

Abstract
Interactions that occur between several tumour necrosis factor (TNF)–TNF receptors that are expressed by T cells and various other immune and non-immune cell types are central to T-cell function. In this Review, I discuss the biology of four different ligand– receptor interactions — OX40 ligand and OX40, 4-1BB ligand and 4-1BB, CD70 and CD27, and TL1A and death receptor 3 — and their potential to be exploited for therapeutic benefit. Manipulating these interactions can be effective for treating diseases in which T cells have an important role, including inflammatory conditions, autoimmunity and cancer. Here, I explore how blocking or inducing the signalling pathways that are triggered by these different interactions can be an effective way to modulate immune responses.
A recognized triumph in immunotherapy has been the development of neutralizing antibodies and Fc fusion proteins that inhibit the binding of tumour necrosis factor (TNF) to one or both of its receptors — TNF receptor 1 (TNFR1; also known as TNFRSF1A) and TNFR2 (also known as TNFRSF1B). As a primary function of the TNF superfamily molecules is to regulate cell survival, inhibition of these interactions prevents the activation of signalling pathways downstream of the TNFRs, thereby minimizing the pro-inflammatory programme they initiate in immune cells and decreasing the pathology of autoimmune and inflammatory diseases. Based on the success of these therapies, increasing attention is now focused on other related molecules in the TNF superfamily, of which there are 19 ligands and 30 receptors. Several of the interactions that occur between TNF molecules and their receptors have gained prominence based on studies of animal models of immune function and disease. These studies indicate that interactions between TNF–TNFR molecules positively regulate T-cell responses and mediate crosstalk between T cells and other cell types. The interactions between OX40 ligand (OX40L; also known as CD252 and TNFSF4) and OX40 (also known as CD134 and TNFRSF4), 4-1BBL (also known as TNFSF9) and 4-1BB (also known as CD137 and TNFRSF9), CD70 (also known as TNFSF7) and CD27 (also known as TNFRSF7), and TL1A (also known as TNFSF15) and death receptor 3 (DR3; also known as TNFRSF25) (TABLE 1), have been the most extensively studied in terms of their direct effects on CD4+ and CD8+ T cells. In support of the importance of these interactions in controlling T-cell function, recent results have shown that blocking or promoting each one of these interactions in animal models of disease markedly affects the outcome of the disease.
Table 1.
Expression profile of some TNF superfamily members
| ligand– receptor pair |
Ligand expressed by | Conditions of ligand expression |
Receptor expressed by |
Conditions of receptor expression |
|---|---|---|---|---|
| CD70–CD27 | APCs (DCs and B cells) | Inducible (triggered by TLR4, TLR9, CD40, Ig or BCR, and IFNγ) |
CD4+ and CD8+ T cells | Constitutive |
| CD4+ and CD8+ T cells | Inducible (triggered by TCR plus CD28 cross-linking, IL-1, IL-2, TNF and IL-12) |
B cells (subset) | Inducible | |
| Mast cells | Inducible (?) | NK cells (subset) | Constitutive | |
| NK cells | Inducible (?) | FOXP3+ TReg cells | Constitutive (mice) or inducible (humans; triggered by TCR cross-linking) |
|
| Smooth muscle cells | Inducible (?) | NKT cells | Constitutive | |
| Thymic epithelial cells | Constitutive | Haematopoietic progenitors |
Constitutive | |
| TL1A–DR3 | APCs (DCs, B cells and macrophages) |
Inducible (triggered by TLR4 and FcγR cross-linking) |
CD4+ T cells | Constitutive or inducible (triggered by TCR plus CD28 cross-linking) |
| CD4+ and CD8+ T cells | Inducible (triggered by TCR cross-linking, IL-12 and IL-18) |
CD8+ T cells | Inducible (triggered by TCR plus CD28 cross-linking) |
|
| Endothelial cells | Inducible (triggered by TNF and IL-1) |
NK cells | Inducible (triggered by IL-12 plus IL-18) | |
| NKT cells | Constitutive | |||
| FOXP3+ TReg cells | Constitutive (mice) or inducible (humans; triggered by TCR cross-linking) |
|||
| LTi cells | Constitutive | |||
| OX40L–OX40 | APCs (DCs, B cells and macrophages) |
Inducible (triggered by TLR2, TLR4, TLR9, CD40, Ig or BCR, TSLP and IL-18) |
CD4+ and CD8+ T cells | Inducible (triggered by TCR plus CD28 cross-linking, IL-1, IL-2 and TNF) |
| CD4+ and CD8+ T cells | Inducible (triggered by TCR cross-linking and IL-12) |
NK cells | Inducible (?) | |
| LTi cells | Inducible (triggered by DR3) | NKT cells | Inducible (triggered by TCR cross-linking) |
|
| NK cells | Inducible (triggered by NKG2D and CD16) |
FOXP3+ TReg cells | Constitutive (mice) or inducible (humans; triggered by TCR cross-linking) |
|
| Mast cells | Inducible (triggered by GM-CSF and FcεRI cross-linking) |
Neutrophils | Inducible (?) | |
| Endothelial cells | Inducible (?) | |||
| Smooth muscle cells | Inducible (?) | |||
| 4-1BBL–4-1BB | APCs (DCs, B cells and macrophages) |
Inducible (triggered by TLR4, TLR9, CD40 and Ig or BCR) |
CD4+ and CD8+ T cells | Inducible (triggered by TCR plus CD28 cross-linking and IL-2) |
| CD4+ and CD8+ T cells | Inducible (triggered by TCR cross-linking) |
NK cells | Inducible (?) | |
| Mast cells | Inducible (triggered by FcεRI cross-linking) |
NKT cells | Inducible (triggered by TCR cross-linking) |
|
| NK cells | Inducible (?) | Mast cells | Inducible (triggered by FcεRI cross-linking) |
|
| Smooth muscle cells | Inducible (?) | Neutrophils | Inducible (?) | |
| Haematopoietic progenitors |
Constitutive | FOXP3+ TReg cells | Constitutive (mouse) or inducible (human; triggered by TCR cross-linking) |
APC, antigen-presenting cell; BCR, B-cell receptor; DC, dendritic cell; DR3, death receptor 3; FcγR, receptor for IgG; FcεRI, high-affinity Fc receptor for IgE; FOXP3, forkhead box P3; GM-CSF, granulocyte/macrophage colony-stimulating factor; IL, interleukin; IFNγ, interferon-γ; Ig, membrane-bound immunoglobulin; LTi, lymphoid-tissue inducer; NK, natural killer; NKG2D, NK group 2, member D; TCR, T-cell receptor; TLR, Toll-like receptor; TNF, tumour necrosis factor; TSLP, thymic stromal lymphopoietin.
In this Review, I focus on the activity of these molecules in regulating the function of T cells and other immune cells and discuss how this relates to studies of inflammation, autoimmunity and cancer, in which these molecules are promising candidates for therapy. There are two main approaches for therapy based on targeting TNF–TNFR interactions: first, to block one or more of these interactions to reduce pathogenic immune responses in autoimmune and inflammatory diseases and, second, to enhance signalling that is triggered by the TNF–TNFR interactions to stimulate a more robust immune response and promote antitumour immunity. The molecules reviewed here can also have important roles in responses to intracellular pathogens and acute viral infection, and persistent expression of these molecules may be related to the pathology that is linked to chronic viral infection. Although this is also a growing area of importance in terms of potential therapies, these roles will not be discussed here.
Overview and signalling
Expression
A feature of the molecules that form these four ligand–receptor pairs is that they are not ubiquitously expressed (TABLE 1). The finding that the expression of several of these molecules is increased following immune-cell activation suggests that they have a central role in modulating immune responses. Indeed, studies of TNFRs expressed by conventional T cells and their ligands expressed by antigen-presenting cells (ApCs) has led to the hypothesis that antigen recognition by T cells results in the engagement and bidirectional activity of the TNF–TNFR pair, which promotes the effector responses of T cells and of other immune cells1-7 Because the expression of OX40 and 4-1BB is induced in response to antigen stimulation, these TNFRs have been proposed to be markers of effector T cells (which are pathogenic in autoimmunity or protective in infection and cancer). Although CD27 and DR3 can be constitutively expressed by conventional T cells, their expression is also strongly upregulated following T-cell activation, possibly in parallel with the upregulation of OX40 and 4-1BB expression. In addition, several stimuli can affect the level and kinetics of expression. The induction or upregulation of OX40, 4-1BB and DR3 expression occurs within 24 hours following the recognition of antigen by and activation of naive T cells, and much more rapidly by memory T cells; the expression of these receptors can last for several hours or even days7-12.
The expression of the TNF ligands CD70, TL1A, OX40L and 4-1BBL is induced in professional ApCs, although the extent to which expression levels vary between dendritic cells (DCs), B cells and macrophages is not clear. various stimuli can promote the expression of the different ligands by ApCs, including innate signals (such as those induced by Toll-like receptor (TLR) ligands) and adaptive signals (such as those induced by interferon-γ (IFNγ))7,12-20. Furthermore, both CD4+ and CD8+ T cells can express both TNF ligands and their receptors, which suggests that T-cell–T-cell interactions can also influence the development of effector T-cell populations21,22.
Importantly, OX40L, 4-1BBL, CD70 and TL1A can also be expressed by non-immune cells (TABLE 1), such as smooth muscle cells and endothelial cells12,23-25, during conditions of inflammation. This implies that TNF–TNFR interactions between T cells and non-immune cells in the periphery can further contribute to the function of effector T cells and also promote an inflammatory response in non-immune cells, which ultimately will propagate tissue inflammation in a disease context. In addition, several mouse studies have shown immunomodulatory roles for CD70–CD27 and 4-1BBL–4-1BB interactions in the bone marrow when expressed by haematopoietic progenitor cells26,27, suggesting that these molecules might contribute to inflammatory conditions that require haematopoiesis.
Finally, other immune-cell types, such as natural killer (NK) and NKT cells, can also express one or more of these molecules, the activation of which can increase their effector function. In addition, it has been reported that regulatory T (TReg) cells can express TNFRs, and some evidence suggests that engaging these receptors on the surface of these cells helps to amplify immune responses by inhibiting the generation and/or activity of this suppressive T-cell subset (see below).
So, there is not a simple set of rules that determines when and where these molecules are expressed, and hence in which context they are important for T-cell responses. These TNF–TNFR interactions occur mainly during an ongoing immune response rather than at the initiation of a response, as their expression in resting or recently activated cells is limited. It is probable that there are instances in which each of the four ligand–receptor interactions are spatially separated (owing to differential expression levels by different cell types) or temporally separated (owing to expression at distinct intervals) during the course of an immune response. Conversely, it is possible that during other types of immune response, T cells or NK and NKT cells can express all four receptors, which would allow simultaneous engagement by the four ligands and an additive or synergistic cellular response. The expression of some of these molecules in normal and disease states has also been described in humans, and several reports suggest a correlation between their expression levels and disease status, and sometimes with therapy outcome (for examples, see REFS 28-30). unfortunately, these studies are too numerous to describe in detail here. Given the diversity in experimental approaches used in some of these analyses, this is an area that warrants much greater attention despite the caveat that human clinical samples are limited throughout the course of any disease. It is probable that there are differences in the expression patterns of these molecules between mice and humans; for example, mouse TReg cells constitutively express several TNFR molecules, whereas the expression of these molecules is inducible on human TReg cells. However, given the hypothesis that the expression of both TNF ligands and receptors will be transient in many cases, understanding when and where they are expressed in humans should aid the diagnosis and the timing of any clinical treatment.
Signalling
The intracellular regions of OX40, 4-1BB, CD27 and DR3 associate with TNFR-associated factors (TRAFs), which are adaptor molecules that link receptor activation to inflammatory signalling pathways. In particular, TRAFs can complex with inhibitor of NF-κB, α subunit (IκBα), IκB kinase-β (IKKβ) and NF-κB-inducing kinase (NIK), thereby allowing activation of both canonical and non-canonical nuclear factor-κB (NF-κB) signalling pathways, which are known to be important for cell survival. Signalling pathways that are triggered by the activation of OX40, 4-1BB and CD27 in CD4+ and CD8+ T cells increase the expression of anti-apoptotic molecules, including BCL-2 (B-cell lymphoma 2), BCL-XL (also known as BCL2L1) and/or BFL1 (also known as BCL2A1)9,31,32, which correlates with the promotion of T-cell survival by these receptors. For OX40 and 4-1BB, this has been linked to the activation of NF-κB, phosphoinositide 3 kinase and protein kinase B (also known as AKT)31,33,34. Another downstream effect of 4-1BB ligation35 that might also be common to the other receptors is inhibition of the expression of the pro-apoptotic molecule BIm (BCL-2-interacting mediator of cell death) through the activation of extracellular-signal-regulated kinase (eRK). CD27 and DR3 are also strong activators of NF-κB12,36-38, and DR3 signalling can result in the increased accumulation of T cells7 and resistance to apoptosis38 (FIG. 1). Indeed, although DR3 was initially named death receptor 3 owing to its intracellular death domain that can lead to apoptosis, more recent functional data suggest that the activity of DR3 is mainly pro-inflammatory.
Figure 1. TnF–TnFR family interactions and molecular targets in T cells and APCs.

Tumour necrosis factor receptors (TNFRs) are characterized by several cysteine-rich domains, and TNF ligands are characterized by a TNF homology domain. Both OX40 ligand (OX40L) and TL1A are homotrimers (that is, three receptor monomers bind to the trimeric ligand), and this molecular arrangement probably applies to interactions between CD70 and CD27, 4-1BBL and 4-1BB, and TL1A and death receptor 3 (DR3). During interactions between T cells and antigen-presenting cells (APCs), the expression of TNF ligands by the APC is probably induced following activating signals from either CD40 (when bound to CD40L expressed by a T cell) or from Toll-like receptor (TLR)-mediated signals. The ligation of cytokine receptors by cytokines such as TNF, interleukin-1 (IL-1), IL-6, IL-12, IL-18 and thymic stromal lymphopoietin (not shown) can also promote TNF ligand expression. The expression of OX40 and 4-1BB can be induced by activation signals from the T-cell receptor (TCR) following recognition of peptide–MHC complexes. The main common downstream signalling event triggered by TNFRs is the activation of nuclear factor-κB 1 (NF-κB1), which leads to cell division and enhanced survival and can contribute to the production of cytokines, such as IL-2, IL-4, IL-5 and interferon-γ (IFNγ). NF-κB2 can also be activated downstream of these TNFRs, although its primary function in cellular responses is not clear. Other signalling molecules that have been described to be activated following TNF-–TNFR interactions include phosphoinositide 3 kinase (PI3K), protein kinase B (PKB), extracellular-signal-regulated kinase (ERK), JUN N-terminal kinase (JNK) (not shown) and nuclear factor of activated T cells (NFAT) (not shown), which also contribute to cell division, survival and cytokine production. Triggering of any TNFR might lead to the expression of other proteins that promote proliferation, including survivin, aurora B kinase, cyclins and cyclin-dependent kinases (CDKs), as well as the expression of anti-apoptotic proteins, including BCL-2 (B-cell lymphoma 2), BCL-XL, BFL1 (BCL-2-related protein A1), and/or the downregulation of the expression of pro-apoptotic proteins, such as BIM (BCL-2-interacting mediator of cell death). Signals downstream of the TNF ligands can promote the secretion of pro-inflammatory cytokines by APCs, such as TNF, IL-1, IL-6 and IL-12, and lead to cellular proliferation.
Signals from OX40, 4-1BB, CD27 and DR3 also synergize with T-cell receptor (TCR)-induced signals to allow cell cycle progression (and thereby promote T-cell division) and cytokine production by T cells. Ligation of OX40 enhances the expression of survivin and aurora B kinase34,39, which function together to promote the activity of cyclin-dependent kinases and allow S phase progression and mitosis in T cells40. Ligation of 4-1BB can also influence the expression of cyclins41. Other events that are reportedly triggered by OX40, 4-1BB, DR3 and CD27 ligation include the activation of JuN N-terminal kinase (JNK) and activator protein 1 (Ap1), the activation of the mitogen-activated protein kinases p38 (REFS 38,42,43) and eRK, and the nuclear accumulation of nuclear factor of activated T cells (NFAT)44. These molecules are involved in promoting the production of cytokines, including interleukin-2 (IL-2), IL-4, IL-5 and IFNγ (FIG. 1). In addition, ligation of TNFRs can lead to the upregulation of cytokine receptor expression, such as IL-2 receptor α-chain (IL-2Rα) and IL-12Rβ, which further amplifies the immune response by increasing the sensitivity of T cells to these growth factors32,45-47.
- Regulatory T (TReg) cell
A specialized T cell that suppresses the effector immune responses of other immune cells and is crucial for the maintenance of peripheral tolerance. One CD4+ TReg-cell subset is characterized by the expression of the transcription factor forkhead box P3 (FOXP3), whereas other types of TReg cell are normally characterized based on their expression of immunosuppressive cytokines such as interleukin-10 and transforming growth factor-β.
The ability of the TNF ligands to activate signalling pathways in professional APCs probably also contributes to their function. In particular, bidirectional signalling affects B-cell function and antibody responses and consequently has a strong effect on this arm of the immune response, although an in-depth discussion of this is outside the scope of this Review. Cross-linking of OX40L and CD70 results in the production of pro-inflammatory cytokines (including TNF, IL-1, IL-6 and IL-12) and the proliferation of DCs and B cells when they are also stimulated through TLRs, CD40 or membrane-expressed immunoglobulin molecules17,48. Binding of 4-1BBL to its receptor can mediate similar effects, such as increased cell division and increased production of TLR-induced pro-inflammatory cytokines by macrophages and DCs49,50. The differentiation of myeloid progenitors that express 4-1BBL is suppressed following ligation of its receptor27, which might be due to the production of cytokines by the cell expressing 4-1BBL that in this case inhibit myeloid-cell development. Whether TL1A can signal has not yet been investigated. Although some of the signalling intermediates that are involved in TNF ligand-induced pathways have been described (FIG. 1), limited data are available to directly link these pathways with distinct cellular responses.
Functional effects in immune cells
Each of the four TNF–TNFR interactions can stimulate conventional T cells and APCs, mediate communication between CD4+ and CD8+ T cells and promote immune responses. These ligand–receptor pairs also mediate interactions between NK cells and T cells, between NKT cells and APCs presenting lipid antigens, and between T cells and other types of immune or tissue cell.
Co-stimulation of CD4+ and CD8+ T cells
Interactions between individual TNF–TNFR pairs control T-cell responses in two ways. First, they regulate the frequency of effector and/or memory CD4+ or CD8+ T cells that can be generated from naive T cells in response to antigen stimulation by providing proliferative and survival signals either directly to the T cells or to the APCs with which they interact. These molecules also regulate the frequency of effector memory T cells that are generated in recall responses. Second, they control T-cell function directly by promoting the production of cytokines such as IL-4 and IFNγ, or indirectly through stimulating the production of pro-inflammatory cytokines, such as IL-1 and IL-12, by professional or non-professional APCs1-7,51-53. It still not clear whether these interactions contribute to the acquisition of cytotoxic function by CD8+ T cells, which has been indicated by some studies11; however, the regulation of T-cell expansion54 might be a more important function to the overall activity of cytotoxic T cells. The interactions between these TNF superfamily members are primarily thought to deliver co-stimulatory signals, as their effects largely depend on antigen recognition and TCR signalling.
It has been directly shown in specific studies of immunity55, or implied in studies of immune-mediated diseases (see below and TABLE 2), that several of these interactions collectively contribute to the overall response of a T cell. However, how the different effects of TNF–TNFR interactions are integrated in the control of T-cell responses is not clear, although a greater understanding of this is important for the design of new therapies that target TNF–TNFR interactions. Several models for how these interactions might contribute to T-cell responses are shown in FIG. 2. In the first scenario, the individual TNFRs act sequentially to promote continued T-cell proliferation, survival and effector functions over the course of an immune response. In this case, CD27, DR3, OX40 and 4-1BB would all sequentially contribute to the generation of large populations of effector T cells. Inhibiting any one of these molecules could then be effective for the treatment of autoimmune or inflammatory conditions, although establishing the optimal stage or time at which to target a given receptor during the disease process would be important. A second, more complex scenario involves the synergistic activity and temporal expression of these molecules. In this scenario, the sustained T-cell response would depend on both a signalling threshold that would be mediated by several different ligand–receptor interactions and multiple signals imparted by TNFRs at distinct time points. Blocking of a single interaction would markedly suppress the response, but the appropriate interaction to target would depend on both the type and stage of disease. Another complex scenario involves a situation in which many antigens (or autoantigens) are presented to several different T-cell populations that express and use different sets of TNFRs. In this case, an effective therapy to block such a heterogenous T-cell response would need to target at least two ligand–receptor interactions to inhibit the associated disease.
Table 2.
Therapeutic targeting of TNF superfamily interactions
| Model | Interaction targeted |
Mice or reagent tested | Effect on disease symptom |
|---|---|---|---|
| EAE | OX40L–OX40 | Toxin-conjugated OX40-specific antibody (depleting) | Substantial inhibition |
| OX40–immunoglobulin fusion protein (neutralizing) | Substantial inhibition | ||
| OX40L-specific antibody (neutralizing) | Substantial inhibition | ||
| Ox40−/−or Ox40l−/− mice | Substantial inhibition | ||
| TL1A–DR3 | Dr3−/− mice | Substantial inhibition | |
| Tl1a−/− mice | Partial inhibition | ||
| CD70–CD27 | CD70-specific antibody (neutralizing) | Substantial inhibition | |
| 4-1BBL–4-1BB | 4-1BB-specific antibody (agonist) | Substantial inhibition | |
| Colitis and IBD | OX40L–OX40 | OX40–immunoglobulin fusion protein (neutralizing and depleting) |
Substantial inhibition |
| OX40L-specific antibody (neutralizing) | Substantial inhibition | ||
| TL1A–DR3 | TL1A-specific antibody (neutralizing) | Partial inhibition | |
| 4-1BBL–4-1BB | 4-1BB-specific antibody (agonist) | Substantial inhibition | |
| Asthma and atopy | OX40L–OX40 | Ox40−/− or Ox40l−/− mice | Substantial inhibition |
| OX40L-specific antibody (neutralizing) | Substantial inhibition | ||
| TL1A–DR3 | Dr3−/− mice | Substantial inhibition | |
| TL1A-specific antibody (neutralizing) | Substantial inhibition | ||
| 4-1BBL–4-1BB | 4-1BB-specific antibody (agonist) | Substantial inhibition | |
| Diabetes | OX40L–OX40 | Ox40l−/− mice | Substantial inhibition |
| OX40L-specific antibody (neutralizing) | Substantial inhibition | ||
| 4-1BBL–4-1BB | 4-1BB-specific antibody (agonist) | Substantial inhibition | |
| Arthritis | OX40L–OX40 | OX40L-specific antibody (neutralizing) | Substantial inhibition |
| Toxin-conjugated OX40-specific antibody (depleting) |
Partial inhibition | ||
| TL1A–DR3 | Dr3−/− mice or TL1A-specific antibody (neutralizing) | Partial inhibition | |
| 4-1BBL–4-1BB | 4-1BB-specific antibody (agonist) | Substantial inhibition | |
| 4-1BBL-specific antibody (neutralizing) | Partial inhibition | ||
| SLE | 4-1BBL–4-1BB | 4-1BB-specific antibody (agonist) | Substantial inhibition |
| Atherosclerosis | OX40L–OX40 | Ox40l−/− mice | Substantial inhibition |
| OX40L-specific antibody (neutralizing) | Substantial inhibition | ||
| Minor MHC transplant mismatch |
OX40L–OX40 | OX40–immunoglobulin fusion protein (neutralizing) |
Substantial inhibition |
| 4-1BBL–4-1BB | 4-1BB–immunoglobulin fusion protein (neutralizing) | Partial inhibition | |
| Major MHC transplant mismatch |
OX40L–OX40 | OX40–immunoglobulin fusion protein (neutralizing) | No effect |
| OX40L-specific antibody (neutralizing) | Substantial inhibition with CD28 or CD28 and CD40L blockade |
||
| CD70–CD27 | CD70-specific antibody (neutralizing) | No effect alone; no effect with CD28 and CD40L blockade; substantial inhibition with CD4 and CD28 blockade |
|
| 4-1BBL–4-1BB | 4-1bbl−/− or 4-1bbl−/− mice | Varying results: no effect or inhibition | |
| 4-1BB–immunoglobulin fusion protein (neutralizing) |
Partial inhibition | ||
| GVHD | OX40L–OX40 | OX40L-specific antibody (neutralising) | Substantial inhibition |
| Ox40−/− mice | Substantial inhibition | ||
| 4-1BBL–4-1BB | 4-1bb−/− mice | Substantial inhibition | |
| 4-1BB-specific antibody (agonist) | Substantial inhibition |
There is an online version of this table that includes references. See Supplementary information S1 (Table). No published reports are available for the interactions that are not mentioned. 4-1BBL, 4-1BB ligand; DR3, death receptor 3; EAE, experimental autoimmune encephalomyelitis; GHVD, graft-versus-host disease; IBD, inflammatory bowel disease; SLE, systemic lupus erythematosus.
Figure 2. Control of T-cell proliferation by cooperative and sequential TNF–TNFR interactions.

A hallmark of T-cell co-stimulation by the tumour necrosis factor receptors (TNFRs) OX40, 4-1BB, CD27 or DR3 is the expansion of the effector T-cell population (during the primary response and/or the secondary and memory response). However, the extent of cooperation between these individual ligand–receptor pairs over the course of most T-cell responses is not clear. Three non-mutually exclusive models that represent what might occur are shown. The involvement of the TNFRs probably varies depending on the inflammatory environment in which antigen recognition takes place and the nature and number of antigens recognized. a| Step-wise involvement of TNFRs, whereby the temporal activity of individual receptors increases and sustains T-cell survival and proliferation. In this case, the interactions of all of the different TNFRs with their ligands are crucial for generating large effector T-cell populations, and inhibiting any one will markedly suppress the response. b| A more complex scenario that involves synergistic action of different TNFRs, whereby several ligand–receptor interactions can function simultaneously, as well as sequentially. In this case, continued T-cell survival and proliferation depend on a threshold level of signalling imparted by multiple receptors. Removing any single interaction would, again, markedly reduce the response. c| A scenario that might explain the involvement of TNFRs in promoting a T-cell response when many antigens or autoantigens are expressed. Several populations that express different TNFRs would be involved in the response (including CD4+ and CD8+ T cells specific for various epitopes). If only one population is involved, blocking a single TNF–TNFR interaction would suppress the response, although which interactions are involved probably varies. However, when several T-cell populations are active, pronounced suppression would require targeting two or more interactions. DR3, death receptor 3.
As discussed below, there are some examples in which blocking individual interactions in the same disease model can inhibit pathology and others in which a therapeutic effect is only observed if more than one interaction is neutralized. These data suggest that, although each molecule could be a good target for blocking a T-cell response, different molecules may be involved in different diseases, or the stage of disease when each molecule is relevant may vary. Systematic approaches that target each of these molecules at various points during disease progression are required to understand which of the models discussed above explain the proliferation and function of T-cell subsets in the context of a given inflammatory condition. It is probable that the involvement of the different TNFRs varies with the nature of the antigen being presented, the presence of pathogen-derived products and the innate inflammatory milieu that results when foreign or self antigen is recognized. Interestingly, most of the studies on CD27 and 4-1BB have examined the regulation of CD8+ T-cell responses by these molecules, whereas more studies on OX40 and DR3 have been carried out with CD4+ T cells. However, it should also be noted that both CD4+ and CD8+ T cells have been reported to express all of the TNFRs, and the perceived dichotomy in the integration of TNF–TNFR interactions referred to above does not translate to all immune responses that have been examined so far. Therefore, there is not a single model describing the use of these molecules that applies to all immune responses.
Amplifying inflammatory responses through non-T cells
The functions of other immune-cell types, in addition to T cells, are also controlled by the TNF–TNFR interactions (FIG. 3, TABLE 1), which probably contribute to the amplification of the immune response. The neutralization of these molecules will therefore compound the effects of therapeutic targeting beyond that of T-cell inhibition. It has been reported that OX40L and OX4056,57, CD70 and CD27 (REF. 58), DR3 (REF. 59) and 4-1BB60 are involved in either directly enhancing NK-cell effector function (that is, cytotoxic ability and cytokine production) or in NK-cell-mediated help for the activation or differentiation of conventional T cells. Similarly, activated NKT cells express OX40 (REF. 61), 4-1BB62 and DR3 (REF. 51), and signals transmitted through these receptors can directly increase NKT-cell activity by promoting either cell expansion or survival and by enhancing cytokine production. Through the production of IFNγ, NKT cells can also promote CD70 expression by DCs that can subsequently prime a conventional T-cell response63, and a similar positive feedback mechanism that involves induction of IFNγ expression by NKT cells probably occurs if other TNFRs expressed by NKT cells are engaged.
Figure 3. TNF–TNFR family interactions regulate many cell types of amplify inflammation.

Effector T cells receive signals for division, survival and cytokine production following the activation of the tumour necrosis factor receptors (TNFRs) OX40, 4-1BB, CD27 and death receptor 3 (DR3) by their ligands. In addition, natural killer (NK) and NKT cells can also receive signals through TNFRs that amplify division, survival and cytokine production. The initiation of inflammatory responses can involve cooperation between NK and NKT cells with effector T cells, which might occur directly or indirectly through antigen-presenting cells (APCs). Feedback mechanisms can occur through NK- or NKT-cell-derived interferon-γ (IFNγ), which enhances APC activation in many ways, including promoting the expression of TNF ligands. In addition, activated mast cells can express many ligands, including OX40 ligand (OX40L) and 4-1BBL, which can co-stimulate effector T cells and NKT cells. The production of pro-inflammatory cytokines, such as IFNγ, interleukin-13 (IL-13) and IL-17, by T cells also can promote the expression of one or several TNF ligands on tissue cells such as endothelial, epithelial and smooth muscle cells. Through additional bidirectional signals with effector T cells, NK cells or NKT cells, these interactions probably further amplify tissue pathology, for example, by inducing the production of additional pro-inflammatory mediators such as leukotrienes and histamine. CTL, cytotoxic T cell; NKG2D, NK group 2, member D; TH, T helper; ULBP3, cytomegalovirus UL16-binding protein.
The expression of CD27, CD70 (REF. 64) and OX40L65 is induced on most B cells and can promote B-cell proliferation and differentiation to antibody-secreting cells. In addition, the expression of OX40, OX40L, 4-1BB, 4-1BBL and CD70 can be induced on activated mast cells. OX40L expressed by mast cells can stimulate conventional T cells66,67, and there is evidence (mainly from studies of 4-1BB) to support the idea that TNFR signalling in mast cells has a role in increasing the production of pro-inflammatory mediators by these cells68. One study also reported that neutrophils express OX40 and 4-1BB, ligation of which might contribute to tissue inflammation by increasing cell survival or the production of pro-inflammatory mediators69. Interestingly, adult lymphoid-tissue inducer cells have been shown to constitutively express DR3, and DR3 stimulation induced the expression of OX40L by these cells. This was proposed to be another amplification loop that might sustain T-cell responses or help to maintain the longevity of memory T cells70,71.
Effects on TReg cells
The expression of OX40, CD27, 4-1BB and DR3 is either constitutive (in mice) or rapidly induced (in humans) on natural and inducible CD4+ or CD8+ TReg cells. Natural TReg cells express forkhead box P3 (FOXP3) and are selected in the thymus, whereas inducible TReg cells can differentiate from naive CD4+ or CD8+ T cells in the periphery in response to antigen and may or may not express FOXP3. Increasing the numbers or activity of TReg cells results in the suppression of immune responses, which is beneficial for the treatment of autoimmune and inflammatory conditions. By contrast, decreasing the numbers and function of TReg cells can enhance both innate and adaptive immune responses, which is beneficial for the treatment of cancer. Studies using mouse TReg cells have shown that ligation of the TNF superfamily members OX40 and 4-1BB affect these T-cell subsets (see below); these studies have been reviewed in detail elsewhere72. Regulation of TReg cells is therefore an important consideration for immunotherapy, although similar studies using human TReg cells are at present limited.
- Lymphoid-tissue inducer cell
A cell that is present in developing lymph nodes, Peyer's patches and nasopharynx-associated lymphoid tissue. Lymphoid-tissue inducer cells are required for the development of these lymphoid organs. Adult lymphoid-tissue inducer cells have been proposed to orchestrate the development of tertiary lymphoid structures during inflammatory responses.
The effects of triggering OX40 and 4-1BB on TReg cells can result in either of two main outcomes that allow greater overall immune responsiveness (FIG. 4). First, signalling triggered by OX40 has been found to inhibit the development of FOXP3+ TReg cells that differentiate from naive CD4+ T cells in the presence of transforming growth factor-β (TGFβ)73-75 and to suppress the differentiation of IL-10-producing (FOXP3−) CD4+ TReg cells from naive CD4+ T cells76. Furthermore, ligation of OX40 negatively affects the stability of these cell populations, as it can lead to the downregulation of FOXP3 and IL-10 expression in newly differentiated inducible TReg cells and consequently result in their conversion to effector T cells (M.C., unpublished observations, and REF. 76). So, ligation of OX40 has a dual effect on promoting T-cell responses: it enhances the proliferation of effector T cells and concomitantly blocks the generation of inducible TReg cells. However, whether signalling through CD27, 4-1BB or DR3 also affects the induction of peripheral TReg cells is not yet known but is of obvious interest. In addition, binding of OX40 and 4-1BB to their ligands has been shown to block the suppressive activity of TReg cells in culture systems that contained both inducible and natural TReg cells. This seems to be due to both a direct effect by blocking TReg-cell function and an indirect effect on effector T cells that renders them resistant to suppression74,75,77-82. Although the mechanistic explanation for both effects is currently unknown, the ability of TNFRs to overcome TReg-cell inhibition is in line with their activating effects on other immune-cell types. No studies on the effect of CD27 and DR3 on TReg-cell activity have been reported.
Figure 4. modulation of TReg-cell development and function by TNF–TNFR interactions.
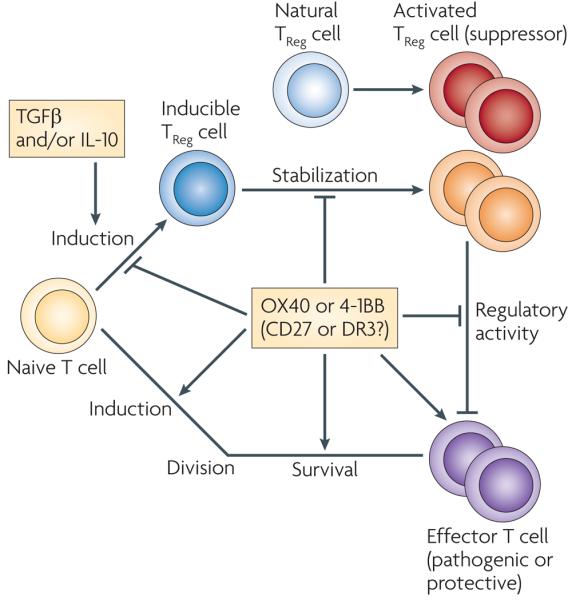
In addition to promoting the activation of effector T cells, the interaction between the tumour necrosis factor receptors (TNFRs) OX40, 4-1BB, CD27 and DR3 and their ligands might further contribute to inflammation by affecting naturally occurring or inducible regulatory T (TReg) cells. To date, only the effect of OX40 or 4-1BB ligation on TReg-cell development and function has been examined, although the fact that the different receptors can use common signalling pathways (including the nuclear factor-κB and protein kinase B pathways) means that it is possible that DR3 and CD27 have similar effects. Signals triggered following the activation of OX40 inhibit the expression of forkhead box P3 (FOXP3) and interleukin-10 (IL-10) by naive CD4+ T cells that are differentiating into TReg cells by an unknown mechanism, which might involve blocking or modulation of the signalling events downstream of transforming growth factor-β receptor (TGFβR), IL-10R or vitamin D receptor (not shown). OX40 can also reduce the stability of TReg cells, as ligation of OX40 can lead to the downregulation of FOXP3 and IL-10 expression in recently differentiated TReg cells. This may occur directly, or indirectly through promoting the production of cytokines by T helper cells, which in turn induce the expression of transcription factors such as GATA-binding protein 3 that prevent FOXP3 and/or IL-10 expression (not shown). Fully differentiated inducible CD4+ and CD8+ TReg cells and natural CD4+ TReg cells also express OX40, 4-1BB, CD27 and DR3. OX40 and 4-1BB signals have been shown to block the suppressive function of these cells, again either directly through effects on the TReg cell itself, or indirectly by promoting the proliferation and survival of effector T cells and by rendering them resistant to TReg-cell-mediated suppression. The combined action of these TNF–TNFR interactions might lead to an increased ratio of effector T cells to TReg cells (that is, too few TReg cells to suppress the inflammatory response) and/or to greater effector T-cell activity through blocking of TReg-cell-mediated suppression. Activation of the TNFRs might also promote the expansion or survival of TReg cells (not shown), as shown in some in vitro systems with agonist stimulation, although studies of knockout animals do not as yet support this expansion as a physiological activity. DR3, death receptor 3.
By contrast, other studies have shown that OX40 and 4-1BB can promote the proliferation or survival of CD4+ and CD8+ TReg cells80,83-86. These results were mainly obtained in experiments that used exogenous stimulation with agonists, so whether this effect occurs in vivo when endogenous ligands bind OX40 or 4-1BB is not clear. Although these results seem to conflict with the studies above on the effect of TNFRs on TReg cells, they are not necessarily mutually exclusive. The enhancement of cell proliferation and survival by TNFRs has been described in some studies of human natural TReg cells in vitro (see below), and this effect could be useful for clinical exploitation, as discussed below.
Therapeutic implications
Inflammatory and autoimmune diseases
To decrease immunopathology and inhibit disease progression, therapies for inflammatory or autoimmune diseases should aim to suppress the immune responses of T cells, ApCs, NK cells and NKT cells. Secondary and complementary to this aim, therapy should ideally allow the function, maintenance or generation of TReg cells to aid the long-term control of disease. Therefore, when considering TNFRs as therapeutic targets, a logical option would be to prevent their interactions with their TNF ligands, which would therefore decrease the expansion and survival of pathogenic cell populations and/or decrease their production of pro-inflammatory cytokines. In support of this concept, analysis of the development of autoimmune and inflammatory diseases in animals that are deficient for individual TNF ligands or TNFRs has revealed that elimination of these interactions decreases the severity of the disease (TABLE 2).
Preclinical studies have analysed the activity of neutralizing antibodies that are specific for TNF ligands, or of Fc fusion proteins that contain a TNFR that binds to the ligand and thereby blocks the endogenous interaction. The effects of blocking each of the four ligand–receptor interactions discussed in this Review have been assessed in models of inflammatory disease (including allergy, asthma, transplantation, graft-versus-host disease (GvHD) and atherosclerosis) and autoimmune disease (including experimental autoimmune encephalomyelitis (EAE), diabetes, colitis, adjuvant- or collagen-induced arthritis, and systemic lupus erythematosus (SLE)) (TABLE 2). These studies have shown that neutralizing any one of these TNF–TNFR interactions can result in strong suppression of disease symptoms, which in many cases is specifically linked to decreased activity of CD4+ or CD8+ T cells, or in some cases to impaired NK- and NKT-cell function. Although most of the TNFRs or their ligands have been shown to be expressed by cells from patients with active autoimmune or inflammatory diseases, the relevance of some of the TNF–TNFR interactions that are being targeted in experimental disease models has not been investigated in human diseases (TABLE 2).
Another therapeutic approach by which to dampen inflammation is through the administration of depleting antibodies. These therapies target the ligand or receptor and directly eliminate the pathogenic cells that express a specific molecule. This approach involves either coupling of specific antibodies to toxins or the development of antibodies that have increased intrinsic mechanisms of antibody-dependent cell-mediated cytotoxicity or complement-dependent cytotoxicity. molecules such as OX40 and 4-1BB, and possibly DR3 and CD70, may be particularly suited to this approach given that their expression is limited and that they might be reliable markers of pathogenic cells. Studies in experimental models of auto-immunity, in which the depletion of OX40-expressing T cells led to a positive outcome, support this idea87,88. Furthermore, as NK and NKT cells might also express these molecules during disease, their depletion could improve the therapeutic outcome. However, it should be noted that a potential side effect of this type of therapy is increased susceptibility to infectious diseases.
Given that an important function of signalling induced by the ligation of OX40, 4-1BB, CD27 and DR3 is to promote cell survival, it follows that neutralizing antibodies that are specific for these molecules could also lead to apoptosis of immune cells (including T cells) that rely on these signals for survival during an immune response. Therefore, neutralization of these receptors should provide the same end result as the direct depletion strategies mentioned above. The finding that both depleting and non-depleting OX40–immunoglobulin fusion proteins were effective in suppressing disease in a model of colitis provides evidence that both approaches can produce the same overall result89. However, an important issue when considering depletion versus neutralization strategies that target TNFRs is whether they affect TReg cells. Accumulating evidence suggests that naturally occurring and inducible TReg cells are essential for the induction of immunological tolerance associated with transplantation and autoimmunity and for the protection against other inflammatory diseases such as asthma. As described above, recent studies of TNFRs suggest that blocking some of these molecules might result in greater differentiation of inducible TReg cells and/or increased functional activity of pre-existing naturally occurring and inducible TReg cells. By contrast, depleting reagents might eliminate both activated TReg cells and pathogenic cells, as the expression of these molecules by human TReg cells is upregulated following activation. Although depletion should still result in strong short-term benefits and inhibition of disease, the lack of TReg-cell activity might lead to re-established disease in the long term, as newly emerging pathogenic cells would not be controlled. Therefore, depletion versus neutralization should be carefully considered before clinical therapy.
- Graft-versus-host disease
(GVHD). A disease that results from the immunological attack by donor allogeneic T cells that are transferred along with the allograft (such as bone marrow, liver or gut allografts) of target recipient organs or tissues (such as the skin and gut). GVHD occurs in graft recipients that cannot eliminate the host-reactive donor T cells owing to immunosuppression, immunological immaturity or tolerance of the recipient.
- Experimental autoimmune encephalomyelitis
(EAE). An animal model of the human autoimmune disease multiple sclerosis. EAE is induced in experimental animals by immunization with myelin or peptides derived from myelin. The animals develop a paralytic disease with inflammation and demyelination in the brain and spinal cord.
- Systemic lupus erythematosus
(SLE). An autoimmune disease in which autoantibodies that are specific for DNA, RNA or proteins associated with nucleic acids form immune complexes that damage small blood vessels, especially in the kidneys. Patients with SLE generally have abnormal B- and T-cell function.
- Antibody-dependent cell-mediated cytotoxicity
A cytotoxic mechanism by which an antibody-coated target cell is directly killed by a leukocyte that expresses Fc receptors, such as a natural killer (NK) cell, macrophage or neutrophil. A specific receptor for the Fc region of IgG is CD16, which is expressed on the surface of most NK cells. Following binding to immunoglobulin, CD16 initiates a signalling cascade that results in the release of cytotoxic granules (containing perforin and granzyme B), which induce apoptosis of the antibody-coated cell.
Another issue is whether targeting a single TNF–TNFR interaction would be effective for the treatment of human diseases. A strong reduction in disease severity has been reported when OX40 or OX40L were absent or neutralized in most of the experimental mouse models of inflammatory and autoimmune disease that have been used; similar results were observed when DR3 or TL1A were absent or neutralized in EAE, asthma, arthritis and colitis models. more encouragingly, several studies have shown that targeting OX40L or TL1A is effective for suppressing ongoing disease in several different pre-clinical studies51,52,87,88,90-98. Collectively, the data suggest that targeting a single TNF–TNFR interaction might yield promising results for the effective treatment of human diseases. Surprisingly, there are limited data on how the absence or neutralization of CD70–CD27 or 4-1BBL–4-1BB interactions affects disease; targeting these interactions suppresses disease in mouse models of EAE, arthritis, GVHD and allograft rejection (TABLE 2), but reports on whether this therapy is effective in other inflammatory and autoimmune conditions have not been published. Given that the manipulation of CD70–CD27 and 4-1BBL–4-1BB interactions has a marked therapeutic effect in tumour models (see later) and that these molecules are involved in T-cell function in many instances99-104, further preclinical studies that analyse the effects of blocking these molecules in other disease models are warranted.
There are, however, some studies that show no effect or only a moderate clinical benefit when a single TNF–TNFR interaction is manipulated. This does not necessarily mean that the interaction is irrelevant or that it is not a good therapeutic target. For example, in the context of transplantation, blocking OX40L alone is effective for limiting the rejection of grafts with minor MHC mismatches105 but has no benefit in preventing the rejection of grafts that are fully MHC mismatched. However, blocking OX40L–OX40 interactions together with CD80/CD86–CD28 and/or CD40L–CD40 interactions is effective for preventing the rejection of fully MHC mismatched grafts75,106,107. Similar results have been obtained by targeting CD70 (REF. 108) or 4-1BBL109 together with CD28 or CD40L in mouse models of transplantation. The fact that blocking multiple TNFRs, or a TNFR and an immunoglobulin superfamily member such as CD28, was more effective than blocking a single TNFR in these studies might indicate either that the T cells that are involved in graft rejection simultaneously express several signalling receptors or that several different subsets of T cells — each regulated by different TNF–TNFR interactions — are involved in the response (FIG. 3). Thus, these data suggest that a significant effect on the immune response in some inflammatory conditions will only occur when multiple interactions are targeted.
Studies of 4-1BB have explored an alternate therapeutic strategy that relies on stimulatory rather than neutralizing reagents to inhibit the activity of pathogenic cells. In this case, agonist antibodies that are specific for 4-1BB suppress disease in mouse models of EAE, asthma, arthritis, SLE, colitis, diabetes and GvHD (TABLE 2). Although there is not yet a consensus on how stimulation of 4-1BB prevents disease, most of the evidence suggests that its effects are related to the expansion of either natural or inducible TReg cells (either CD4+ or CD8+ T cells), or to the hyperactivation of IFNγ-producing CD8+ T cells that acquire regulatory capacity110-114. However, this approach has not been tested extensively and there are no reports indicating that reagents which stimulate OX40, DR3 or CD27 have similar inhibitory effects on inflammatory disease. This raises the question of whether 4-1BB is fundamentally different from other TNFRs in terms of the signalling pathways it induces or in terms of its expression profile. Alternatively, it is possible that the antibody used in these studies has specific properties, such as the ability to act as a superagonist, thereby mediating the suppression of disease.
Despite these promising results, it is counter-intuitive to suggest that reagents which enhance signalling through a co-stimulatory receptor can also suppress inflammatory and autoimmune diseases. Therefore, caution is needed when considering the use of a 4-1BB-specific agonist antibody to treat inflammatory diseases, as pathogenic effector T cells might be stimulated in addition to TReg cells. Indeed, a clinical trial that tested a superagonist antibody specific for CD28 that was expected to promote the expansion of the TReg-cell population without augmenting the activity of other T cells had disastrous results115. However, the finding that stimulatory TNFR reagents can expand TReg cells might be useful for cellular therapy approaches. Adoptive transfer of TReg cells is being considered as a treatment for autoimmune disease, although a limiting factor is that it is difficult to obtain sufficient numbers of functional TReg cells to suppress pathogenic cells in vivo. Reagents that expand TReg cells in vitro, such as TNFR-specific agonists and ApCs that are engineered to express OX40L, 4-1BBL and CD70, are currently being tested as strategies to achieve this; however, an important outstanding question with this approach is whether the TReg cells will retain their suppressive function once they have been adoptively transferred to a patient85,86,116.
- Complement-dependent cytotoxicity
A mechanism by which a monoclonal antibody binds complement, leading to direct cell toxicity and complement-mediated killing of the cell to which the antibody is bound. The result is a membrane attack complex that makes a hole within the cell membrane, causing cell lysis and death.
- Tolerance
A term that denotes lymphocyte non-responsiveness to antigen, but implies an active process, not simply a passive lack of response.
Studies of cancer
Therapy for cancer should aim to promote the antitumour activity of T cells, ApCs, NK cells and NKT cells, to kill existing tumour cells and to promote immunological memory that protects against recurring tumours. In addition, antitumour therapy should ideally prevent the generation and/or function of TReg cells. Numerous mouse studies have investigated the effectiveness of agonists or stimulatory Fc fusion proteins that express the extracellular portion of TNF ligands and cross-link TNFRs to trigger productive signalling117,118 (TABLE 3). As with most tumour studies, the level of protection achieved with the various approaches is highly variable, largely depending on the tumour model used. In most reports, protective antitumour responses are associated with increased effector activity of CD4+ and/or CD8+ T cells, as well as NK and NKT cells. Because the potential effect of stimulating TNFRs on TReg cells has only recently been recognized, it is not clear whether the approaches used in these studies also modulate TReg cells. However, a recent report of OX40 suggests that a proportion of the antitumour effect is probably mediated through the inhibition of TReg cells81. The effects of targeting DR3 in the context of cancer have yet to be investigated.
Table 3.
Therapeutic targeting of TNF superfamily members in cancer
| Mode of therapy | Target | Combination treatment | Tumour type |
|---|---|---|---|
| Stimulatory antibody or ligand Fc protein |
OX40 | NA | Sarcoma, melanoma, glioma, colon carcinoma, mammary carcinoma, thymoma and renal-cell carcinoma |
| Adoptive transfer of CTLs | Sarcoma, thymoma and prostate tumour | ||
| Administration of IL-12 and 4-1BB-specific antibody |
Colon carcinoma | ||
| Administration of GM-CSF | Colon and breast carcinoma | ||
| Tumour transfection with CD80 | B-cell lymphoma | ||
| Administration of DC vaccine and 4-1BB-specific antibody |
Breast carcinoma | ||
| Administration of GM-CSF and tumour antigen vaccine |
Breast tumour | ||
| Administration of IL-12 | Sarcoma and prostate tumor | ||
| 4-1BB | NA | Sarcoma, mastocytoma, glioma, colon carcinoma and B-cell lymphoma |
|
| Administration of IL-12 | Colon carcinoma and melanoma | ||
| Adoptive transfer of CTLs | Plasmacytoma | ||
| FLT3L-mediated DC mobilization | Fibrosarcoma | ||
| HLA-DR- and CD40-specific antibodies | Renal carcinoma and mammary carcinoma | ||
| 5-fluorouracil | Renal carcinoma | ||
| CD27 | NA | B-cell lymphoma | |
| Transfection of tumour cells |
OX40L | Administration of GM-CSF | Melanoma, lung carcinomathymoma and colon carcinoma |
| 4-1BBL | CD80 co-transfection | Sarcoma and colon carcinoma | |
| CD80 and CD86 co-transfection | B-cell lymphoma | ||
| IL-12 co-transfection | Colon carcinoma | ||
| CD80 | Squamous-cell carcinoma | ||
| CD80, CD40L and CD48 co-transfection | T-cell lymphoma | ||
| Adoptive transfer of LAK cells and NK cells |
Adenocarcinoma | ||
| TRANCE, CD95L and CCL21 co-transfection |
T-cell lymphoma | ||
| Soluble PD1 co-transfection | Hepatocarcinoma | ||
| Single-chain Fv fragments specific for 4-1BB |
NA | Melanoma and mammary carcinoma | |
| CD70 | NA | Sarcoma, mastocytoma, colon carcinoma, thymoma, lymphoma, mammary adenocarcinoma and glioma |
|
| CD80 co-transfection | Melanoma and mammary adenocarcinoma | ||
| CD40L co-transfection | Melanoma | ||
| Transfection of DCs | OX40L | NA | Melanoma, thymoma and melanoma |
| 4-1BBL | NA | Colon carcinoma and adenocarcinoma | |
| CD70 | NA | Thymoma | |
| Stimulatory RNA aptamer |
OX40 | Administration of DC vaccine | Melanoma |
| 4-1BB | NA | Mastocytoma | |
| Depleting antibody | CD70 | NA | B-cell lymphoma, renal carcinoma and non-Hodgkin lymphoma |
There is an online version of this table that includes references. See Supplementary information S1 (Table). 4-1BBL, 4-1BB ligand; CCL21, CC-chemokine ligand 21; CD95L, CD95 ligand; CTL, cytotoxic T lymphocyte; DC, dendritic cell; DR3, death receptor 3; FLT3, FMS-related tyrosine kinase 3; GM-CSF; granulocyte/macrophage colony-stimulating factor; IL, interleukin; LAK, lymphokine activated killer; NA, not applicable; NK, natural killer; PD1, programmed cell death 1; TNF, tumour necrosis factor; TRANCE, TNF-related activation-induced cytokine.
An important issue in antitumour therapy is determining which type of agonist will be most effective. Studies that use more stringent (that is, less immunogenic) tumour models have indicated that treatment with a single stimulatory agonist is not effective and that co-administration of a second agonist or another factor that stimulates T-cell function, such as granulocyte/macrophage colony-simulating factor (Gm-CSF) or IL-12 (REFS 119-121), results in significantly greater antitumour reactivity. Furthermore, Fc fusion proteins may have better activity than antibodies122. The development of reagents that are hexameric123,124 and/or directly linked to antibody fragments that specifically target the tumour or tumour site124,125, or the development of RNA aptamers126,127, may lead to more effective therapeutic options. In addition, these reagents may have fewer safety issues as they would be less likely to induce a robust immune response against the reagent or to cause sustained immune responses that lead to side effects such as a cytokine storm. Although the development of auto-immune disease symptoms is an inherent risk of tumour immunotherapy, the benefits will probably outweigh the risks for most patients with cancer. As the expression of OX40 and 4-1BB is mainly restricted to activated immune cells, targeting these molecules is less likely to induce autoimmune side effects than targeting molecules such as CD27, which is constitutively expressed.
Additional antitumour strategies involve the transduction of tumour cells with CD70, OX40L or 4-1BBL using viral vectors that express the genes encoding these molecules. This can be carried out ex vivo, followed by re-implantation of the tumour cells into the host, or in vivo by directly injecting the viral vector into the tumour. This approach causes the tumour to become more immunogenic and leads to the stimulation of an immune response. A combined approach, such as the transduction of a TNF ligand together with Gm-CSF, IL-12 or CD80 into the tumour has also been found to be effective128-130. Cellular therapy with DCs engineered to express TNF ligands to boost antitumour T-cell responses is another strategy that has shown some success in mouse models131,132. Although cellular therapy approaches can be more complicated than treatment that involves a simple injection of an agonist, they have the potential to be more specific owing to the ability of DCs to present tumour antigens and to thereby target T cells that are specific for the tumour.
Finally, strategies to directly kill tumour cells by targeting them with antibodies specific for CD70 are currently being developed for clinical testing. In contrast to the limited expression of this molecule by normal cells, many tumours express CD70, and targeting CD70+ tumours with depleting or drug-conjugated CD70-specific antibodies has shown promising results in experimental models of cancer133,134. However, similar to other depletion strategies, there is the inherent potential that activated DCs, B cells and T cells may also be depleted, thereby putting the patient at risk of infection.
In summary, targeting any of the four TNF–TNFR interactions discussed here has great therapeutic potential based on the concept that the continued activity of an effector cell (pathogenic or protective) requires signals from these molecules to maintain cell division, allow survival and promote cytokine production. The fact that several of the TNF superfamily members are inducibly expressed during the active phase (or phases) of disease or inflammation means that these molecules might be better therapeutic targets than more broadly expressed cell surface molecules.
Concluding remarks
Accumulating evidence has shown that many TNF superfamily molecules have a central role in immune regulation, immune-mediated diseases and cancer. The four interactions that are discussed here are only a part of the bigger superfamily; other co-stimulatory interactions, such as those of CD30, TNFR2, Hvem (herpes-virus entry mediator; also known as TNFRSF14), GITR (glucorticoid-induced TNFR-related protein; also known as TNFRSF18), TACI (also known as TNFRSF13B), CD40 and lymphotoxin-β receptor with their ligands, are equally noteworthy. It is clear that the interactions between OX40, 4-1BB, CD27 or DR3 with their ligands have important roles in controlling T-cell function and the interactions of T cells with other immune cells. In addition, preclinical studies in mouse models indicate that the therapeutic targeting of these interactions might have the same potential as targeting TNF, which has proven so successful.
- RNA aptamer
An oligonucleotide sequence that has the ability to recognize virtually any class of target molecules with high affinity and specificity. RNA aptamers are emerging as a class of molecules that rival antibodies in terms of therapeutic and diagnostic applications. They provide some advantages over antibodies, as they can be produced by chemical synthesis, have better storage properties and are less immunogenic.
- Cytokine storm
A sudden surge in the circulating levels of pro-inflammatory cytokines, such as interleukin-1 (IL-1), IL-6, tumour necrosis factor and interferon-γ.
There are still great gaps in our knowledge regarding the activity, expression characteristics and involvement of these different molecules at various stages of the immune response and over the course of disease. Importantly, it is still unresolved how similar or dissimilar each of these receptors are to one another in terms of the signalling complexes they form, the level of signalling that each one triggers and whether different TNF–TNFR interactions induce different cellular responses. In the case of therapy, it is not clear which molecule will be the best target; this is a particularly hard question to address given the difficulty in translating preclinical mouse studies to humans. To better resolve these issues, further studies in animal models of disease are warranted that mimic specific stages of human disease more closely, and targeting combinations of these family members should be carried out to determine the extent of cooperation and overlap between these four ligand–receptor pairs. So far, clinical trials are in progress to test agonist antibodies that are specific for OX40 and 4-1BB in patients with cancer, OX40L-specific neutralizing antibodies in patients with asthma and CD70-specific depleting antibodies in patients with autoimmune disease and cancer. Further research will provide new information regarding the potential usefulness of these molecules in different clinical applications.
Supplementary Material
Acknowledgements
M.C. is supported by grants AI67341, CA91837, AI49453 and AI070535 from the National Institutes of Health.
Footnotes
DATABASES
Entrez Gene: http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?db=gene 4-1BB | 4-1BBL | CD27 | CD70 | DR3 | OX40 | OX40L | TL1A | TNF | TNFR1 | TNFR2
FURTHER INFORMATION
Michael Croft's homepage: http://www.liai.org/pages/faculty-croft
SUPPLEMENTARY INFORMATION
See online article: S1 (table)
ALL LINKS ARE ACTIVE IN THE ONLINE PDF
References
- 1.Croft M. Co-stimulatory members of the TNFR family: Keys to effective T-cell immunity? Nature Rev. Immunol. 2003;3:609–620. doi: 10.1038/nri1148. [DOI] [PubMed] [Google Scholar]
- 2.Croft M. Costimulation of T cells by OX40, 4-1BB, and CD27. Cytokine Growth Factor Rev. 2003;14:265–273. doi: 10.1016/s1359-6101(03)00025-x. [DOI] [PubMed] [Google Scholar]
- 3.Sugamura K, Ishii N, Weinberg AD. Therapeutic targeting of the effector T-cell co-stimulatory molecule OX40. Nature Rev. Immunol. 2004;4:420–431. doi: 10.1038/nri1371. [DOI] [PubMed] [Google Scholar]
- 4.Borst J, Hendriks J, Xiao Y. CD27 and CD70 in T cell and B cell activation. Curr. Opin. Immunol. 2005;17:275–281. doi: 10.1016/j.coi.2005.04.004. [DOI] [PubMed] [Google Scholar]
- 5.Watts TH. TNF/TNFR family members in costimulation of T cell responses. Annu. Rev. Immunol. 2005;23:23–68. doi: 10.1146/annurev.immunol.23.021704.115839. [DOI] [PubMed] [Google Scholar]
- 6.Vinay DS, Kwon BS. Immunotherapy targeting 4-1BB and its ligand. Int. J. Hematol. 2006;83:23–28. doi: 10.1532/IJH97.05125. [DOI] [PubMed] [Google Scholar]
- 7.Meylan F, et al. The TNF-family receptor DR3 is essential for diverse T cell-mediated inflammatory diseases. Immunity. 2008;29:79–89. doi: 10.1016/j.immuni.2008.04.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gramaglia I, Weinberg AD, Lemon M, Croft M. OX-40 ligand: a potent costimulatory molecule for sustaining primary CD4 T cell responses. J. Immunol. 1998;161:6510–6517. [PubMed] [Google Scholar]
- 9.Rogers PR, Song J, Gramaglia I, Killeen N, Croft M. OX40 promotes Bcl-xL and Bcl-2 expression and is essential for long-term survival of CD4 T cells. Immunity. 2001;15:445–455. doi: 10.1016/s1074-7613(01)00191-1. [DOI] [PubMed] [Google Scholar]
- 10.Dawicki W, Bertram EM, Sharpe AH, Watts TH. 4-1BB and OX40 act independently to facilitate robust CD8 and CD4 recall responses. J. Immunol. 2004;173:5944–5951. doi: 10.4049/jimmunol.173.10.5944. [DOI] [PubMed] [Google Scholar]
- 11.Lee SW, et al. Functional dichotomy between OX40 and 4-1BB in modulating effector CD8 T cell responses. J. Immunol. 2006;177:4464–4472. doi: 10.4049/jimmunol.177.7.4464. [DOI] [PubMed] [Google Scholar]
- 12.Migone TS, et al. TL1A is a TNF-like ligand for DR3 and TR6/DcR3 and functions as a T cell costimulator. Immunity. 2002;16:479–492. doi: 10.1016/s1074-7613(02)00283-2. [DOI] [PubMed] [Google Scholar]
- 13.Futagawa T, et al. Expression and function of 4-1BB and 4-1BB ligand on murine dendritic cells. Int. Immunol. 2002;14:275–286. doi: 10.1093/intimm/14.3.275. [DOI] [PubMed] [Google Scholar]
- 14.Prehn JL, et al. The T cell costimulator TL1A is induced by FcγR signaling in human monocytes and dendritic cells. J. Immunol. 2007;178:4033–4038. doi: 10.4049/jimmunol.178.7.4033. [DOI] [PubMed] [Google Scholar]
- 15.Taraban VY, Rowley TF, Tough DF, Al-Shamkhani A. Requirement for CD70 in CD4+ Th cell-dependent and innate receptor-mediated CD8+ T cell priming. J. Immunol. 2006;177:2969–2975. doi: 10.4049/jimmunol.177.5.2969. [DOI] [PubMed] [Google Scholar]
- 16.Sanchez PJ, McWilliams JA, Haluszczak C, Yagita H, Kedl RM. Combined TLR/CD40 stimulation mediates potent cellular immunity by regulating dendritic cell expression of CD70 in vivo. J. Immunol. 2007;178:1564–1572. doi: 10.4049/jimmunol.178.3.1564. [DOI] [PubMed] [Google Scholar]
- 17.Ohshima Y, et al. Expression and function of OX40 ligand on human dendritic cells. J. Immunol. 1997;159:3838–3848. [PubMed] [Google Scholar]
- 18.Murata K, et al. Impairment of antigen-presenting cell function in mice lacking expression of OX40 ligand. J. Exp. Med. 2000;191:365–374. doi: 10.1084/jem.191.2.365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Maxwell JR, et al. IL-18 bridges innate and adaptive immunity through IFN-γ and the CD134 pathway. J. Immunol. 2006;177:234–245. doi: 10.4049/jimmunol.177.1.234. [DOI] [PubMed] [Google Scholar]
- 20.Ito T, et al. TSLP-activated dendritic cells induce an inflammatory T helper type 2 cell response through OX40 ligand. J. Exp. Med. 2005;202:1213–1223. doi: 10.1084/jem.20051135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Agematsu K, et al. Direct cellular communications between CD45R0 and CD45RA T cell subsets via CD27/CD70. J. Immunol. 1995;154:3627–3635. [PubMed] [Google Scholar]
- 22.Soroosh P, Ine S, Sugamura K, Ishii N. OX40–OX40 ligand interaction through T cell–T cell contact contributes to CD4 T cell longevity. J. Immunol. 2006;176:5975–5987. doi: 10.4049/jimmunol.176.10.5975. [DOI] [PubMed] [Google Scholar]
- 23.Burgess JK, et al. Detection and characterization of OX40 ligand expression in human airway smooth muscle cells: a possible role in asthma? J. Allergy Clin. Immunol. 2004;113:683–689. doi: 10.1016/j.jaci.2003.12.311. [DOI] [PubMed] [Google Scholar]
- 24.Souza HS, Elia CC, Spencer J, MacDonald TT. Expression of lymphocyte–endothelial receptor-ligand pairs, α4β7/MAdCAM-1 and OX40/OX40 ligand in the colon and jejunum of patients with inflammatory bowel disease. Gut. 1999;45:856–863. doi: 10.1136/gut.45.6.856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Seko Y, et al. Expression of tumor necrosis factor ligand superfamily costimulatory molecules CD27L, CD30L, OX40L and 4-1BBL in the heart of patients with acute myocarditis and dilated cardiomyopathy. Cardiovasc. Pathol. 2002;11:166–170. doi: 10.1016/s1054-8807(02)00101-1. [DOI] [PubMed] [Google Scholar]
- 26.Nolte MA, et al. Immune activation modulates hematopoiesis through interactions between CD27 and CD70. Nature Immunol. 2005;6:412–418. doi: 10.1038/ni1174. [DOI] [PubMed] [Google Scholar]
- 27.Lee SW, et al. Identification of regulatory functions for 4-1BB and 4-1BBL in myelopoiesis and the development of dendritic cells. Nature Immunol. 2008;9:917–926. doi: 10.1038/ni.1632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ochsenbein AF, et al. CD27 expression promotes long-term survival of functional effector-memory CD8+ cytotoxic T lymphocytes in HIV-infected patients. J. Exp. Med. 2004;200:1407–1417. doi: 10.1084/jem.20040717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wang X, et al. Positional identification of TNFSF4, encoding OX40 ligand, as a gene that influences atherosclerosis susceptibility. Nature Genet. 2005;37:365–372. doi: 10.1038/ng1524. [DOI] [PubMed] [Google Scholar]
- 30.Olofsson PS, et al. CD137 is expressed in human atherosclerosis and promotes development of plaque inflammation in hypercholesterolemic mice. Circulation. 2008;117:1292–1301. doi: 10.1161/CIRCULATIONAHA.107.699173. [DOI] [PubMed] [Google Scholar]
- 31.Lee HW, et al. 4-1BB promotes the survival of CD8+ T lymphocytes by increasing expression of Bcl-xL and Bfl-1. J. Immunol. 2002;169:4882–4888. doi: 10.4049/jimmunol.169.9.4882. [DOI] [PubMed] [Google Scholar]
- 32.van Oosterwijk MF, et al. CD27–CD70 interactions sensitise naive CD4+ T cells for IL-12-induced Th1 cell development. Int. Immunol. 2007;19:713–718. doi: 10.1093/intimm/dxm033. [DOI] [PubMed] [Google Scholar]
- 33.Song J, et al. The costimulation-regulated duration of PKB activation controls T cell longevity. Nature Immunol. 2004;5:150–158. doi: 10.1038/ni1030. [DOI] [PubMed] [Google Scholar]
- 34.Song J, So T, Croft M. Activation of NF-κB1 by OX40 contributes to antigen-driven T cell expansion and survival. J. Immunol. 2008;180:7240–7248. doi: 10.4049/jimmunol.180.11.7240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sabbagh L, Pulle G, Liu Y, Tsitsikov EN, Watts TH. ERK-dependent Bim modulation downstream of the 4-1BB–TRAF1 signaling axis is a critical mediator of CD8 T cell survival in vivo. J. Immunol. 2008;180:8093–8101. doi: 10.4049/jimmunol.180.12.8093. [DOI] [PubMed] [Google Scholar]
- 36.Chinnaiyan AM, et al. Signal transduction by DR3, a death domain-containing receptor related to TNFR-1 and CD95. Science. 1996;274:990–992. doi: 10.1126/science.274.5289.990. [DOI] [PubMed] [Google Scholar]
- 37.Ramakrishnan P, Wang W, Wallach D. Receptor-specific signaling for both the alternative and the canonical NF-κB activation pathways by NF-κB-inducing kinase. Immunity. 2004;21:477–489. doi: 10.1016/j.immuni.2004.08.009. [DOI] [PubMed] [Google Scholar]
- 38.Wen L, Zhuang L, Luo X, Wei P. TL1A-induced NF-κB activation and c-IAP2 production prevent DR3-mediated apoptosis in TF-1 cells. J. Biol. Chem. 2003;278:39251–39258. doi: 10.1074/jbc.M305833200. [DOI] [PubMed] [Google Scholar]
- 39.Song J, So T, Cheng M, Tang X, Croft M. Sustained survivin expression from OX40 costimulatory signals drives T cell clonal expansion. Immunity. 2005;22:621–631. doi: 10.1016/j.immuni.2005.03.012.This report, together with references 9, 33 and 34, defines the main events downstream of OX40 activation that regulate T-cell division and survival.
- 40.Song J, Salek-Ardakani S, So T, Croft M. The kinases aurora B and mTOR regulate the G1–S cell cycle progression of T lymphocytes. Nature Immunol. 2007;8:64–73. doi: 10.1038/ni1413. [DOI] [PubMed] [Google Scholar]
- 41.Lee HW, Nam KO, Park SJ, Kwon BS. 4-1BB enhances CD8+ T cell expansion by regulating cell cycle progression through changes in expression of cyclins D and E and cyclin-dependent kinase inhibitor p27kip1. Eur. J. Immunol. 2003;33:2133–2141. doi: 10.1002/eji.200323996.Together with references 31 and 35, this paper defines how 4-1BB promotes the survival and proliferation of CD8+ T cells.
- 42.Akiba H, et al. CD27, a member of the tumor necrosis factor receptor superfamily, activates NF-κB and stress-activated protein kinase/c-Jun N-terminal kinase via TRAF2, TRAF5, and NF-κB-inducing kinase. J. Biol. Chem. 1998;273:13353–13358. doi: 10.1074/jbc.273.21.13353. [DOI] [PubMed] [Google Scholar]
- 43.Cannons JL, Choi Y, Watts TH. Role of TNF receptor-associated factor 2 and p38 mitogen-activated protein kinase activation during 4-1BB-dependent immune response. J. Immunol. 2000;165:6193–6204. doi: 10.4049/jimmunol.165.11.6193. [DOI] [PubMed] [Google Scholar]
- 44.So T, Song J, Sugie K, Altman A, Croft M. Signals from OX40 regulate nuclear factor of activated T cells c1 and T cell helper 2 lineage commitment. Proc. Natl Acad. Sci. USA. 2006;103:3740–3745. doi: 10.1073/pnas.0600205103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tan JT, et al. Analysis of expression and function of the costimulatory molecule 4-1BB in alloimmune responses. Transplantation. 2000;70:175–183. [PubMed] [Google Scholar]
- 46.Williams CA, Murray SE, Weinberg AD, Parker DC. OX40-mediated differentiation to effector function requires IL-2 receptor signaling but not CD28, CD40, IL-12Rβ2, or T-bet. J. Immunol. 2007;178:7694–7702. doi: 10.4049/jimmunol.178.12.7694. [DOI] [PubMed] [Google Scholar]
- 47.Ruby CE, Montler R, Zheng R, Shu S, Weinberg AD. IL-12 is required for anti-OX40-mediated CD4 T cell survival. J. Immunol. 2008;180:2140–2148. doi: 10.4049/jimmunol.180.4.2140. [DOI] [PubMed] [Google Scholar]
- 48.Arens R, et al. Signaling through CD70 regulates B cell activation and IgG production. J. Immunol. 2004;173:3901–3908. doi: 10.4049/jimmunol.173.6.3901. [DOI] [PubMed] [Google Scholar]
- 49.Langstein J, et al. CD137 (ILA/4-1BB), a member of the TNF receptor family, induces monocyte activation via bidirectional signaling. J. Immunol. 1998;160:2488–2494. [PubMed] [Google Scholar]
- 50.Kang YJ, et al. Cell surface 4-1BBL mediates sequential signaling pathways ‘downstream’ of TLR and is required for sustained TNF production in macrophages. Nature Immunol. 2007;8:601–609. doi: 10.1038/ni1471.Together with reference 27, this work provides new insights into the modulatory role of 4-1BBL in controlling diverse aspects of an immune response.
- 51.Fang L, Adkins B, Deyev V, Podack ER. Essential role of TNF receptor superfamily 25 (TNFRSF25) in the development of allergic lung inflammation. J. Exp. Med. 2008;205:1037–1048. doi: 10.1084/jem.20072528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Takedatsu H, et al. TL1A (TNFSF15) regulates the development of chronic colitis by modulating both T-helper 1 and T-helper 17 activation. Gastroenterology. 2008;135:552–567. doi: 10.1053/j.gastro.2008.04.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Pappu BP, et al. TL1A-DR3 interaction regulates Th17 cell function and Th17-mediated autoimmune disease. J. Exp. Med. 2008;205:1049–1062. doi: 10.1084/jem.20071364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hendriks J, et al. CD27 is required for generation and long-term maintenance of T cell immunity. Nature Immunol. 2000;1:433–440. doi: 10.1038/80877. [DOI] [PubMed] [Google Scholar]
- 55.Hendriks J, et al. During viral infection of the respiratory tract, CD27, 4-1BB, and OX40 collectively determine formation of CD8 memory T cells and their capacity for secondary expansion. J. Immunol. 2005;175:1665–1676. doi: 10.4049/jimmunol.175.3.1665. [DOI] [PubMed] [Google Scholar]
- 56.Zingoni A, et al. Cross-talk between activated human NK cells and CD4+ T cells via OX40–OX40 ligand interactions. J. Immunol. 2004;173:3716–3724. doi: 10.4049/jimmunol.173.6.3716. [DOI] [PubMed] [Google Scholar]
- 57.Liu C, et al. Plasmacytoid dendritic cells induce NK cell-dependent, tumor antigen-specific T cell cross-priming and tumor regression in mice. J. Clin. Invest. 2008;118:1165–1175. doi: 10.1172/JCI33583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kelly JM, et al. Induction of tumor-specific T cell memory by NK cell-mediated tumor rejection. Nature Immunol. 2002;3:83–90. doi: 10.1038/ni746. [DOI] [PubMed] [Google Scholar]
- 59.Papadakis KA, et al. TL1A synergizes with IL-12 and IL-18 to enhance IFN-γ production in human T cells and NK cells. J. Immunol. 2004;172:7002–7007. doi: 10.4049/jimmunol.172.11.7002. [DOI] [PubMed] [Google Scholar]
- 60.Wilcox RA, Tamada K, Strome SE, Chen L. Signaling through NK cell-associated CD137 promotes both helper function for CD8+ cytolytic T cells and responsiveness to IL-2 but not cytolytic activity. J. Immunol. 2002;169:4230–4236. doi: 10.4049/jimmunol.169.8.4230. [DOI] [PubMed] [Google Scholar]
- 61.Zaini J, et al. OX40 ligand expressed by DCs costimulates NKT and CD4+ Th cell antitumor immunity in mice. J. Clin. Invest. 2007;117:3330–3338. doi: 10.1172/JCI32693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kim DH, et al. 4-1BB engagement costimulates NKT cell activation and exacerbates NKT cell ligand-induced airway hyperresponsiveness and inflammation. J. Immunol. 2008;180:2062–2068. doi: 10.4049/jimmunol.180.4.2062. [DOI] [PubMed] [Google Scholar]
- 63.Taraban VY, et al. Invariant NKT cells promote CD8 cytotoxic T cell responses by inducing CD70 expression on dendritic cells. J. Immunol. 2008;180:4615–4620. doi: 10.4049/jimmunol.180.7.4615.This work, as well as references 51, 61 and 62, provides new insights into the role of TNF superfamily molecule interactions that occur between APCs, NKT cells and T cells.
- 64.Xiao Y, Hendriks J, Langerak P, Jacobs H, Borst J. CD27 is acquired by primed B cells at the centroblast stage and promotes germinal center formation. J. Immunol. 2004;172:7432–7441. doi: 10.4049/jimmunol.172.12.7432. [DOI] [PubMed] [Google Scholar]
- 65.Morimoto S, et al. CD134L engagement enhances human B cell Ig production: CD154/CD40, CD70/CD27, and CD134/CD134L interactions coordinately regulate T cell-dependent B cell responses. J. Immunol. 2000;164:4097–4104. doi: 10.4049/jimmunol.164.8.4097. [DOI] [PubMed] [Google Scholar]
- 66.Kashiwakura J, Yokoi H, Saito H, Okayama Y. T cell proliferation by direct cross-talk between OX40 ligand on human mast cells and OX40 on human T cells: comparison of gene expression profiles between human tonsillar and lung-cultured mast cells. J. Immunol. 2004;173:5247–5257. doi: 10.4049/jimmunol.173.8.5247. [DOI] [PubMed] [Google Scholar]
- 67.Nakae S, et al. Mast cells enhance T cell activation: importance of mast cell costimulatory molecules and secreted TNF. J. Immunol. 2006;176:2238–2248. doi: 10.4049/jimmunol.176.4.2238. [DOI] [PubMed] [Google Scholar]
- 68.Nishimoto H, et al. Costimulation of mast cells by 4-1BB, a member of the tumor necrosis factor receptor superfamily, with the high-affinity IgE receptor. Blood. 2005;106:4241–4248. doi: 10.1182/blood-2005-04-1358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Baumann R, et al. Functional expression of CD134 by neutrophils. Eur. J. Immunol. 2004;34:2268–2275. doi: 10.1002/eji.200424863. [DOI] [PubMed] [Google Scholar]
- 70.Kim MY, et al. CD4+ CD3− accessory cells costimulate primed CD4 T cells through OX40 and CD30 at sites where t cells collaborate with B cells. Immunity. 2003;18:643–654. doi: 10.1016/s1074-7613(03)00110-9. [DOI] [PubMed] [Google Scholar]
- 71.Kim MY, et al. Neonatal and adult CD4+ CD3− cells share similar gene expression profile, and neonatal cells up-regulate OX40 ligand in response to TL1A (TNFSF15) J. Immunol. 2006;177:3074–3081. doi: 10.4049/jimmunol.177.5.3074. [DOI] [PubMed] [Google Scholar]
- 72.So T, Lee SW, Croft M. Immune regulation and control of regulatory T cells by OX40 and 4-1BB. Cytokine Growth Factor Rev. 2008;19:253–262. doi: 10.1016/j.cytogfr.2008.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.So T, Croft M. Cutting Edge: OX40 inhibits TGF-β- and antigen-driven conversion of naive CD4 T cells into CD25+Foxp3+ T cells. J. Immunol. 2007;179:1427–1430. doi: 10.4049/jimmunol.179.3.1427. [DOI] [PubMed] [Google Scholar]
- 74.Vu MD, et al. OX40 costimulation turns off Foxp3+ Tregs. Blood. 2007;110:2501–2510. doi: 10.1182/blood-2007-01-070748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Chen M, Xiao X, Demirci G, Li XC. OX40 controls islet allograft tolerance in CD154 deficient mice by regulating FOXP3+ Tregs. Transplantation. 2008;85:1659–1662. doi: 10.1097/TP.0b013e3181726987. [DOI] [PubMed] [Google Scholar]
- 76.Ito T, et al. OX40 ligand shuts down IL-10-producing regulatory T cells. Proc. Natl Acad. Sci. USA. 2006;103:13138–13143. doi: 10.1073/pnas.0603107103.References 73, 74 and 76 show that OX40 blocks the differentiation of inducible TReg cells.
- 77.Takeda I, et al. Distinct roles for the OX40–OX40 ligand interaction in regulatory and nonregulatory T cells. J. Immunol. 2004;172:3580–3589. doi: 10.4049/jimmunol.172.6.3580. [DOI] [PubMed] [Google Scholar]
- 78.Choi BK, et al. 4-1BB-dependent inhibition of immunosuppression by activated CD4+ CD25+ T cells. J. Leukoc. Biol. 2004;75:785–791. doi: 10.1189/jlb.1003491. [DOI] [PubMed] [Google Scholar]
- 79.Valzasina B, et al. Triggering of OX40 (CD134) on CD4+ CD25+ T cells blocks their inhibitory activity: a novel regulatory role for OX40 and its comparison with GITR. Blood. 2005;105:2845–2851. doi: 10.1182/blood-2004-07-2959. [DOI] [PubMed] [Google Scholar]
- 80.Kroemer A, et al. OX40 controls functionally different T cell subsets and their resistance to depletion therapy. J. Immunol. 2007;179:5584–5591. doi: 10.4049/jimmunol.179.8.5584. [DOI] [PubMed] [Google Scholar]
- 81.Piconese S, Valzasina B, Colombo MP. OX40 triggering blocks suppression by regulatory T cells and facilitates tumor rejection. J. Exp. Med. 2008;205:825–839. doi: 10.1084/jem.20071341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Robertson SJ, et al. CD137 costimulation of CD8+ T cells confers resistance to suppression by virus-induced regulatory T cells. J. Immunol. 2008;180:5267–5274. doi: 10.4049/jimmunol.180.8.5267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Myers L, Takahashi C, Mittler RS, Rossi RJ, Vella AT. Effector CD8 T cells possess suppressor function after 4-1BB and Toll-like receptor triggering. Proc. Natl Acad. Sci. USA. 2003;100:5348–5353. doi: 10.1073/pnas.0837611100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Zheng G, Wang B, Chen A. The 4-1BB costimulation augments the proliferation of CD4+ CD25+ regulatory T cells. J. Immunol. 2004;173:2428–2434. doi: 10.4049/jimmunol.173.4.2428. [DOI] [PubMed] [Google Scholar]
- 85.Elpek KG, et al. Ex vivo expansion of CD4+ CD25+ FoxP3+ T regulatory cells based on synergy between IL-2 and 4-1BB signaling. J. Immunol. 2007;179:7295–7304. doi: 10.4049/jimmunol.179.11.7295. [DOI] [PubMed] [Google Scholar]
- 86.Golovina TN, et al. CD28 costimulation is essential for human T regulatory expansion and function. J. Immunol. 2008;181:2855–2868. doi: 10.4049/jimmunol.181.4.2855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Weinberg AD, et al. Selective depletion of myelin-reactive T cells with the anti-OX-40 antibody ameliorates autoimmune encephalomyelitis. Nature Med. 1996;2:183–189. doi: 10.1038/nm0296-183.This report was the first to show that depletion of cells expressing a TNFR was effective for blocking autoimmunity.
- 88.Boot EP, et al. CD134 as target for specific drug delivery to auto-aggressive CD4+ T cells in adjuvant arthritis. Arthritis Res. Ther. 2005;7:R604–R615. doi: 10.1186/ar1722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Taylor L, et al. In vitro and in vivo activities of OX40 (CD134)–IgG fusion protein isoforms with different levels of immune-effector functions. J. Leukoc. Biol. 2002;72:522–529. [PubMed] [Google Scholar]
- 90.Weinberg AD, Wegmann KW, Funatake C, Whitham RH. Blocking OX-40/OX-40 ligand interaction in vitro and in vivo leads to decreased T cell function and amelioration of experimental allergic encephalomyelitis. J. Immunol. 1999;162:1818–1826. [PubMed] [Google Scholar]
- 91.Higgins LM, et al. Regulation of T cell activation in vitro and in vivo by targeting the OX40–OX40 ligand interaction: amelioration of ongoing inflammatory bowel disease with an OX40–IgG fusion protein, but not with an OX40 ligand–IgG fusion protein. J. Immunol. 1999;162:486–493. [PubMed] [Google Scholar]
- 92.Yoshioka T, et al. Contribution of OX40/OX40 ligand interaction to the pathogenesis of rheumatoid arthritis. Eur. J. Immunol. 2000;30:2815–2823. doi: 10.1002/1521-4141(200010)30:10<2815::AID-IMMU2815>3.0.CO;2-#. [DOI] [PubMed] [Google Scholar]
- 93.Salek-Ardakani S, et al. OX40 (CD134) controls memory T helper 2 cells that drive lung inflammation. J. Exp. Med. 2003;198:315–324. doi: 10.1084/jem.20021937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Obermeier F, et al. OX40/OX40L interaction induces the expression of CXCR5 and contributes to chronic colitis induced by dextran sulfate sodium in mice. Eur. J. Immunol. 2003;33:3265–3274. doi: 10.1002/eji.200324124. [DOI] [PubMed] [Google Scholar]
- 95.Totsuka T, et al. Therapeutic effect of anti-OX40L and anti-TNF-α MAbs in a murine model of chronic colitis. Am. J. Physiol. Gastrointest. Liver Physiol. 2003;284:G595–G603. doi: 10.1152/ajpgi.00450.2002. [DOI] [PubMed] [Google Scholar]
- 96.Pakala SV, Bansal-Pakala P, Halteman BS, Croft M. Prevention of diabetes in NOD mice at a late stage by targeting OX40/OX40 ligand interactions. Eur. J. Immunol. 2004;34:3039–3046. doi: 10.1002/eji.200425141. [DOI] [PubMed] [Google Scholar]
- 97.Seshasayee D, et al. In vivo blockade of OX40 ligand inhibits thymic stromal lymphopoietin driven atopic inflammation. J. Clin. Invest. 2007;117:3868–3878. doi: 10.1172/JCI33559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Bull. MJ, et al. The death receptor 3-TNF-like protein 1A pathway drives adverse bone pathology in inflammatory arthritis. J. Exp. Med. 2008;205:2457–2464. doi: 10.1084/jem.20072378.Together with references 7, 51 and 53, this report shows that a deficiency in the expression of DR3 or TL1A strongly impairs the development of several inflammatory diseases.
- 99.Rowley TF, Al-Shamkhani A. Stimulation by soluble CD70 promotes strong primary and secondary CD8+ cytotoxic T cell responses in vivo. J. Immunol. 2004;172:6039–6046. doi: 10.4049/jimmunol.172.10.6039. [DOI] [PubMed] [Google Scholar]
- 100.Laouar A, et al. CD70+ antigen-presenting cells control the proliferation and differentiation of T cells in the intestinal mucosa. Nature Immunol. 2005;6:698–706. doi: 10.1038/ni1212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Seo SK, et al. Blocking 4-1BB/4-1BB ligand interactions prevents herpetic stromal keratitis. J. Immunol. 2003;171:576–583. doi: 10.4049/jimmunol.171.2.576. [DOI] [PubMed] [Google Scholar]
- 102.Lee SC, et al. 4-1BB (CD137) is required for rapid clearance of Listeria monocytogenes infection. Infect. Immun. 2005;73:5144–5151. doi: 10.1128/IAI.73.8.5144-5151.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Bertram EM, Lau P, Watts TH. Temporal segregation of 4-1BB versus CD28-mediated costimulation: 4-1BB ligand influences T cell numbers late in the primary response and regulates the size of the T cell memory response following influenza infection. J. Immunol. 2002;168:3777–3785. doi: 10.4049/jimmunol.168.8.3777. [DOI] [PubMed] [Google Scholar]
- 104.Keller AM, Schildknecht A, Xiao Y, van den Broek M, Borst J. Expression of costimulatory ligand CD70 on steady-state dendritic cells breaks CD8+ T cell tolerance and permits effective immunity. Immunity. 2008;29:934–946. doi: 10.1016/j.immuni.2008.10.009.References 15, 16, 28, 54, 99, 100 and 104 show the important role for CD27 and CD70 in controlling the priming of CD8+ T cells.
- 105.Curry AJ, et al. OX40 (CD134) blockade inhibits the co-stimulatory cascade and promotes heart allograft survival. Transplantation. 2004;78:807–814. doi: 10.1097/01.tp.0000131670.99000.54. [DOI] [PubMed] [Google Scholar]
- 106.Yuan X, et al. The role of the CD134–CD134 ligand costimulatory pathway in alloimmune responses in vivo. J. Immunol. 2003;170:2949–2955. doi: 10.4049/jimmunol.170.6.2949. [DOI] [PubMed] [Google Scholar]
- 107.Demirci G, et al. Critical role of OX40 in CD28 and CD154-independent rejection. J. Immunol. 2004;172:1691–1698. doi: 10.4049/jimmunol.172.3.1691. [DOI] [PubMed] [Google Scholar]
- 108.Yamada A, et al. CD70 signaling is critical for CD28- ndependent CD8+ T cell-mediated alloimmune responses in vivo. J. Immunol. 2005;174:1357–1364. doi: 10.4049/jimmunol.174.3.1357. [DOI] [PubMed] [Google Scholar]
- 109.DeBenedette MA, et al. Analysis of 4-1BB ligand (4-1BBL)-deficient mice and of mice lacking both 4-1BBL and CD28 reveals a role for 4-1BBL in skin allograft rejection and in the cytotoxic T cell response to influenza virus. J. Immunol. 1999;163:4833–4841. [PubMed] [Google Scholar]
- 110.Seo SK, et al. 4-1BB-mediated immunotherapy of rheumatoid arthritis. Nature Med. 2004;10:1088–1094. doi: 10.1038/nm1107. This study shows that administration of an agonist to 4-1BB can prevent autoimmune disease
- 111.Lee J, et al. Administration of agonistic anti-4-1BB monoclonal antibody leads to the amelioration of inflammatory bowel disease. Immunol. Lett. 2005;101:210–216. doi: 10.1016/j.imlet.2005.06.001. [DOI] [PubMed] [Google Scholar]
- 112.Choi BK, Asai T, Vinay DS, Kim YH, Kwon BS. 4-1BB-mediated amelioration of experimental autoimmune uveoretinitis is caused by indoleamine 2, 3-dioxygenase-dependent mechanisms. Cytokine. 2006;34:233–242. doi: 10.1016/j.cyto.2006.04.008. [DOI] [PubMed] [Google Scholar]
- 113.Polte T, et al. CD137-mediated immunotherapy for allergic asthma. J. Clin. Invest. 2006;116:1025–1036. doi: 10.1172/JCI23792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Irie J, Wu Y, Kachapati K, Mittler RS, Ridgway WM. Modulating protective and pathogenic CD4+ subsets via CD137 in type 1 diabetes. Diabetes. 2007;56:186–196. doi: 10.2337/db06-0793. [DOI] [PubMed] [Google Scholar]
- 115.Schraven B, Kalinke U. CD28 superagonists: what makes the difference in humans? Immunity. 2008;28:591–595. doi: 10.1016/j.immuni.2008.04.003. [DOI] [PubMed] [Google Scholar]
- 116.Hippen KL, et al. Umbilical cord blood regulatory T-cell expansion and functional effects of tumor necrosis factor receptor family members OX40 and 4-1BB expressed on artificial antigen-presenting cells. Blood. 2008;112:2847–2857. doi: 10.1182/blood-2008-01-132951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Melero I, et al. Monoclonal antibodies against the 4-1BB T-cell activation molecule eradicate established tumors. Nature Med. 1997;3:682–685. doi: 10.1038/nm0697-682. [DOI] [PubMed] [Google Scholar]
- 118.Weinberg AD, et al. Engagement of the OX-40 receptor in vivo enhances antitumor immunity. J. Immunol. 2000;164:2160–2169. doi: 10.4049/jimmunol.164.4.2160. Together with reference 117, this work set the precedent for many future studies that targeted 4-1BB and OX40 for the treatment of cancer
- 119.Pan P, Zang Y, Weber K, Meseck M, Chen S. OX40 ligation enhances primary and memory cytotoxic T lymphocyte responses in an immunotherapy for hepatic colon metastases. Mol. Ther. 2002;6:528–536. doi: 10.1006/mthe.2002.0699. [DOI] [PubMed] [Google Scholar]
- 120.Cuadros C, et al. Vaccination with dendritic cells pulsed with apoptotic tumors in combination with anti-OX40 and anti-4-1BB monoclonal antibodies induces T cell-mediated protective immunity in Her-2/neu transgenic mice. Int. J. Cancer. 2005;116:934–943. doi: 10.1002/ijc.21098. [DOI] [PubMed] [Google Scholar]
- 121.Murata S, et al. OX40 costimulation synergizes with GM-CSF whole-cell vaccination to overcome established CD8+ T cell tolerance to an endogenous tumor antigen. J. Immunol. 2006;176:974–983. doi: 10.4049/jimmunol.176.2.974. [DOI] [PubMed] [Google Scholar]
- 122.Sadun RE, et al. Fc–mOX40L fusion protein produces complete remission and enhanced survival in 2 murine tumor models. J. Immunother. 2008;31:235–245. doi: 10.1097/CJI.0b013e31816a88e0. [DOI] [PubMed] [Google Scholar]
- 123.Morris NP, et al. Development and characterization of recombinant human Fc:OX40L fusion protein linked via a coiled-coil trimerization domain. Mol. Immunol. 2007;44:3112–3121. doi: 10.1016/j.molimm.2007.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Muller N, Wyzgol A, Munkel S, Pfizenmaier K, Wajant H. Activity of soluble OX40 ligand is enhanced by oligomerization and cell surface immobilization. FEBS J. 2008;275:2296–2304. doi: 10.1111/j.1742-4658.2008.06382.x. [DOI] [PubMed] [Google Scholar]
- 125.Zhang N, et al. Targeted and untargeted CD137L fusion proteins for the immunotherapy of experimental solid tumors. Clin. Cancer Res. 2007;13:2758–2767. doi: 10.1158/1078-0432.CCR-06-2343. [DOI] [PubMed] [Google Scholar]
- 126.McNamara JO, et al. Multivalent 4-1BB binding aptamers costimulate CD8+ T cells and inhibit tumor growth in mice. J. Clin. Invest. 2008;118:376–386. doi: 10.1172/JCI33365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Dollins CM, et al. Assembling OX40 aptamers on a molecular scaffold to create a receptor-activating aptamer. Chem. Biol. 2008;15:675–682. doi: 10.1016/j.chembiol.2008.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Martinet O, et al. Immunomodulatory gene therapy with interleukin 12 and 4-1BB ligand: long- term remission of liver metastases in a mouse model. J. Natl Cancer Inst. 2000;92:931–936. doi: 10.1093/jnci/92.11.931. [DOI] [PubMed] [Google Scholar]
- 129.Douin-Echinard V, et al. The expression of CD70 and CD80 by gene-modified tumor cells induces an antitumor response depending on the MHC status. Cancer Gene Ther. 2000;7:1543–1556. doi: 10.1038/sj.cgt.7700268. [DOI] [PubMed] [Google Scholar]
- 130.Gri G, Gallo E, Di Carlo E, Musiani P, Colombo MP. OX40 ligand-transduced tumor cell vaccine synergizes with GM-CSF and requires CD40–APC signaling to boost the host T cell antitumor response. J. Immunol. 2003;170:99–106. doi: 10.4049/jimmunol.170.1.99. [DOI] [PubMed] [Google Scholar]
- 131.Dannull J, et al. Enhancing the immunostimulatory function of dendritic cells by transfection with mRNA encoding OX40 ligand. Blood. 2004;105:3206–3213. doi: 10.1182/blood-2004-10-3944. [DOI] [PubMed] [Google Scholar]
- 132.Yurkovetsky ZR, et al. Comparative analysis of antitumor activity of CD40L, RANKL, and 4-1BBL in vivo following intratumoral administration of viral vectors or transduced dendritic cells. J. Gene Med. 2006;8:129–137. doi: 10.1002/jgm.834. [DOI] [PubMed] [Google Scholar]
- 133.Law CL, et al. Lymphocyte activation antigen CD70 expressed by renal cell carcinoma is a potential therapeutic target for anti-CD70 antibody-drug conjugates. Cancer Res. 2006;66:2328–2337. doi: 10.1158/0008-5472.CAN-05-2883. [DOI] [PubMed] [Google Scholar]
- 134.McEarchern JA, et al. Engineered anti-CD70 antibody with multiple effector functions exhibits in vitro and in vivo antitumor activities. Blood. 2007;109:1185–1192. doi: 10.1182/blood-2006-07-034017.Together with reference 133, this paper shows that targeting CD70 to directly deplete tumour cells could be a promising new avenue for therapy.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.