

Abstract
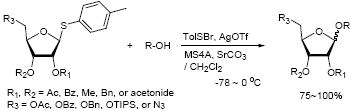
A wide variety of p-tolyl thioriboside donors are examined for O-ribosylations of primary and secondary alcohols. p-Tolylsulfenyl trifluoromethanesulfonate (p-TolSOTf) is very effective in promoting O-ribosylations with p-tolyl thioriboside; all reactions are completed within 1~15 min. to provide the desired products in good yield with reliable α/β selectivity. A wide range of functional groups are tolerated under these conditions. The described O-ribosylation conditions are very useful for the generation of ribosamino-uridine library molecules in solution or on polymer-support.
The 5-deoxy-5-aminoribose moiety of ribosamino-uridine antibiotics (i.e. muraymycin, liposidomycin, caprazamycin, and FR-900493) has been considered as an important functional group to exhibit excellent antibacterial activities.1 These antibiotics are known to interfere with the enzyme involved in the committed step of peptidoglycan biosynthesis, MraY which catalyzes the transformation of UDP-N-acylmuramyl-l-alanyl-γ-d-glutamyl-meso-diaminopimelyl-d-alanyl-d-alanine (Park’s nucleotide) to prenylpyrophosphoryl-N-acylmuramyl-l-Ala-γ-d-glu-meso-DAP2-d-Ala-d-Ala (lipid I), and are non-toxic or less toxic in vivo. Thus, MraY has been a target of interest for the discovery of novel antimycobacterial agents.3 However, medicinal chemistry programs of these natural products with an aim to improve the efficacy including pharmacokinetics are hampered by a limited number of modification reactions which can selectively modify the desired positions of such complex molecules.4 In addition, one of the drawbacks of performing medicinal chemistry with these complex antibiotics is the requirement for the fermentation of the natural product starting materials.5
In our ongoing effort to define the minimum structure requirement of ribosamino-uridine MraY inhibitors of natural product origin to exhibit equal biological activity,6 we envisioned synthesizing a library of ribosamino-uridine molecules in solution or on polymer-support.7 Because structural diversity of ribosamino-uridine antibiotics are observed not only in the lipid moiety (R group in Figure 1) but also at the 2-position of aminoribose moiety (i.e. methyl or sulfonyl group), it is desired to develop a versatile and robust ribosylation method which also amenable to glycosylations on polymer-support.8
FIGURE 1.
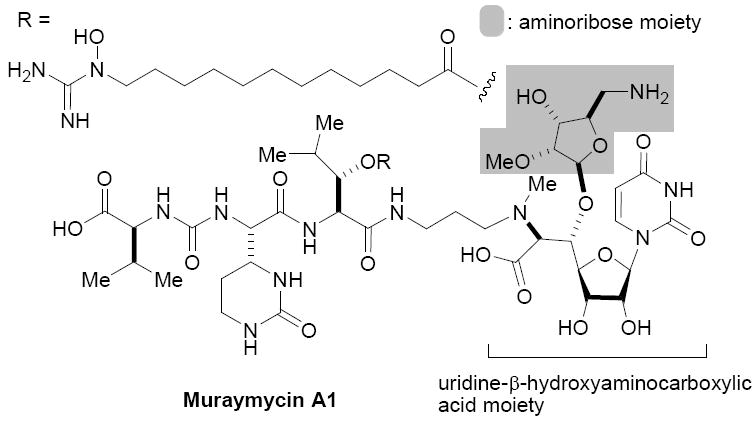
Representative Structure of Ribosamino-uridine Antibiotics.
Although numerous examples of N-glycosylations of purine or pyrimidine bases with furanose derivatives have been reported,9 O-ribosylation has rarely discussed in literatures. Recently, Merrer and co-workers reported O-ribosylations of primary alcohols using bromo- or fluoro-ribosides with the established promoters such as AgOTf or SnCl2/AgClO4.10 Reaction rates for O-ribosylations with 1-halo riboside are very slow (6~36h), and in our hands under these conditions O-ribosylations of the secondary alcohols resulted in unsatisfactory yields of 15~65%. In addition, because of the limited accessibility of 1-halo ribosides it is difficult to diversify ribosamino-uridine library molecules by O-ribosylations with these methods. In this note, we wish to report expeditious O-ribosylations of primary and secondary alcohols with a wide variety of p-tolyl thioriboside donors and p-TolSOTf that would be very useful to synthesize a library of ribosamino-uridine molecules.11
Because thioglycosides 1) can conveniently be synthesized via acid catalyzed thioglycosylations of 1-O-acetylglycoside, and 2) exhibit excellent chemical stabilities against acids and bases, the anomeric thioethers have widely been utilized as a temporary protecting group of anomeric positions as well as glycosyl donors.12 However, a choice of promoter requires careful consideration when use less electrophilic thioglycosides; in some cases no activation or very slow reaction rate was observed using conventional thiophilic reagents.13 In order to identify an effective thiophilic reagent for O-ribosylations, we first screened thiophilic reagents using an anomeric mixture (α/β = 1/6.6) of p-tolyl tribenzoyl thioglycosides 1a and the immobilized propargyl alcohol on the polymer-resin i14. As a result of extensive reaction screenings it was found that p-TolSOTf in-situ generated from p-TolSBr15 and AgOTf provided the β-O-ribosylated product ii in near quantitative yield within 15 min. at 0 °C;16 ii was characterized after cleavage from the polymer-resin with 20% TFA. Significantly, even in the presence of a large excess of the promoters (>10 equiv) all functional groups (acetal, alkyne, alkene, and BOM protecting group) were intact. It is worthy while mentioning that the other conditions tested (i.e. NIS, NIS/TfOH, NIS/AgOTf, and NIS/AgBF4)17 provided ii in 0~15 % yields together with a significant amount of byproducts.18
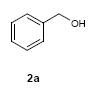
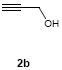
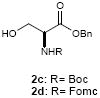
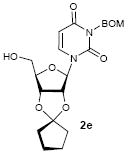
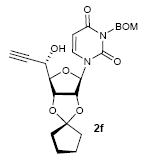
To investigate how effectively p-TolSOTf can promote glycosylations of a wide range of p-tolyl thioribosides we synthesized the donor ribosides 1b~n and the conditions developed for an O-ribosylation on the polymer-resin (Scheme 1) were applied to primary and secondary alcohols in solution. All reactions were conducted with donor (2 equiv against acceptor), p-TolSOTf (1 equiv against donor), and MS4Å (2 times the weight of donor)19 in CH2Cl2 in the presence or absence of SrCO320. The representative O-ribosylations using these conditions were summarized in Table 1. The reactions of the primary alcohols 2a~e with tri-O-acyl riboside 1a or 1b at 0 °C provided the corresponding β-glycosides 3a~f exclusively with greater than 85% yield within 1~5 min. (entries 1~6). It was realized that p-TolSOTf, which colours blue in CH2Cl2, could be generated even at -78 °C21 and could also activate 1a and 1b at the same temperature without noticeable decrease in the reaction rate. Because chemical modifications of the 5-position of 5-deoxy-5-aminoribose in ribosamino-uridine antibiotics is important to improve in vitro and in vivo efficacy, glycosylations of the acceptor 2e with the 5-TIPS-protected thioribose 1c and 5-azido-5-deoxy-thioriboside 1d, whose 5-positions can easily be diversified after glycosylations, were evaluated. Under the buffered conditions (condition B in Table 1) the azide and silyl groups in the ribosyl donors were intact (entries 7~8). The primary and secondary donor alcohols (i.e. alkanols, alkynols, and homoallylic alcohols)22 were ribosylated with the donors 1a~d to furnish the β-glycosides in greater than 80% yield; limited examples of the reactions of the secondary alcohols (i.e 2f) were shown in Table 1 (entries 19~21).22 However, primary and secondary allylic alcohols (i.e. 5-phenylpent-1-en-3-ol) were not applicable to these conditions due probably to its low nucleophilicity. Ribosylations of propargyl alcohol with the ether protected thioribosides 1e~g (entries 9~11) provided a mixture of α/β glycosides (5~6.6/1) in 75~90% yield. Thus, synthetically useful level of α-selectivity in O-ribosylations was observed by using the ether protecting groups. Glycosylations of propargyl alcohol with the acetonide protected thioribosides 1h~j (entries 12~14) provided a 1:1.1 (α/β) mixture of the corresponding products regardless of structure of the 5-position of 1h~j. As seen in muraymycins (Figure 1), an alkylation is observed at the 2-position of aminoribose unit. To study the effect of 3-O-acyl group in the 2-O-methylated thioglycosides, glycosylations of propargyl alcohol with 1k~n (entries 15~18) were examined. The 3-O-acetyl group in 1k~n were effective in reversing the selectivity; the reactions with 1k or 1l gave a 1:3 mixture of the α- and β-ribosides. The 3-O-benzoyl protected donors 1m or 1n improved α/β -selectivity (α/β = 1:5.5). These selectivities observed at 0 °C were not dramatically enhanced by lowering the reaction temperatures (i.e. -78 °C); the α/β selectivity was increased to 1:6.5 for 1m. The glycosylations of 2f, a versatile intermediate for the generation of ribosamino-uridine libraries, with 1m or 1n gave identical selectivities (α/β = 1:5.5) observed for the primary alcohol. Similarly, the reactions of 2f with the 3-O-acetate, 1k or 1l, provided an anomeric mixture (α/β = 1:2.5) of the glycosides (Scheme 2). Mechanistically, the ribosyl carbenium ion generated by p-TolSOTf would first be stabilized by the formation of an intimate ion pair23 with triflate ion, and undergoes a pseudo 5-membered ring formation when 2-O-acyl ribosyl donor is applied. These processes must proceed prior to the glycosylation step. Although an anchimeric assistance by the 3-O-acyl group to form a pseudo 6-membered ring (iii in Scheme 2) has not been proven theoretically, the data summarized in Table 1 (e.g. Entry 11 vs. 17) clearly indicate that the 3-O-acyl carbonyl group is responsible for β-selective glycosylations. It is speculated that the formation of a pseudo 6-membered ring is a slower process than that of a 5-membered ring. Thus, non-selective glycosylation of an intermediate iv may compete with β-selective glycosylation (through iii) to provide an α/β anomeric mixture. The 3-O-benzoyl group seems to be stereoelectronically more favoured than the 3-O-acetyl group to participate in anchimeric assistance in ribosylations (Scheme 2).24
SCHEME 1.
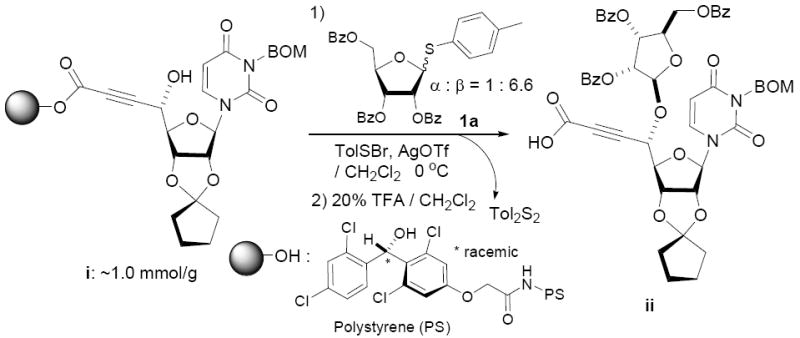
Screening of a Promoter for 1 Using the Immobilized Propargyl Alcohol on the Polymer Resin i.
TABLE 1.
Representative O-Ribosylations of Alcohols Using p-Tolyl thioglycosides and p-TolSOTf.a
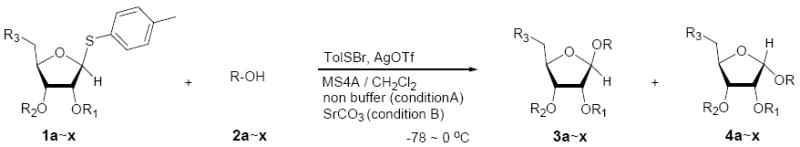 | |||||||
|---|---|---|---|---|---|---|---|
| Entry | Donor | Acceptor | Temperature (°C) | Condition | Major Product | Yield(%) | α/β Selectivity (4/3)b |
| 1 | 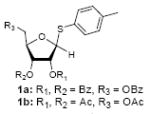 |
 |
-78~0 | A | 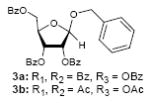 |
98 | 0/1 |
| 2 | 98 | 0/1 | |||||
| 3 | 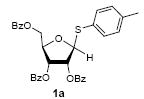 |
 |
-78~0 | A | 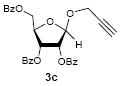 |
100 | 0/1 |
| 4 | |
 |
-78~0 | B | 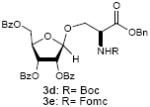 |
95 | 0/1 |
| 5 | 98 | 0/1 | |||||
| 6 | 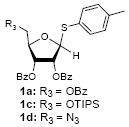 |
 |
0 | B | 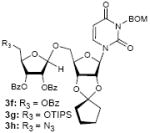 |
98 | 0/1 |
| 7 | 95 | 0/1 | |||||
| 8 | 85 | 0/1 | |||||
| 9 | 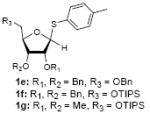 |
|
0 | B | 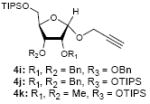 |
90 | 6.6/1 |
| 10 | 75 | 6.0/1 | |||||
| 11 | 78 | 5.0/1 | |||||
| 12 | 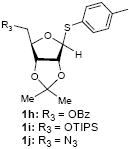 |
|
0 | B | 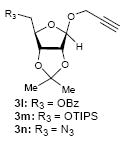 |
95 | 1/1.1 |
| 13 | 85 | 1/1.1 | |||||
| 14 | 80 | 1/1.1 | |||||
| 15 | 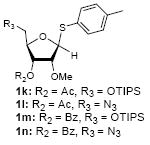 |
|
0 | B | 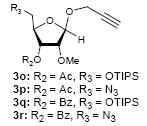 |
85 | 1/3.0 |
| 16 | 90 | 1/3.0 | |||||
| 17 | 85 | 1/5.5 | |||||
| 18 | 90 | 1/5.5 | |||||
| 19 | 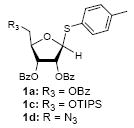 |
 |
0 | B | 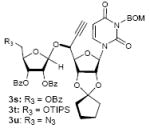 |
90 | 0/1 |
| 20 | 80 | 0/1 | |||||
| 21 | 90 | 0/1 | |||||
2 equivalents of donors against acceptor were used in all reactions.
ratio was established by 1H-NMR analysis or based on isolated yield.
SCHEME 2.
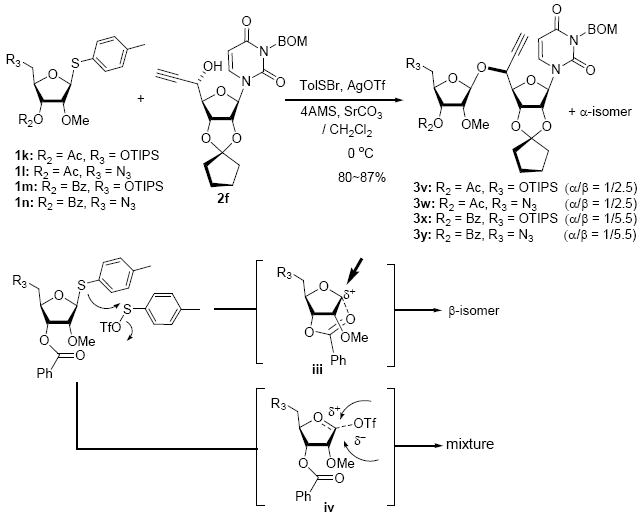
Glycosylations of 2f with the thioribosides 1k~n.
In conclusion, thioribosides were, for the first time, generalized for O-ribosylations using p-TolSOTf.25 p-TolSOTf can be generated by a mixing p-TolSBr and AgOTf in CH2Cl2 at -78~0 °C, and has a short half-life (ca. 15 min.) at 0 °C. However, the reaction rate of O-ribosylations under these conditions is very fast; all reactions summarized in Table 1 were completed within 1~15 min. The 2-O-acyl group is an important factor to obtain β-selective ribosylation product exclusively. The anchimeric assistance of 3-O-acyl group may attribute to furnish β-selective ribosylations with 2-O-alkyl thioribosides. The ether protected thioglycosides provided useful level of α-selectivities (i.e. α/β = 6.6/1 for benzyl ether). The 2,3-acetonide protected thioglycosides give rise to a 1:1.1 (α/β) anomeric mixture of ribosides. Most importantly, p-TolSOTf is applicable to substrates possessing a wide variety of functional groups such as alkyne, alkene, and commonly utilized protecting groups in multiple step syntheses (i.e. silyl, ketal, and ether protecting groups, and N3 group). Moreover, the conditions reported here are applicable to O-ribosylations on the polymer-supported resin (Scheme 1). The 2- and 5-positions of aminoribose moiety of ribosamino-uridine library molecules can be diversified by using the thioriboside donors summarized in Table 1 and their related structure of molecules. The generation of diverse structures of ribosamino-uridine libraries and biological evaluations of these library molecules will be reported elsewhere.
Experimental Section
Typical Procedure for O-Ribosylation with 1a
A mixture of 1a (2.0 equiv), alcohols (1.0 equiv), MS4Å, AgOTf (2.0 equiv), and SrCO3 (4.0 equiv; Condition B) in CH2Cl2 (0.1~0.2 M) was stirred at rt for 15 min. The reaction mixture was cooled 0 °C and p-TolSBr (2.0 M in ClCH2CH2Cl, 2.0 equiv) was added. After 1~15 min. the reaction mixture was quenched with Et3N (5.0 equiv), and the resulting mixture was filtered through a SiO2 plug. Purification by silica gel chromoatography (hexanes:EtOAc) provided the desired products. SrCO3 was excluded for Condition A.
Propargyl 5-azido-5-deoxyl-2,3-O-isopropylidene-β-D-ribofuranoside (3n and 4n)
3n (β isomer):[α]20D = +78.4 (c 0.5 in CHCl3); IR (film) 2102, 1077 cm-1; 1H NMR (300 MHz, CDCl3) δ 5.30 (s, 1H), 4.65 (dd, J = 6.0 Hz, 18.9 Hz, 2H), 4.32 (t, J = 7.2 Hz, 1H), 4.26 (dd, J = 1.2, 2.4 Hz, 1H), 4.24 (dd, J = 1.2, 2.1 Hz, 1H), 3.46 (dd, J = 7.8, 12.6 Hz, 1H), 3.30 (dd, J = 6.9, 12.6 Hz, 1H), 2.46 (m, 1H), 1.50 (s, 3H), 1.33 (s, 3H); 13C NMR (75 MHz, CDCl3) δ 113.1, 107.1, 85.9, 85.4, 82.2, 75.2, 54.8, 53.8, 26.6, 25.1; HRMS (ESI) Calcd. for C11H15N3NaO4 (M+Na)+. 276.0960; found: 276.0960. 4n (α isomer): [α] 20D = −16.0 (c 0.1 in CHCl3); 1H NMR (300 MHz, CDCl3) δ 5.23 (d, J = 4.8 Hz, 1H), 4.73 (dd, J = 4.8, 7.2 Hz, 1H), 4.55 (dd, J = 3.6 Hz, 7.2 Hz, 1H), 4.32 (dd, J = 2.4, 4.2 Hz, 2H), 4.27 (m, 1H), 3.55 (dd, J = 4.0, 13.2 Hz, 1H), 3.36 (dd, J = 4.4, 16.0 Hz, 1H), 2.39 (t, J = 2.4 Hz, 1H), 1.55 (s, 3H), 1.33 (s, 3H); 13C NMR (75 MHz, CDCl3) δ116.2, 99.8, 81.2, 81.2, 80.7, 75.0, 55.1, 52.6, 26.2, 26.1; HRMS (ESI) Calcd. for C11H15N3NaO4 (M+Na)+. 276.0960; found: 276.0962.
Supplementary Material
Experimental procedures, full characterization data for new compounds, and copies of NMR spectra. This material is available free of charge via the Internet at http://pubs.acs.org.
Acknowledgments
This paper is dedicated to the memory of Professor Albert I. Meyers, an inspirational scientist. We thank the National Institutes of Health and Colorado State University for generous financial supports.
REFERENCES AND NOTES
- 1.Dini C. Curr Top Med Chem. 2005;5:1221. doi: 10.2174/156802605774463042. [DOI] [PubMed] [Google Scholar]
- 2.DAP: (2R,6S)-2,6-diaminoheptanedioic acid.
- 3.van Heijenoort J. Glycobiology. 2001;11:25R. doi: 10.1093/glycob/11.3.25r. [DOI] [PubMed] [Google Scholar]
- 4.a) Corre LL, Gravier-Pelletier C, Merrer YL. Eur J Org Chem. 2007:5386. [Google Scholar]; b) Hirano S, Ichikawa S, Matsuda A. Angew Chem Int Ed. 2005;44:1854. doi: 10.1002/anie.200462439. [DOI] [PubMed] [Google Scholar]; c) Hotoda H, Daigo M, Furukawa M, Murayama K, Hasegawa CA, Kaneko M, Muramatsu Y, Ishii MM, Miyakoshi S, Takatsu T, Inukai M, Kakuta M, Abe T, Fukuoka T, Utsui Y, Ohya S. Bioorg Med Chem lett. 2003;13:2833. doi: 10.1016/s0960-894x(03)00597-3. [DOI] [PubMed] [Google Scholar]
- 5.No efficient chemical synthesis of ribosamino-uridine natural products which can be performed medicinal chemistry has been reported.
- 6.For synthetic effort towards identification of the minimum structure requirement of liposidomycins. see, Dini C, Drochon N, Feteanu S, Guillot JC, Peixoto C, Aszodi J. Bioorg Med Chem Lett. 2001;11:529. doi: 10.1016/s0960-894x(00)00715-0.
- 7.Kurosu M, Narayanasamy P, Crick DC. Heterocycles. 2007;72:339. [Google Scholar]
- 8.In mycobacterial growth inhibitory and enzymatic assays against MraY of representative uridine-β-hydroxyaminocarboxilic acid derivative, it was realized that the molecules which do not possess the aminoribose group significantly lowered mycobacterial growth inhibitory activity. Thus, it is important to retain the aminoribose (highlighted in Figure 1) or its equivalent in MraY inhibitors of uridine-β-hydroxyaminocaroxylic acid analogs.
- 9.(a) Ferrero M, Gotor V. Chem Rev. 2000;100:4319. doi: 10.1021/cr000446y. [DOI] [PubMed] [Google Scholar]; (b) Townsend LB. In: Nucleoside Analogues Chemistry Biology and Medical Applications. Walker RT, De Clercq E, Eckstein F, editors. Plenum Press; New York: 1979. p. 193. [Google Scholar]; (c) Reese CB. Tetrahedron. 1978;34:3143. [Google Scholar]
- 10.Ginistry M, Gravier-Pelletier C, Merrer YL. Tetrahedron Asymmetry. 2006;17:142. [Google Scholar]
- 11.Other potential glycosylations with ribose derivatives, see. Garcia BA, Poole JA, Gin DY. J Am Chem Soc. 1997;119:7597.Knapp S, Shieh W-C. 1991;32:3627.
- 12.a) Ekelof K, Garegg PJ, Olsson L, Oscarson S. Pure and Appl Chem. 1997;69:1847. [Google Scholar]; b) Garegg PJ. Advances in Carbohyd Chem and Biochem. 1997;52:179. doi: 10.1016/s0065-2318(08)60091-8. [DOI] [PubMed] [Google Scholar]
- 13.For an example of the nonsusceptible thioglycoside against NIS/TfOH, see. Li K, Kurosu M. Heterocycles. 2007 doi: 10.3987/com-09-s(s)24. in press.
- 14.Kurosu M, Biswas K, Crick DC. Org Lett. 2007;9:1141. doi: 10.1021/ol070150f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.P-TolSBr was generated by mixing a 1:1 ratio of (p-TolS)2 and Br2 in ClCH2CH2Cl, and this was stable over a month at rt.
- 16.The utility of p-TolSCl/AgOTf for glycosylations with the thiopyranosides, see. Huang X, Huang L, Wang H, Ye X-S. Angew Chem Int Ed. 2004;43:5221. doi: 10.1002/anie.200460176.
- 17.Kaeothip S, Pornsuriyasak P, Demchenko AV. Tetrahedron Lett. 2008;49:1542. doi: 10.1016/j.tetlet.2007.12.105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.We also examined the effect of MeSOTf for this reaction. However, trace amounts of the desired product were isolated. see, Kurosu M, Kitagawa I. J Carbohydr Chem. 2006;25:427.Dasgupta F, Garegg PJ. Carbohydr Res. 1988;177:C13. doi: 10.1016/0008-6215(90)84082-6.
- 19.4Å MS is not indispensable for these reactions. In order not to obtain inconsistent results caused by adventitious water, two times weight of MS was added in all reactions.
- 20.SrCO3 was applied as a buffer of the reactions. Addition of SrCO3 was effective especially in the reaction with the silyl group containing donors. 2,6-Di-ter-butylpyridine was also effective, but reaction rate was diminished.
- 21.There is a short half-life (ca. 5~15 min) of p-TolSOTf at rt.
- 22.For other examples of ribosylations with primary and secondary alcohols, see, Supporting Information.
- 23.Winstein S, Clippinger E, Fainberg AH, Heck R, Robinson GC. J Am Chem Soc. 1956;78:328. [Google Scholar]
- 24.For the other mechanistic considerations, see. Smith DM, Woerpel KA. Org Biomol Chem. 2006;4:1195. doi: 10.1039/b600056h.
- 25.For applications of thiohlycoside for arabinosylation and galactofuranosylation reactions. see, Pathak AK, Pathak V, Seitz L, Gurcha SS, Besra GS, Riordan JM, Reynolds RC. Bioorg Med Chem. 2007;15:5629. doi: 10.1016/j.bmc.2007.04.012.Joe M, Nacario RC, Lowary TL. J Am Chem Soc. 2007;129:9885. doi: 10.1021/ja072892+.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Experimental procedures, full characterization data for new compounds, and copies of NMR spectra. This material is available free of charge via the Internet at http://pubs.acs.org.