

Abstract
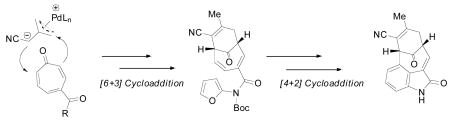
A concise approach to the core skeleton of the welwitindolinone alkaloids was developed based on sequential cycloaddition reactions. First, a palladium catalyzed enantioselective [6 + 3] trimethylenemethane cycloaddition onto a tropone nucleus was used to generate the requisite bicyclo[4.3.1]decadiene. Subsequent modifications to the cycloadduct allowed for an intramolecular [4 + 2] cycloaddition to generate the oxindole and complete the core of the natural product family.
The palladium catalyzed trimethylenemethane (Pd-TMM) cycloaddition reaction represents a highly effective tool for the rapid synthesis of complex carbocycles.1 A rather useful extension to the widely studied [3 + 2] cycloaddition to electron deficient olefins is a [6 + 3] cycloaddition to a tropone nucleus, providing access to functionalized bicyclo[4.3.1]decadienes.2 Building upon our disclosures of enantioselective [3 + 2] Pd-TMM cycloadditions controlled using phosphoramidite ligands,3 we have recently rendered the [6 + 3] cycloaddition enantioselective as well, thus enabling access to stereodefined bicyclo[4.3.1]decadienes in an efficient manner (Scheme 1).4
Scheme 1.

Enantioselective Pd-TMM [6 + 3] Cycloadditions
The advent of such methodology opens the door for a unique, enantioselective, synthesis of bioactive molecules possessing the [4.3.1] bicyclic motif. Of these, we chose to initiate a program to develop a synthesis of the welwitindolinone B & C class of marine alkaloids (1-7; Figure 1).5 These particular compounds are characterized by a highly functionalized [4.3.1] bicyclic carbon skeleton containing an oxindole, two quaternary stereocenters, and multiple sensitive functional groups. While the bioactivity of these molecules varies, the more potent of these, N-methylwelwitindolinone C isothiocyanate (1), acts as a powerful antagonist for the over expression of P-glycoprotein, offering a potential therapeutic benefit against multiple drug resistant tumors.6
Figure 1.

Selected Welwitindolinone B & C Alkaloids
Although a total synthesis of any member of this class of compounds has yet to be accomplished,7 several approaches to a core structure have been reported.8 The majority of these syntheses rely on using an intact oxindole or indole moiety as a starting point, followed by stepwise construction of the bicyclo[4.3.1]decane core. In contrast, we envisioned a novel approach that would rely on a series of sequential cycloadditions to rapidly build the common core structure 8 (Scheme 2). Central to this theme was an enantioselective [6 + 3] cycloaddition that would rapidly construct the bicyclic fragment 10 from a suitable tropone (11 or 12) and the cyano TMM donor 13. Based on work by Padwa,9 an intramolecular [4 + 2] cycloaddition reaction between a pendent amidofuran and the endocyclic olefin would then be used to generate the oxindole core 8 in a single operation. This core structure could conceivably be elaborated to any of the natural products 1-7.
Scheme 2.

Retrosynthetic Analysis
Ideally, a tropone system bearing a 2-amino-5-ester substitution pattern, as in compound 12, would provide the most straightforward synthetic approach. However, in order to attain high levels of regioselectivity for the TMM cycloaddition, the unusual isophthalimide group was required.4 Unfortunately, this unstable protecting group led to numerous difficulties in carrying out further synthetic transformations. As a result, our attention turned to the potential of a late stage installation of the bridgehead amine using an intramolecular nitrenoid insertion into the C11-H bond.10 This, in turn, enabled the use of a tropone lacking the 2-amino group (11), known to be highly successful in [6 + 3] cyclodadditions.4
Studies began with the construction of several 4-substituted tropones beginning from cycloheptatriene 14 (Scheme 3); easily prepared by the cyclopropanation/ring expansion of anisole.11 Barium hydroxide mediated hydrolysis delivered acid 15, which could be readily functionalized as desired.12 Ideally, a tropone bearing the imidofuran ring (19) would offer the most straightforward approach. However, oxidation of the corresponding cycloheptatriene 16 to tropone 19 proved difficult. Use of a more electron deficient amidofuran possessing a methyl ester9b was then explored. Standard amide coupling conditions and N-methylation gave intermediate 17, which was oxidized13 to the tropone 20 in 54% overall yield. While the methyl ester would require eventual removal to generate the natural product series, it was thought that such an amidofuran would offer a method to access synthetic analogues, as well as providing an additional handle for the proposed nitrenoid insertion at C11. Nonetheless, to expand our synthesis options to allow for a later stage installation of the unsubstituted amidofuran, tropone 21 bearing the easily cleaved p-methoxybenzyl group (PMB) was also prepared in good yield from cycloheptatriene 18.
Scheme 3.

Preparation of the Tropone Intermediates
Palladium catalyzed [6 + 3] cycloadditions of both tropones 20 and 21 were conducted using the (bis)biphenyl pyrrolidine ligand L114 (see Scheme 1). Tropone 20 reacted to give the desired cycloadduct 22 as a single regio- and diastereomer in 60% yield and high (94% ee) enantioselectivity (Scheme 4). Concurrently, the 4-PMB ester tropone 21 delivered the cycloadduct 23 in better yield (80%) and comparable enantioselectivity. Both cycloadducts 22 and 23 were independently carried forward to the natural product core to illustrate the effectiveness of our synthetic approach.
Scheme 4.

Asymmetric [6 + 3] Cycloadditions
Already possessing the amidofuran, cycloadduct 22 was poised to undergo the anticipated [4 + 2] cycloaddition to generate the oxindole core. However, as predicted, a facile [3,3] sigmatropic rearrangement occurred upon heating.4 To avoid this, chemoselective derivatizations of the exocyclic olefin, such as oxidation, were attempted yet remain a challenge for this synthetic route. Fortunately, isomerization of the double bond to the endocyclic position using catalytic DMAP proved facile, giving compound 24 in high yield (Scheme 5). Gratifyingly, heating this intermediate under microwave conditions promoted the intramolecular [4 + 2] cycloaddition to provide alcohol 25 as a single diastereomer,15 albeit in moderate yield. While it was hoped that dehydration to the oxindole core would be spontaneous, treatment of the unstable intermediate with the dehydrating agent developed by Burgess16 proved necessary to give the completed core structure 26.17
Scheme 5.

Elaboration of Adduct 22
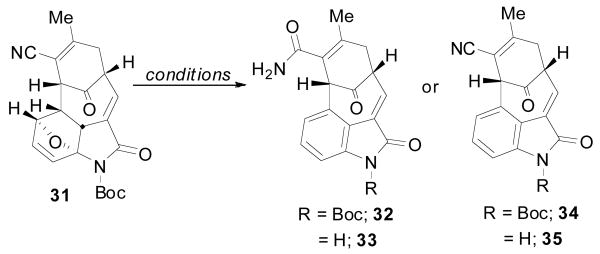
In considering the conversion of TMM adduct 23 to the core structure, the next stage of the synthesis called for installation of the amidofuran and a thermal [4 + 2] cycloaddition to generate the oxindole. As before, isomerization of the exocyclic olefin was readily accomplished with catalytic DMAP to give the α, β-unsaturated nitrile 27 in excellent yield (Scheme 6). Removal of the PMB group followed by coupling with N-Boc amidofuran 299c gave the Diels-Alder precursor 30 in good yield. Heating of imidofuran 30 in toluene at reflux temperature promoted the intramolecular cycloaddition to give oxabicycle 31 as a single diastereomer in almost quantitative yield. Not surprisingly, this cycloaddition occured at much lower temperature and with greatly improved yield as compared to the more electron deficient amidofuran system discussed above. The configuration was tentatively assigned as shown using 1H NMR analysis. Somewhat surprisingly, however, this compound remained as the oxabicycle 31 and did not undergo dehydration to the oxindole even after prolonged reaction times.
Scheme 6.

Elaboration of Adduct 23
As previously demonstrated by Padwa,9d the electron withdrawing properties of the Boc group were likely preventing the spontaneous opening and dehydration of the oxabicycle. Thus, removal of the Boc group was expected to lead directly to the formation of the desired oxindole. Interestingly, while confirming the hypothesis, the common removal technique employing TFA also led to hydrolysis of the nitrile to provide primary amides 32 and 33 (Table 1). Several alternate reagents (BF3·OEt2, BCl3, bromocatechol borane, [Rh(COD)Cl]29d were also examined without much success. Ultimately, two useful conditions were identified; Burgess reagent could be used to dehydrate and leave the Boc group intact to generate oxindole 34 and, alternatively, use of catalytic Yb(OTf)3 could deliver the N-H oxindole core 35 as a single component in moderate yield.
Table 1.
Completion of Oxindole Core
 | ||
|---|---|---|
| reagents | compound | yield |
| TFA | 32 & 33 | not determined |
| BF3 · OEt3 | 34 & 35 | 18% & 34% |
| Burgess Reagent | 34 | 48% |
| Yb(OTf)3 | 35 | 50% |
In summary, a particularly concise strategy for the synthesis of the core of several welwitindolinone alkaloids derives from the combination of the asymmetric [6 + 3] Pd-TMM cycloaddition to form the bridged [4.3.1]bicycle with the [4 + 2] cycloaddition-dehydration to form the oxindole bicycle. The examples presented provided the complete tetracyclic ring system in less than 10 steps beginning with anisole. Efforts are underway to elaborate these intermediates and complete the synthesis of several of the more bioactive members in this group of natural products.
Supplementary Material
Acknowledgments
We thank the NSF and NIH (Grant GM 33049) for generous support of our programs. PJM thanks the American Cancer Society for a Postdoctoral Fellowship.
Footnotes
Supporting Information Available Full characterization and NMR spectra of all new compounds is available free of charge at http://pubs.acs.org.
References
- 1.(a) Trost BM. Angew Chem Int Ed Engl. 1986;25:1–20. [Google Scholar]; (b) Trost BM. Pure Appl Chem. 1988;60:1615–1626. [Google Scholar]
- 2.Trost BM, Seoane PR. J Am Chem Soc. 1987;109:615–617. [Google Scholar]
- 3.(a) Trost BM, Stambuli JP, Silverman SM, Schworer U. J Am Chem Soc. 2006;128:13328–13329. doi: 10.1021/ja0640750. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Trost BM, Cramer N, Silverman SM. J Am Chem Soc. 2007;129:12396–12397. doi: 10.1021/ja075335w. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Trost BM, Silverman SM, Stambuli JP. J Am Chem Soc. 2007;129:12398–12399. doi: 10.1021/ja0753389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Trost BM, McDougall PJ, Hartmann O, Wathen PT. J Am Chem Soc. 2008;130:14960–14961. doi: 10.1021/ja806979b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.(a) Stratmann K, Moore RE, Bonjouklian R, Deeter JB, Patterson GML, Shaffer S, Smith CD, Smitka TA. J Am Chem Soc. 1994;116:9935–9942. [Google Scholar]; (b) Jimenez JI, Huber U, Moore RE, Patterson GM. L J Nat Prod. 1999;62:569–572. doi: 10.1021/np980485t. [DOI] [PubMed] [Google Scholar]
- 6.(a) Smith CD, Zilfou JT, Stratmann K, Patterson GML, Moore RE. Mol Pharmacol. 1995;47:241–247. [PubMed] [Google Scholar]; (b) Zhang X, Smith C. D Mol Pharmacol. 1996;49:288–294. [PubMed] [Google Scholar]
- 7.Two total syntheses of the biosynthetically related welwitindolinone A isonitrile have been reported: Baran PS, Richter JM. J Am Chem Soc. 2005;127:15394–15396. doi: 10.1021/ja056171r.Reisman SE, Ready JM, Hasuoka A, Smith CJ, Wood JL. J Am Chem Soc. 2006;128:1448–1449. doi: 10.1021/ja057640s.
- 8.Reviews: Avendaño C, Menéndez JC. Curr Org Synth. 2004;1:65–82.Brown LE, Konopelski JP. Org Prep Proced Int. 2008;40:411, 445.Recent progress: Boissel V, Simpkins NS, Bhalay G. Tetrahedron Lett. 2009;50:3283–3286.Tian X, Huters AD, Douglas CJ, Garg NK. Org Lett. 2009;11:2349–2351. doi: 10.1021/ol9007684.
- 9.(a) Padwa A, Brodney MA, Dimitroff M. J Org Chem. 1998;63:5304–5305. doi: 10.1021/jo010020z. [DOI] [PubMed] [Google Scholar]; (b) Padwa A, Dimitroff M, Waterson AG, Wu T. J Org Chem. 1997;62:4088–4096. [Google Scholar]; (c) Lynch SM, Bur SK, Padwa A. Org Lett. 2002;4:4643–4645. doi: 10.1021/ol027024q. [DOI] [PubMed] [Google Scholar]; (d) Padwa A, Wang Q. J Org Chem. 2006;71:3210–3220. doi: 10.1021/jo060238r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Espino CG, Wehn PM, Chow J, Du Bois J. J Am Chem Soc. 2001;123:6935–6936. [Google Scholar]
- 11.Anciaux AJ, Demonceau A, Noels AF, Hubert AJ, Warin R, Teyssie P. J Org Chem. 1981;46:873–876. [Google Scholar]
- 12.See Supporting Information for details.
- 13.Bartels-Keith JR, Johson AW, Langeman A. J Chem Soc. 1952:4461–4466. [Google Scholar]
- 14.A commercially available phosphoramidite (Aldrich Chemical Co. Inc., Catalog #665290) could also be used, although it gave low ee. See Supporting Information.
- 15.Due to the unstable nature of intermediate 25, the configuration was not rigourously established.
- 16.Burgess EM, Penton HR, Taylor EA, Williams WM. Org Synth Coll. 1988;6:788. [Google Scholar]
- 17.A single step conversion of intermediate 24 to oxidole 26 was also possible using the Burgess reagent at high temperature, however, purification of the product was difficult.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.