. Our current findings, therefore, do not support a role for these repeats as a class of universal gene expression regulators. A more comprehensive evaluation of the relationship between these repeats and gene expression, potentially in other tissues, may be necessary to illustrate their roles in gene regulation in the future.
Keywords: CA repeat, intron, polymorphism, gene expression, pathway
Introduction
Microsatellite sequences that consist of repeating units of two to five base pairs with different lengths and complexities are dispersed along the human genome and exhibit extensive length polymorphism, a feature widely used in genetic mapping [1-3]. Among the dinucleotide repeats, (TG/CA)n (CA-) repeats represent the most common class of microsatellites in vertebrates, including humans [4]. Because of their structure of alternating purine/pyrimidine sequences, CA- repeats have a tendency to form Z-DNA under physiological conditions, suggesting a possible role for such Z elements in chromatin activation or genome rearrangements [5]. Biochemically, this feature of CA-repeats may affect the movement of RNA polymerases, thus modulating gene expression levels [6]. It has also been shown that intronic CA sequences constitute novel and widespread regulatory elements of alternative splicing [7], which, in turn, adds another layer of complexity in the human genome [8].
The abundance of intronic CA-repeats and gene expression has been found to be inversely correlated for some highly expressed housekeeping genes, indicating a putative global mechanism of gene expression [9]. This observation raises the question of whether CA-repeats in intronic regions are functional as a gene regulation mechanism for other human genes as well. In other words, does this feature represent a unique class of gene regulators for the whole genome? Interestingly, the relationship between the length polymorphisms of CA-repeats located in the first introns or intron 1 regions (I1Rs) and gene expression has recently been reported in several cancer- or disease-related genes, including the genes encoding the epidermal growth factor receptor (EGFR) [10], hydroxysteroid (11-beta) dehydrogenase 2 (HSD11B2)[11] and interferon-gamma (IFNG) [12]. These studies indicated a correlation between gene expression level and the polymorphic status of CA-repeats in the I1Rs. While gene expression could be regulated by various genetic or non-genetic factors -- such as the presence of cis- or trans- regulatory elements [13-15], copy number variants (CNVs)[16] and DNA methylation [17], -- a genome-wide search for genes with similar structure to these genes could potentially provide evidence that this particular feature (I1 repeats) represents a class of universal gene regulation mechanism.
Structurally, CA- repeats can be divided into categories by length, which may reflect their biological properties [18]. For example, short repeats (6 ≤ n < 12 units) have a very low propensity for polymorphisms, while long repeats (n ≥ 12 units) are more likely to be polymorphic and functional [19,20]. More than 90 per cent of the CA-repeats of n ≥ 12 units were found to display length polymorphisms and may act as cis-regulators of transcription [21]. In general, CA- repeats of n ≥ 12 units in highly expressed housekeeping genes show a down-regulatory effect on transcription, which suggests an inverse correlation with the proportion of repeats in introns [9]. Therefore, we sought to explore the relationship between gene expression and relatively long repeats (n ≥ 12 units) located within the I1Rs. In particular, using a public database of small repeats and the RefSeq-supported [22] genes, we searched for genes with I1 (TG/CA)n repeats in the proximity of their neighbouring exons in the human genome. Since genes within the same biological pathways are potentially under similar levels of evolutionary pressure, our next goal was to evaluate if there were any enriched or over-represented known pathways and biological processes in the genes with this particular feature. Known functional annotations, such as those maintained at the Gene Ontology (GO)[23] and Kyoto Encyclopaedia of Genes and Genomes (KEGG)[24] databases, were searched for the classification of the genes. Statistically significant pathways and biological processes were found to be enriched in the genes with I1 (TG/CA)n repeats. Genotyping of the I1 repeats showed that the majority of these repeats were polymorphic in a panel of human lymphoblastoid cell lines (LCLs) derived from apparently healthy Caucasian individuals of northern and western European ancestry from the International HapMap Project [7,25] (CEU samples; ie Caucasians from Utah, USA). We then tried to evaluate experimentally the relationship between mRNA expression and the polymorphisms of the I1 repeats in these LCLs.
Materials and methods
Dataset
(TG/CA)n (where n ≥ 6) repeats in introns were identified in the human genome using Satellog (http://satellog.bcgsc.ca) [26], which is a database for the identification and prioritisation of pure repeat unit microsatellite repeats. Satellog runs on v. 34 of the human genome (version hg16, July 2003) and provides the ability to identify repeats based on user-specified characteristics, such as repeat unit, period, length and genomic coordinates. To collect our analysis set of I1 repeats, we mapped the identified intronic repeats from Satellog to the same version of the human genome reference using the exonic positions of the RefSeq-supported genes (curated non-redundant genes) retrieved by PipHelper (http://pipmaker.bx.psu.edu/cgi-bin/piphelper/) [27]. The final analysis dataset comprised I1 (TG/CA)n repeats within the 2.5 kilobase (kb) windows either downstream or upstream of neighbouring exons (Figure 1). We limited our analysis to those introns bigger than 2.5 kb.
Figure 1.
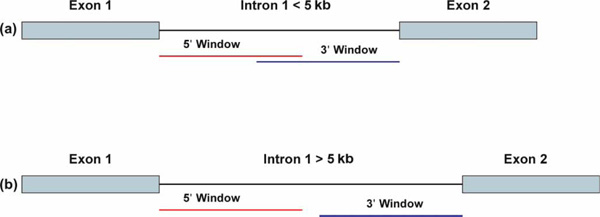
Schematic of the intron 1 regions covered in the analysis. The 5' windows (2.5 kb) and 3' windows (2.5 kb) of the intron 1 regions were included in the analysis. (a) The intron 1 region is smaller than 5 kb. (b) The intron 1 region is larger than 5 kb.
Chromosomal distribution of the genes containing I1 (TG/CA)n repeats
Distribution of the genes containing I1 (TG/CA)n repeats across the human genome were tested against the null chromosomal distribution of the human genome (version hg16), which has 18,299 RefSeq-supported [22] genes. Over- or under-represented chromosomes were determined using binomial tests. A false discovery rate (FDR) of 5 per cent after the Benjamini-Hochberg (BH) correction [28] was used as the cut-off for statistical significance.
Gene ontology and pathway analyses
We used the Database for Annotation, Visualization and Integrated Discovery (DAVID)[29,30] (http://david.abcc.ncifcrf.gov) to identify enriched GO [23] (http://www.geneontology.org) or Protein ANalysis THrough Evolutionary Relationships (PANTHER)[31] (http://www.pantherdb.org/pathway/) biological processes, as well as known pathways such as those maintained in the KEGG [24] (http://www.genome.jp/kegg/), Biocarta (http://www.biocarta.com) and PANTHER [31] databases among the genes containing long I1 (TG/CA)n repeats. Biological processes or pathways over-represented relative to the whole human genome (version hg16) were identified using Fisher's exact test (five hits or more, FDR < 0.05 after BH correction [28]).
Genotyping the I1 (TG/CA)n repeats
To evaluate if the I1 (TG/CA)n repeats were polymorphic, we genotyped a list of genes with relatively long (n ≥ 12) repeats in the 59 unrelated HapMap CEU samples. The gene expression levels of these samples previously had been measured using the Affymetrix GeneChip ® Human Exon 1.0ST array (Affymetrix, Santa Clara, CA, USA) and are publicly available through the Gene Expression Omnibus (GEO; http://www.ncbi.nlm.nih.gov/geo/), accession GSE7851 [15]. Ten genes with the top-ranking variability and associated with enriched pathways or biological processes were included in the genotyping (Table 1). DNA samples were extracted from LCLs purchased from the Coriell Institute for Medical Research (Camden, NJ, USA). Polymerase chain reaction (PCR), in combination with fluorescently labelled oligonucleotide primers, was used to amplify the I1Rs containing (TG/CA)n repeats. Primers were designed to amplify the repeat region [32]. One primer was labelled with 6-carboxyfluorescein at the 5' end. The primer sequences are listed in Table 1. The PCR reaction mix was prepared with 1 unit of HotStart Taq Polymerase (Qiagen, Valencia, CA, USA), 1.5 mM MgCl2 (Qiagen, Valencia, CA, USA), 250 nM of each primer (IDT, San Jose, CA, USA) and 20 ng of genomic DNA. PCR amplification was performed in a thermal cycler (Bio-Rad, Hercules, CA, USA) with an initial step of 12 minutes at 95°C, followed by 35 cycles of 10 seconds at 95°C, 20 seconds at 56°C and 40 seconds at 72°C. PCR products were separated in the ABI 3700 DNA sequencer (Applied Biosystems, Foster City, CA, USA), and the genotypes were read using GeneMapper® software v4.0 (Applied Biosystems). The Hardy-Weinberg equilibrium (HWE) was tested for the frequencies of each polymorphism. There lative lengths of the repeats were used in further analyses. We set the location of the first peak to appear in the GeneMapper® software as 1. The following peaks were read based on the distance from the first peak. Three random homozygous samples of two genes (the genes encoding the insulin receptor [INSR] and calmin [CLMN]) were sequenced to validate the genotyping.
Table 1.
Primers used in genotyping and real-time PCR
| Experiment | Affymetrix transcript cluster IDa | Gene | Description | Direction | Primer |
|---|---|---|---|---|---|
| Genotyping | 3930360 | RUNX | Runt-related transcription factor 1 | Forward | 5'-CCTCTGCTGCCAGTAAAGAAG-3' |
| Reverse | 5'-Fam-CCGGCCACTTTATCACAGC-3' | ||||
| 2719656 | CD38 | CD38 molecule | Forward | 5'-CCTGTCCAGATACTTGATACTTTC-3' | |
| Reverse | 5'-Fam-AGTCCAGAGTGCTGCCAGT-3' | ||||
| 3577940 | CLMN | Calmin | Forward | 5'-Fam-TGCCTCGAAGGTACTTAGGAAGT-3' | |
| Reverse | 5'-AGAAGTTTGTACGTGGGCTCTTG-3' | ||||
| 3160175 | VLDLR | Very low density lipoprotein receptor | Forward | 5'-AGCACACTGTACTCCTAATGCCTG-3' | |
| Reverse | 5'-Fam-AGGACAGGCATCAGCATTTTC-3' | ||||
| 2370433 | CACNA1E | Calcium channel, voltage-dependent, R type, alpha 1E subunit | Forward | 5'-Fam-AATGTTGTGTGCTGTCCCAA-3' | |
| Reverse | 5'-TTGCATACAGTGGGCACACA-3' | ||||
| 2991233 | AHR | Aryl hydrocarbon receptor | Forward | 5'-Fam-AGTTGTACAGACAACCTGGAGA-3' | |
| Reverse | 5'-CAGTGATGTATGTGGAGAAGTAAT-3' | ||||
| 2878074 | NRG2 | Neuregulin 2 | Forward | 5'-TTCCGGCCTAAAGAACTAGCTC-3' | |
| Reverse | 5'-Fam-AGACTGGCCTGGGATGTGT-3' | ||||
| 3284596 | PARD3 | Par-3 partitioning defective 3 homolog | Forward | 5'-GCTTTGGACACACTAGTGTCTGC-3' | |
| Reverse | 5'-Fam-ATGAGGATCCAGAGCAGGAAA-3' | ||||
| 3848243 | INSR | Insulin receptor | Forward | 5'-GAAGTCTTTAGATGGCTTCTCTCTG-3' | |
| Reverse | 5'-Fam-GCCTTATATTCTAGGTGCTGAGG-3' | ||||
| 3721010 | IGFBP4 | Insulin-like growth factor binding protein 4 | Forward | 5'-Fam-GGTGGTAGAAAAGTCTTGGCTC-3' | |
| Reverse | 5'-GTGTAAGAATGAGGTTAGGGCAA-3' | ||||
| Real-time PCR | 3848243 | INSR | Insulin receptor | Forward | 5'-AAGACCATCGACTCGGTGAC-3' |
| Reverse | 5'-GGATCGGCGGATTTTTAGAT-3' | ||||
| 3577940 | CLMN | Calmin | Forward | 5'-GGTTCTTGGGCTGATATGGA-3' | |
| Reverse | 5'-GGCCTTGATAGCCTTCCTCT-3' | ||||
aFrom GSE7851.
Real-time PCR
We quantified the transcriptional expression levels of two genes, INSR and CLMN, by real-time PCR in the 59 unrelated CEU samples. For each cell line, 3 μg of total RNA was used and was reversely transcribed into cDNA with the High Capacity cDNA Reverse Transcription Kit (Applied Biosystems). The MxPro - Mx3000P QPCR System (Stratagene, La Jolla, CA, USA) was used to quantify the expression levels of each gene. The reactions were performed in a 96-well plate, in a final volume of 25 μl containing iQ™ SYBR® Green Supermix (Bio-Rad), 500 nM of forward and reverse primers, respectively, and cDNA template corresponding to 4.5 ng of total RNA. The thermal cycling conditions were 10 minutes at 95°C, followed by 40 cycles of 30 seconds at 95°C, 1 minute at 55°C and 30 seconds at 72°C, completing with one cycle of 1 minute at 95°C, 30 seconds at 55°C and 30 seconds at 95°C. A housekeeping gene, the gene encoding FK-506-binding protein 1A (FKBP1A), was included as the internal control. Primer sequences are listed in Table 1.
Association of the repeat genotypes with gene expression
Samples were grouped into classes based on their relative repeat genotypes. For example, the samples can be grouped into either 'low category' (with relatively low numbers of repeats '1/1') or 'high category' (with other genotypes such as '2/5' or '1/7'). Other combinations, such as 'samples with allele 1' versus 'other samples' and 'sum of alleles in genotype A' versus 'sum of alleles in genotype B', were also tested. Gene expression levels from realtime PCR were then evaluated for correlation with the repeat genotypes in the 59 unrelated CEU samples. Linear regression was performed using the stat::lm function in the R Statistical Package [33].
Results
Identifying genes with I1 (TG/CA)n repeats
A total of 5,424 (TG/CA)n (where n ≥ 6) repeats in all introns were identified in the human genome (version hg16) using Satellog. By mapping the repeats to the human genome, a total of 1,137 repeats were found to be located in the I1Rs (Table S1 in the supplementary material), corresponding to 839 unique human genes. We grouped these genes according to whether they contained short (six to 11 units) repeats (704 repeats and 536 genes) or long (≥12 units) repeats (433 repeats and 388 genes). We further limited our analysis dataset to the simple repeats within the 2.5 kb windows either downstream of exon 1 or upstream of exon 2, as well as to I1Rs larger than 2.5 kb (Figure 1). In total, our final analysis set comprised 371 long (TG/CA)n repeats (n ≥12) (corresponding to 330 genes) in either the 5' (205 repeats and 187 genes) or 3' (194 repeats and 187 genes) 2.5 kb windows (Table 2 and Table S2 in the supplementary material). Among these, 44 genes had long repeats in both the 5' and 3' windows.
Table 2.
Summary of the (TG/CA)n repeats in intron 1 regions (version hg 16)
| All (n ≥ 6)a | Short (6 ≤ n < 12)a | Long (n ≥ 12)a | Long (n ≥ 12)b | Long (5' windowb) | Long (3' windowa) | |
|---|---|---|---|---|---|---|
| Number of repeats | 1137 | 704 | 433 | 371 | 205 | 194 |
| Number of genes | 839 | 536 | 388 | 330 | 187 | 187 |
aAll intron 1 regions.
bIntron 1 regions larger than 2.5 kb.
Chromosomal distribution of the genes containing I1 (TG/CA)n repeats
The chromosomal distribution of the 330 genes containing long I1 (TG/CA)n repeats was compared against the null distribution of the human genome (version hg16). Figure 2 shows the chromosomal distribution of these genes. At FDR < 5 per cent, chromosome 11 was found to be underrepresented relative to the whole genome (eight genes; p = 8.3 × 10-4; FDR = 0.02). No other chromosomes were found to be either over-represented or under-represented relative to the human genome reference.
Figure 2.
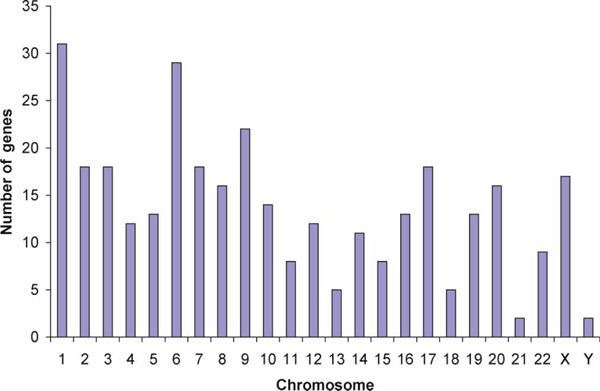
Chromosomal distribution of genes containing long (TG/CA)n repeats (where n ≥ 12). In total, 330 genes containing long repeats were identified in the 5' and/or 3' 2.5 kb windows of the intron I regions in the human genome. Chromosome II was under-represented relative to the human genome reference (version hg16).
Enriched biological processes and pathways
Using the DAVID web application [29,30], four GO [23] and 17 PANTHER [31] biological processes were found to be enriched among the 330 gene-containing long I1 (TG/CA)n repeats. The most significant enriched biological process was the PANTHER [31] term for cation transport (97 genes; p = 6.20 × 10-11; pc = 1.40 × 10-8). One KEGG [24] pathway -- the calcium signalling pathway (16 genes; p = 2.1 × 10-11, FDR = 0.041) -- was also enriched among these genes. Table 3 lists these enriched biological processes and pathways (FDR < 0.05).
Table 3.
Enriched biological processes and pathways in the genes containing II (TG/CA)n repeats
| Category | Database | Database ID | Name | Count | P | FDR |
|---|---|---|---|---|---|---|
| Pathway | KEGG | hsa:04020 | Calcium signalling pathway | 16 | 2.10E-04 | 4.10E-02 |
| Biological process | GO | GO:0030001 | Metal ion transport | 25 | 1.20E-05 | 3.10E-02 |
| GO:0030182 | Neurone differentiation | 18 | 1.40E-05 | 1.80E-02 | ||
| GO:0006812 | Cation transport | 28 | 2.20E-05 | 1.90E-02 | ||
| GO:0015672 | Monovalent inorganic cation transport | 21 | 2.50E-05 | 1.60E-02 | ||
| PANTHER | BP00143 | Cation transport | 97 | 6.20E-11 | 1.40E-08 | |
| BP00142 | Ion transport | 57 | 4.10E-07 | 4.50E-05 | ||
| BP00044 | mRNA transcription regulation | 160 | 8.90E-07 | 6.50E-05 | ||
| BP00071 | Proteolysis | 90 | 4.60E-05 | 2.50E-03 | ||
| BP00040 | mRNA transcription | 105 | 8.50E-05 | 3.70E-03 | ||
| BP00102 | Signal transduction | 75 | 1.20E-04 | 4.50E-03 | ||
| BP00141 | Transport | 34 | 3.40E-04 | 1.10E-02 | ||
| BP00019 | Lipid, fatty acid and steroid metabolism | 24 | 4.10E-04 | 1.10E-02 | ||
| BP00112 | Calcium-mediated signalling | 15 | 9.50E-04 | 2.10E-02 | ||
| BP00063 | Protein modification | 53 | 9.90E-04 | 2.00E-02 | ||
| BP00284 | Haematopoiesis | 6 | 1.10E-03 | 2.10E-02 | ||
| BP00124 | Cell adhesion | 18 | 1.20E-03 | 2.10E-02 | ||
| BP00199 | Neurogenesis | 25 | 1.80E-03 | 2.80E-02 | ||
| BP00286 | Cell structure | 59 | 2.40E-03 | 3.50E-02 | ||
| BP00064 | Protein phosphorylation | 43 | 2.90E-03 | 3.90E-02 | ||
| BP00111 | Intracellular signalling cascade | 24 | 3.20E-03 | 4.00E-02 | ||
| BP00060 | Protein metabolism and modification | 54 | 3.40E-03 | 4.10E-02 | ||
Genotyping of the I1 repeats in the CEU samples
Ten I1-repeats-containing genes (the genes encoding runt-related transcription factor [RUNX], CD38 molecule [CD38], CLMN, very low-density lipoprotein receptor [VLDLR], calcium channel, voltage-dependent, R type, alpha 1E subunit [CACNA1E], aryl hydrocarbon receptor [AHR], neuregulin 2 [NRG2], par-3 partitioning defective 3 homolog [PARD3], INSR and insulin-like growth factor binding protein 4 [IGFBP4]) with highly variable gene-level expression based on a public microarray expression dataset [15] in the unrelated CEU samples were genotyped. Table S3 in the supplementary material shows the relative lengths of repeats of the 59 unrelated CEU samples. The data from microsatellite analysis showed that the I1 repeats of these genes were polymorphic. Table 4 shows the summary of the genotyping results. RUNX and IGFBP4 did not follow the HWE and are therefore not listed in Table 4. All the other genes followed the HWE (Fisher's exact test, p < 0.05). Three PCR products of INSR and CLMN were randomly selected for sequencing validation. The sequencing results were consistent with the genotyping data (data not shown).
Table 4.
Polymorphic I1 repeats in the unrelated CEU samples
| Gene | Chromosome | Repeat lengtha |
|---|---|---|
| INSR | 19 | 1-8 |
| NRG2 | 5 | 1-6 |
| PARD3 | 10 | 1-5 |
| CD38 | 4 | 1-5 |
| CACNA1E | 1 | 1-8 |
| AHR | 7 | 1-4 |
| CLMN | 14 | 1-6 |
| VLDLR | 9 | 1-11 |
aRelative repeat length.
Evaluation of the relationship between repeat length polymorphism and gene expression
The relationship between genotype and gene expression level was evaluated in the remaining eight genes whose repeat genotypes were consistent with the HWE (Table 4). Real-time PCR was performed to measure their gene expression levels (Table S4 in the supplementary material). When grouping the samples based on repeat length genotypes, among the eight genes we tested, no statistically significant associations were observed using combinations of genotypes, including 'samples with allele 1' versus 'other samples', 'sum of alleles in genotype A' versus 'sum of alleles in genotype B' and 'low category' (with relatively low numbers of repeats '1/1') versus 'other genotypes'. The two most significant relationships were INSR (r = -0.23; p = 0.088) and CLMN (r = -0.19; p = 0.15) for the 'low category' (genotype 1/1) versus 'other genotypes' comparison.
Discussion
Understanding gene regulation is critical to understanding the underlying mechanisms of health-related phenotypes, such as the risks of common diseases. Except for a few classes of elements, such as the promoter regions [34], the putative role of most non-coding elements (eg simple tandem repeats) in gene regulation, however, has not been comprehensively investigated. In this work, we explored the relationship between gene expression and a class of dinucleotide repeats in the intron 1 regions -- that is, I1 (TG/CA)n repeats.
We focused on relatively long repeats because they are more likely to be polymorphic [21], and therefore potentially more likely to have a role in gene regulation. We further limited our analysis set of repeats to the flanking regions of 2.5 kb windows (Figure 1). This allowed us to focus on the relationship between gene expression and neighbouring repeats, which also were the targets of a couple of previous studies of EGFR [10], HSD11B2 [11] and IFNG [12]. Overall, approximately 2 per cent of known human genes have this particular feature based on our criteria. The 330 genes containing long I1 repeats were generally distributed evenly across the human genome, although chromosome 11 was found to be under-represented. This under-representation could be due to the arbitrary cut-offs (ie the 2.5 kb windows and n ≥ 12) we chose to use in the final analysis set. We confirmed that relatively long (TG/CA)n repeats in the I1Rs are universal across the human genome and exist in hundreds of human genes. Furthermore, a couple of biological processes were found to be enriched among these 330 genes using different annotation databases. For example, the GO and PANTHER biological processes related to metal ion transport, as well as the KEGG and PANTHER pathways related to calcium signalling, were found to be enriched, indicating that these repeats may play a role in the function or regulation of certain signalling pathways. Interestingly, the PANTHER biological processes BP00040: mRNA transcription and BP00044: mRNA transcription regulation were enriched among these genes, hinting at a potential relationship between I1 repeats and transcriptional gene expression.
By genotyping the I1 repeats of ten highly variable genes expressed in the unrelated HapMap CEU samples [15], we demonstrated that the majority of repeats in these LCL samples were polymorphic (Table 4). Because the functional models of these repeats are not yet known, various classifications of the repeat polymorphisms, including 'low category' (with relatively low numbers of repeats '1/1') versus 'other genotypes' were tested for association with gene expression measured by real-time PCR. No statistically significant associations (p < 0.05) were observed between various repeat genotype classes and gene expression in these LCLs, however. The two top-ranking relationships were identified in INSR and CLMN when comparing the repeat genotypes between 'low category' and 'others'. Overall, these current findings do not appear to support a putative role for I1 repeats as a novel class of universal gene regulation mechanism which contributes to variation in transcript abundance. Because of the limited number of genes we tested experimentally, however, a much more comprehensive investigation might be necessary to confirm finally whether this relationship is a universal mechanism for other genes. Due to the fact that only approximately 50 per cent of genes are believed to be reliably expressed in the LCLs [15,35], and that gene regulation can be tissue specific [36], it could be interesting to carry out the same investigation in other tissue types, to illustrate the potential role of I1 repeats in gene regulation. Furthermore, since population-level gene expression variation has been observed in [15,35,37,38] humans, a future survey, using different human populations, might be also worthwhile for illustrating the relationship between I1 repeats and gene expression.
Acknowledgments
This Pharmacogenetics of Anticancer Agents Research (PAAR) Group (http://www.pharmacogenetics.org) study was supported by NIH/NIGMS grant U01GM61393. Data will be deposited into PharmGKB (supported by NIH/NIGMS Pharmacogenetics Research Network and Database grant U01GM61374, http://www.pharmgkb.org/).
References
- Weissenbach J. Microsatellite polymorphisms and the genetic linkage map of the human genome. Curr Opin Genet Dev. 1993;3:414–417. doi: 10.1016/0959-437X(93)90114-5. [DOI] [PubMed] [Google Scholar]
- Cullis CA. The use of DNA polymorphisms in genetic mapping. Genet Eng (NY) 2002;24:179–189. doi: 10.1007/978-1-4615-0721-5_8. [DOI] [PubMed] [Google Scholar]
- Science. Vol. 258. NIH/CEPH Collaborative Mapping Group; 1992. A comprehensive genetic linkage map of the human genome. NIH/CEPH Collaborative Mapping Group; pp. 67–86. [PubMed] [Google Scholar]
- Toth G, Gaspari Z, Jurka J. Microsatellites in different eukaryotic genomes: Survey and analysis. Genome Res. 2000;10:967–981. doi: 10.1101/gr.10.7.967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nordheim A, Rich A. Proc Natl Acad Sci. Vol. 80. USA; 1983. The sequence (dC-dA)n X (dG-dT)n forms left-handed Z-DNA in negatively supercoiled plasmids; pp. 1821–1825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peck LJ, Wang JC. Transcriptional block caused by a negative supercoiling induced structural change in an alternating CG sequence. Cell. 1985;40:129–137. doi: 10.1016/0092-8674(85)90316-2. [DOI] [PubMed] [Google Scholar]
- Hui J, Hung LH, Heiner M. et al. Intronic CA-repeat and CA-rich elements: A new class of regulators of mammalian alternative splicing. EMBO J. 2005;24:1988–1998. doi: 10.1038/sj.emboj.7600677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W, Duan S, Bleibel WK. et al. Identification of common genetic variants that account for transcript isoform variation between human populations. Hum Genet. 2008;125:81–93. doi: 10.1007/s00439-008-0601-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma VK, Kumar N, Brahmachari SK. et al. Abundance of dinucleotide repeats and gene expression are inversely correlated: A role for gene function in addition to intron length. Physiol Genomics. 2007;31:96–103. doi: 10.1152/physiolgenomics.00183.2006. [DOI] [PubMed] [Google Scholar]
- Gebhardt F, Zanker KS, Brandt B. Modulation of epidermal growth factor receptor gene transcription by a polymorphic dinucleotide repeat in intron 1. J Biol Chem. 1999;274:13176–13180. doi: 10.1074/jbc.274.19.13176. [DOI] [PubMed] [Google Scholar]
- Agarwal AK, Giacchetti G, Lavery G. et al. CA-repeat polymorphism in intron 1 of HSD11B2: Effects on gene expression and salt sensitivity. Hypertension. 2000;36:187–194. doi: 10.1161/01.HYP.36.2.187. [DOI] [PubMed] [Google Scholar]
- Dufour C, Capasso M, Svahn J. et al. Homozygosis for (12) CA repeats in the first intron of the human IFN-gamma gene is significantly associated with the risk of aplastic anaemia in Caucasian population. Br J Haematol. 2004;126:682–685. doi: 10.1111/j.1365-2141.2004.05102.x. [DOI] [PubMed] [Google Scholar]
- Zhang W, Ratain MJ, Dolan ME. The HapMap resource is providing new insights into ourselves and its application to pharmacogenomics. Bioinform Biol Insights. 2008;2:15–23. doi: 10.4137/bbi.s455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duan S, Huang RS, Zhang W. et al. Genetic architecture of transcript-level variation in humans. Am J Hum Genet. 2008;82:1101–1113. doi: 10.1016/j.ajhg.2008.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W, Duan S, Kistner EO. et al. Evaluation of genetic variation contributing to differences in gene expression between populations. Am J Hum Genet. 2008;82:631–640. doi: 10.1016/j.ajhg.2007.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stranger BE, Forrest MS, Dunning M. et al. Relative impact of nucleotide and copy number variation on gene expression pheno-types. Science. 2007;315:848–853. doi: 10.1126/science.1136678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W, Huang RS, Dolan ME. Integrating epigenomics into pharmacogenomic studies. Pharmacogenom Person Med. 2008;1:7–14. doi: 10.2147/pgpm.s4341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma VK, Brahmachari SK, Ramachandran S. (TG/CA)n repeats in human gene families: Abundance and selective patterns of distribution according to function and gene length. BMC Genomics. 2005;6:83. doi: 10.1186/1471-2164-6-83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fondon JW, Mele GM, Brezinschek RI, Proc Natl Acad Sci. Vol. 95. USA; 1998. Computerized polymorphic marker identification: Experimental validation and a predicted human polymorphism catalog; pp. 7514–7519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wren JD, Forgacs E, Fondon JW. et al. Repeat polymorphisms within gene regions: Phenotypic and evolutionary implications. Am J Hum Genet. 2000;67:345–356. doi: 10.1086/303013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dib C, Faure S, Fizames C. et al. A comprehensive genetic map of the human genome based on 5,264 microsatellites. Nature. 1996;380:152–154. doi: 10.1038/380152a0. [DOI] [PubMed] [Google Scholar]
- Pruitt KD, Tatusova T, Maglott DR. NCBI reference sequences (RefSeq): A curated non-redundant sequence database of genomes, transcripts and proteins. Nucleic Acids Res. 2007;35:D61–D65. doi: 10.1093/nar/gkl842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashburner M, Ball CA, Blake JA. et al. Gene ontology: Tool for the unification of biology. The Gene Ontology Consortium. Nat Genet. 2000;25:25–29. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanehisa M, Goto S, Kawashima S. et al. The KEGG resource for deciphering the genome. Nucleic Acids Res. 2004;32:D277–D280. doi: 10.1093/nar/gkh063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- International_HapMap_Consortium. The International HapMap Project. Nature. 2003;426:789–796. doi: 10.1038/nature02168. [DOI] [PubMed] [Google Scholar]
- Missirlis PI, Mead CL, Butland SL. et al. Satellog: A database for the identification and prioritization of satellite repeats in disease association studies. BMC Bioinformatics. 2005;6:145. doi: 10.1186/1471-2105-6-145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz S, Zhang Z, Frazer KA. et al. PipMaker -- A web server for aligning two genomic DNA sequences. Genome Res. 2000;10:577–586. doi: 10.1101/gr.10.4.577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benjamini Y, Hochberg Y. Controlling the false discovery rate: A practical and powerful approach to multiple testing. J R Stat Soc B. 1995;57:289–300. [Google Scholar]
- Dennis G Jr, Sherman BT, Hosack DA. et al. DAVID: Database for annotation, visualization, and integrated discovery. Genome Biol. 2003;4:P3. doi: 10.1186/gb-2003-4-5-p3. [DOI] [PubMed] [Google Scholar]
- Huang da W, Sherman BT, Tan Q. et al. The DAVID Gene Functional Classification Tool: A novel biological module-centric algorithm to functionally analyze large gene lists. Genome Biol. 2007;8:R183. doi: 10.1186/gb-2007-8-9-r183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas PD, Campbell MJ, Kejariwal A. et al. PANTHER: A library of protein families and subfamilies indexed by function. Genome Res. 2003;13:2129–2141. doi: 10.1101/gr.772403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chi DD, Hing AV, Helms C. et al. Two chromosome 7 dinucleotide repeat polymorphisms at gene loci epidermal growth factor receptor (EGFR) and pro alpha 2 (I) collagen (COL1A2) Hum Mol Genet. 1992;1:135. doi: 10.1093/hmg/1.2.135. [DOI] [PubMed] [Google Scholar]
- R: A language and environment for statistical computing. R Development Core Team; 2005. http://www.R-project.org [Google Scholar]
- Kim TH, Barrera LO, Zheng M. et al. A high-resolution map of active promoters in the human genome. Nature. 2005;436:876–880. doi: 10.1038/nature03877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spielman RS, Bastone LA, Burdick JT. et al. Common genetic variants account for differences in gene expression among ethnic groups. Nat Genet. 2007;39:226–231. doi: 10.1038/ng1955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W, Liu W, Innocenti F. et al. Searching for tissue-specific expression pattern-linked nucleotides of UGT1A isoforms. PLoS ONE. 2007;2:e396. doi: 10.1371/journal.pone.0000396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W, Dolan ME. Ancestry-related differences in gene expression: Findings may enhance understanding of health disparities between populations. Pharmacogenomics. 2008;9:489–492. doi: 10.2217/14622416.9.5.489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stranger BE, Nica AC, Forrest MS. et al. Population genomics of human gene expression. Nat Genet. 2007;39:1217–1224. doi: 10.1038/ng2142. [DOI] [PMC free article] [PubMed] [Google Scholar]