

Abstract
Cardiac ATP-sensitive K+ (KATP) channels, gated by cellular metabolism, are formed by association of the inwardly rectifying potassium channel Kir6.2, the potassium conducting subunit, and SUR2A, the ATP-binding cassette protein that serves as the regulatory subunit. Kir6.2 is the principal site of ATP-induced channel inhibition, while SUR2A regulates K+ flux through adenine nucleotide binding and catalysis. The ATPase-driven conformations within the regulatory SUR2A subunit of the KATP channel complex have determinate linkage with the states of the channel’s pore. The probability and life-time of ATPase-induced SUR2A intermediates, rather than competitive nucleotide binding alone, defines nucleotide-dependent KATP channel gating. Cooperative interaction, instead of independent contribution of individual nucleotide binding domains within the SUR2A subunit, serves a decisive role in defining KATP channel behavior. Integration of KATP channels with the cellular energetic network renders these channel/enzyme heteromultimers high-fidelity metabolic sensors. This vital function is facilitated through phosphotransfer enzyme-mediated transmission of controllable energetic signals. By virtue of coupling with cellular energetic networks and the ability to decode metabolic signals, KATP channels set membrane excitability to match demand for homeostatic maintenance. This new paradigm in the operation of an ion channel multimer is essential in providing the basis for KATP channel function in the cardiac cell, and for understanding genetic defects associated with life-threatening diseases that result from the inability of the channel complex to optimally fulfill its physiological role.
Keywords: KATP channel, Kir6.2, SUR2A, Eneregetics, Creatine kinase, Adenylate kinase, Glycolysis, Sulfonylurea receptor, Phosphotransfer, Nucleotide, ATP-binding cassette, Action potential, Heart failure, Potassium channel opener, Knock-out
1. Introduction
ATP-sensitive K+ (KATP) channels are molecular combinations of an inwardly rectifying K+ channel, Kir6.x, and a regulatory module, the sulfonylurea receptor SUR [1–3]. Biogenesis of the heteromultimeric KATP channel occurs through combinations of subunit isoforms that define the intrinsic properties and tissue specificity of the channel complex [3,4]. Physical association of the Kir6.2 and SUR2A isoforms generates cardiac KATP channels that are expressed in high density at the sarcolemma [5,6]. By virtue of their integration with cellular energetic networks and their ability to decode metabolic signals, KATP channels set membrane excitability to match demand for homeostatic maintenance [7–11]. Recent progress in the understanding of KATP channel structure and function has been founded on the dissection of channel subunit properties, mapping of channel coupling with cellular energetics and definition of the metabolic sensing role in both healthy and diseased cells.
Under conditions of metabolic surplus, the cardiac KATP channel responds by closure while metabolic challenge provokes channel opening with consequent K+ efflux, action potential shortening, and limitation of potentially damaging intracellular Ca2+ loading [10,12,13]. The basic gating of the KATP channel that underlies the channel’s metabolic response occurs in reaction to the balance at the channel site of inhibitory and stimulatory nucleotides, ATP and ADP, respectively [1,14]. The way in which the cellular metabolic state is “read” incorporates generation and delivery of nucleotide signals to the KATP channel subunits, and nucleotide interactions with specialized channel domains that ultimately secure signal processing and translation into pore gating [15,16].
In this way, KATP channels mediate a homeostatic membrane response to the metabolic insults of ischemia or hypoxia contributing to a cardioprotective outcome [17,18]. Recent studies indicate an even broader function for cardiac KATP channels in the tolerance of cardiomyocytes to numerous acute and chronic metabolic challenges, including sympathetic surge, and physical training [10,19]. Furthermore, the concept of KATP channel-mediated myocardial protection has been expanded to include balancing increased performance to meet augmented demands of stress while avoiding an excessive response that could result in cellular injury and/or arrhythmia [10,19–21]. The homeostatic role of KATP channels is underscored by studies of altered channel behavior in heart disease. Channel gene mutations that disrupt KATP channel function [14] and/or defects in signaling pathways proximal to the channel site [20] compromise the channel’s ability to optimally respond to metabolic challenge. Thus, proper metabolic gating of KATP channels is vital in limiting acute adverse myocardial outcomes under stress, and in evading injury that precipitates the development or progression of chronic heart disease [10,11,14,19,20].
2. Kir6.2 pore-forming subunit: site of KATP channel ATP inhibition
Tetramers of Kir6.2 subunits comprise the pore of KATP channel complexes [3,22]. The pore-forming Kir6.2 subunits cannot readily traffic to the plasma membrane alone, without the regulatory SUR module, due to a C-terminal RKR endoplasmic reticulum retention signal [23,24]. When engineered to be expressed independently of SUR through truncation of the C-terminal amino acids, the Kir6.2 subunit was identified as critical for KATP channel inhibition by intracellular ATP [25,26]. Although a conventional nucleotide binding motif has not been identified within the Kir6.2 sequence, photoaffinity labeling and scanning with sulfhydryl group reagents accomplished by mutagenesis identified that both N- and C-termini may contribute to recognition of ATP [27,28,29]. The most recently developed model implicates two Kir6.2 subunits in coordination of one ATP molecule [30]. While K185 and R201 in the C-terminus of one subunit and R50 in the N-terminus of another Kir6.2 have been directly implicated in the interaction with the γ- and β-phosphate of ATP, respectively, the adenine ring of ATP interacts with E179 and R301 of the second subunit. Mutations that impact the ATP-responsiveness of Kir6.2 alter cardiac mechanical properties at rest and produce a poor myocardial response to ischemic and metabolic challenge [31], and in non-cardiac tissues are associated with disease due to deficient metabolic coupling [32,33].
Assembly of Kir6.2 with SUR enhances ATP-induced inhibition of the channel pore [25], and defines the tissue-specific burst kinetics of KATP channel behavior [5,34,35]. Furthermore, fundamental KATP channel properties, including stimulation by MgADP and potassium channel openers as well as inhibition by sulfonylurea drugs that are absent in truncated Kir6.2 channels, are rescued by co-expression of Kir6.2 with SUR [25,36].
3. SUR regulatory module: nucleotide binding and catalysis
SUR, the regulatory subunit of the KATP channel, incorporates two bundles of six hydrophobic transmembrane-spanning domains (TMD) that are fused to hydrophilic nucleotide binding domains (NBD) also known as the ATP-binding cassettes (ABC). By virtue of structure and sequence homology, SUR belongs to the ABCC subfamily of ABC proteins (http://www.gene.ucl.ac.uk/nomenclature/genefamily/abc.html), that includes the multidrug resistance-associated protein (MRP1 or ABCC1) and the cystic fibrosis transmembrane regulator (CFTR or ABCC7), that generally exist as dimers with two ABC modules and two TMDs (TMD-ABC-TMD-ABC [37]). SUR (TMD0-TMD-ABC-TMD-ABC), in addition to its unique property of association with a distinct protein subunit, Kir6.2, also contains an additional bundle of five TMDs (TMD0), proposed to anchor SUR to the Kir6.2 channel pore [38]. Typical for ABCs, two highly conserved motifs, Walker A (GXXXXGKS/T) and Walker B (four hydrophobic residues followed by aspartate) frame a signature LSGGQ linker motif [39,40]. The crystal structure of homologous proteins indicates that the ε-amino acid and/or the main chain nitrogen of the invariant lysine of the Walker A motif participates in the binding of the β- and γ-phosphate of ATP whereas the Walker B aspartate coordinates Mg2+ through hydrogen bonding to a water molecule [41]. Together with a histidine switch region, postulated to polarize the attached water molecule for hydrolysis, as well as the Q-loop (between the Walker A and the signature motifs) that interacts with the γ-phosphate of ATP through a water bond, Walker motifs form sites for nucleotide binding and hydrolysis within SUR module (Fig. 1).
Fig. 1.
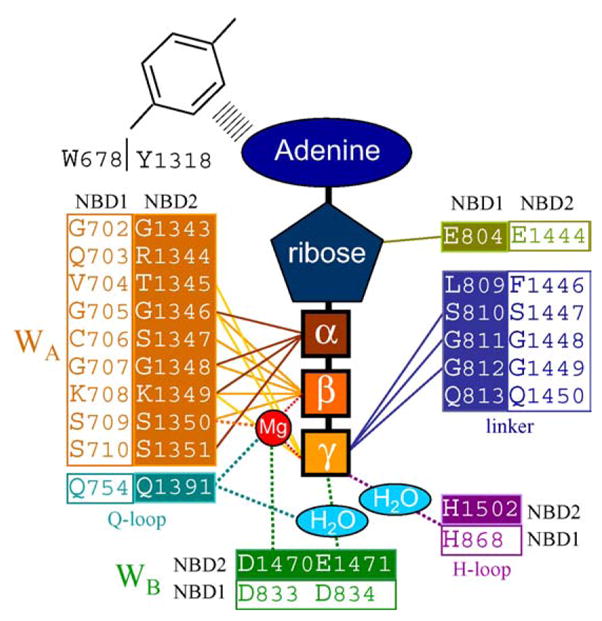
Hypothetical coordination of the ATP molecule within SUR. Internal circle (filled elements) symbolizes the first binding pocket formed by the Walker A motif, Q-loop, Walker B motif, H-loop of NBD2 and accomplished with the linker region of NBD1. External circle (clear elements) represents the second binding pocket comprised by the counterparts of NBD1 and NBD2. The scheme is developed based on sequence alignment between NBDs of SUR and MJ0796 protein [67,77].
Testing of nucleotide binding and hydrolytic properties of SUR with photoaffinity labeling has revealed that NBD1 can be labeled with 8-azido-ATP even in the absence of Mg2+ and γ-phosphate analogs (e.g. orthovanadate) which is usually applied in order to prolong the life-time of nucleotide-NBD complexes securing the probability for covalent bonding of azido-nucleotides in a binding pocket [42,43]. Thus in contrast to certain ABC proteins NBD1 of SUR apparently does not hydrolyze ATP but possesses relatively stable nucleotide binding. Indeed, in the presence of Mg2+, NBD1 but not NBD2 of SUR could be labeled with 8-azido[γ-32P]ATP suggesting that at least one hydrolytic cycle occurred at the second domain prior to labeling leaving the possibility only for tagging of NBD2 with of 8-azido-[α-32P]ATP [44,45]. Asymmetrical properties of nucleotide interactions with NBDs correlate with the different structural homology between NBDs. Specifically, in the cardiac KATP channel, the two NBDs of SUR2A share a 28% sequence homology, such that they are more similar to their counterparts in CFTR than they are to each other (i.e. ~40% sequence homology of SUR2A and CFTR NBD1s, as well as ~40% between the NBD2s of these two ABC proteins). In the SUR close relatives MRP1 and CFTR NBD2 also typically more catalytically active than NBD1 [44–46].
The rate of the catalytic reaction of SUR was directly probed in immunoprecipitates of SUR2A and in purified constructs containing either NBD1 or NBD2 fused to maltose binding protein MBD [47]. In fact, [32P]Pi generation from γ-labeled [32P]ATP was catalyzed by purified MBD-NBD2 and to a lesser extent if at all by MBD-NBD1, confirming primary catalytic activity at NBD2 [47]. Magnesium-dependent NBD2 ATPase activity was within the range reported for other ABC proteins [9,47]. Site-directed mutagenesis of the lysine residue in the Walker A motif of NBD2 or mutation in the Walker B aspartate suppressed this ATPase activity, and produced KATP channels with a higher sensitivity to ATP compared to wild-type Kir6.2/SUR2A [47,48] indicating that the hydrolytically-driven transition from the MgATP- to the MgADP-liganded conformations of SUR influences the gating properties of KATP channels.
4. SUR catalysis-mediated Kir6,2 channel gating
While SUR2A shares the major property of ATP interaction and hydrolysis with other ABCC proteins, no transport function coupled to catalysis, typical for MRP1 protein, has so far been identified. Rather, in the SUR/Kir6.2 complex, similarly to CFTR that also functions as a channel [45,49–51], an intrinsic catalysis is not required for passive ion permeation down the elecrtochemical gradient but could be involved in allosteric regulation of pore gating. Specifically, coupling of discrete conformations in the SUR catalytic cycle with channel gating was solved using nucleotide trapping procedures in conjunction with on-line channel recording when intrinsic enzymatic activity was enhanced by elevating temperature [9]. Capture of ATPase intermediates was achieved using γ-phosphate analogs, orthovanadate (Vi) and beryllium-fluoride (BeFx), which stabilize the catalytic cycle in distinct conformations [49]. Orthovanadate forms a pentavalent pyramidal structure, like γ-phosphate in ATP undergoing hydrolysis, and stabilizes post-hydrolytic states whereas pre-hydrolytic intermediates can be distinguished using beryllium-fluoride which forms a tetrahedral structure, and mimics γ-phosphate in ATP prior to hydrolysis. Although both Vi and BeFx in the presence of nucleotides arrest NBD2ATPase activity, the outcome of the distinct catalytic intermediates induced by these γ-phosphate analogs on the KATP channel regulation is opposite [9]. Trapping the prehydrolytic MgATP-bound state at the SUR ATPase with beryllium-fluoride (SUR*MgADP·BeFx) translates into pore closure or negative channel gating. Recruitment of a posthydrolytic MgADP-bound state by orthovanadate (SUR**MgADP·Vi), promotes opening of ATP-inhibited KATP channels or positive channel gating. Trapping the SUR ATPase by beryllium-fluoride and orthovanadate requires divalent cations, hydrolyzable ATP or externally applied ADP, and the structural intactness of NBD2, underscoring the involvement of operational ATP hydrolysis in channel regulation.
Elevated levels of intracellular MgADP, as it occurs under metabolic stress [15,52–55] effectively decelerate ATPase cycling increasing the probability of the post-hydrolytic conformation associated with channel opening [9,14]. In this way, nucleotide exchange between the SUR ATPase and intracellular metabolic pathways could couple KATP channel function with cellular energetics. In the cell, the high-rate of the major phosphotransfer reaction, catalyzed by creatine kinase, scavenges the ATPase product and facilitates disengagement of posthydrolytic conformations promoting SUR ATPase cycling. Indeed, the rate of ATP hydrolysis at NBD2 can be doubled by creatine kinase, reversing MgADP-induced KATP channel opening highlighting a critical role for phosphotransfer reactions in setting the balance between the lifetime/probability of MgADP versus MgATP-bound conformational intermediates in the SUR ATPase cycle [9,14]. As a consequence of metabolic stress, a drop of creatine kinase flux compromises ADP removal from ATPase sites and suspends the ATPase cycle in a MgADP-bound state producing channel opening [12,15,16].
Expanding on the conventional nucleotide competition model of KATP channel gating, such mechanism underscores a critical role for the regulation of substrate/product-dependent engagement of SUR into ATP/ADP-bound intermediates during the ATPase cycle. Stopped-flow spectroscopy, in conjunction with steady-state characteristics of NBD2 catalysis, has allowed determination of the reaction kinetic constants defining the ATPase cycle of SUR2A [14]. The derived values indicated that the rate-limiting step of the NBD2 ATPase cycle is the actual dissociation of ADP. A normally progressing MBP-NBD2 ATPase catalysis is characterized by a relatively long lifetime of the NBD2·ADP intermediate state in comparison with shorter lifetimes in other intermediates NBD2·ATP and NBD2·ADP·Pi. Thus, the probability that NBD2 will adopt the ADP-bound state through the ATPase cycle will be higher than the probability that this state will form through simple competitive binding in the absence of ATPase activity [14]. Hence, the SUR catalytic activity provides conformational changes that would otherwise occur only rarely, as has also been suggested for gating of CFTR channels [51] with analogy to GTP hydrolysis by G-proteins [56].
Structural characteristics of SUR2A suggest specialized domains that particularly influence the ATPase cycle and associated nucleotide-dependent KATP channel gating [57]. A high resolution three-dimensional model of SUR2A NBD2, obtained with 1 fs time step molecular dynamics simulation, identifies a β-strand, within the SUR tissue-specific carboxy-terminal tail, that may interact with the WalkerA motif due to the spatial geometry of the folded molecule [14]. Mutations flanking the carboxy-terminal β-strand do not affect ATP-binding but dramatically change the rate and kinetics of the ATPase reaction resulting in altered nucleotide-dependent KATP channel regulation [14]. This finding underscores that SUR-mediated gating of the channel pore depends not only upon nucleotide binding to NBDs as classically proposed, but also on the predominant conformational intermediate adopted by the intrinsic ATPase cycle. Furthermore, differences in the pattern of probabilities and lifetimes of individual SUR conformations, in addition to distinct ATPase rates, may contribute to the tissue-specificity of nucleotide-dependent KATP channel regulation [57,58].
While activation of ATP-inhibited KATP channels following stabilization of the post-hydrolytic state is the well-established mechanism of MgADP-dependent positive channel gating, the outcome of the pre-hydrolytic MgATP-bound SUR conformation represents a previously unrecognized mode of negative nucleotide-dependent channel gating. Although KATP channel inhibition is usually attributed exclusively to interaction of ATP with the channel pore, negative channel gating apparently could also be induced following binding of sulfonylurea drugs to SUR [48]. Whereas traditionally the effect of sulfonylurea is believed to be nucleotide-independent, binding of glyburide to SUR has been shown to modulate the nucleotide interaction with SUR [59], suggesting the involvement of ATPase-related intermediates in sulfonylurea-induced KATP channel inhibition [60,61].
The regulation of KATP channels by potassium channel openers (KCOs) also relies on the NBD2 ATPase-driven conformations within SUR2A. Arrest of the ATPase in a prehydrolytic, but not posthydrolytic, state prevents potassium channel openers from activating KATP channels [9]. The effect of potassium channel openers could be abolished by non-hydrolyzable ATP analogs, and rescued in the absence of active hydrolysis by recruitment of a posthydrolytic conformation with MgADP [9]. Disengagement of the posthydrolytic conformation by ADP scavenging through the creatine kinase system reverses the effect of potassium channel openers [9,47]. Thus, entry of SUR into a posthydrolytic conformation plays a permissive role in mediating the action of potassium channel openers. By affecting the ATPase activity [47], potassium channel openers stabilize the posthydrolytic MgADP-bound conformation [9]. This is in accord with the finding that ATP-inhibited KATP channels, when activated by potassium channel openers, exhibit single-channel kinetic behavior indistinguishable from that measured in the presence of MgADP [35,47]. Further, in other SUR isoforms, potassium channel openers maintain MgADP binding, thereby prolonging channel activity following removal of the nucleoside diphosphate [62]. Conversely, intermediates in the ATPase cycle may define the affinity of bound ligands to SUR, as proposed for ATP hydrolysis in other ABC proteins [63,64]. Specifically, the slow off-rate of potassium channel openers in the presence of magnesium nucleotides [65] suggests that engagement in the posthydrolytic conformation may in turn stabilize potassium channel opener binding. Taken together these data support the hypothesis that KATP channel regulation by Mg2+-nucleotides and drugs recruits a common allosteric transduction pathway that originates from conformational inter-conversion of NBDs in SUR and culminates with structural “adaptation” of the channel pore.
Although such allosteric coupling implicates a determinate linkage between the states of the channel pore and the set of NBD2 ATPase conformations, an individual intermediate of NBD2 ATPase cycle may not directly translate into regulation of the channel pore. Indeed, during a normally operating ATPase, NBD2 during each single cycle unavoidably adopts the ADP-bound conformation that potentially could activate KATP channels. Paradoxically, ATP-regenerating systems that facilitate ATPase cycling and therefore promote transit through the MgADP-bound intermediate state, do not activate KATP channels [9,15,16,45]. It is only through arrest of the SUR ATPase in a pre- or post-hydrolytic conformation that specific channel outcome, pore closure or antagonism of ATP-induced pore inhibition, can be achieved [9]. In other words, antagonism of ATP-induced channel inhibition can only be secured through prolongation of the lifetime or increase in the probability of posthydrolytic intermediates [9,14]. In turn, this would indicate that conformational transitions of NBD2 in SUR are not directly translated into a specific state of the channel pore, but rather mediated by an intervening structural rearrangement with the time characteristics slower than the life-time of the post-hydrolytic state. An additional slow-rate conformational rearrangement that transmits the information provided by catalytic intermediates of SUR to the channel pore may rely on dimerization of the two NBDs as has recently been established for a number of ABC proteins [29,66].
5. NBD dimerization and KATP channel gating
In the structure of the NBD2 monomer of SUR2A [14] as well as other ABC members, the ATP-binding site is exposed, suggesting that the catalytic site can be completed by interaction with another domain [51,66,67]. Evidence of physical proximity of NBDs [68] has hinted that one NBD monomer could complete the binding pocket of the adjacent one.
In the KATP channel complex cooperative interaction, rather than separate contributions of each of the NBD in SUR2A, is critical for coupling NBD2 ATP intermediates and the functional state of the KATP channel pore [69,70]. A conformational/functional association of the two NBDs of SUR is supported by biochemical studies of cooperative nucleotide binding. Specifically, MgADP, either by direct binding or as a product of ATP hydrolysis at NBD2, facilitates ADP/ATP-binding to NBD1, an effect abolished by mutations in the Walker A and Walker B motifs of NBD2 [42,43,59]. Furthermore, mutation of the Walker A lysine of NBD1 is crucial for KATP channel activation induced by stabilization of NBD2·ADP·Vi or inhibition by NBD2·ADP·BeFx complexes, indicating a joint action of NBD1 and NBD2 on channel gating [69]. Cooling of membrane patches, which prevents dissociation of pre-bound nucleotides and “freezes” the conformational intermediates of SUR, activates cardiac KATP channels in the presence of inhibitory ATP following fast reheating [69]. Such an observation indicates a relatively long-lived cooling-induced conformational rearrangement characterized by barricade of nucleotides in the binding pockets presumably following association of NBDs. Pre-incubation of membrane patches either with MgADP alone or with non-hydrolyzable analogs of ATP prevents KATP channel activation after reheating. Mutation of the Walker A lysine that impairs nucleotide binding to NBD1 also abolishes cooling/reheating-induced KATP channel activation, as do mutations in the Walker A lysine and Walker B aspartate critical for hydrolysis in NBD2. Thus, ATP-binding to NBD1, cooperatively supported by hydrolysis at NBD2, is a necessary step in securing the proper structural arrangement of SUR that translates into positive gating of KATP channels. This paradigm finds support where cooperative binding of two ATP molecules has been demonstrated in other ABC proteins, such as histidine permease [21], maltose transporter [71], MRP1 [72] and P-glycoprotein [73], where cooperative nucleotide binding provides a kinetic control for the formation of a closed NBD dimer [74].
The determination of crystal structures of several bacterial ABC proteins provides direct evidence that the NBD of ABC proteins physically interact, forming a NBD:NBD dimer [41,75,76]. However, this finding implicates a different topology of signature and Walker motifs within a NBD dimer, making it difficult to generalize a single dimer structure to all ABC proteins [66]. Furthermore, in the presence of nucleotide, the biochemical or structural evidence of dimer formation was obtained only for Rad50 but not for ABC transporters [41,67]. Implementation of Rad50 architecture as a template for modeling of other ABC structures is limited, since in contrast to conventional ABC transporters Rad50 lacks membrane spanning domains and interacts with nucleotides via regions that are not conserved among ABC proteins [41,67]. The most recent crystal structure of MJ0796 obtained with the highest resolution of any NBD nucleotide-sandwich dimer allowed modeling of the SUR1 NBD dimer on the basis of sequence alignment [77]. In this structure, an ATP molecule is “sandwiched” between the Walker A motif of the first NBD and the linker motif of the second NBD (Fig. 1) [29,41]. Thus, one ATP molecule interacts with portions of both NBD.
Evidence for NBD dimerization along with the hydrolytic activity within NBDs, provides the basis for various mechanisms of substrate transport by ABC proteins. It has been suggested that dimerization of the NBDs, induced by ATP-binding, represents a key conformational change ultimately leading to dislocation of the signature motifs, ATP hydrolysis and substrate transport, providing the rationale for the existence of two NBD components in all ABC transporters. To this end, hydrolysis would administrate a catalytic passage to dissociation of NBDs aimed to reset the molecular transport machinery to an initial state following the liberation of inorganic phosphate and ADP [41,78]. Remarkably, biochemical studies as well as crystal structures of intact ABC transporters did not reveal significant structural modification induced by the transition of NBDs from the ATP- to the ADP-bound state [74,79,80] indicating that individual reorientation of each NBD is not sufficient for altering of the whole protein structure. Therefore, it appears that it is through dimerization of the NBDs that a more generic structural rearrangement—“power stroke” [74]—can be accomplished to allosterically couple intrinsic catalysis with an associated protein function.
6. Allosteric regulation of the KATP channel complex
In this regard, the allosteric regulation of the KATP channel complex seems unique among enzymatic systems since it implicates not only structural coupling and cooperativity, induced by nucleotide interaction within the regulatory channel module, but also transmission of such modification to the associated pore-forming subunit aimed at gating otherwise passive ion permeation. Indeed, the classical allosteric model was successfully applied to nucleotide-dependent KATP channel gating [15] which presumed that four identical binding sites for ATP and ADP co-exist within the octameric stoichiometry of the KATP channel complex [22]; binding of ATP to the pore-forming Kir6.2 subunit inhibits channel opening [25,26], whereas binding of ADP to the corresponding regulatory SUR subunit antagonizes ATP-binding to Kir6.2 [43,81]. Although this classic allosteric model of channel regulation was in agreement with experimentally defined nucleotide-dependent gating (i.e. ATP concentration-dependence, saturation of MgADP-induced channel activation), it only postulated intersubunit communication and provided neither the molecular mechanism nor the structural determinants underlying SUR-Kir6.2 interaction.
Regulation of the KATP channel complex seems to require a complex allosteric hierarchy since the effects of non-nucleotide channel regulators (potassium channel openers, sulfonylureas) also rely on the nucleotide-dependent state of both NBDs in the SUR regulatory module. This suggests that conformational rearrangements of NBDs induced by exogenous (openers and sulfonylurea) and endogenous (Mg-nucleotides) ligands are coupled with the open or closed state of the channel pore through a common, integrating allosteric pathway. Such a suggestion stems from a number of experimental observations. Specifically, endogenous as well as exogenous channel regulators (sulfonylurea and KCO) produced the same kinetic fingerprint for channel behavior significantly modulating the probability of burst termination with a similar pattern of changes in interburst kinetics [35,47]. Activation of KATP channels by potassium channel openers apparently involves cooperative interaction of NBDs since their effect/binding is dependent on intracellular adenine nucleotides and Mg2+, requires the intactness of both NBDs, and relies on post-hydrolytic SUR conformations [40,47,65,82]. Biochemical studies have demonstrated that binding of a sulfonylurea to SUR modulates the cooperative interaction between NBDs inducing ATP dissociation from NBD1 [59]. Neutralization of a number of positively charged arginine moieties in Kir6.2 produced a KATP channel phenotype insensitive to both MgADP-induced activation and sulfonylurea-induced inhibition [83]. Thus, a structural component within SUR2A to be considered as a candidate common transducer of conformational rearrangements from the regulatory sub-unit to the Kir6.2 channel pore may bear a significant negative charge complementary to positively charged residues in Kir6.2. In this regard a 15 amino acid stretch of negatively charged aspartate and glutamate residues located in SUR downstream of NBD1 in cytoplasmic loop 6 (CL6) between TMD1 and TMD2 is strategically suitable to facilitate read-out of cooperative interactions between NBDs.
7. The KATP channel complex as a component of the cellular energetic network
The established homeostatic role for KATP channels in providing metabolic sensing and adjusting membrane excitability under physiologic or pathologic stress implies an integration of channel gating with the cellular energetic network. However, channel sensing of bulk nucleotide levels is limited since the effect of MgADP reaching saturation (at > 100 μM) shifts the range for ATP inhibition (IC50 from ~30 to ~300 μM) which is still far below intracellular ATP levels (6–10 mM), implying that MgADP-dependent KATP channel regulation is insufficient for channel gating at the normally high cytosolic concentrations of ATP [15,54,84,85]. This indicates that the bulk cytosolic nucleotide composition cannot be the sole determinant of KATP channel function. Rather, KATP channels could sense local nucleotide concentrations set by ATPases in the submembrane space at a level distinct from that of the “bulk” cytosol [52,54,86], provided that significant diffusional limitations within the cell exist to establish distinct cellular compartments [15,87–89]. However, such cellular compartmentalization would obstruct proper energetic sensing by KATP channels, as channel gating would be distorted by local fluctuations of nucleotides, remaining weakly dependent on the cellular metabolic status. Therefore, energetic signaling to the channel site must be managed by systems capable of shunting diffusional barriers transmitting nucleotide signals in order to secure beneficial channel activity in accord with cellular energetic demand.
Cells with high and fluctuating energy demands, such as cardiomyocytes, possess well-defined phosphotransfer systems that facilitate energetic signaling between sites of ATP production and ATP utilization or sensing [7,90]. Structural and functional interactions of KATP channel proteins with the phosphotransfer enzymes, adenylate kinase (AK: 2ADP ↔ ATP + AMP) and creatine kinase (CK: ADP + CrP ↔ ATP + Cr), are assured by the wide distribution of these enzymes in distinct cellular compartments, including membranes, and their physical association with KATP channel sub-units [8,15,91]. Creatine phosphate, the substrate for creatine kinase, facilitates ATP-induced KATP channel inhibition, a regulation absent in knockout mice lacking the creatine kinase gene M-CK [15]. Analogously, AMP, the substrate for adenylate kinase, antagonizes ATP-induced KATP channel closure, an effect lost in cardiomyocytes from mice lacking the adenylate kinase AK1 gene [8]. Thus, phosphotransfer reactions regulate KATP channels providing a mechanism for coupling channel behavior with the cellular energetic state.
In principle, KATP channel function can be regulated by phosphotransfer enzymes based on product/substrate exchange in the channel microenvironment or through phosphotransfer-dependent modification of the channel’s catalytic properties. While the first mechanism is intuitive, the second possibility stems from experimental data indicating alterations in the Michaelis–Menten constant and the maximal rate of the ATPase reaction of purified NBD2 in the presence of phosphotransfer systems. These experiments indicate that creatine or pyruvate kinases significantly increase (~50%) the maximal ATPase reaction rate (Selivanov et al., unpublished data) that would not be possible if phosphotransfer enzymes would solely regulate product/substrate exchange.
Theoretical and experimental analysis of cooperative interaction between the two prototypic phosphotransfer enzymes, creatine kinase (CK) and adenylate kinase (AK) systems, in a compartmentalized cellular environment reveals unique properties of these enzymes in transmission of energetic signals to sarcolemmal KATP channels [8,15,16]. Specifically, signal transmission from the cytosol to the submembrane compartment is limited due to restricted diffusion of nucleotides, creatine (Cr) and creatine phosphate (CrP). However, facilitated diffusion provided by CK and AK phosphotransfer systems that could bridge diffusional barriers essentially dissipates nucleotide gradients imposed by membrane AT-Pases and diffusional restrictions. Under cardiomyocyte stress, phosphotransfer flux through CK is significantly damped [92–94] such that with only a moderate drop in bulk cytosolic ATP, CK could no longer effectively dissipate nucleotide gradients precipitating a significant fall in submembrane CrP, and generation of an amplified nucleotide response at the KATP channel site (Fig. 2) [16]. According to the CK reaction equilibrium, substitution by creatine of 9.95 out of the total 10.25 mM of creatine phosphate, a 50 μM drop of bulk ATP (from 0.3 to 0.25 mM) and concomitant increase of bulk ADP can be expected. Experimentally it has been demonstrated that under the active submembrane Na/K-ATPase this minor drop in bulk ATP, which by itself is insufficient to induce channel opening, was amplified by the altered creatine kinase flux into significant nucleotide changes in the submembrane space manifested by vigorous activation of KATP channels [16].
Fig. 2.
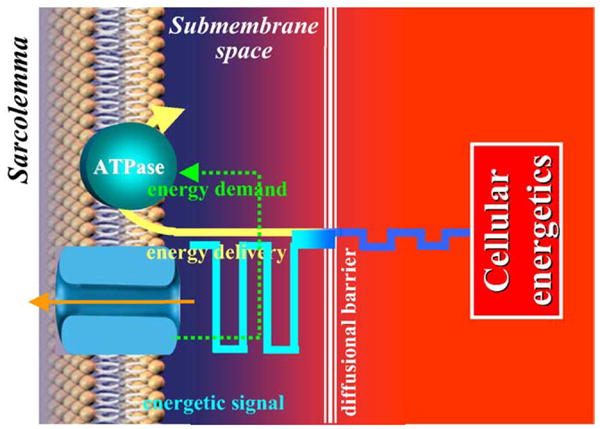
In the compartmentalized cellular environment changes in cellular energetics can be transmitted over diffusion barriers, amplified and decoded as a signal by membrane metabolic sensor which triggers adaptive response adjusting energy demand and preserving, thereby, cellular well-being.
CK-dependent amplification of the nucleotide response can be tuned by the AK system capable of modulating changes in the ATP/ADP ratio in the submembrane compartment, securing transmission of controllable metabolic signals to KATP channels. Under severe metabolic challenge, provided that a local regenerating system maintains submembrane ATP levels (i.e. glycolysis or an external source of ATP), bulk AK catalysis could elevate AMP flux into the submembrane compartment and promote the generation of submembrane ADP (ATP + AMP → 2~ADP) facilitating KATP channel opening [8,16]. Thus, energetic signals generated in the cytosol are processed by CK and AK systems that are capable of synchronizing KATP channel function with changes in the cellular energetic state (Fig. 2).
Integration with other cellular systems also provides for potential energetic regulation of the KATP channel. There are several contributors including physical interactions between the channel and the actin cytoskeleton [95–99], as well as additional metabolic systems such as lactate dehydrogenase (LDH) that, like CK and AK has also been shown to physical associate with KATP channel proteins [8,91,100]. LDH catalyzes the reaction NADH + pyruvate ↔ lactate + NAD. Lactate modulates the ATP sensitivity of KATP channels and would accumulate in the vicinity of LDH, and thus of cardiac KATP channels, under anaerobic conditions [100]. Other examples include direct channel effects of G protein subunits [101,102], changes in the composition of membrane phospholipids that interact with Kir6.2 to modulate pore function [103,104], activation of the protein kinase systems PKC and PKA with channel effects through phosphorylation/dephosphorylation [105–108], and pH effects through protonation of channel proteins [109].
8. KATP channel regulation in cardiac disease
Assigning to the channel catalytic module a role in integrating ion permeation with intracellular metabolic pathways identifies a novel principle in the regulation of cellular excitability. In principle, defects in the function of channel proteins themselves, disruption of intracellular metabolic networks, and/or disturbed communication between KATP channels and the energetic network can all be envisioned as molecular mechanisms contributing to cardiac disease.
In heart failure, cardiomyocytes undergo extensive remodeling including diminished mitochondrial respiration, suppressed creatine kinase flux, decreased energy storage, and cytoskeletal disruption, although bulk cytosolic ATP levels are preserved [20,92,93]. These changes impact metabolic signal generation, signal trafficking through phosphotransfer and diffusional effects by altered cellular architecture, among other factors, that affect the ability of cellular distress to be properly communicated to KATP channels. KATP channels from such hearts display normal basic physical properties and function when the channels are studied in isolation, however channel function within intact cells, when analyzed at both the cellular and organ level, demonstrates a significantly blunted response to stress [20]. Specifically, KATP channels from failing hearts display an improper response to ATP and CK, a reduced recognition of metabolic distress, and do not provide for appropriate action potential shortening in hearts exposed to hypoxia. Thus the KATP channels in myopathic hearts are uncoupled from the cellular metabolic state resulting in compromised regulation of membrane excitability. Consequently such hearts are excessively vulnerable to calcium loading and myocyte necrosis under stress that can be largely ameliorated by pharmacologic restoration of KATP channel opening with KATP channel opening drugs, such as nicorandil and pinacidil. The observations that KATP channels from failing hearts function normally in isolation and that protective effects of channel function can be restored by KATP channel openers indicates that the defect in KATP channel function in failing hearts is not within the channel itself but within the cellular signaling systems with which the channel is integrated and upon which the channel depends. The resulting inability of the KATP channel to fulfill its physiologic role under these conditions renders failing hearts more susceptible to injury and dysfunction under stress that could further contribute to the progression of disease by setting up a downward spiral through additional disruption of cellular energetic signaling cascades.
Derangement of metabolic sensing by cardiac KATP channels can also occur by mutation of channel proteins such as has been discovered in cases of dilated cardiomyopathy in humans [14]. Scanning of genomic DNA identified two mutations in ABCC9, which encodes the regulatory SUR2A sub-unit of the cardiac KATP channel, in individuals with heart failure and rhythm disturbances due to idiopathic dilated cardiomyopathy but not in normal control patients. Three dimensional modeling indicates these missense and frameshift mutations occur in evolutionarily conserved residues that flank the carboxy-terminal β-strand of SUR2A in close proximity to the Walker A motif required for coordination of nucleotides in the catalytic pocket of ABC proteins. Based on this modeling, the mutations are predicted to disrupt protein folding and thus the integrity of NBD2. Indeed, the two mutations do not significantly affect ATP-binding but do reduce the SUR2A ATPase activity and alter the characteristic reaction kinetics, translating into an abnormal distribution of conformations within the NBD2ATPase cycle and an inability of CK to effectively regulate the ATPase activity of the mutant channels. These aberrances in catalysis generate defective KATP channel phenotypes that are characterized by abnormal responses to both ATP and ADP. Suboptimal tolerance to stress and a propensity towards cardiomyopathy and arrhythmia in mice with genetic disruption of cardiac KATP channels [10,19,21], supports the implication that the significant metabolic sensing deficit demonstrated in humans with cardiac KATP channel mutations contributes to development of heart failure and thus further establishes the importance of proper SUR2A NBD2 ATPase kinetics in the metabolic signal decoding that permits the cardiac KATP channel to fulfill its physiologic role.
Acknowledgments
Supported by National Institutes of Health (HL64822, HL07111), Marriott Program for Heart Disease Research, Marriott Foundation, Ted Nash Long Life Foundation, Ralph Wilson Medical Research Foundation, Miami Heart Research Institute, and Mayo-Dubai Healthcare City Research Project.
References
- 1.Noma A. ATP-regulated K+ channels in cardiac muscle. Nature. 1983;305:147–8. doi: 10.1038/305147a0. [DOI] [PubMed] [Google Scholar]
- 2.Aguilar-Bryan L, Nichols CG, Wechsler SW, Clement JP, 4th, Boyd AE, 3rd, Gonzalez G, et al. Cloning of the beta cell high-affinity sulfonylurea receptor: a regulator of insulin secretion. Science. 1995;268:423–6. doi: 10.1126/science.7716547. [DOI] [PubMed] [Google Scholar]
- 3.Inagaki N, Gonoi T, Clement JP, Namba N, Inazawa J, Gonzales G, et al. Reconstitution of I KATP: an inward rectifier subunit plus the sulfonylurea receptor. Science. 1995;270:1166–70. doi: 10.1126/science.270.5239.1166. [DOI] [PubMed] [Google Scholar]
- 4.Seino S, Miki T. Physiological and pathophysiological roles of ATP-sensitive K+ channels. Prog Biophys Mol Biol. 2003;81:133–76. doi: 10.1016/s0079-6107(02)00053-6. [DOI] [PubMed] [Google Scholar]
- 5.Inagaki N, Gonoi T, Clement JP, Wang CZ, Aguilar-Bryan L, Bryan J, et al. A family of sulfonylurea receptors determines the pharmacological properties of ATP-sensitive K_ channels. Neuron. 1996;16:1011–7. doi: 10.1016/s0896-6273(00)80124-5. [DOI] [PubMed] [Google Scholar]
- 6.Lorenz E, Terzic A. Physical association between recombinant cardiac ATP-sensitive K+ channel subunits Kir6.2 and SUR2A. J Mol Cell Cardiol. 1999;31:425–34. doi: 10.1006/jmcc.1998.0876. [DOI] [PubMed] [Google Scholar]
- 7.O’Rourke B, Ramza BM, Marban E. Oscillations of membrane current and excitability driven by metabolic oscillations in heart cells. Science. 1994;265:962–6. doi: 10.1126/science.8052856. [DOI] [PubMed] [Google Scholar]
- 8.Carrasco AJ, Dzeja PP, Alekseev AE, Pucar D, Zingman LV, Abraham MR, et al. Adenylate kinase phosphotransfer communicates cellular energetic signals to ATP-sensitive potassium channels. Proc Natl Acad Sci USA. 2001;98:7623–8. doi: 10.1073/pnas.121038198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zingman LV, Alekseev AE, Bienengraeber M, Hodgson DM, Karger AB, Dzeja PP, et al. Signaling in channel/enzyme multimers: ATPase transitions in SUR module gate ATP-sensitive K+ conductance. Neuron. 2001;31:233–45. doi: 10.1016/s0896-6273(01)00356-7. [DOI] [PubMed] [Google Scholar]
- 10.Zingman LV, Hodgson DM, Bast PH, Kane GC, Perez-Terzic C, Gumina RJ, et al. Kir6.2 is required for adaptation to stress. Proc Natl Acad Sci USA. 2002;99:13278–83. doi: 10.1073/pnas.212315199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zingman LV, Hodgson DM, Alekseev AE, Terzic A. Stress without distress: homeostatic role for KATP channels. Mol Psychiatry. 2003;8:253–4. doi: 10.1038/sj.mp.4001323. [DOI] [PubMed] [Google Scholar]
- 12.Nichols CG, Lederer WJ. Adenosine triphosphate-sensitive potassium channels in the cardiovascular system. Am J Physiol. 1991;261:H1675–H1686. doi: 10.1152/ajpheart.1991.261.6.H1675. [DOI] [PubMed] [Google Scholar]
- 13.Terzic A, Jahangir A, Kurachi Y. Cardiac ATP-sensitive K+ channels: regulation by intracellular nucleotides and K+ channel-opening drugs. Am J Physiol. 1995;269:C525–C545. doi: 10.1152/ajpcell.1995.269.3.C525. [DOI] [PubMed] [Google Scholar]
- 14.Bienengraeber M, Olson TM, Selivanov VA, Kathmann EC, O’Coclain F, Gao F, et al. Terzic. ABCC9 mutations identified in human dilated cardiomyopathy disrupt catalytic KATP channel gating. Nat Genet. 2004;36:382–7. doi: 10.1038/ng1329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Abraham MR, Selivanov VA, Hodgson DM, Pucar D, Zingman LV, Wieringa B, et al. Coupling of cell energetics with membrane metabolic sensing. Integrative signaling through creatine kinase phosphotransfer disrupted by M-CK gene knock-out. J Biol Chem. 2002;277:24427–34. doi: 10.1074/jbc.M201777200. [DOI] [PubMed] [Google Scholar]
- 16.Selivanov V, Alekseev AE, Hodgson DM, Dzeja PP, Terzic A. Nucleotide-gated KATP channels integrated with creatine and adenylate kinases: amplification, tuning and sensing of energetic signals in the compartmentalized cellular environment. Mol Cell Biochem. 2004;256:243–56. doi: 10.1023/b:mcbi.0000009872.35940.7d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Suzuki M, Sasaki N, Miki T, Sakamoto N, Ohmoto-Sekine Y, Tamagawa M, et al. Role of sarcolemmal K(ATP) channels in cardioprotection against ischemia/reperfusion injury in mice. J Clin Invest. 2002;109:509–16. doi: 10.1172/JCI14270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gumina RJ, Pucar D, Bast P, Hodgson DM, Kurtz CE, Dzeja PP, et al. Knockout of Kir6.2 negates ischemic preconditioning-induced protection of myocardial energetics. Am J Physiol. 2003;284:H2106–H2113. doi: 10.1152/ajpheart.00057.2003. [DOI] [PubMed] [Google Scholar]
- 19.Kane GC, Behfar A, Yamada S, Perez-Terzic C, O’Cochlain F, Reyes S, et al. ATP-sensitive K+ channel knockout compromises the metabolic benefit of exercise training, resulting in cardiac deficits. Diabetes. 2004;53:S169–75. doi: 10.2337/diabetes.53.suppl_3.s169. [DOI] [PubMed] [Google Scholar]
- 20.Hodgson DM, Zingman LV, Kane GC, Perez-Terzic C, Bienengraeber M, Ozcan C, et al. Cellular remodeling in heart failure disrupts KATP channel-dependent stress tolerance. EMBO J. 2003;22:1732–42. doi: 10.1093/emboj/cdg192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liu XK, Yamada S, Kane GC, Alekseev AE, Hodgson DM, O’Cochlain F, et al. Genetic disruption of Kir6.2, the pore-forming subunit of ATP-sensitive K+ channel, predisposes to catecholamine-induced ventricular dysrhythmia. Diabetes. 2004;53:S165–8. doi: 10.2337/diabetes.53.suppl_3.s165. [DOI] [PubMed] [Google Scholar]
- 22.Clement JP, 4th, Kunjilwar K, Gonzalez G, Schwanstecher M, Panten U, Aguilar-Bryan L, et al. Association and stoichiometry of K(ATP) channel subunits. Neuron. 1997;18:827–38. doi: 10.1016/s0896-6273(00)80321-9. [DOI] [PubMed] [Google Scholar]
- 23.Zerangue N, Schwappach B, Jan YN, Jan LY. A new ER trafficking signal regulates the subunit stoichiometry of plasma membrane KATP channels. Neuron. 1999;22:537–48. doi: 10.1016/s0896-6273(00)80708-4. [DOI] [PubMed] [Google Scholar]
- 24.Schwappach B, Zerangue N, Jan YN, Jan LY. Molecular basis for K(ATP) assembly: transmembrane interactions mediate association of a K+ channel with an ABC transporter. Neuron. 2000;26:155–67. doi: 10.1016/s0896-6273(00)81146-0. [DOI] [PubMed] [Google Scholar]
- 25.Tucker SJ, Gribble FM, Zhao C, Trapp S, Ashcroft FM. Truncation of Kir6.2 produces ATP-sensitivie K+ channels in the absence of the sulphonylurea receptor. Nature. 1997;387:179–83. doi: 10.1038/387179a0. [DOI] [PubMed] [Google Scholar]
- 26.Drain P, Geng X, Li L. Concerted gating mechanism underlying KATP channel inhibition by ATP. Biophys J. 2004;86:2101–12. doi: 10.1016/S0006-3495(04)74269-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tanabe K, Tucker SJ, Matsuo M, Proks P, Ashcroft FM, Seino S, et al. Direct photoaffinity labeling of the Kir6.2 subunit of the ATP-sensitive K+ channel by 8-azido-ATP. J Biol Chem. 1999;274:3931–3. doi: 10.1074/jbc.274.7.3931. [DOI] [PubMed] [Google Scholar]
- 28.Trapp S, Haider S, Jones P, Sansom MS, Ashcroft FM. Identification of residues contributing to the ATP binding site of Kir6.2. EMBO J. 2003;22:2903–12. doi: 10.1093/emboj/cdg282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Antcliff JF, Haider S, Proks P, Sansom MS, Ashcroft FM. Functional analysis of a structural model of the ATP-binding site of the K(ATP) channel Kir6.2 subunit. EMBO J. 2005;24:229–39. doi: 10.1038/sj.emboj.7600487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Antcliff JF, Haider S, Proks P, Sansom MS, Ashcroft FM. Functional analysis of a structural model of the ATP-binding site of the K(ATP) channel Kir6.2 subunit. EMBO J. 2005 Jan;24:13. doi: 10.1038/sj.emboj.7600487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rajashree R, Koster JC, Markova KP, Nichols CG, Hofmann PA. Contractility and ischemic response of hearts from transgenic mice with altered sarcolemmal K(ATP) channels. Am J Physiol. 2002;283:H584–H590. doi: 10.1152/ajpheart.00107.2002. [DOI] [PubMed] [Google Scholar]
- 32.Koster JC, Marshall BA, Ensor N, Corbett JA, Nichols CG, Koster JC, et al. Targeted overactivity of beta cell K(ATP) channels induces profound neonatal diabetes. Cell. 2000;100:645–54. doi: 10.1016/s0092-8674(00)80701-1. [DOI] [PubMed] [Google Scholar]
- 33.Gloyn AL, Pearson ER, Antcliff JF, Proks P, Bruining GJ, Slingerland AS, et al. Activating mutations in the gene encoding the ATP-sensitive potassium-channel subunit Kir6.2 and permanent neonatal diabetes. N Engl J Med. 2004;350:1838–49. doi: 10.1056/NEJMoa032922. [DOI] [PubMed] [Google Scholar]
- 34.Lorenz E, Alekseev AE, Krpavinsky GP, Carrasco AJ, Clapham DE, Terzic A. Evidence for direct physical interaction between KATP channel (Kir6.2) and an ATP-binding cassette protein (SUR1) which affects cellular distribution and kinetic behavior of an ATP-sensitive K+ channel. Mol Cell Biol. 1998;18:1652–9. doi: 10.1128/mcb.18.3.1652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Alekseev AE, Brady PA, Terzic A. Ligand-insensitive state of cardiac ATP-sensitive K+ channels. Basis for channel opening. J Gen Physiol. 1998;111:381–94. doi: 10.1085/jgp.111.2.381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Moreau C, Jacquet H, Prost AL, D’hahan N, Vivaudou M. The molecular basis of the specificity of action of K(ATP) channel openers. EMBO J. 2000;19:6644–51. doi: 10.1093/emboj/19.24.6644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lorkowski S, Cullen P. ABCG subfamily of human ATP-binding cassette proteins. Pure Appl Chem. 2002;74:2057–81. [Google Scholar]
- 38.Babenko AP, Bryan J. Sur domains that associate with and gate KATP pores define a novel gatekeeper. J Biol Chem. 2003;278:41577–80. doi: 10.1074/jbc.C300363200. [DOI] [PubMed] [Google Scholar]
- 39.Walker JE, Saraste M, Runswick MJ, Gay NJ. Distantly related sequences in the alpha- and beta-subunits of ATP synthase, myosin, kinases and other ATP-requiring enzymes and a common nucleotide binding fold. EMBO J. 1982;1:945–51. doi: 10.1002/j.1460-2075.1982.tb01276.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gribble FM, Tucker SJ, Ashcroft FM. The essential role of the Walker A motifs of SUR1 in K-ATP channel activation by Mg-ADP and diazoxide. EMBO J. 1997;16:1145–52. doi: 10.1093/emboj/16.6.1145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hopfner KP, Karcher A, Shin DS, Craig L, Arthur LM, Carney JP, et al. Structural biology of Rad50 ATPase: ATP-driven conformational control in DNA double-strand break repair and the ABC-ATPase superfamily. Cell. 2000;101:789–800. doi: 10.1016/s0092-8674(00)80890-9. [DOI] [PubMed] [Google Scholar]
- 42.Matsuo M, Kioka N, Amachi T, Ueda K. ATP binding properties of the nucleotide-binding folds of SUR1. J Biol Chem. 1999;274:37479–82. doi: 10.1074/jbc.274.52.37479. [DOI] [PubMed] [Google Scholar]
- 43.Matsuo M, Tanabe K, Kioka N, Amachi T, Ueda K. Different binding properties and affinities for ATP and ADP among sulfonylurea receptor subtypes, SUR1, SUR2A, and SUR2B. J Biol Chem. 2000;275:28757–63. doi: 10.1074/jbc.M004818200. [DOI] [PubMed] [Google Scholar]
- 44.Gao M, Cui HR, Loe DW, Grant CE, Almquist KC, Cole SP, et al. Comparison of the functional characteristics of the nucleotide binding domains of multidrug resistance protein 1. J Biol Chem. 2000;275:13098–108. doi: 10.1074/jbc.275.17.13098. [DOI] [PubMed] [Google Scholar]
- 45.Aleksandrov L, Aleksandrov AA, Chang XB, Riordan JR. The First nucleotide binding domain of cystic fibrosis transmembrane conductance regulator is a site of stable nucleotide interaction, whereas the second is a site of rapid turnover. J Biol Chem. 2002;277:15419–25. doi: 10.1074/jbc.M111713200. [DOI] [PubMed] [Google Scholar]
- 46.Takada Y, Yamada K, Taguchi Y, Kino K, Matsuo M, Tucker SJ, et al. Non-equivalent cooperation between the two nucleotide-binding folds of P-glycoprotein. Biochim Biophys Acta. 1998;1373:131–6. doi: 10.1016/s0005-2736(98)00099-6. [DOI] [PubMed] [Google Scholar]
- 47.Bienengraeber M, Alekseev AE, Abraham MR, Carrasco AJ, Moreau C, Vivaudou M, et al. ATPase activity of the sulfonylurea receptor: a catalytic function for the KATP channel complex. FASEB J. 2000;14:1943–52. doi: 10.1096/fj.00-0027com. [DOI] [PubMed] [Google Scholar]
- 48.Gribble FM, Tucker SJ, Haug T, Ashcroft FM. MgATP activates the beta cell KATP channel by interaction with its SUR1 subunit. Proc Natl Acad Sci USA. 1998;95:7185–90. doi: 10.1073/pnas.95.12.7185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Senior AE, Gadsby DC. ATP hydrolysis cycles and mechanism in p-glycoprotein and CFTR. Semin Cancer Biol. 1997;8:143–50. doi: 10.1006/scbi.1997.0065. [DOI] [PubMed] [Google Scholar]
- 50.Baukrowitz T, Hwang TC, Gadsby DC, Nairn AC. Coupling of CFTR Cl− channel gating to an ATP hydrolysis cycle. Neuron. 1994;12:473–82. doi: 10.1016/0896-6273(94)90206-2. [DOI] [PubMed] [Google Scholar]
- 51.Vergani P, Nairn AC, Gadsby DC. On the mechanism of MgATP-dependent gating of CFTR Cl− channels. J Gen Physiol. 2003;121:17–36. doi: 10.1085/jgp.20028673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lederer WJ, Nichols CG. Nucleotide modulation of the activity of rat heart ATP-sensitive K+ channels in isolated membrane patches. J Physiol. 1989;419:193–211. doi: 10.1113/jphysiol.1989.sp017869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Weiss JN, Venkatesh N, Lamp ST. ATP-sensitive K+ channels and cellular K+ loss in hypoxic and ischaemic mammalian ventricle. J Physiol. 1992;447:649–73. doi: 10.1113/jphysiol.1992.sp019022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Weiss JN, Venkatesh N. Metabolic regulation of cardiac ATP-sensitive K+ channels. Cardiovasc Drugs Ther. 1993;7:499–505. doi: 10.1007/BF00877614. [DOI] [PubMed] [Google Scholar]
- 55.Terzic A, Jahangir A, Kurachi Y. Cardiac ATP-sensitive K+ channels: regulation by intracellular nucleotides and K+ channel-opening drugs. Am J Physiol. 1995;269:C525–C545. doi: 10.1152/ajpcell.1995.269.3.C525. [DOI] [PubMed] [Google Scholar]
- 56.Manavalan P, Dearborn DG, McPherson JM, Smith AE. Sequence homologies between nucleotide binding regions of CFTR and G-proteins suggest structural and functional similarities. FEBS Lett. 1995;366:87–91. doi: 10.1016/0014-5793(95)00463-j. [DOI] [PubMed] [Google Scholar]
- 57.Matsushita K, Kinoshita K, Matsuoka T, Fujita A, Fujikado T, Tano Y, et al. Intramolecular interaction of SUR2 subtypes for intracellular ADP-induced differential control of K(ATP) channels. Circ Res. 2002;90:554–61. doi: 10.1161/01.res.0000012666.42782.30. [DOI] [PubMed] [Google Scholar]
- 58.Reimann F, Gribble FM, Ashcroft FM. Differential response of K(ATP) channels containing SUR2A or SUR2B subunits to nucleotides and pinacidil. Mol Pharmacol. 2000;58:1318–25. doi: 10.1124/mol.58.6.1318. [DOI] [PubMed] [Google Scholar]
- 59.Ueda K, Komine J, Matsuo M, Seino S, Amachi T. Cooperative binding of ATP and MgADP in the sulfonylurea receptor is modulated by glibenclamide. Proc Natl Acad Sci USA. 1999;96:1268–72. doi: 10.1073/pnas.96.4.1268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Virag L, Furukawa T, Hiraoka M. Modulation of the effect of glibenclamide on KATP channels by ATP and ADP. Mol Cell Biochem. 1993;119:209–15. doi: 10.1007/978-1-4615-3078-7_28. [DOI] [PubMed] [Google Scholar]
- 61.Brady PA, Alekseev AE, Terzic A. Operative condition-dependent response of cardiac ATP-sensitive K+ channels toward sulfonylureas. Circ Res. 1998;82:272–8. doi: 10.1161/01.res.82.2.272. [DOI] [PubMed] [Google Scholar]
- 62.Satoh E, Yamada M, Kondo C, Repunte VP, Horio Y, Iijima T, et al. Intracellular nucleotide-mediated gating of SUR/Kir6.0 complex potassium channels expressed in a mammalian cell line and its modification by pinacidil. J Physiol. 1998;11:663–74. doi: 10.1111/j.1469-7793.1998.663bg.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Nagata K, Nishitani M, Matsuo M, Kioka N, Amachi T, Ueda K. Nonequivalent nucleotide trapping in the two nucleotide binding folds of the human multidrug resistance protein MRP1. J Biol Chem. 2000;275:17626–30. doi: 10.1074/jbc.M000792200. [DOI] [PubMed] [Google Scholar]
- 64.Sauna ZE, Ambudkar SV. Characterization of the catalytic cycle of ATP hydrolysis by human P-glycoprotein. The two ATP hydrolysis events in a single catalytic cycle are kinetically similar but affect different functional outcomes. J Biol Chem. 2001;276:11653–61. doi: 10.1074/jbc.M011294200. [DOI] [PubMed] [Google Scholar]
- 65.Ashcroft FM, Gribble FM. New windows on the mechanism of action of K(ATP) channel openers. Trends Pharmacol Sci. 2000;21:439–45. doi: 10.1016/s0165-6147(00)01563-7. [DOI] [PubMed] [Google Scholar]
- 66.Kerr ID. Structure and association of ATP-binding cassette transporter nucleotide-binding domains. Biochim Biophys Acta. 2002;1561:47–64. doi: 10.1016/s0304-4157(01)00008-9. [DOI] [PubMed] [Google Scholar]
- 67.Smith PC, Karpowich N, Millen L, Moody JE, Rosen J, Thomas PJ, et al. ATP binding to the motor domain from an ABC transporter drives formation of a nucleotide sandwich dimer. Mol Cell. 2002;10:139–49. doi: 10.1016/s1097-2765(02)00576-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Qu Q, Chu JW, Sharom FJ. Transition state P-glycoprotein binds drugs and modulators with unchanged affinity, suggesting a concerted transport mechanism. Biochemistry. 2003;42:1345–53. doi: 10.1021/bi0267745. [DOI] [PubMed] [Google Scholar]
- 69.Zingman LV, Hodgson DM, Bienengraeber M, Karger AB, Kathmann EC, Alekseev AE, et al. Tandem function of nucleotide binding domains confers competence to sulfonylurea receptor in gating ATP-sensitive K+ channels. J Biol Chem. 2002;277:14206–10. doi: 10.1074/jbc.M109452200. [DOI] [PubMed] [Google Scholar]
- 70.Matsuo M, Dabrowski M, Ueda K, Ashcroft FM. Mutations in the linker domain of NBD2 of SUR inhibit transduction but not nucleotide binding. EMBO J. 2002;21:4250–8. doi: 10.1093/emboj/cdf419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Davidson AL, Sharma S. Mutation of a single MalK subunit severely impairs maltose transport activity in Escherichia coli. J Bacteriol. 1997;179:5458–64. doi: 10.1128/jb.179.17.5458-5464.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Hou YX, Riordan JR, Chang XB. ATP binding, not hydrolysis, at the first nucleotide-binding domain of multidrug resistance-associated protein MRP1 enhances ADP. Vi trapping at the second domain. J Biol Chem. 2003;278:3599–605. doi: 10.1074/jbc.M210480200. [DOI] [PubMed] [Google Scholar]
- 73.Hrycyna CA, Ramachandra M, Ambudkar SV, Ko YH, Pedersen PL, Pastan I, et al. Mechanism of action of human P-glycoprotein ATPase activity. Photochemical cleavage during a catalytic transition state using orthovanadate reveals cross-talk between the two ATP sites. J Biol Chem. 1998;273:16631–4. doi: 10.1074/jbc.273.27.16631. [DOI] [PubMed] [Google Scholar]
- 74.Higgins CF, Linton KJ. The ATP switch model for ABC transporters. Nat Struct Mol Biol. 2004;11:918–26. doi: 10.1038/nsmb836. [DOI] [PubMed] [Google Scholar]
- 75.Hung LW, Wang IX, Nikaido K, Liu PQ, Ames GFL, Kim SH. Crystal structure of the ATP-binding subunit of an ABC transporter. Nature. 1998;396:703–7. doi: 10.1038/25393. [DOI] [PubMed] [Google Scholar]
- 76.Diederichs K, Diez J, Greller G, Muller C, Breed J, Schnell C, et al. Crystal structure of MalK, the ATPase stubunit of the trehalose/maltose ABC transporter of the archaeon Thermococcus litoralis. J Biol Chem. 2000;19:5951–61. doi: 10.1093/emboj/19.22.5951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Campbell JD, Sansom MS, Ashcroft FM. Potassium channel regulation. EMBO Rep. 2003;4:1038–42. doi: 10.1038/sj.embor.7400003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Janas E, Hofacker M, Chen M, Gompf S, Van der Does C, Tampe R. The ATP hydrolysis cycle of the nucleotide-binding domain of the mitochondrial ATP-binding cassette transporter Mdl1p. J Biol Chem. 2003;278:26862–9. doi: 10.1074/jbc.M301227200. [DOI] [PubMed] [Google Scholar]
- 79.Revington M, Holder TM, Zuiderweg ER. NMR study of nucleotide-induced changes in the nucleotide binding domain of Thermus thermophilus Hsp70 chaperone DnaK: implications for the allosteric mechanism. J Biol Chem. 2004;279:33958–67. doi: 10.1074/jbc.M313967200. [DOI] [PubMed] [Google Scholar]
- 80.Altenberg GA. Structure of multidrug-resistance proteins of the ATP-binding cassette (ABC) superfamily. Curr Med Chem Anti-Canc Agents. 2004;4:53–62. doi: 10.2174/1568011043482160. [DOI] [PubMed] [Google Scholar]
- 81.Shyng SL, Ferrigni T, Nichols CG. Regulation of channel KATP activity by diazoxide and MgADP: distinct functions of the two nucleotide binding folds of the sulfonylurea receptor. J Gen Physiol. 1997;110:643–54. doi: 10.1085/jgp.110.6.643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Schwanstecher M, Sieverding C, Dorschner H, Gross I, Aguilar-Bryan L, Schwanstecher C, et al. Potassium channel openers require ATP to bind to and act through sulfonylurea receptors. EMBO J. 1998;17:5529–35. doi: 10.1093/emboj/17.19.5529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.John SA, Weiss JN, Ribalet B. Regulation of cloned ATP-sensitive K+ channels by adenine nucleotides and sulfonylureas: interactions between SUR1 and positively charged domains on Kir6.2. J Gen Physiol. 2001;118:391–405. doi: 10.1085/jgp.118.4.391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Bittl JA, DeLayre J, Ingwall JS. Rate equation for creatine kinase predicts the in vivo reaction velocity: 31P NMR surface coil studies in brain, heart, and skeletal muscle of the living rat. Biochemistry. 1987;26:6083–90. doi: 10.1021/bi00393a021. [DOI] [PubMed] [Google Scholar]
- 85.Saks VA, Aliev MK. Is there the creatine kinase equilibrium in working heart cells? Biochem Biophys Res Commun. 1996;227:360–7. doi: 10.1006/bbrc.1996.1513. [DOI] [PubMed] [Google Scholar]
- 86.Weiss JN, Lamp ST. Glycolysis preferentially inhibits ATP-sensitive K+ channels in isolated guinea pig cardiac myocytes. Science. 1987;238:67–9. doi: 10.1126/science.2443972. [DOI] [PubMed] [Google Scholar]
- 87.Lederer WJ, Niggli E, Hadley RW. Sodium–calcium exchange in excitable cells: fuzzy space. Science. 1990;248:283. doi: 10.1126/science.2326638. [DOI] [PubMed] [Google Scholar]
- 88.Kaasik A, Veksler V, Boehm E, Novotova M, Minajeva A, Ventura-Clapier R. Energetic crosstalk between organelles: architectural integration of energy production and utilization. Circ Res. 2001;89:153–9. doi: 10.1161/hh1401.093440. [DOI] [PubMed] [Google Scholar]
- 89.Sasaki N, Sato T, Marban E, O’Rourke B. ATP consumption by uncoupled mitochondria activates sarcolemmal K(ATP) channels in cardiac myocytes. Am J Physiol. 2001;280:H1882–H1888. doi: 10.1152/ajpheart.2001.280.4.H1882. [DOI] [PubMed] [Google Scholar]
- 90.Dzeja PP, Terzic A. Phosphotransfer reactions in the regulation of ATP-sensitive K+ channels. FASEB J. 1998;12:523–9. doi: 10.1096/fasebj.12.7.523. [DOI] [PubMed] [Google Scholar]
- 91.Crawford RM, Ranki JH, Botting CH, Budas GR, Jovanovic A. Creatine kinase is physically associated with the cardiac ATP-sensitive K+ channel in vivo. FASEB J. 2002;16:102–4. doi: 10.1096/fj.01-0466fje. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Nascimben L, Ingwall JS, Pauletto P, Friedrich J, Gwathmey JK, Saks V, et al. Creatine kinase system in failing and nonfailing human myocardium. Circulation. 1996;94:1894–901. doi: 10.1161/01.cir.94.8.1894. [DOI] [PubMed] [Google Scholar]
- 93.Dzeja PP, Pucar D, Redfield MM, Burnett JC, Terzic A. Reduced activity of enzymes coupling ATP-generating with ATP-consuming processes in the failing myocardium. Mol Cell Biochem. 1999;201:33–40. doi: 10.1023/a:1007016703229. [DOI] [PubMed] [Google Scholar]
- 94.Dzeja PP, Redfield MM, Burnett JC, Terzic A. Failing energetics in failing hearts. Curr Cardiol Rep. 2000;2:212–7. doi: 10.1007/s11886-000-0071-9. [DOI] [PubMed] [Google Scholar]
- 95.Brady PA, Alekseev AE, Aleksandrova LA, Gomez LA, Terzic A. A disrupter of actin microfilaments impairs sulfonylurea-inhibited gating of cardiac KATP channels. Am J Physiol. 1996;271:H2710–H2716. doi: 10.1152/ajpheart.1996.271.6.H2710. [DOI] [PubMed] [Google Scholar]
- 96.Yokoshiki H, Datsube Y, Sunugawa M, Seki T, Sperelakis N. Disruption of actin cytoskeleton attenuates sulfonylurea inhibition of cardiac ATP-sensitive K+ channels. Pflug Arch. 1997;434:203–5. doi: 10.1007/s004240050384. [DOI] [PubMed] [Google Scholar]
- 97.Furukawa T, Yamane T, Terai T, Katayama Y, Hiraoka M. Functional linkage of the cardiac ATP-sensitive K+ channel to the actin cytoskeleton. Pflug Arch. 1996;431:504–12. doi: 10.1007/BF02191896. [DOI] [PubMed] [Google Scholar]
- 98.Terzic A, Kurachi Y. Actin microfilament disrupters enhance K(ATP) channel opening in patches from guinea-pig cardiomyocytes. J Physiol. 1996;492:395–404. doi: 10.1113/jphysiol.1996.sp021316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Korchev YE, Negulyaev YA, Edwards CR, Vodyanoy I, Lab MJ. Functional localization of single active ion channels on the surface of a living cell. Nat Cell Biol. 2000;2:616–9. doi: 10.1038/35023563. [DOI] [PubMed] [Google Scholar]
- 100.Crawford RM, Budas GR, Jovanovic S, Ranki HJ, Wilson TJ, Davies AM, et al. M-LDH serves as a sarcolemmal KATP channel subunit essential for cell protection against ischemia. EMBO J. 2002;21:3936–48. doi: 10.1093/emboj/cdf388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Sanchez JA, Gonoi T, Inagaki N, Katada T, Seino S. Modulation of reconstituted ATP-sensitive K(+)-channels by GTP-binding proteins in a mammalian cell line. J Physiol. 1998;507:315–24. doi: 10.1111/j.1469-7793.1998.315bt.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Terzic A, Tung RT, Inanobe A, Katada T, Kurachi Y. G proteins activate ATP-sensitive K+ channels by antagonizing ATP-dependent gating. Neuron. 1994;12:885–93. doi: 10.1016/0896-6273(94)90340-9. [DOI] [PubMed] [Google Scholar]
- 103.Baukrowitz T, Schulte U, Oliver D, Herlitze S, Krauter T, Tucker SJ, et al. PIP2 and PIP as determinants for ATP inhibition of KATP channels. Science. 1998;282:1141–4. doi: 10.1126/science.282.5391.1141. [DOI] [PubMed] [Google Scholar]
- 104.Shyng SL, Nichols CG. Membrane phospholipid control of nucleotide sensitivity of KATP channels. Science. 1998;282:1138–41. doi: 10.1126/science.282.5391.1138. [DOI] [PubMed] [Google Scholar]
- 105.Beguin P, Nagashima K, Nishimura M, Gonoi T, Seino S. PKA-mediated phosphorylation of the human K(ATP) channel: separate roles of Kir6.2 and SUR1 subunit phosphorylation. EMBO J. 1999;18:4722–32. doi: 10.1093/emboj/18.17.4722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Lin YF, Jan YN, Jan LY. Regulation of ATP-sensitive potassium channel function by protein kinase A-mediated phosphorylation in transfected HEK293 cells. EMBO J. 2000;19:942–55. doi: 10.1093/emboj/19.5.942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Hu K, Huang CS, Jan YN, Jan LY. ATP-sensitive potassium channel traffic regulation by adenosine and protein kinase C. Neuron. 2003;38:417–32. doi: 10.1016/s0896-6273(03)00256-3. [DOI] [PubMed] [Google Scholar]
- 108.Light PE, Bladen C, Winkfein RJ, Walsh MP, French RJ. Molecular basis of protein kinase C-induced activation of ATP-sensitive potassium channels. Proc Natl Acad Sci USA. 2000;97:9058–63. doi: 10.1073/pnas.160068997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Vivaudou M, Forestier C. Modification by protons of frog skeletal muscle KATP channels: effects on ion conduction and nucleotide inhibition. J Physiol. 1995;486:629–45. doi: 10.1113/jphysiol.1995.sp020840. [DOI] [PMC free article] [PubMed] [Google Scholar]