

Abstract
In β-cells of the pancreas, the glucose transporter (GLUT)-2 facilitative glucose transporter protein is localized to the plasma membrane and functions as part of the glucose sensing mechanism for the stimulation of insulin secretion. We observed that expressed GLUT2 protein in the cultured Min6B1 cell line undergoes enhanced endocytosis at high extracellular glucose concentrations that stimulate insulin secretion. Moreover, the internalized GLUT2 protein undergoes rapid degradation induced by chronic high-glucose or arginine stimulation but does not undergo plasma membrane recycling or accumulation in any microscopically apparent intracellular membrane compartment. The rapid degradation of GLUT2 was prevented by lysosomal inhibition (chloroquine) concomitant with the accumulation of GLUT2 in endomembrane structures. In contrast, neither endocytosis nor the lack of internal membrane localized GLUT2 remained completely unaffected by proteosomal inhibition (lactacystin) or an heat shock protein-90 inhibitor (geldanamycin). Moreover, the endocytosis and degradation of GLUT2 was specific for β-cells because expression of GLUT2 in 3T3L1 adipocytes remained cell surface localized and did not display a rapid rate of degradation. Together, these data demonstrate that hyperglycemia directly affects β-cell function and activates a trafficking pathway that results in the rapid endocytosis and degradation of the cell surface GLUT2 glucose transporter.
Hyperglycemia activates a β-cell trafficking pathway, resulting in the rapid endocytosis and degradation of the cell surface GLUT2 glucose transporter.
In the postprandial state, increased plasma levels of glucose as well as other nutrients stimulate pancreatic β-cells to secrete insulin, thereby suppressing hepatic glucose output and increasing peripheral tissue (muscle and adipose) to take up glucose (1,2,3). One of the hallmarks of type 2 diabetes is a marked impairment of glucose-stimulated insulin secretion (GSIS) (4,5,6,7). GSIS is dependent on a metabolic pathway that couples glucose uptake to metabolism generating increased levels of ATP (8,9,10,11). The subsequent changes in the ATP to ADP ratio results in the closure of potassium channels, inducing cellular depolarization and activation of voltage-dependent calcium channels (12). The rise in intracellular calcium provides the signal for the initial fusion of prime/predocked insulin-containing granules with the plasma membrane (13). The initiating event in this process is the cellular uptake of glucose via the cell surface facilitative glucose transporter protein (GLUT)-2 that functions to transport glucose in proportion to circulating levels (14). Thus, GLUT2 is considered part of the β-cell glucose sensing mechanisms.
Loss of pancreatic β-cell GLUT2 expression in humans is associated with hyperglycemia and impaired GSIS (4). In several rodent models of type 2 diabetes, the loss of GSIS directly correlates with decreased expression of the β-cell GLUT2 protein (4). For example, pancreatic β-cell GLUT2 knockout mice display a near complete absence of first phase glucose-stimulated insulin secretion demonstrating that the GLUT2 transporter is essential for GSIS (15,16,17). Similarly, high-fat diet or glucocorticoid treatment to induce a diabetic phenotype results in decreased GLUT2 protein levels that are directly responsible for the loss of GSIS (18,19). Although the mechanism for GLUT2 protein loss has not been identified, recent studies suggested that alterations in GLUT2 N-linked glycosylation can markedly decrease the cell surface level of GLUT2 (4). To investigate the trafficking of GLUT2, we expressed single and/or dual exofacial myc and endofacial green fluorescent protein (GFP) epitope tagged GLUT2 reporters in the glucose-responsive cultured β-cell line Min6B1. Our data demonstrate that after endocytosis GLUT2 does not accumulate in endosomes compartments but undergoes a very rapid lysosomal-mediated degradation that is glucose dependent.
Materials and Methods
Materials
Wild-type rat GLUT2 cDNA in pDNR-LIB vector was purchased from American Type Tissue Collection (ATCC clone MGC:93555 IMAGE:7127751; Manassas, VA). Full-length GLUT2 cDNA was amplified by PCR and subcloned in pcDNA3 vector (Invitrogen, Carlsbad, CA). Enhanced GFP fragment from pEGFP-C2 vector (CLONTECH, Palo Alto, CA) was next fused in frame at the C-terminal end of GLUT2 cDNA giving a GLUT2-GFP vector. Three repetitions of the Myc epitope were next inserted within the first extracellular loop of GLUT2 (position +150), generating a Myc-GLUT2 and a Myc-GLUT2-GFP vector. All constructs were checked by DNA sequencing.
Cell culture
Min6B1 (transformed mouse) β-cells (20) were grown at 37 C in 5% CO2 in DMEM supplemented with 10% fetal bovine serum containing penicillin-streptomycin (100 U/ml, 100 μg/ml) and 71 μm 2-mercaptoethanol. Murine 3T3L1 preadipocytes were purchased from American Type Tissue Collection. Cells were cultured at 37 C and 8% CO2 and differentiated into adipocytes as previously described (21).
Transfection
For confocal examination and endocytosis assay, the Min6B1 cells were transfected with 2 μg of various GLUT2 constructs or cotransfected with the transferrin receptor (Tfr) using Lipofactamine 2000 according to the manufacturer’s instructions (Invitrogen).
Electroporation
Fully differentiated 3T3L1 adipocytes were transiently transfected by electroporation under low-voltage conditions (160 V, 950 μF) with 50 μg each of Myc-GLUT4 and Myc-GLUT2 DNA plasmids as described previously (22).
Western blot
For the Western blot analysis, Min6B1 cells were transfected with myc-GLUT2 construct and after overnight incubation, the cells were incubated with or without cycloheximide (10 μg/ml; Sigma-Aldrich, St. Louis, MO) and/or chloroquine (50 μm; Sigma) as the time indicated. Thirty micrograms of total protein (protein assay kit; Pierce, Rockford, IL) were separated by 10% SDS-PAGE and subsequently transferred to a polyvinylidene difluoride membrane. Western blotting was performed using the monoclonal (clone E10) c-Myc mouse antibody (Santa Cruz Biotechnology, Santa Cruz, CA) or rabbit polyclonal GLUT2 antibody (Millipore, Bedford, MA).
Endocytosis assay
Min6B1 cells were plated in a six-well plate with coverslips and transfected with 2 μg Myc-GLUT2 construct per well. Twenty-four hours later, the cells were cooled to 4 C and then labeled with a c-Myc mouse antibody (1:50) with or without 50 μm chloroquine (Sigma-Aldrich), 20 μm lactacystin (Calbiochem, La Jolla, CA), or 1 μm geldanamycin (Sigma-Aldrich) for 1 h on ice. The cells were washed to remove the excess (nonbound) c-Myc antibody and returned to 37 C for various times to initiate endocytosis. The cells were then fixed in 4% paraformaldehyde at room temperature for 10 min, blocked with 5% donkey serum, and incubated with Texas Red-conjugated antimouse secondary antibody (1:100) for 45 min at 37 C under permeabilizing conditions. The slides were then mounted in Vectashield medium (Vector Laboratories, Burlingame, CA) and examined by confocal-laser scanning microscopy. Endocytosis was quantified by dividing the plasma membrane surface Texas Red fluorescent intensity by the total cell content of Texas Red fluorescent intensity in 10 cells for each time point per experiment. The mean value was calculated from the average of three independent experiments.
GLUT2 stability assay
35S cell labeling-Min6B1 cells were plated in a six-well plate and transfected with Myc-GLUT2 and PCDNA 3.1 vector. Twenty-four hours later, the cells were rinsed twice with 1.5 ml prewarmed labeling medium (DMEM without cysteine and methionine; Life Technologies, Inc., Grand Island, NY) containing 10% fetal bovine serum supplemented with glutamine. Cells were then labeled for 1 h at 37 C with the addition of 20–30 μl of [35S]EXPRE35S35S (NEG) methionine/cysteine mix (∼250 μCi/well). Cells were quickly washed with 1.5 ml prewarmed chase medium (sterile DMEM, 10% fetal bovine serum + 500 μg/ml cysteine + 500 μg methionine, with or without 50 μm chloroquine). Cells are incubated for 0, 2, or 4 h of chase at 37 C.
Immunoprecipitation and autoradiography
After the chase time, cells are immediately placed on ice, rinsed three times with ice-cold PBS, and lysed with 250 μl of radioimmunoprecipitation assay buffer [150 mm NaCl, 50 mm Tris, 1 mm EDTA, 1% Nonidet P-40, 0.5% sodium deoxycholate, and 0.1%SDS (pH 7.4)] supplemented with a 1:1000 dilution of a protease inhibitors cocktail (Sigma). After 10 min on ice, cells were scrapped and the lysates were transferred and centrifuged for 20 min at 15,000 × g at 4 C. The supernatant was transferred into a clean tube and protein concentration determined by bicinchoninic assay protein assay. Protein levels were equalized before immunoprecipitation and 1 μg of c-myc mouse antibody was added to the lysate and incubated for 1 h at 4 C.
Fifty microliters of protein A/G agarose beads (Santa Cruz) were then added and gently rocked for 20 min at 4 C. Beads were centrifuged at 800 × g for 2 min and then washed with ice-cold radioimmunoprecipitation assay buffer. One hundred microliters of SDS-PAGE sample buffer were directly added to pellets. Samples were heated at 70 C for 10 min and centrifuged at 15,000 × g, and 10 μl of each sample (∼20 μg) were loaded onto a NuPAGE 4–12% SDS-PAGE gel (Invitrogen). After electrophoresis, the gels were fixed with 8% acetic acid and 20% methanol for 30 min and rinsed three times, each 10 min in water and then soaked in Amplify (Amersham Biosciences, Piscataway, NJ) solution for 20 min. The gels were vacuum dried at 50 C for 1 h and exposed in an autoradiography cassette with reflective screens for 24 h using Hyperfilm (Amersham Biosciences) at −80 C. The bands corresponding to GLUT2 were cut from the dry gel, solubilized in scintillation liquid, and counted for 10 min in a β-counter (Beckman Coulter, Fullerton, CA).
Sucrose gradient fractionation
Min6B1 cells were incubated for 24 h with 5 or 25 mm glucose in the presence or absence of 50 μm chloroquine. The cells were homogenized and then loaded onto a 10–50% (wt/wt) sucrose gradient and centrifuged for 16 h at 37, 500 rpm at 4 C in a SW40 Ti rotor. After centrifugation nine fractions were collected and analyzed by Western blotting for GLUT2 as described above.
Results
GLUT2 does not accumulate in intracellular membrane compartments
GLUT2 is a multimembrane-spanning protein with 12-transmembrane helices with both the amino and carboxyl termini oriented toward the cytoplasm (23). To address the subcellular distribution and trafficking of GLUT2, we initially transfected the glucose-responsive β-cell line Min6B1 with a GLUT2-GFP, myc-GLUT2 (Fig. 1A), and myc-GLUT2-GFP reporter constructs (Fig. 1B). Surprisingly although GLUT2 is a multimembrane-spanning protein, 24–48 h after expression, GLUT2 was primarily detected at the plasma membrane with a surprisingly low level of intracellular membrane distribution.
Figure 1.

GLUT2 does not accumulated in the intracellular membrane compartments. Min6B1 cells were transfected with 2 μg of the GLUT2 cDNA. Forty-eight hours later, the transfected cells were fixed and subjected to either immunostaining with anti-myc monoclonal antibody or direct confocal fluorescent microscopy analysis. A, GLUT2-GFP (a), Myc-GLUT2 (b). B, Myc-GLUT2-GFP (a–c). Green represents GFP expression, and red is anti-myc monoclonal antibody staining.
Because GLUT2 would be expected to undergo endomembrane trafficking after biosynthesis and transport to the plasma membrane, we next compared the localization of GLUT2 with that of the related glucose transporter proteins, GLUT1 and GLUT4, after various times of expression in the Min6B1 cells (Fig. 2). As expected, 6 h after transfection of the GLUT1-GFP reporter, there was relatively strong labeling of the perinuclear region indicative of the Golgi apparatus with a relatively lower level of plasma membrane labeling along with a diffuse intracellular labeling indicative of the endoplasmic reticulum (Fig. 2a). As a function of expression time 12–72 h, the diffuse endoplasmic reticulum labeling rapidly declined with robust labeling both in the Golgi and plasma membrane (Fig. 2, b–e). By 96 h the transfected plasmid was probably no longer expressing because the endomembrane (Golgi) was no longer labeled, whereas the plasma membrane localized GLUT1 remained (Fig. 2f). In contrast, 6 h after transfection with GLUT2-GFP, there was a pronounced diffuse labeling (endoplasmic reticulum) with a relatively low level in the perinuclear (Golgi) compartment (Fig. 2g). Over the time course from 12 to 48 h, GLUT2 was primarily localized to the plasma membrane with only low levels in intracellular membrane compartments (Fig. 2, h and i). Moreover, by 72–96 h, there was essentially no intracellular labeling and the majority of expressed GLUT2 was confined to the plasma membrane (Fig. 2, j and k). Interestingly, the localization of GLUT4 in the Min6B1 cells was initially (between 6 and 48 h) very similar to that of GLUT1 (Fig. 2, m–p). However, at later time points (72–96 h), there was a pronounced loss of GLUT4 from the cell surface with a concomitant marked increase in the localization of GLUT4 in intracellular granules and/or endosome structures (Fig. 2, q and r). These data indicate that each of the glucose transporter proteins undergo distinct trafficking itineraries within the same cell context. Importantly, GLUT2 appears to rapidly transient through and/or is relatively excluded from the endomembrane system.
Figure 2.

Comparison of intracellular localization of GLUT1, GLUT2, and GLUT4. Min6B1 cells were transfected with 2 μg of GLUT1 (panels a–f), GLUT2 (panels g–l), and GLUT4 (panels m–r). After transfection, the cells were fixed at different time points such as 6 (panels a, g, and m), 12 (panels b, h, and n), 24 (panels c, I, and o), 48 (panels d, j, and p), 72 (panels e, k, and q), and 96 (panels f, l, and r) h, respectively. The fixed cells were then examined by confocal fluorescent microscopy. These are the represented images selected from two independent experiments.
Because one would expect GLUT2 to undergo multiple rounds of endocytosis and exocytosis yet we could not observe any significant intracellular membrane localization, we next expressed the myc-GLUT2 and labeled the exposed plasma membrane exofacial myc epitope with a myc antibody at 4 C (Fig. 3A). As expected, the cell surface myc-GLUT2 was predominantly labeled (Fig. 3A, a). However, after warming the cells to 37 C for 2 or 4 h, there was again no significant accumulation of the myc label in intracellular membrane compartments (Fig. 3A, b and c). Instead, there was a clear time-dependent decrease in the myc fluorescent signal. In contrast, the cell surface transferrin receptor was also labeled with an exofacial antibody at 4 C, but in this case warming the cells to 37 C for 2 h resulted in a redistribution of the cell surface transferrin receptor into intracellular membrane compartments (Fig. 3B, a and b). These data suggest that although the transferrin receptor can undergo normal plasma membrane endocytosis and endomembrane cycling, the GLUT2 transporter either is directly degraded at the plasma membrane or undergoes endocytosis coupled to a rapid degradation process.
Figure 3.

Myc-GLUT2 and TfR endocytosis in Min6B1 cells. A, Min6B1 cells were transfected with myc-GLUT2 (a–c). Twenty-four hours after transfection, the cells were cooled to 4 C, and the surface exposed myc epitope was labeled with myc-antibody for 1 h. The cells were then either fixed right after 4 C labeling (time 0, panel a) or warmed to 37 C for various time points (b and c) followed by fixation and subjected to confocal fluorescent microscopy. B, In parallel, the Min6B1 cells were transfected with 2 μg TfR, and cells were labeled with Texas Red-conjugated transferrin at 4 C. The cells were either fixed right after 4 C labeling (time 0, a) or warmed to 37 C for 2 h (b) after fixation and subjected to confocal fluorescent microscopy. These are representative images taken from two to three independent experiments.
The plasma membrane internalized GLUT2 protein undergoes rapid lysosomal degradation
There are two general pathways that can be involved in membrane protein degradation. To determine whether GLUT2 was undergoing a proteosome and/or lysosomal-mediated trafficking and degradation pathway, we surface labeled the myc-GLUT2 reporter with the myc antibody at 4 C and examined the effects of several inhibitors (supplemental Fig. 1S, published as supplemental data on The Endocrine Society’s Journals Online web site at http://endo.endojournals.org). As previously observed, predominant surface labeling was obtained at 4 C, and after warming to 37 C for 2 or 4 h, there was no appreciable intracellular membrane labeling, although there was a clear decrease in fluorescent signal (supplemental Fig. 1, a, e, and i). Treatment with the proteasome inhibitor lactacystin had no significant on the intracellular distribution or decrease in myc-GLUT2 fluorescent (supplemental Fig. 1, b, f, and g). Similarly, the heat shock protein-90 inhibitor geldanamycin that increases the intracellular accumulation and degradation of the oncogenic ErbB2 receptor (24) was also without effect (supplemental Fig. 1, c, g, and k). However, treatment of the Min6B1 cells with the lysosomal inhibitor, chloroquine stabilized the fluorescent myc-GLUT2 signal and resulted in an apparent intracellular accumulation (supplemental Fig. 1, d, h, and l). In addition, identical results were obtained using another lysosomal inhibitor bafilomycin (data not shown).
Because the cell surfaced-labeled myc-GLUT2 reporter accumulated in intracellular membrane compartments in the presence of chloroquine, these data suggest a relatively rapid lysosomal degradation. To directly determine the rate of degradation of GLUT2, we first transfected Min6B1 cells with myc-GLUT2 and either left them untreated or treated with cycloheximide to block protein synthesis, chloroquine, or cycloheximide plus chloroquine for various times (Fig. 4a). In untreated cells, there was no significant decrease in total cell GLUT2 protein levels because the cells reached an equilibrium steady state between the rate of new synthesis and degradation. In contrast, treatment with cycloheximide to block new synthesis resulted in a time-dependent loss of GLUT2 protein levels. However, chloroquine treatment prevented the effect of cycloheximide to decrease the myc-GLUT2 levels. To establish that the endogenous GLUT2 protein undergoes lysosomal-mediated degradation, Min6B1 cells were pulsed labeled with 35S-methionine and chased with unlabeled methionine and cysteine (Fig. 4B). After the pulse labeling, two specific bands were apparent in GLUT2 immunoprecipitates, probably reflecting the fully mature and unglycosylated GLUT2 proteins. Two hours of chase resulted in the presence of the higher molecular form that displayed a time-dependent reduction. In contrast, the addition of chloroquine during the chase prevented the loss of the labeled endogenous GLUT2 protein. Quantification of the 35S-methionine pulse-chase labeling indicated an approximately 3.5 h half-life for the newly synthesized GLUT2 protein, whereas in the presence of chloroquine, there was no detectable loss of the labeled GLUT2 protein over this time frame (Fig. 4C). Similarly, quantification of the cell surface myc-antibody-labeled GLUT2 by confocal microscopy indicated an essentially identical half-life in the absence of chloroquine and that was completely stabilized in the presence of chloroquine (Fig. 4D). The fact that these curves are essentially identical demonstrates that the degradation of GLUT2 is primarily resulting from plasma membrane endocytosis and trafficking to lysosomes and that the decay of the cell surface myc-GLUT2 reporter label provides a quantitative measure of GLUT2 degradation.
Figure 4.
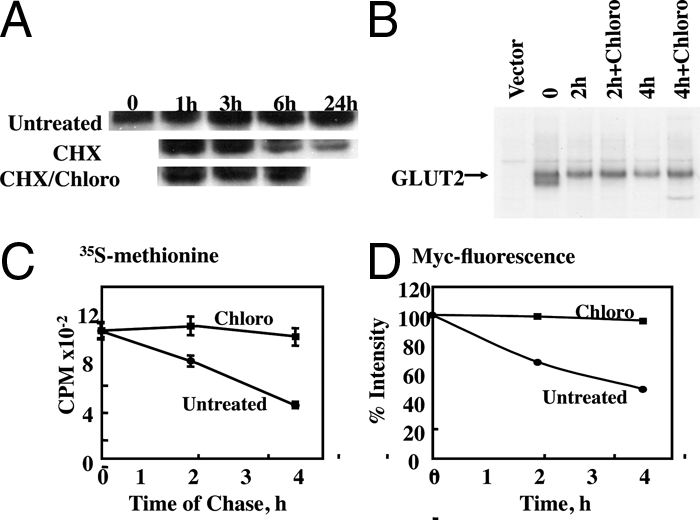
Determination of GLUT2 degradation rate in Min6B1 cells. A, Min6B1 cells were transfected with myc-GLUT2, 24 h later either left untreated or treated with 10 μg/ml cycloheximide (CHX), 50 μm chloroquine (Chloro), or cycloheximide plus chloroquine for various time points as indicated. The cell lysates were collected and subjected to SDS-PAGE and blot with anti-myc monoclonal antibody as described in Materials and Methods. This is a representative image from two independent experiments. B, Min6B1 cells were plated in a six-well plate and transfected with Myc-GLUT2 and PCDNA 3.1 vector. Twenty-four hours later, the cells were pulsed labeled with 35S-methionine and chased with unlabeled methionine and cysteine in the presence or absence of 50 μm chloroquine for various time points as indicated. The cell lysates were collected and subjected to immunoprecipitation and SDS-PAGE. The gel was exposed in an autoradiography cassette with reflective screens for at least 24 h on Hyperfilm (Amersham Biosciences) at −80 C as described Materials and Methods. This is a representative image from two independent experiments. C, The band intensities in B were quantified by radioactivity counting. D, Min6B1 cells were transfected with the Myc-GLUT2 construct. Twenty-four hours later, the cells were cooled to 4 C and then labeled with a myc-antibody for 1 h in the presence or absence of 50 μm chloroquine; cells were then returned to 37 C to initiate endocytosis. Cells were examined by confocal fluorescent microscopy. Endocytosis was quantified by dividing the plasma membrane surface Texas Red fluorescent intensity by the total cell content of Texas Red fluorescent intensity in 10 cells for each time point per experiment. The mean value was calculated from the average of three independent experiments.
GLUT2 lysosomal degradation is β-cell specific and secretagogue dependent
The endocytosis and trafficking of other facilitative glucose transporters has been examined and undergo expected endosome sorting/recycling and are relative stable proteins (23,25). Thus, the rapid lysosomal degradation of GLUT2 could be either intrinsic to the GLUT2 protein itself or occurs as a consequence of cell context. To address this issue, we transfected differentiated 3T3L1 adipocytes with an exofacial myc-GLUT4, myc-GLUT1, and myc-GLUT2 reporters and labeled the cell surface proteins with the myc antibody at 4 C (supplemental Fig. 2S). In adipocytes, GLUT4 is well established to be stored in intracellular insulin-responsive vesicle compartments and to undergo insulin-stimulated translocation to the cell surface. As previously observed (21), after insulin stimulation GLUT4 translocates to the plasma membrane and the exofacial myc epitope can be readily labeled with the myc antibody (supplemental Fig. 2SA, a). Warming the cells to 37 C for 2 or 4 h resulted in clearance of the surface GLUT4 protein and its accumulation back into intracellular membrane compartments (supplemental Fig. 2SA, b and c). The endocytosis and intracellular distribution of GLUT4 was not significantly affected by chloroquine treatment (supplemental Fig. 2SA, d and e). In contrast, GLUT1 has a high basal steady-state distribution to the plasma membrane although undergoes a smaller degree of insulin-stimulated translocation (22). In this case, cell surface myc-GLUT1 was readily labeled with the myc antibody in the presence of insulin at 4 C (supplemental Fig. 2SB, a). Warming of the cell surface-labeled GLUT1 resulted in the intracellular accumulation of GLUT1, but a large proportion remained at the cell surface consistent with a reduced degree of GLUT1 recycling (supplemental Fig. 2SB, b and c). Similarly, chloroquine did not significantly affect the intracellular accumulation of GLUT1 (supplemental Fig. 2SB, d and e).
Similar to GLUT1, GLUT2 also displayed a higher plasma membrane steady-state distribution in 3T3L1 adipocytes than GLUT4. In any case, surface labeling of myc-GLUT2 at 4 C, followed by warming to 37 C resulted in the appearance of internalized GLUT2 in intracellular membrane compartments to a similar extent as that seen for GLUT1 (supplemental Fig. 2SC, b and c). Moreover, the localization to these intracellular membranes was again chloroquine insensitive (supplemental Fig. 2SC, d and e). Together these data indicate that the rapid trafficking of internalized GLUT2 to the lysosome and degradation is specific for β-cells and not intrinsic to the GLUT2 protein itself.
Because the loss of cell surface GLUT2 in vivo occurs in rodent models of type 2 diabetes, we assessed the effects of ambient glucose concentrations to mediate the degradation of GLUT2 (Fig. 5). The data presented so far were performed on Min6B1 cells maintained in culture medium containing high glucose (25 mm) that is physiologically a hyperglycemic state. Thus, we transfected Min6B1 cells with the myc-GLUT2 reporter and labeled the exofacial myc epitope at 4 C. The cells were then warmed to 37 C in culture medium containing 5 or 25 mm glucose in the presence and absence of chloroquine (Fig. 5A). Consistent with our previous findings, cells incubated in 25 mm glucose displayed a time-dependent loss of GLUT2 protein that was prevented by treatment with chloroquine. However, the GLUT2 protein was remarkably stable when the cells were incubated in 5 mm glucose whether or not chloroquine was present. To determine whether the increased rate of GLUT2 degradation was specific for high glucose concentrations, we also examined the effect of another insulin secretagogue, l-arginine (Fig. 5B). As shown in Fig. 6A, GLUT2 was relatively stable at 5 mm glucose and chloroquine treatment had no effect (Fig. 5B). In contrast, GLUT2 underwent a more rapid rate of degradation in the presence of 20 mm arginine that was again prevented by treatment with chloroquine. Together, these data demonstrate the instability of the GLUT2 protein is not only cell context dependent but also regulated by agonist-induced secretion and is not a glucose-specific response.
Figure 5.
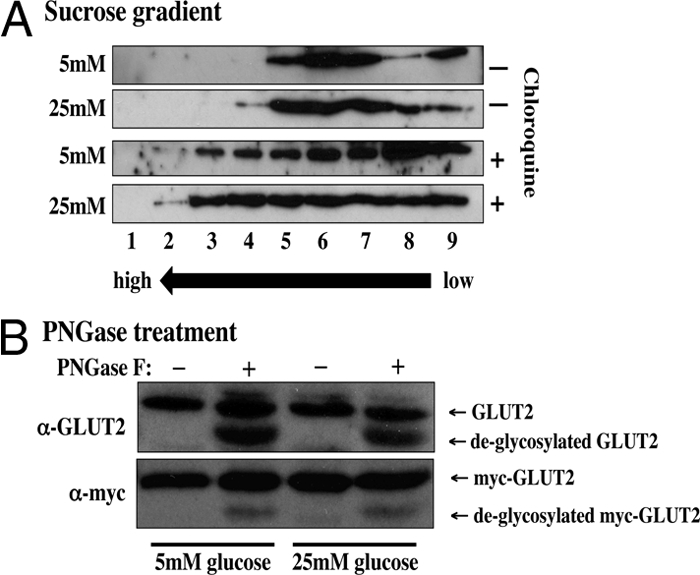
Intracellular GLUT2 distribution and glycosylation. A, Min6B1 cells were incubated for 24 h with 5 or 25 mm glucose (Glu) in the presence or absence of 50 μm chloroquine (Chloro). The cells were homogenized and subjected to sucrose gradient centrifugation as described in Materials and Methods. The gradient fractions were then extracts with Laemmli sample buffer and immunoblotted for the presence of the endogenous GLUT2 protein. B, Min6B1 cells were transfected with 2 μg of Myc-GLUT2 plasmid and incubated 24 h with 5 or 25 mm glucose. Cell extracts were then prepared and treated with and without PGNase F. The samples were then subjected to SDS-PAGE and immunoblotted for the endogenous GLUT2 protein or the exogenous Myc-GLUT2 protein. PNGase, N-glycopeptidase.
Figure 6.

GLUT2 lysosomal degradation is secretogogue dependent. Min6B1 cells were plated in six-well plates and transfected with 2 μg Myc-GLUT2 construct per well. A, The cells were cultured in 5 mm glucose for 24 h, and the surface myc-GLUT2 was labeled at 4 C with or without 50 μm chloroquine. The cells were then warmed to 37 C in culture medium containing either 5 or 25 mm glucose in the presence and absence of 50 μm chloroquine. B, The Myc-GLUT2 transfected cells were cultured in 5 mm glucose for 24 h, and the surface myc-GLUT2 was labeled at 4 C with or without 50 μm chloroquine. The cells were then warmed to 37 C in culture medium containing either 5 mm glucose or 20 mm arginine in the presence and absence of 50 μm chloroquine. Endocytosis was quantified by dividing the plasma membrane surface Texas Red fluorescent intensity by the total cell content of Texas Red fluorescent intensity in 10 cells for each time point per experiment using confocal fluorescence microscopy. The mean value was calculated from the average of three independent experiments.
Chloroquine alters the intracellular membrane distribution of GLUT2
If chloroquine prevents the trafficking of GLUT2 to the lysosomal compartment, then we would expect that the intracellular distribution of GLUT2 would be different in cells treated with and without chloroquine. To test this prediction, we examined the intracellular distribution of the endogenous GLUT2 protein by sucrose gradient fractionation. In the absence of chloroquine, GLUT2 migrated in the lower density region of the gradient, and this distribution was not significantly different between cells incubated with 5 or 25 mm glucose (Fig. 6A). In contrast, chloroquine treatment resulted in a more broad distribution toward the denser region of the gradient. These data indicate that although glucose treatment does not alter the intracellular steady-state distribution of GLUT2, chloroquine had a significant shift in the intracellular distribution. These data suggest that intracellular trafficking of GLUT2 is relatively rapid and thus accumulation in intracellular compartments is relatively low. However, in the presence of chloroquine, GLUT2 gets trapped in these intracellular compartments consistent with a shift to greater density membrane fractions. Please note that the Western blots were exposed to equal intensity to allow for a direct visual comparison of GLUT2 protein distribution.
Because different glucose concentrations could potentially alter the glycosylation state of GLUT2, we also examined the effect of N-glycopeptidase F treatment on both the endogenous and transfected myc-GLUT2 protein (Fig. 6B). Again there was no discernible change in apparent molecular weight of GLUT2 from Min6B1 cells incubated with 5 or 25 mm glucose. Moreover, deglycosylation with of N-linked sugars with N-glycopeptidase resulted in increased electrophoretic mobility, but there was no difference between 5 and 25 mm glucose-treated cells. Although these data do not distinguish between changes in individual monosaccharides, they are consistent with both glucose and arginine stimulation increasing GLUT2 degradation independent of glycosylation.
Discussion
It is well established that the initial phase of insulin release from pancreatic β-cells results from a series of complex events that involve metabolic/nutrient sensing that is coupled to cellular depolarization through changes in the cellular ATP to ADP ratio (10,26,27,28). Although cells have numerous pathways and mechanisms to maintain intracellular energy status, insulin-secreting β-cells express the two highly restricted isoforms that are necessary to sense circulating glucose levels, the GLUT2 facilitative glucose transporter and the hexokinase isoform glucokinase (29,30,31). In the postprandial state, elevation of circulating glucose is sensed by the coupling of facilitative glucose transport with glucokinase-dependent phosphorylation and conversion to glucose-1-phosphate, the rate-limiting step in β-cell glycolysis (29). Thus, due to their kinetic properties, the coupling of these two specific isoforms allows for the cellular ATP to ADP ratio to change in proportion to the extracellular ambient glucose concentration (10). In this regard, numerous mutations in the glucokinase gene that either alter its activity and/or expression markedly affect the ability of β-cells to secrete insulin in response to changes in glucose levels (32,33,34).
Although few naturally occurring mutations in GLUT2 have been identified, several states of impaired glucose-stimulated insulin secretion result from a decrease in GLUT2 expression levels. For example, loss of GSIS in aging and states of hyperglycemia have been associated with a decline in GLUT2 expression levels, whereas high-fat diet-induced insulin resistance results in impaired plasma membrane targeting of GLUT2 (4,19,35). The essential nature of GLUT2 has been demonstrated in GLUT2 null mice that are hyperglycemic and hypoinsulinemic with a concomitant loss of first-phase GSIS (14). Interestingly, these β-cells still displayed second-phase GSIS and with a decreased glucose dose-dependent sensitivity (16). In any case, lentivirus reexpression of GLUT2 in the GLUT2 null β-cells restored normal GSIS (16).
Based on these data, we examined the intracellular trafficking properties of expressed GLUT2 reporter constructs in a culture murine β-cell line, Min6B1. Although the Min6B1 cells are a culture model system, they display similar GSIS, both first and second phase similar to that found in isolated rodent islets (4). Moreover, the glucose-dependent down-regulation of GLUT2 that has been observed in vivo (4) is recapitulated in the Min6B1 cells. Thus, the Min6B1 cells appear to be a reasonable heterologous model assay system to examine the trafficking properties of GLUT2 that cannot be performed in vivo. Nevertheless, it is important to recognize that cultured β-cells may not necessarily reflect the same biological process that occur in vivo and thus can be considered only a model system.
In any case, our data demonstrate that GLUT2 undergoes an usual biosynthetic and membrane sorting in Min6B1 cells compared with two other highly related facilitative glucose transporter isoforms, GLUT1 and GLUT4. GLUT1 is typically considered as the constitutive glucose transporter being highly expressed in numerous cell types and predominantly plasma membrane localized (22). In contrast, GLUT4 displays a highly cell type-restricted expression and is predominantly localized to intracellular membrane compartments but undergoes a dramatic insulin-dependent translocation to the plasma membrane in adipocytes and striated muscle (25). As expected, GLUT1 and GLUT4 when expressed in β-cells appears to undergo a normal time-dependent endomembrane processing from the endoplasmic reticulum, accumulating in the perinuclear regions, most likely the Golgi apparatus, and eventually residing at the plasma membrane. Unexpectedly, GLUT2 did not appreciably accumulate in the perinuclear region but appeared diffuse throughout the cytoplasm (characteristic of the endoplasmic reticulum) and at the plasma membrane. These data suggest that although GLUT1 and GLUT4 undergo a relatively slow processing through the endomembrane system, GLUT2 more rapidly transits these membranes structures en route to the plasma membrane.
More surprisingly, at steady state there were relatively low levels of intracellular membrane GLUT2 compared with that of GLUT1 or GLUT4 in the Min6B1 β-cells. The absence of intracellular GLUT2 compared with GLUT1 or GLUT4 was not intrinsic to GLUT2 itself but was cell context dependent because a typical intracellular membrane distribution was observed for GLUT2 in 3T3L1 adipocytes. The lack of intracellular GLUT2 in Min6B1 cells apparently results from a rapid lysosomal-mediated degradation after GLUT2 plasma membrane endocytosis. We base this conclusion on the ability of chloroquine, but not lactacystin or geldanamycin, to induce accumulation of the cell surface GLUT2 into intracellular compartments. Moreover, a direct measurement of the GLUT2 half-life by 35S-methionine/cysteine pulse chase indicated a 3.5-h half-life, which is substantially less than that reported for other facilitative glucose transporters (36).
At present we do not know the structural basis for the different trafficking patterns between the GLUT1, GLUT2, and GLUT4 isoforms. Previous studies demonstrated that specific intracellular domains (both amino and carboxyl terminal domains) in the GLUT4 protein are required for the intracellular sequestration and insulin responsiveness in adipocytes (37,38). Because these regions are not conserved in either the GLUT1 or GLUT2 proteins, it is likely that unique sequences within the cytoplasmic domains of GLUT2 are responsible for its unique trafficking properties in β-cells.
We also observed that the rapid lysosomal mediated degradation of GLUT2 was glucose concentration dependent. Typically, β-cells in culture are maintained in 25 mm d-glucose, and under these conditions the degradation rate of GLUT2 was approximately 3.5 h. However, switching to a more physiological glucose concentration (5 mm) resulted in a dramatic stabilization of the GLUT2 protein. These data are consisted with a recent study demonstrating that the down-regulation of GLUT2 protein after high-fat feeding-induced insulin resistance and hyperglycemia was due to an accelerated rate of degradation (4). However, our data also demonstrate that this down-regulation of GLUT2 is not glucose specific because the secretagogue arginine also induced an increase in GLUT2 degradation that was chloroquine sensitive.
In summary, we have observed that after initial biosynthesis the GLUT2 protein undergoes a relatively rapid processing through the endomembrane system en route to the plasma membrane compared with other facilitative glucose transporters. Once at the cell surface, GLUT2 is further processed by lysosomal degradation, whereas the other glucose transporters have a significantly greater rate of membrane recycling. The net effect results in a low level of intracellular GLUT2 protein. The dynamics of a rapid cell surface delivery coupled with degradation in the absence of membrane recycling allows for an acute regulation of GLUT2 protein levels. The degradation of GLUT2 is dependent on secretagogue activation of insulin secretion, suggesting that in vivo, chronic states of hyperinsulin secretion may be sufficient to impair subsequent β-cell glucose-dependent insulin secretion. This model of β-cell desensitization is consistent with the acquired loss of β-cell function in type 2 diabetes (39,40,41).
Supplementary Material
Footnotes
This work was supported by Research Grants DK55811, DK33823, and DK20541 from the National Institutes of Health.
Disclosure Summary: The authors have nothing to disclose.
First Published Online May 28, 2009
Abbreviations: GFP, Green fluorescent protein; GLUT, glucose transporter; GSIS, glucose-stimulated insulin secretion; TfR transferrin receptor.
References
- Bryant NJ, Govers R, James DE 2002 Regulated transport of the glucose transporter GLUT4. Nat Rev Mol Cell Biol 3:267–277 [DOI] [PubMed] [Google Scholar]
- Ducluzeau PH, Fletcher LM, Vidal H, Laville M, Tavaré JM 2002 Molecular mechanisms of insulin-stimulated glucose uptake in adipocytes. Diabetes Metab 28:85–92 [PubMed] [Google Scholar]
- Watson RT, Kanzaki M, Pessin JE 2004 Regulated membrane trafficking of the insulin-responsive glucose transporter 4 in adipocytes. Endocr Rev 25:177–204 [DOI] [PubMed] [Google Scholar]
- Ohtsubo K, Takamatsu S, Minowa MT, Yoshida A, Takeuchi M, Marth JD 2005 Dietary and genetic control of glucose transporter 2 glycosylation promotes insulin secretion in suppressing diabetes. Cell 123:1307–1321 [DOI] [PubMed] [Google Scholar]
- Orci L, Ravazzola M, Baetens D, Inman L, Amherdt M, Peterson RG, Newgard CB, Johnson JH, Unger RH 1990 Evidence that down-regulation of β-cell glucose transporters in non-insulin-dependent diabetes may be the cause of diabetic hyperglycemia. Proc Natl Acad Sci USA 87:9953–9957 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Del Prato S, Tiengo A 2001 The importance of first-phase insulin secretion: implications for the therapy of type 2 diabetes mellitus. Diabetes Metab Res Rev 17:164–174 [DOI] [PubMed] [Google Scholar]
- Del Guerra S, Lupi R, Marselli L, Masini M, Bugliani M, Sbrana S, Torri S, Pollera M, Boggi U, Mosca F, Del Prato S, Marchetti P 2005 Functional and molecular defects of pancreatic islets in human type 2 diabetes. Diabetes 54:727–735 [DOI] [PubMed] [Google Scholar]
- Prentki M, Matschinsky FM 1987 Ca2+, cAMP, and phospholipid-derived messengers in coupling mechanisms of insulin secretion. Physiol Rev 67:1185–1248 [DOI] [PubMed] [Google Scholar]
- Newgard CB, Lu D, Jensen MV, Schissler J, Boucher A, Burgess S, Sherry AD 2002 Stimulus/secretion coupling factors in glucose-stimulated insulin secretion: insights gained from a multidisciplinary approach. Diabetes 51(Suppl 3):S389–S393 [DOI] [PubMed] [Google Scholar]
- Deeney JT, Prentki M, Corkey BE 2000 Metabolic control of β-cell function. Semin Cell Dev Biol 11:267–275 [DOI] [PubMed] [Google Scholar]
- Kowluru A 2008 Emerging roles for protein histidine phosphorylation in cellular signal transduction: lessons from the islet β-cell. J Cell Mol Med 12:1885–1908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berggren PO, Larsson O 1994 Ca2+ and pancreatic B-cell function. Biochem Soc Trans 22:12–18 [DOI] [PubMed] [Google Scholar]
- Barg S 2003 Mechanisms of exocytosis in insulin-secreting B-cells and glucagon-secreting A-cells. Pharmacol Toxicol 92:3–13 [DOI] [PubMed] [Google Scholar]
- Thorens B, Guillam MT, Beermann F, Burcelin R, Jaquet M 2000 Transgenic reexpression of GLUT1 or GLUT2 in pancreatic β cells rescues GLUT2-null mice from early death and restores normal glucose-stimulated insulin secretion. J Biol Chem 275:23751–23758 [DOI] [PubMed] [Google Scholar]
- Guillam MT, Hümmler E, Schaerer E, Yeh JI, Birnbaum MJ, Beermann F, Schmidt A, Deriaz N, Thorens B 1997 Early diabetes and abnormal postnatal pancreatic islet development in mice lacking Glut-2. Nat Genet 17:327–330 [DOI] [PubMed] [Google Scholar]
- Guillam MT, Dupraz P, Thorens B 2000 Glucose uptake, utilization, and signaling in GLUT2-null islets. Diabetes 49:1485–1491 [DOI] [PubMed] [Google Scholar]
- Thorens B 2003 A gene knockout approach in mice to identify glucose sensors controlling glucose homeostasis. Pflugers Arch 445:482–490 [DOI] [PubMed] [Google Scholar]
- Kim Y, Iwashita S, Tamura T, Tokuyama K, Suzuki M 1995 Effect of high-fat diet on the gene expression of pancreatic GLUT2 and glucokinase in rats. Biochem Biophys Res Commun 208:1092–1098 [DOI] [PubMed] [Google Scholar]
- Reimer MK, Ahren B 2002 Altered β-cell distribution of pdx-1 and GLUT-2 after a short-term challenge with a high-fat diet in C57BL/6J mice. Diabetes 51(Suppl 1):S138–S143 [DOI] [PubMed] [Google Scholar]
- Lilla V, Webb G, Rickenbach K, Maturana A, Steiner DF, Halban PA, Irminger JC 2003 Differential gene expression in well-regulated and dysregulated pancreatic β-cell (MIN6) sublines. Endocrinology 144:1368–1379 [DOI] [PubMed] [Google Scholar]
- Capilla E, Suzuki N, Pessin JE, Hou JC 2007 The glucose transporter 4 FQQI motif is necessary for Akt substrate of 160-kilodalton-dependent plasma membrane translocation but not Golgi-localized γ-ear-containing Arf-binding protein-dependent entry into the insulin-responsive storage compartment. Mol Endocrinol 21:3087–3099 [DOI] [PubMed] [Google Scholar]
- Khan AH, Capilla E, Hou JC, Watson RT, Smith JR, Pessin JE 2004 Entry of newly synthesized GLUT4 into the insulin-responsive storage compartment is dependent upon both the amino terminus and the large cytoplasmic loop. J Biol Chem 279:37505–37511 [DOI] [PubMed] [Google Scholar]
- Brown GK 2000 Glucose transporters: structure, function and consequences of deficiency. J Inherit Metab Dis 23:237–246 [DOI] [PubMed] [Google Scholar]
- Pedersen NM, Madshus IH, Haslekås C, Stang E 2008 Geldanamycin-induced down-regulation of ErbB2 from the plasma membrane is clathrin dependent but proteasomal activity independent. Mol Cancer Res 6:491–500 [DOI] [PubMed] [Google Scholar]
- Hou JC, Pessin JE 2007 Ins (endocytosis) and outs (exocytosis) of GLUT4 trafficking. Curr Opin Cell Biol 19:466–473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreno-Sánchez R 1985 Regulation of oxidative phosphorylation in mitochondria by external free Ca2+ concentrations. J Biol Chem 260:4028–4034 [PubMed] [Google Scholar]
- Aguilar-Bryan L, Bryan J, Nakazaki M 2001 Of mice and men: K(ATP) channels and insulin secretion. Recent Prog Horm Res 56:47–68 [DOI] [PubMed] [Google Scholar]
- Henquin JC 2000 Triggering and amplifying pathways of regulation of insulin secretion by glucose. Diabetes 49:1751–1760 [DOI] [PubMed] [Google Scholar]
- Newgard CB, McGarry JD 1995 Metabolic coupling factors in pancreatic β-cell signal transduction. Annu Rev Biochem 64:689–719 [DOI] [PubMed] [Google Scholar]
- Iynedjian PB 1993 Mammalian glucokinase and its gene. Biochem J 293(Pt 1):1–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Vos A, Heimberg H, Quartier E, Huypens P, Bouwens L, Pipeleers D, Schuit F 1995 Human and rat β cells differ in glucose transporter but not in glucokinase gene expression. J Clin Invest 96:2489–2495 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magnuson MA 1990 Glucokinase gene structure. Functional implications of molecular genetic studies. Diabetes 39:523–527 [DOI] [PubMed] [Google Scholar]
- Matschinsky FM 1990 Glucokinase as glucose sensor and metabolic signal generator in pancreatic β-cells and hepatocytes. Diabetes 39:647–652 [DOI] [PubMed] [Google Scholar]
- Matschinsky F, Liang Y, Kesavan P, Wang L, Froguel P, Velho G, Cohen D, Permutt MA, Tanizawa Y, Jetton TL, Niswender, K, Magnuson MA 1993 Glucokinase as pancreatic β cell glucose sensor and diabetes gene. J Clin Invest 92:2092–2098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laybutt DR, Sharma A, Sgroi DC, Gaudet J, Bonner-Weir S, Weir GC 2002 Genetic regulation of metabolic pathways in β-cells disrupted by hyperglycemia. J Biol Chem 277:10912–10921 [DOI] [PubMed] [Google Scholar]
- Sargeant RJ, Paquet MR 1993 Effect of insulin on the rates of synthesis and degradation of GLUT1 and GLUT4 glucose transporters in 3T3-L1 adipocytes. Biochem J 290(Pt 3):913–919 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watson RT, Pessin JE 2001 Subcellular compartmentalization and trafficking of the insulin-responsive glucose transporter, GLUT4. Exp Cell Res 271:75–83 [DOI] [PubMed] [Google Scholar]
- Lalioti V, Vergarajauregui S, Sandoval IV 2001 Targeting motifs in GLUT4. Mol Membr Biol 18:257–264 [DOI] [PubMed] [Google Scholar]
- Poitout V, Robertson RP 2008 Glucolipotoxicity: fuel excess and β-cell dysfunction. Endocr Rev 29:351–366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujimoto Y, Torres TP, Donahue EP, Shiota M 2006 Glucose toxicity is responsible for the development of impaired regulation of endogenous glucose production and hepatic glucokinase in Zucker diabetic fatty rats. Diabetes 55:2479–2490 [DOI] [PubMed] [Google Scholar]
- Lindmark S, Buren J, Eriksson JW 2006 Insulin resistance, endocrine function and adipokines in type 2 diabetes patients at different glycaemic levels: potential impact for glucotoxicity in vivo. Clin Endocrinol (Oxf) 65:301–309 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.