

Abstract

Various 1,1-disubstituted terminal olefins have been investigated for asymmetric epoxidation using chiral ketone catalysts. Up to 88% ee has been achieved with a lactam ketone, and a planar transition state is likely to be a major reaction pathway.
Keywords: asymmetric epoxidation; chiral dioxirane; 1,1-disubstituted terminal olefins
Introduction
Chiral dioxiranes have recently been shown to be effective for asymmetric epoxidation of olefins, and a number of laboratories have extensively investigated chiral ketones of various structures.1 In our own studies, we found that fructose-derived ketone 1 is a very effective catalyst for the epoxidation of trans- and trisubstituted olefins,2 and oxazolidinone-bearing ketones 2 can give high ee’s for olefins such as conjugated aromatic cis-olefins,3a,c,d,e,g conjugated cis-dienes3k and enynes,3a,c,l styrenes,3b,c,d,f certain trisubstituted,3h,j and tetrasubstituted olefins3i,j which had not been effective with ketone 1 (Figure 1). Studies have shown that the enantioselectivity afforded by ketone 2 results from an apparent attractive interaction between the Rπ group of the olefin and the spiro oxazolidinone of the ketone catalyst.3
Figure 1.

Among six classes of olefins (Figure 2), 1,1-disubstituted terminal olefins (VI) have generally been challenging for asymmetric epoxidation.4–8 Epoxidation of α-methylstyrene and α-isopropylstyrene with ketone 2a gave (S)-α-methylstyrene oxide in 30% ee and α- isopropylstyrene oxide in 58% ee.3c Several possible spiro and planar transition states for epoxidation with ketone 2 are shown in Figure 3.2b,3c Spiro transition states (A-D) are generally favored stereoelectronically as a result of the stabilizing interaction of an oxygen lone pair with the π* orbital of the alkene.1,9,10 However, planar transition states E and G appear to be sterically more favored as compared to spiro transition states. Planar transition states F and H are disfavored both electronically and sterically, thus are unlikely to be significant contributors. Between the two planar transition states E and G, E is likely to be favored over G due to the associative interaction between the phenyl group of the olefin and the oxazolidinone of the ketone catalyst. We hypothesized that planar E might be the major transition state for the epoxidation of α-methylstyrene based on the S configuration of the resulting epoxide obtained with ketone 2a. A higher ee obtained by with α-isopropylstyrene could be due to disfavoring competing spiro D by a larger isopropyl group.3c Based on these observations, we decided to search for ketone catalysts that can further favor planar E-like transition state to enhance the enantionselectivity for the epoxidation of 1,1-disubstituted terminal olefins. We have found that lactam ketones 3 provide very promising results (Figure 1). Herein we wish to report our studies on this subject.
Figure 2.

Figure 3.

The proposed spiro and planar transition states for the epoxidation of 1,1- disubstituted terminal olefins
Results and Discussion
The synthesis of lactam ketone 3 is outlined in Schemes 1 and 2. Diol 4, prepared from D-glucose as previously reported,11 was treated with BrCH2COBr to form compound 5, which was then converted to ketone 3a after cyclization and oxidation. Upon introduction of a Boc or Ac group, ketone 3a was converted to ketones 3b and 3c (Scheme 1). Ketones 3d-h were prepared from D-glucose in four steps by Amadori rearrangement,12 ketalization,3d formation of the six-membered lactam, and subsequent oxidation (Scheme 2). The X-ray structure of ketone 3d is shown in Figure 4. An overlay of ketones 2b and 3d is shown in Figure 5. In contrast to ketone 2b,3d the N-phenyl group and the lactam carbonyl group in 3d are not coplanar.
Scheme 1.

Scheme 2.

Figure 4.

The X-ray structure of ketone 3d (stereoview)
Figure 5.

Crystal structure overlay of ketones 2b and 3d (stereoview)
Initial studies on the epoxidation of α-isopropylstyrene with ketone 3d showed that 1,4-dioxane was among the best solvents, giving 94% conversion and 84% ee (Table 1, entry 4). The enantioselectivity was also affected by the N-substituents of ketone catalysts with ketones 3a, 3d, 3e, 3f, and 3h giving the highest enantioselectivity (82-84% ee) (Table 1, entries 7, 4, 10, 11, and 13). Ketone 3d, readily synthesized from inexpensive starting materials, was subsequently investigated for the epoxidation of 1,1-disubstituted terminal olefins. As shown in Table 2, a variety of aryl-substituted 1,1-disubstituted olefins can be effectively epoxidized in good enantioselectivities (62-88% ee). Generally speaking, substrates with bulky alkyl groups at α positions of olefins produce epoxides with higher enantioselectivity than those with small groups. The substituents on the phenyl groups of olefins also have some effects on the enantioselectivities (74-88% ee) (Table 2, entries 7–14). Allylic, homoallylic, and bishomoallylic alcohols are also effective substrates (Table 2, entries 16–21). Up to 88% ee was obtained for 1,1-dialkyl-2-aryl allylic alcohols (Table 2, entries 19–21). A reasonable enantioselectivity (60% ee) was also obtained for a non-aromatic allylic alcohol (Table 2, entry 22).
Table 1.
Asymmetric Epoxidation of α-Isopropylstyrene with Ketones 3a
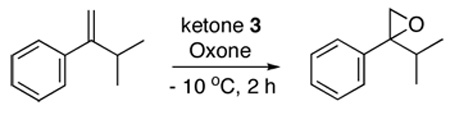 | ||||
|---|---|---|---|---|
| entry | ketone | solvent | conv. (%)b | ee (%)b |
| 1 | 3d | CH3CN/DMM (1/2) | 97 | 71 |
| 2 | DME | 94 | 81 | |
| 3 | DME/n-BuOH | 99 | 76 | |
| 4 | 1,4-dioxane | 94 | 84 | |
| 5 | 1,4-dioxane/DME (2/1) | 99 | 80 | |
| 6 | 1,4-dioxane/n-BuOH (1/1) | 100 | 78 | |
| 7 | 3a | 91 | 82 | |
| 8 | 3b | 69 | 71 | |
| 9 | 3c | 10 | nd | |
| 10 | 3e | 100 | 83 | |
| 11 | 3f | 98 | 83 | |
| 12 | 3g | 80 | 52 | |
| 13 | 3h | 99 | 84 | |
All epoxidations were carried out with the olefin (0.2 mmol), ketone 3 (0.06 mmol), Oxone (0.32 mmol), and K2CO3 (1.344 mmol) in organic solvent (3 mL) and buffer (0.1 M K2CO3/AcOH, pH 9.3; 2 mL) at −10 °C for 2 h.
The conversion and ee were determined by chiral GC (B-DM column).
Table 2.
Asymmetric Epoxidation of 1,1-Disubstituted Terminal Olefins with Ketone 3da
| entry | substrate | yield (%)b | ee (%) | config.e |
|---|---|---|---|---|
| 1 | R = Me | 60 | 62c | (+)−(S)13a |
| 2 | R = Et | 71 | 78d | (+)–(S)13a |
| 3 | R = n-Pr | 90 | 75d | (+) |
| 4 | R = i-Bu | 54 | 74d | (+) |
| 5 | R = c-C6H11 | 62 | 77c | (+) |
| 6 | R = t-Bu | 43 | 86d | (+) |
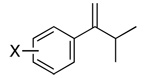 |
||||
| 7 | X = H | 71 | 84d | (+) |
| 8 | X = p-i-Pr | 51 | 82c | (+) |
| 9 | X = p-MeO | 94 | 84c | (+) |
| 10 | X = p-F | 78 | 74d | (+) |
| 11 | X = p-Br | 68 | 78d | (+) |
| 12 | X = m-Me | 57 | 82c | (+) |
| 13 | X = m-F | 74 | 81d | (+) |
| 14 | X = o-F | 72 | 88d | (+) |
| 15 | 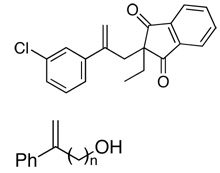 |
51 | 66c | (−)–(S)13b |
| 16 | n = 1 | 93 | 77c | (+)–(R)13c |
| 17 | n = 2 | 47 | 72c | (+) |
| 18 | n = 3 | 62 | 74c | (+) |
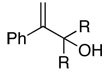 |
||||
| 19 | R = Me | 76 | 87c | (+)–(S)13a |
| 20 | R = Et | 85 | 87d | (+) |
| 21 | R,R = (CH2)4 | 86 | 88d | (+)–(S)13a |
| 22 | 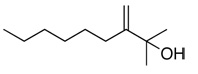 |
78 | 60d | (+) |
All epoxidations were carried out with the olefin (0.2 mmol), ketone 3d (0.06 mmol), Oxone (0.32 mmol), and K2CO3 (1.344 mmol) in 1,4-dioxane (3 mL), and buffer (0.1 M K2CO3/AcOH, pH 9.3; 2 mL) at −10 °C for 2 h (4 h for entries 6, 11, 13, and 14).
Isolated yield except entry 7 which is crude yield.
The ee was determined by chiral HPLC (Chiracel OD column).
The ee was determined by chiral GC (B-DM column).
The absolute configurations were determined by comparing the measured optical rotations and HPLC trace with reported ones.
Ketone 3d gave a similar level of enantioselectivity to ketone 2 for epoxidation of cis-olefins (Table 3, entries 1 and 2), indicating that there still exists an electronic attraction between the amide moiety and the phenyl group of the olefin in spiro transition state I (Figure 6). When 1-phenylcyclohexene was epoxidized with ketone 3d, the (S,S)-epoxide derived from planar L (Figure 7) was obtained with 80% ee while the epoxidation with ketones 2a and 2b gave 43% ee of the (S,S)-epoxide3c and 25% ee of the (R,R)-epoxide,3d respectively, suggesting that the six-membered lactam moiety provides a more favorable environment for the attraction between the lactam moiety of the ketone and the phenyl group of the olefin in the planar transition state as compared to ketones 2a and 2b. In the case of 1-phenyl-3,4-dihydronaphthalene, the epoxide resulting from the planar transition state was obtained in as high as 90% ee (Table 3, entry 4), further illustrating the aforementioned attraction in the planar transition state.
Table 3.
Asymmetric Epoxidation of cis- and Trisubstituted Olefins by Ketone 3da
| entry | substrate | conv. (yield) (%)b | ee (%) | config.g |
|---|---|---|---|---|
| 1 | 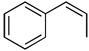 |
100c(60) | 85e | (−)-(1R,2S)3a |
| 2 | 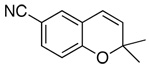 |
89d(87) | 84f | (+)-(3R,4R)3a |
| 3 | 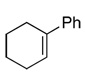 |
99c(89) | 80e | (−)-(S,S)2b,3c,d |
| 4 | 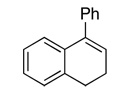 |
88d(56) | 90f | (+)-(1R, 2S)2b |
All reactions were carried out with substrate (0.2 mmol), ketone 3d (0.06 mmol for entry 1, 0.04 mmol for entries 2, 3, and 4), Oxone (0.32 mmol), and K2CO3 (1.344 mmol) in DME/DMM (3:1, v/v; 3.0 mL) and buffer (0.1 M K2CO3-AcOH in 4 × 10−4 M aqueous EDTA, pH 9.3; 2 mL); For entries 1, 3, and 4, the reaction was carried out at −10 °C for 4 h; For entry 2, the reaction was carried out at 0 °C for 12 h.
Isolated yield.
The conversion was determined by GC (B-DM column).
The conversion was determined by 1H NMR.
The ee was determined by chiral GC (B-DM column).
The ee was determined by chiral HPLC (Chiracel OD column).
The absolute configurations were determined by comparing the measured optical rotations and GC trace with reported ones.
Figure 6.

The proposed competing spiro transition states for the epoxidation of cis-olefins with ketone 3
Figure 7.

The proposed competing spiro and planar transition states for the epoxidation of 1-phenylcyclohexene with ketone 3
The known absolute configurations of selected epoxides (Table 2, entries 1, 2, 15, 16, 19, and 21) are consistent with the notion that the epoxidation proceed mainly via planar transition state P (Figure 8). A bulky R substituent on the olefin disfavors spiro O, thus resulting in higher ee’s as observed. Further improvement of the enantioselectivity will require further disfavoring spiro N and/or planar Q transition states.
Figure 8.

The proposed competing transition states for the epoxidation of 1,1-disubstituted terminal olefins with ketone 3
In summary, a variety of 1,1-disubstituted terminal olefins can be enantioselectively epoxidized using lactam ketone 3d as catalyst and Oxone as oxidant, giving up to 88% ee. Studies indicate that the epoxidation of 1,1-disubstituted terminal olefins with ketone 3 proceeds mainly via a planar transition state. Ketone 3 provides a promising lead for further improvement of the enantioselectivity for this challenging class of olefins.
Experimental
Representive Ketone Synthesis
To a solution of amino alcohol 7d (prepared from D-glucose in two steps)3d (3.09 g, 10.0 mmol) and Et3N (1.11 g, 1.54 mL, 11.0 mmol) in dry THF (50 mL), a solution of 2-bromoacetyl bromide (2.22 g, 0.95 mL, 11.0 mmol) in dry THF (10 mL) was added dropwise at rt over 2 h. After the resulting mixture was stirred at rt for 3 h, NaH (95 %, 0.6 g, 23.7 mmol) was added into the reaction mixture carefully. Upon stirring at rt for 0.5 h, the reaction mixture was quenched with MeOH (0.25 mL) and filtered. The filtrate was concentrated and purified by flash chromatography (silica gel, hexane /EtOAc = 1/6) to give lactam 8d as a white solid (1.42 g, 41% yield): mp 198–199 °C; [α]D25 = −144.6 (c 1.0, CHCl3); IR (film) 3410, 1661 cm−1; 1H NMR (400 MHz, CDCl3) δ 7.21-7.14 (m, 4H), 4.40-4.36 (m, 1H), 4.30-4.21 (m, 4H), 4.12 (d, J = 13.2 Hz, 1H), 3.96 (dd, J = 13.2, 2.8 Hz, 1H), 3.62-3.59 (m, 1H), 3.53-3.48 (m, 1H), 3.10-2.88 (m, 1H), 2.33 (s, 3H), 1.51 (s, 3H), 1.37 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 165.6, 138.4, 137.4, 130.1, 125.8, 109.7, 96.2, 76.5, 73.4, 71.7, 62.7, 60.5, 54.2, 28.2, 26.2, 21.2; HRMS Calcd for C18H24O6N (M+H): 350.1604; Found: 350.1607.
AcOH (0.15 mL) was added to a slurry of lactam 8d (4.8 g, 13.76 mmol), PDC (10.3 g, 27.5 mmol), and 3Å MS (6.5 g) in CH2Cl2 (300 mL). Upon stirring at rt for 3 d (no SM left as judged by TLC), the reaction mixture was filtered through a pad of silica gel, and the filter cake was washed with EtOAc. The filtrate was concentrated and purified by flash chromatography (silica gel, hexane/EtOAc = 3/1) to give ketone 3d as a white solid (4.5 g, 95% yield): mp 184–185 °C; [α]D25 = −86.5 (c 1.0, CHCl3); IR (film) 1753, 1674 cm−1; 1H NMR (300 MHz, CDCl3) δ 7.24-7.18 (m, 4H), 4.86 (d, J = 5.7 Hz, 1H), 4.66-4.64 (m, 1H), 4.49-4.23 (m, 5H), 3.64 (d, J = 13.8 Hz, 1H), 2.36 (s, 3H), 1.47 (s, 3H), 1.43 (s, 3H); 13C NMR (75 MHz, CDCl3) δ 197.7, 165.2, 138.2, 137.7, 130.2, 125.8, 111.0, 96.1, 78.4, 75.7, 63.2, 59.9, 51.9, 27.3. 26.2, 21.3; HRMS Calcd for C18H22NO6 (M+H): 348.1447; Found: 348.1447; Anal. Calcd. For C18H21NO6: C, 62.24; H, 6.09. Found: C, 62.02; H, 6.01.
Representative Epoxidation Procedure (Table 2, Entry 19)
To a solution of the olefin (0.324 g, 0.20 mmol), tetrabutylammonium hydrogen sulfate (0.0038 g, 0.010 mmol), and ketone 3d (0.0208 g, 0.06 mmol) in dioxane (3 mL) was added buffer (0.1 M K2CO3-AcOH in 4 × 10−4 M aqueous EDTA, pH = 9.3; 2 mL) with stirring. After the mixture was cooled to −10 °C (bath temperature), a solution of Oxone (0.20 M in 4 × 10−4 M aqueous EDTA, 1.6 mL) (0.197 g, 0.32 mmol) and a solution of K2CO3 (0.84 M in 4 × 10−4 M aqueous EDTA, 1.6 mL) (0.185 g, 1.344 mmol) were added separately and simultaneously via a syringe pump over a period of 2 h. The reaction mixture was quenched with hexane, extracted with EtOAc, dried over Na2SO4, filtered, concentrated, and purified by flash chromatography (silica gel was buffered with 1% Et3N in organic solvent; hexane/Et2O = 5/1 as eluent) to give the epoxide as white solid (0.027 g, 76% yield, 87% ee).
Supplementary Material
Supporting Information Available: The synthesis and characterization of ketones 3a-h, the epoxidation procedure and characterization of epoxides, and the X-ray structures of ketones 3c, 3d, and 3g along with the data for the determination of the enantiomeric excess of the epoxides obtained with ketone 3d (126 pages).
Acknowledgements
We are grateful to the generous financial support from the General Medical Sciences of the National Institutes of Health (GM59705-08). We thank Dr. Pingzhen Wang for obtaining the crystal structure of ketone 2b.
References
- 1.For leading reviews, see: Denmark SE, Wu Z. Synlett. 1999:847. Frohn M, Shi Y. Synthesis. 2000:1979. Shi Y. Acc. Chem. Res. 2004;37:488. doi: 10.1021/ar030063x. Yang D. Acc. Chem. Res. 2004;37:497. doi: 10.1021/ar030065h. Wong OA, Shi Y. Chem. Rev. 2008;108 doi: 10.1021/cr068367v. ASAP.
- 2.(a) Tu Y, Wang Z-X, Shi Y. J. Am. Chem. Soc. 1996;118:9806. [Google Scholar]; (b) Wang Z-X, Tu Y, Frohn M, Zhang J-R, Shi Y. J. Am. Chem. Soc. 1997;119:11224. [Google Scholar]; (c) Shu L, Shi Y. Tetrahedron. 2001;57:5213. [Google Scholar]
- 3.(a) Tian H, She X, Shu L, Yu H, Shi Y. J. Am. Chem. Soc. 2000;122:11551. [Google Scholar]; (b) Tian H, She X, Xu J, Shi Y. Org. Lett. 2001;3:1929. doi: 10.1021/ol010066e. [DOI] [PubMed] [Google Scholar]; (c) Tian H, She X, Yu H, Shu L, Shi Y. J. Org. Chem. 2002;67:2435. doi: 10.1021/jo010838k. [DOI] [PubMed] [Google Scholar]; (d) Shu L, Wang P, Gan Y, Shi Y. Org. Lett. 2003;5:293. doi: 10.1021/ol020229e. [DOI] [PubMed] [Google Scholar]; (e) Shu L, Shi Y. Tetrahedron Lett. 2004;45:8115. [Google Scholar]; (f) Goeddel D, Shu L, Yuan Y, Wong OA, Wang B, Shi Y. J. Org. Chem. 2006;71:1715. doi: 10.1021/jo0520285. [DOI] [PubMed] [Google Scholar]; (g) Wong OA;, Shi Y. J. Org. Chem. 2006;71:3973. doi: 10.1021/jo0604179. [DOI] [PubMed] [Google Scholar]; (h) Shen Y-M, Wang B, Shi Y. Angew. Chem. Int. Ed. 2006;45:1429. doi: 10.1002/anie.200501520. [DOI] [PubMed] [Google Scholar]; (i) Shen Y-M, Wang B, Shi Y. Tetrahedron Lett. 2006;47:5455. [Google Scholar]; (j) Wang B, Shen Y-M, Shi Y. J. Org. Chem. 2006;71:9519. doi: 10.1021/jo061341j. [DOI] [PubMed] [Google Scholar]; (k) Burke CP, Shi Y. Angew. Chem. Int. Ed. 2006;45:4475. doi: 10.1002/anie.200600840. [DOI] [PubMed] [Google Scholar]; (l) Burke CP, Shi Y. J. Org. Chem. 2007;72:4093. doi: 10.1021/jo070205r. [DOI] [PubMed] [Google Scholar]; (m) Burke CP, Shi Y. J. Org. Chem. 2007;72:6320. doi: 10.1021/jo0708644. [DOI] [PubMed] [Google Scholar]
- 4.For a leading review on asymmetric epoxidation, see: Xia Q-H, Ge H-Q, Ye C-P, Liu Z-M, Su K-X. Chem. Rev. 2005;105:1603. doi: 10.1021/cr0406458.
- 5.For leading references on asymmetric epoxidation of 1,1-disubtituted terminal olefins directed by hydroxyl groups, see: Johnson RA, Sharpless KB. Chapter 4.1. In: Ojima I, editor. Catalytic Asymmetric Synthesis. New York: VCH; 1993. Katsuki T, Martin VS. Org. React. 1996;48:1. Barlan AU, Zhang W, Yamamoto H. Tetrahedron. 2007;63:6075. doi: 10.1016/j.tet.2007.03.071.
- 6.For examples of asymmetric epoxidation of 1,1-disubstituted terminal olefins with chiral metal catalysts, see: Zhang W, Loebach JL, Wilson SR, Jacobsen EN. J. Am. Chem. Soc. 1990;112:2801. Halterman RL, Jan S-T, Nimmons HL, Standlee DJ, Khan MA. Tetrahedron. 1997;53:11257. Kim G-J, Shin J-H. Catal. Lett. 1999;63:83. Tanaka H, Kuroboshi M, Taked H, Kanda H, Torii S. J. Electroanal. Chem. 2001;507:75. Zhang R, Yu W-Y, Sun H-Z, Liu W-S, Che C-M. Chem. Eur. J. 2002;8:2495. doi: 10.1002/1521-3765(20020603)8:11<2495::AID-CHEM2495>3.0.CO;2-G. Zhang H, Xiang S, Li C. Chem. Commun. 2005:1209. doi: 10.1039/b417041e. Fristrup P, Dideriksen BB, Tanner D, Norrby PO. J. Am. Chem. Soc. 2005;127:13672. doi: 10.1021/ja051851f. Zhang H, Zhang Y, Li C. Tetrahedron: Asymmetry. 2005;16:2417. Yu K, Lou L-L, Ding F, Wang S, Wang Z, Liu S. Catal. Commun. 2006;7:170. Sun Y, Tang N. J. Mol. Catal. A: Chem. 2006;255:171. Lou LL, Yu K, Ding F, Zhou W, Peng X, Liu S. Tetrahedron Lett. 2006;47:6513.
- 7.For examples of asymmetric epoxidation of 1,1-disubstituted terminal olefins with chiral dioxiranes, see: Yang D, Yip Y-C, Tang M-W, Wong M-K, Zheng J-H, Cheung K-K. J. Am. Chem. Soc. 1996;118:491. ref. 2b. Wang Z-X, Shi Y. J. Org. Chem. 1997;62:8622. doi: 10.1021/jo962392r. Yang D, Wong M-K, Yip Y-C, Wang X-C, Tang M-W, Zheng J-H, Cheung K-K. J. Am. Chem. Soc. 1998;120:5943. Wang ZX, Miller SM, Anderson OP, Shi Y. J. Org. Chem. 1999;64:6443. doi: 10.1021/jo001343i. refs. 3b,c. Armstrong A, Moss WO, Reeves JR. Tetrahedron: Asymmetry. 2001;12:2779. Armstrong A, Ahmed G, Dominguez-Fernandez B, Hayter BR, Wailes JS. J. Org. Chem. 2002;67:8610. doi: 10.1021/jo026322y. Chan W-K, Yu W-Y, Che C-M, Wong M-K. J. Org. Chem. 2003;68:6576. doi: 10.1021/jo034296d. Bez G, Zhao C-G. Tetrahedron Lett. 2003;44:7403. Bortolini O, Fantin G, Fogagnolo M, Mari L. Tetrahedron: Asymmetry. 2004;15:3831. Armstrong A, Tsuchiya T. Tetrahedron. 2006;62:257. Armstrong A, Dominguez-Fernandez B, Tsuchiya T. Tetrahedron. 2006;62:6614.
- 8.For examples of asymmetric epoxidation of 1,1-disubstituted terminal olefins with oxaziridinium salts, see: Page PCB, Rassias GA, Barros D, Bethell D, Schilling MB. J. Chem. Soc. Perkin Trans. 2000;1:3325. Page PCB, Rassias GA, Barros D, Ardakani A, Buckley B, Bethell D, Smith TAD, Slawin AMZ. J. Org. Chem. 2001;66:6926. doi: 10.1021/jo010258n. Page PCB, Rassias GA, Barros D, Ardakani A, Bethell D, Merifield E. Synlett. 2002:580. Page PCB, Barros D, Buckley BR, Ardakani A, Marples BA. J. Org. Chem. 2004;69:3595. doi: 10.1021/jo035820j. Page PCB, Buckley BR, Rassias GA, Blacker AJ. Eur. J. Org. Chem. 2006:803.
- 9.(a) Baumstark AL, McCloskey CJ. Tetrahedron Lett. 1987;28:3311. [Google Scholar]; (b) Baumstark AL, Vasquez PC. J. Org. Chem. 1988;53:3437. [Google Scholar]
- 10.(a) Bach RD, Andrés JL, Owensby AL, Schlegel HB, McDouall JJW. J. Am. Chem. Soc. 1992;114:7207. [Google Scholar]; (b) Houk KN, Liu J, DeMello NC, Condroski KR. J. Am. Chem. Soc. 1997;119:10147. [Google Scholar]; (c) Jenson C, Liu J, Houk KN, Jorgensen WL. J. Am. Chem. Soc. 1997;119:12982. [Google Scholar]; (d) Deubel DV. J. Org. Chem. 2001;66:3790. doi: 10.1021/jo010127m. [DOI] [PubMed] [Google Scholar]; (e) Singleton DA, Wang Z. J. Am. Chem. Soc. 2005;127:6679. doi: 10.1021/ja0435788. [DOI] [PubMed] [Google Scholar]
- 11.Shu L, Shen Y-M, Burke C, Goeddel D, Shi Y. J. Org. Chem. 2003;68:4963. doi: 10.1021/jo0206770. [DOI] [PubMed] [Google Scholar]
- 12.Hodge JE, Fisher BE. Methods Carbohydr. Chem. 1963;2:99. [Google Scholar]
- 13.(a) Capriati V, Florio S, Luisi R, Salomone A. Org. Lett. 2002;4:2445. doi: 10.1021/ol026212d. [DOI] [PubMed] [Google Scholar]; (b) Tanaka K, Yoshida K, Sasaki C, Osano YT. J. Org. Chem. 2002;67:3131. doi: 10.1021/jo010816y. [DOI] [PubMed] [Google Scholar]; (c) Adam W, Alsters PL, Neumann R, Saha-Möller CR, Seebach D, Zhang R. Org. Lett. 2003;5:725. doi: 10.1021/ol027498p. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information Available: The synthesis and characterization of ketones 3a-h, the epoxidation procedure and characterization of epoxides, and the X-ray structures of ketones 3c, 3d, and 3g along with the data for the determination of the enantiomeric excess of the epoxides obtained with ketone 3d (126 pages).