

Abstract
Vascular endothelial growth factor (VEGF) binding induces phosphorylation of VEGF receptor-2 (VEGFR2) in tyrosine, which is followed by disruption of VE-cadherin-mediated cell-cell contacts of endothelial cells (ECs), thereby stimulating EC proliferation and migration to promote angiogenesis. Tyrosine phosphorylation events are controlled by the balance of activation of protein tyrosine kinases and protein tyrosine phosphatases (PTPs). Little is known about the role of endogenous PTPs in VEGF signaling in ECs. In this study, we found that PTP1B expression and activity are markedly increased in mice hindlimb ischemia model of angiogenesis. In ECs overexpression of PTP1B, but not catalytically inactive mutant PTP1B-C/S, inhibits VEGF-induced phosphorylation of VEGFR2 and ERK1/2 as well as EC proliferation, while knockdown of PTP1B by siRNA enhances these responses, suggesting that PTP1B negatively regulates VEGFR2 signaling in ECs. VEGF-induced p38MAP kinase phosphorylation and EC migration are not affected by PTP1B overexpression or knockdown. In vivo dephosphorylation and co-transfection assays reveal that PTP1B binds to VEGFR2 cytoplasmic domain in vivo, and directly dephosphorylates activated VEGFR2 immunoprecipitates from HUVECs. Overexpression of PTP1B stabilizes VE-cadherin-mediated cell-cell adhesions by reducing VE-cadherin tyrosine phosphorylation, while PTP1B siRNA causes opposite effects with increasing endothelial permeability as measured by transendothelial electrical resistance. In summary, PTP1B negatively regulates VEGFR2 receptor activation via binding to the VEGFR2 as well as stabilizes cell-cell adhesions through reducing tyrosine phosphorylation of VE-cadherin. Induction of PTP1B by hindlimb ischemia may represent important counter-regulatory mechanism that blunts over-activation of VEGFR2 during angiogenesis in vivo.
Keywords: Protein tyrosine phosphatase 1B, vascular endothelial growth factor, endothelial cell, cell-cell adhesions, angiogenesis
Introduction
Vascular endothelial growth factor (VEGF) induces angiogenesis by stimulating endothelial cell (EC) migration and proliferation primarily through the VEGF type2 receptor (VEGFR2, KDR/Flk-1)1. VEGF binding initiates autophosphorylation of VEGFR2, which in turn creates docking sites for Src homology domain-2 (SH2) domain containing adaptor molecules. This event is followed by activation of diverse key angiogenic enzymes such as ERK1/2 and p38 MAP kinase which are involved in EC proliferation and migration, respectively 2, 3. One of the initial responses of quiescent ECs to induce angiogenesis is the loss of established cell-cell contacts, which is followed by EC migration, proliferation and formation of capillary tube networks. The molecule primarily responsible for cell-cell adhesions of ECs is the transmembrane homophilic adhesion molecule, vascular endothelial (VE)-cadherin whose tyrosine phosphorylation is important for disruption of cell-cell contacts 4. Thus, tyrosine phosphorylation of VEGFR2 and VE-cadherin is important initial signaling events by which VEGF stimulates angiogenesis in ECs. Tyrosine phosphorylation events are controlled by the balance of activation of protein tyrosine kinases and protein tyrosine phosphatases (PTPs). PTP inhibitors accelerate neovascularization in rat ischemia hindlimb models 5, 6; however, little is known about role of endogenous PTPs in VEGF signaling in ECs.
In preliminary studies to identify PTPs involved in angiogenesis in vivo, we identified PTP1B as one of the major PTP whose expression is dramatically induced during angiogenesis in a mouse ischemia hindlimb model. PTP1B is the first PTP isolated in homogenous form, and is ubiquitously expressed enzymes 7. Studies using PTP1B-deficient mice show that PTP1B is a key negative regulator of insulin and leptin signaling and a therapeutic target for the type 2 diabetes and obesty 8, 9. PTP1B is also implicated in growth factor- 10 and integrin- 11 mediated signaling. Furthermore, PTP1B binds to E- and N-cadherin and regulates the cell-cell adhesions in neural retina cells and epithelial cells, respectively 12, 13. However, the specific role of PTP1B in VEGF signaling and VE-cadherin-mediated cell-cell adhesions in ECs is unknown.
We performed the present study to test the hypothesis that PTP1B may play an important role in VEGF signaling linked to angiogenesis. Using overexpression and knockdown strategies, we demonstrate that PTP1B negatively regulates VEGFR2 receptor autophosphorylation via binding to the VEGFR2, which in turn inhibits downstream ERK1/2 phosphorylation and EC proliferation. We also found that PTP1B associates with VE-cadherin, thereby reducing its tyrosine phosphorylation, which may contribute to stabilizing cell-cell adhesions. PTP1B expression and activity are markedly increased in a mouse hindlimb ischemia model, which may represent important counter-regulatory mechanism that blunts over-activation of VEGFR2 during angiogenesis in vivo.
Materials and Methods
Materials, cell culture, immunoprecipitation and immunoblotting, transient transfection of CHO cells, Adenovirus transduction, siRNA transfection, PTPase activity assay, In vivo receptor dephosphorylation assays, cell proliferation and migration assays, confocal immunofluorescence microscopy, transendothelial electrical resistance (TER) measurement, mouse ischemic hindlimb model and statistical analyses are described in the Material and Methods section in the online data supplement.
Results
PTP1B overexpression inhibits while PTP1B knockdown enhances VEGF-induced autophosphorylation of VEGFR
To determine the function of PTP1B in VEGF signaling, we examined the effect of PTP1B overexpression on VEGFR2 autophosphorylation in HUVECs. Figure 1 shows that transduction of adenovirus expressing wild-type PTP1B (Ad.PTP1B-WT) significantly increased PTP1B protein expression and activity (Fig. 1A), and inhibited VEGF-induced VEGFR2 tyrosine phosphorylation without affecting VEGFR2 expression (Fig. 1B). In contrast, Ad.PTP1B-C/S (catalytically inactive C215S mutant), which increased PTP1B protein expression at similar extent as PTP1B-WT but had no effect on PTP1B activity (Fig. 1A), significantly enhanced VEGF-induced phosphorylation of VEGFR2 (Fig. 1B). These results suggest that overexpression of PTP1B inhibits VEGFR2 phosphorylation in a catalytic activity-dependent manner, and that PTP1B-C/S protects from dephosphorylation by endogenous PTP1B. In contrast, Figure 2 shows that knockdown of endogenous PTP1B protein and specific activity with siRNA (Fig. 2A) enhanced VEGF-induced VEGFR2 tyrosine phosphorylation (Fig. 2B). These results suggest that PTP1B functions as negative regulator for VEGFR2 autophosphorylation.
Figure 1. PTP1B overexpression inhibits VEGF-induced autophosphorylation of VEGFR2.
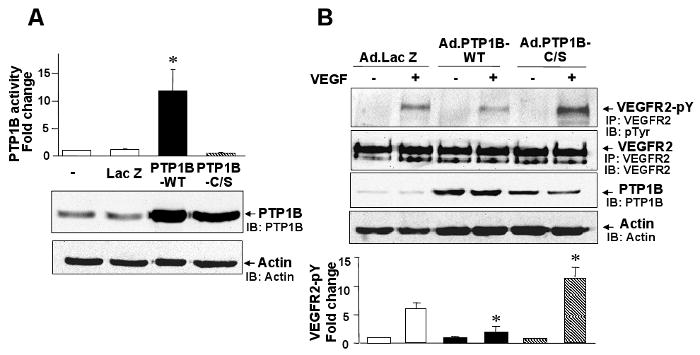
HUVECs were infected with Ad.LacZ (control), Ad.PTP1B-WT, or Ad.PTP1B-C/S. A, PTP1B activity (upper graph) and PTP1B protein expression (lower blot) measured by western analysis with anti-PTP1B or actin antibody (loading control). PTP1B activity was expressed as fold change over basal (the ratio in untreated cells was set to 1). * P<0.05 for the increase induced by PTP1B overexpression. B, Cells were stimulated with VEGF (20 ng/ml) for 5 min, and lysates were immunoprecipitated with anti-VEGFR2 antibody, followed by immunoblotting with anti-phosphotyrosine (pTyr) antibody. Bottom panel shows averaged data, expressed as fold change over basal (means ± S.E., n=3). *P<0.05 vs. Ad.LacZ.
Figure 2. Knockdown of endogenous PTP1B by siRNA enhances VEGFR2 autophosphorylation.
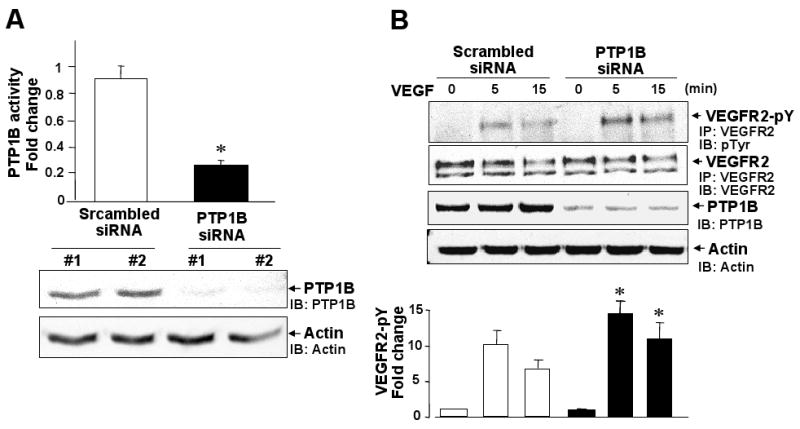
HUVECs were transfected with scrambled or PTP1B siRNA. A, PTP1B activity (upper graph) and PTP1B protein expression (lower blot). PTP1B activity was expressed as fold change from control (the ratio in untransfected cells was set to 1). * P<0.05 vs. Scrambled siRNA. B, Cells were stimulated with VEGF (20 ng/ml) and lysates were immunoprecipitated with anti-VEGFR2 antibody, followed by immunoblotting with anti-pTyr or VEGFR2 antibody. Bottom panel shows averaged data, expressed as fold change of phosphorylation over basal (means ± S.E., n=3). *P<0.05 vs. Scrambled siRNA at each time point.
PTP1B dephosphorylates and associates with VEGFR2
To determine if VEGFR2 can serve as a substrate for PTP1B, we performed in vivo dephosphorylation assay. Figure 3A shows that recombinant active PTP1B protein dephosphorylated VEGFR2 obtained from VEGFR2 immunoprecipitates of VEGF-stimulated HUVEC lysates, in a dose-dependent manner. To assess the mechanism by which PTP1B negatively regulates VEGFR2 phosphorylation, we examined whether PTP1B physically interacts to VEGFR2. Figure 3B shows that PTP1B was co-immunoprecipitated with VEGFR2 in HUVECs infected with Ad.PTP1B-WT, but not with Ad.LacZ, in basal state and after VEGF stimulation. These results suggest that PTP1B associates with VEGFR2 in a phosphotyrosine-independent manner, and that endogenous PTP1B interaction with VEGFR2 is unstable to be detected by co-precipitation. To confirm further their interaction in vivo, GST-tagged PTP1B-WT or substrate-trapping mutants PTP1B-D/A (D181A) or PTP1B-C/S, and Myc-tagged entire intracellular domain of human VEGFR2 (VEGFR2cyto, residues 790-1356) 14 were co-transfected in CHO cells. Figure 3C shows that either PTP1B-WT or PTP1B-D/A or PTP1B-C/S, but not empty vector alone, co-precipitated with Myc-VEGFR2cyto. These results suggest that PTP1B associates with VEGFR2 kinase domain, independent of phosphorylation status of the receptor and phosphatase activity.
Figure 3. PTP1B dephosphorylates and associates with VEGER2 in vivo.
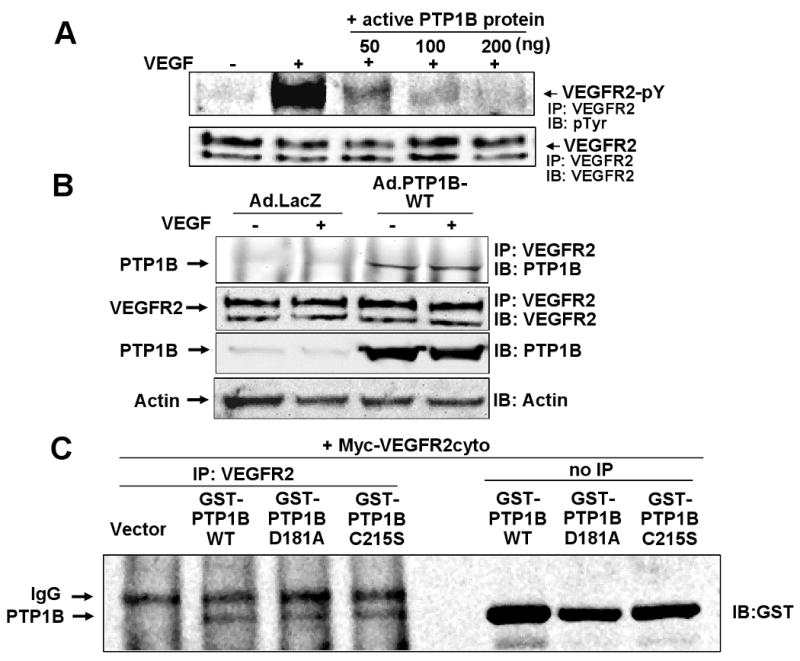
A, In vivo receptor dephosphorylation assay. VEGFR2 immunoprecipitates obtained from HUVECs stimulated with VEGF for 5 min were incubated with active recombinant PTP1B protein at 30 °C for 10 min. Samples were immunoblotted with anti-pTyr or VEGFR2 antibody. B, HUVECs infected with Ad.LacZ (control) or Ad.PTP1B-WT were stimulated with VEGF (20 ng/mL) for 5 min. Lysates were immunoprecipitated with anti-VEGFR2 antibody, followed by immunoblotting with anti-PTP1B or VEGFR2 antibody. Bottom blot shows western analysis with anti-PTP1B antibody. C, CHO cells were co-transfected with Myc-tagged VEGFR2 cytoplasmic domain (myc-VEGFR2cyto) and GST-tagged various mutants of PTP1B (GST-PTP1B-WT, D181A, C215A) or empty vector. Lysates immunoprecipitated with VEGFR2 antibody (IP: VEGFR2) or without immunoprecipitation (no IP) were immunoblotted with anti-GST antibody.
PTP1B overexpression inhibits while PTP1B knockdown enhances VEGF-induced phosphorylation of ERK1/2, but not p38MAPK
To characterize further the effect of PTP1B on VEGF signaling, we examined the effect of overexpression of PTP1B on phosphorylation of downstream signaling such as ERK1/2 and p38MAPK, which are involved in endothelial proliferation and migration, respectively 2, 3. Figure 4A shows that Ad.PTP1B-WT significantly inhibited, while Ad.PTP1B-C/S slightly enhanced, VEGF-induced ERK1/2 phosphorylation without affecting p38MAPK phosphorylation. Conversely, PTP1B siRNA significantly enhanced VEGF-induced phosphorylation of ERK1/2 without affecting p38MAPK phosphorylation (Fig. 4B). We also found that PTP1B siRNA has no effect on Akt phosphorylation (data not shown). These results suggest that PTP1B is endogenous negative regulator for VEGFR2-ERK1/2 pathway. This notion is further supported by the observation that overexpression of PTP1B-WT inhibited, but PTP1B siRNA enhanced, VEGF-stimulated phosphorylation of VEGFR2 at Tyr1175 and PLCγ, which are upstream of ERK1/2 2 (Supplemental Figure I).
Figure 4. PTP1B overexpression inhibits while PTP1B knockdown enhances VEGF-induced phosphorylation of ERK1/2, but not p38MAPK.
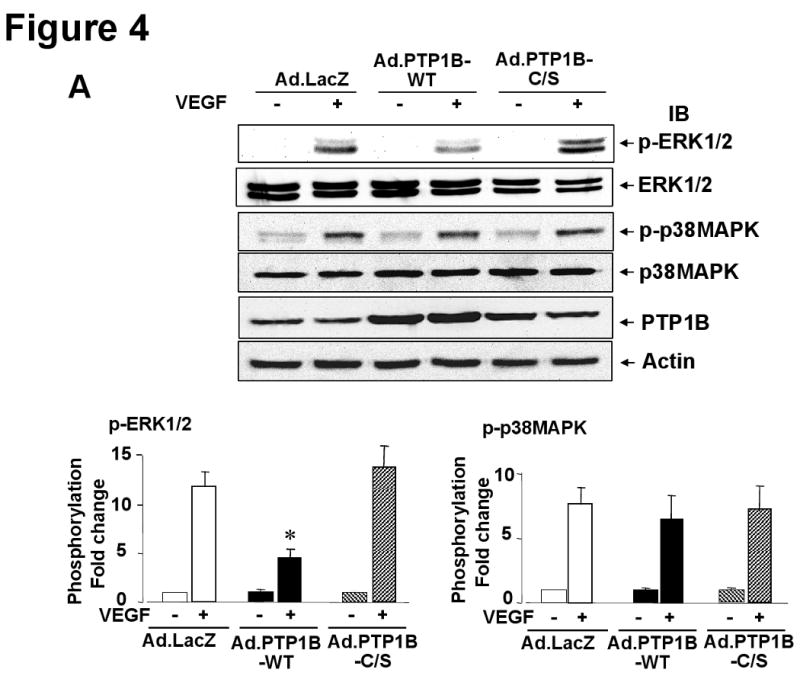
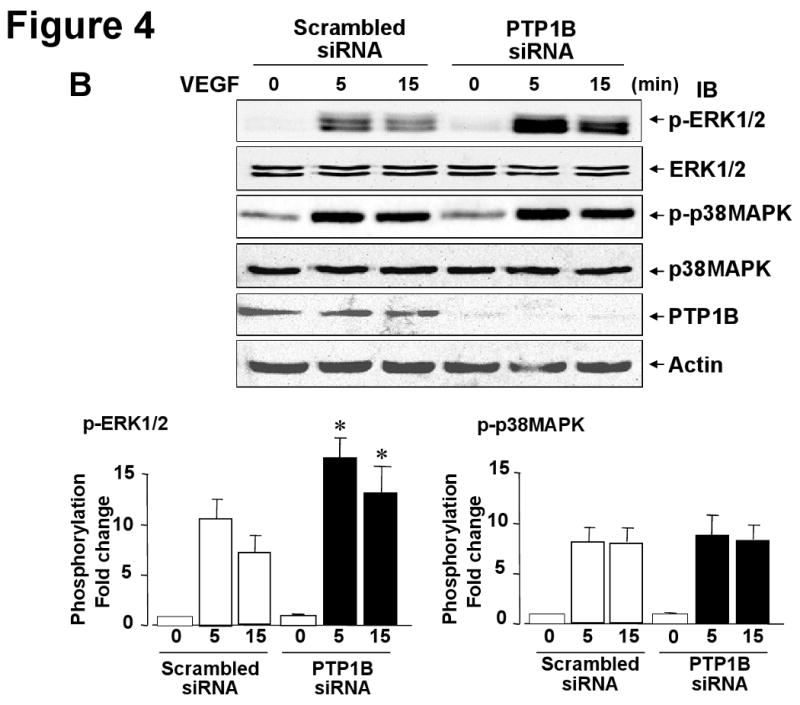
HUVECs infected with Ad.LacZ (control) or Ad.PTP1B-WT (A) or transfected with scrambled or PTP1B siRNAs (B) were stimulated with VEGF (20 ng/mL) for 5 min. Lysates were immunoblotted with phospho-ERK1/2, ERK1/2, phospho-p38MAPK, p38MAPK, PTP1B or actin antibody. Representative blots for ERK1/2 and actin are reprobe from p-ERK1/2 membrane. The representative p-p38MAPK blot is derived from different lysates used for p-ERK1/2 blot, and is reprobed for p38MAPK blot. Bottom panels show averaged data obtained from more than 3 independent experiments, expressed as fold change of phosphorylation over basal (means ± S.E., n=3). *P < 0.05 vs. Ad.LacZ (A) or Scrambled siRNA (B) at each time point.
PTP1B negatively regulates VEGF-stimulated EC proliferation
Since VEGF-induced ERK1/2 is involved in EC proliferation 2, we examined the effects of overexpression of PTP1B and PTP1B siRNA on this response using MTT assay. Figure 5A shows that Ad.PTP1B-WT, but not either LacZ or PTP1B-C/S, significantly inhibited VEGF-stimulated EC proliferation. Either treatment did not affect on basal proliferation. Conversely, PTP1B siRNA significantly enhanced VEGF-stimulated EC proliferation without affecting basal response (Fig. 5B). Similar responses are obtained by cell number measurement (data not shown). In contrast, both PTP1B overexpression and knockdown had no effects on VEGF-mediated migration (Supplemental Figure II) and cell survival (data not shown), which are dependent on p38MAPK 3 and Akt, respectively.
Figure 5. PTP1B negatively regulates VEGF-induced cell proliferation.

HUVECs infected with Ad.LacZ (control) or Ad.PTP1B-WT or Ad.PTP1B-C/S (A) or transfected with scrambled or PTP1B siRNA (B) were used for measurement of cell proliferation. Values are the mean ± SE for 3 independent duplicate experiments. *P < 0.05 vs. Ad.LacZ (A) or scrambled siRNA (B).
PTP1B stabilizes VE-cadherin-mediated cell-cell adhesion
Disruption of cell-cell contacts by reducing VE-cadherin from the cell-cell adhesion sites or tyrosine phosphorylation of VE-cadherin is important for initiating angiogenesis 4. We thus examined the role of PTP1B in localization of VE-cadherin at adherence junctions in confluent monolayer of ECs. Figure 6A shows that Ad.PTP1B-WT increased VE-cadherin staining at sites of cell-cell contact, while Ad.PTP1B-C/S markedly reduced it, resulting in small gaps between adjacent cells in basal state. VEGF stimulation reduced VE-cadherin staining at sites of cell-cell contact in LacZ-infected cells, which was prevented by PTP1B-WT while further decreased by PTP1B-C/S. Furthermore, basal and VEGF-induced tyrosine phosphorylation of VE-cadherin was significantly reduced by overexpression of PTP1B-WT, while enhanced by PTP1B-C/S (Fig. 6B and Supplemental Figure IIIA). Conversely, PTP1B siRNA markedly reduced VE-cadherin staining at cell-cell contacts and significantly enhanced basal and VEGF-induced tyrosine phosphorylation of VE-cadherin (Fig. 7AB and Supplemental Figure IIIB). We also determined transendothelial electric resistance (TER), a measure of intercellular adhesion between ECs and found that PTP1B-WT significantly increased, while PTP1B-C/S and PTP1B siRNA significantly decreased TER (Fig. 6C and 7C). Either Ad.PTP1B-WT or Ad.PTP1B-C/S or PTP1B siRNA did not affect protein expression of VE-cadherin (data not shown). These results suggest that PTP1B activity stabilizes VE-cadherin-mediated cell-cell adhesion in ECs.
Figure 6. Overexpression of PTP1B stabilizes VE-cadherin-mediated cell-cell adhesion and reduces VE-cadherin tyrosine phosphorylation.
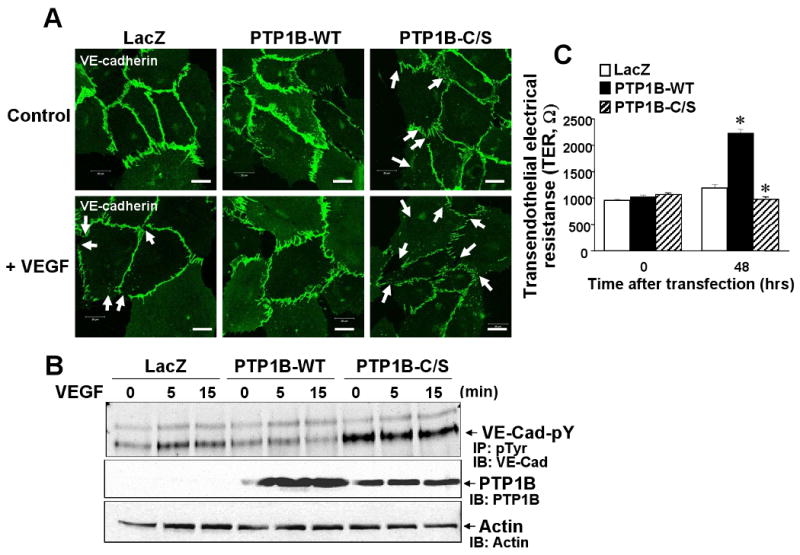
HUVECs infected with Ad.LacZ, PTP1B-WT or PTP1B-C/S were stimulated with VEGF (50 ng/mL). A, Cells were permeabilized, fixed and stained with anti-VE-cadherin antibody, followed by FITC-conjugated goat anti-rabbit antibody. White arrows show VEGF-induced decrease in VE-cadherin staining at cell-cell contact with formation of small gaps. Images were taken at 5 different fields/well, and representative of more than 3 independent experiments. B, Lysates were immunoprecipitated with anti-pTyr antibody, followed by immunoblotting with VE-cadherin antibody (Upper). Lysates without immunoprecipitation were immunoblotted with anti-PTP1B antibody (bottom). Results are representative of 3 independent experiments. C, Real-time measurement of transendothelial electrical resistance (TER). Values are the mean ± SE for 3 independent duplicate experiments. *P < 0.05 vs. Ad.LacZ.
Figure 7. PTP1B knockdown reduces VE-cadherin-mediated cell-cell adhesion and increases VE-cadherin tyrosine phosphorylation.
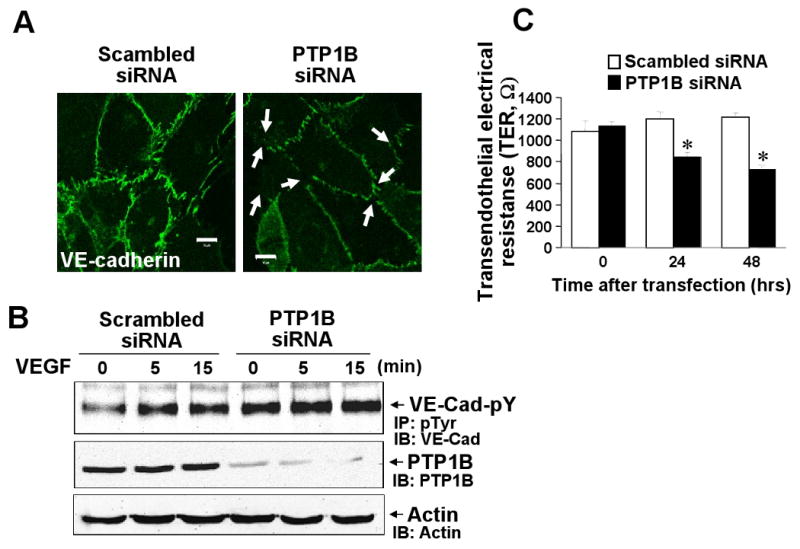
HUVECs transfected with scrambled or PTP1B siRNA were used for VE-cadherin staining (A), VE-cadherin tyrosine phosphorylation analysis (B) and real-time measurement of transendothelial electrical resistance (TER)(C). A, White arrows show decrease in VE-cadherin staining at cell-cell contacts with formation of small gaps. B, Cells stimulated with VEGF (20 ng/ml) were used for measurement of VE-cadherin tyrosine phosphorylation and PTP1B protein expression as described in Figure 6 legend. Results are representative of 3 independent experiments. C, Values are the mean ± SE for 8 independent duplicate experiments. *P < 0.05 vs. scrambled siRNA.
We also found that PTP1B was co-immunoprecipitated with VE-cadherin in unstimulated and VEGF-stimulated confluent HUVECs infected with Ad.LacZ, Ad.PTP1B-WT or Ad.PTP1B-C/S (Supplemental Figure IIIC). Moreover, confocal analysis with HUVEC transiently transfected with GFP-PTP1B-D181A (D/A) revealed that PTP1B colocalized with VE-cadherin at cell-cell contacts but majority of PTP1B was found at perinucleus as reported 15, 16 (data not shown). Colocalization of GFP-PTP1B-WT with VE-cadherin was weaker than the D/A mutant, indicating that stabilization of enzyme-substrate complex is necessary for visualization of PTP1B-VE-cadherin complex. These results suggest that PTP1B associates with VE-cadherin at cell-cell junctions and dephosphorylates VE-cadherin in confluent ECs, thereby stabilizing cell-cell adhesions.
Increase of PTP1B expression and activity in mouse ischemic hindlimb model of angiogenesis
To assess the role of PTP1B in angiogenesis in vivo, we confirmed the expression of PTP1B and examined its activity in a mouse hindlimb ischemia model. Figure 8A shows that hindlimb blood flow recovery after femoral artery ligation was markedly decreased immediately after surgery (day 0) and recovered to the level of that of the nonischemic limb by day 7. Western analysis shows that PTP1B protein was robustly increased in the ischemic hindlimb at 7 days after operation compared with nonischemic legs (Fig. 8C), which was associated with the increase in PTP1B activity (Fig. 8B). Of note, their increase was time-dependent with a peak at day 7, and there was no significant difference between day 0 and 1 (data not shown). Furthermore, immunofluorescence analysis demonstrated that PTP1B expression was increased at lectin-positive capillary ECs and skeletal myocyte at 3 and 7 days after ischemic injury, in a time-dependent manner (Supplemental Figure IV). In contrast, expression of SHP-2 was not changed after hindlimb ischemia (Fig. 8C). We also found that SHP-2 siRNA had no effect on VEGFR2 autophosphorylation and ERK1/2 phosphorylation in HUVECs (data not shown). Given the functional role of PTP1B in VEGF signaling and cell-cell adhesion in ECs, these data suggest that PTP1B may be involved in the process by which new blood vessels are formed in vivo.
Figure 8. Increase of PTP1B expression and activity in mouse ischemic hindlimb model of angiogenesis.
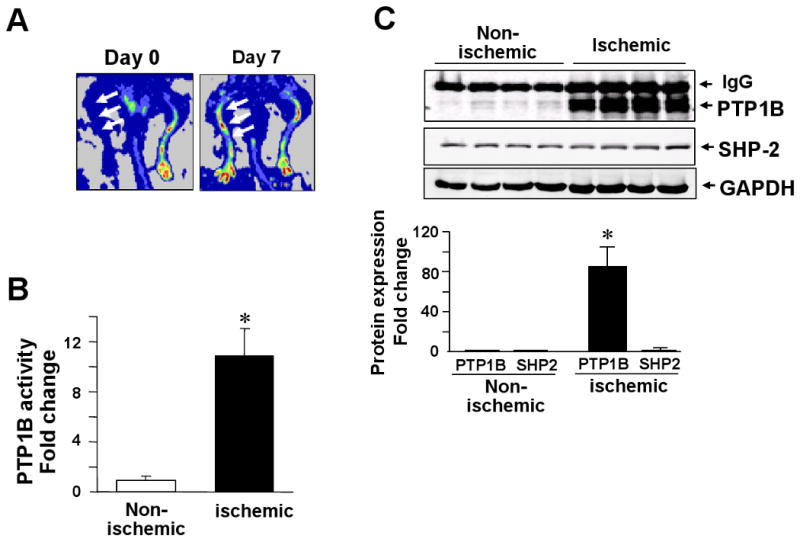
Hindlimb ischemia was induced by the right femoral artery ligation. A, Laser Doppler blood flow analysis: Arrows indicate a low perfusion signal (dark blue) at immediately after operation (day 0) and a high perfusion signal (yellow to red) detected on day 7 in the ischemic hindlimbs. B, PTP1B specific activity in nonischemic and ischemic tissue at 7 day after ischemia. C, Upper blots show PTP1B and SHP2 protein expression in nonischemic and ischemic tissues at 7 days after ischemia. Each lane indicates different mouse. For B and C, bar graph shows averaged data, expressed as fold change over basal (the value in nonischemic tissue was set to 1). Values are the mean ± SE (n = 4). *P < 0.05 vs. hindlimb ischemia.
Discussion
Many studies have focused on mechanism of VEGFR2 signal activation as opposed to signal termination, which is much less studied. The present study provides evidence that: 1) Overexpression of PTP1B inhibits VEGFR2 autophosphorylation, ERK1/2 phosphorylation and EC proliferation, while knockdown of PTP1B using siRNA significantly enhances these responses; 2) PTP1B binds to VEGFR2 in HUVEC overexpressing PTP1B as well as in CHO cells co-transfected with PTP1B and VEGFR2 cytoplasmic domain; 3) Recombinant active PTP1B protein dephosphorylates VEGFR2 immunoprecipitates of VEGF-stimulated HUVECs; 4) Overexpression of PTP1B stabilizes endothelial cell-cell junction by reducing VE-cadherin tyrosine phosphorylation and increasing TER, while PTP1B siRNA causes opposite effects; 5) PTP1B physically associates with VE-cadherin in HUVECs; 6) PTP1B expression and activity are dramatically increased in ischemic hindlimb model.
Previous studies demonstrated that several PTPs including SHP-1, SHP-2 and HCPTPA bind to VEGFR2 directly or indirectly 1. However, their endogenous role in VEGFR2 autophosphorylation under physiological condition remains unclear. SHP-1 is involved in TNFα-mediated prevention of VEGFR2 phosphorylation 17, while SHP-2 inhibits tyrosine phosphorylation of VEGFR2 in ECs when they are cultured only on type I collagen 18. HCPTPA overexpression attenuates VEGFR2 autophosphorylation, but this has not been studied with a loss of function approach 19. Using overexpression and knockdown approaches as well as co-precipitation and co-transfection assays, the present study demonstrates that PTP1B functions as a negative regulator for VEGFR2 autophosphorylation in ECs via binding to the VEGFR2 cytoplasmic kinase domain. Of note, VEGFR2 is co-precipitated with PTP1B in HUVEC overexpressing PTP1B-WT as well as in CHO cells transfected with PTP1B-WT or substrate-trapping mutants of PTP1B (PTP-D/A and PTP1B-C/S), suggesting that PTP1B binds to VEGFR2 in a phosphorylation-independent manner, as demonstrated for its binding to p130Cas 20. We could not detect PTP1B association with VEGFR2 in LacZ-infected cells, which may due to the unstable complex formation of endogenous PTP1B with its substrate 10. In addition, VEGFR2 dephosphorylation assay demonstrates that ligand-induced, activated VEGFR2 is dephosphorylated by PTP1B. Consistent with our findings, PTP1B has been shown to bind to and dephosphorylate PDGF receptor 21, EGF receptor (EGFR) 10 and insulin receptor 7, 11, indicating that PTP1B is a biologically relevant receptor tyrosine kinase phosphatase. Our study cannot rule out the possibility that PTP1B-VEGFR2 interaction is indirect, perhaps through intermediate proteins, which is required for PTP1B-induced dephosphorylation of VEGFR2.
VEGFR2 autophosphorylation is required for activation of diverse signaling pathways such as ERK1/2 and p38MAPK which are involved in EC proliferation and migration, respectively 2, 3. The downstream signaling events that derive from the autophosphorylated tyrosines within VEGFR2 as well as dephosphorylation of RTKs by PTPs are highly site-specific. Previous reports show that phosphorylation of Y1175 within VEGFR2 is essential for activation of PLCγ, and subsequent ERK phosphorylation and cell proliferation 2, while pY1214 is involved in VEGF-stimulated p38MAPK activation and cell migration 3. The present study demonstrates that PTP1B siRNA enhances, while overexpression of PTP1B inhibits, phosphorylation of Y1175 of VEGFR2, PLCγ, ERK and EC proliferation without affecting phosphorylation of p38MAPK or Akt as well as EC migration. Consistent with our result, Milarski et al. 22 reported that PTP1B specifically dephosphorylates EGFR pY992 that is specific binding site for PLCγ. On the other hand, using porcine aortic ECs transfected with chimeric murine VEGFR2, Holmqvist et al. 23 and Dayanir et al. 24 showed that VEGFR2-pY1175/1173 is required for PI3 kinase activation which is involved in EC migration and proliferation, respectively. Moreover, PTP1B inhibitors enhance phosphorylation of Akt and eNOS, and improve peripheral endothelial dysfunction in chronic heart failure model of mice 25. This conflicting evidence may be attributable to differences in experimental conditions (cell culture conditions or transfection system with chimeric murine VEGFR2 or cell types or species). It is possible that PTP1B may preferentially bind to the VEGFR2-PLCγ complex, but not to the VEGFR2-PI3 kinase complex, which may form appropriate conformation or localize at PTP1B-associated subcellular compartments. Furthermore, PTP1B may affect other VEGFR2 phosphorylation sites in addition to pY1175 of the receptor. Nevertheless, our data clearly demonstrate that PTP1B negatively regulates EC proliferation at least in part by selectively inhibiting VEGFR2-pY1175-PLCγ-ERK pathway in intact human ECs. Detailed analysis using mutant Y1175F-VEGFR2, characterization of the interaction of PTP1B and VEGFR2 and their localization, and identification of the dephosphorylation site(s) of VEGFR2 by PTP1B are objective of future studies.
VE-cadherin-mediated cell-cell adhesion is important for preserving endothelial integrity of ECs. Tyrosine phosphorylation of VE-cadherin is associated with a loss of cell-cell contacts in confluent monolayer of ECs and an increase in endothelial permeability, which is initial events to promote angiogenesis 4. PTPs activity also regulates the integrity of cell-cell junctions 26. We found that PTP1B binds to and colocalizes with VE-cadherin at cell-cell contacts in basal state. Overexpression of PTP1B stabilizes endothelial cell-cell junction by reducing VE-cadherin tyrosine phosphorylation and increasing TER, while PTP1B siRNA causes opposite effects. Thus, PTP1B functions to maintain endothelial barrier integrity. Consistent with our results, PTP1B is shown to localize at cell-cell junctions in confluent rat corneal ECs 27, and binds directly to E-cadherin 12 or N-cadherin 13 in other systems, which are involved in stabilizing intercellular junctions. Given that a fraction of VEGFR2 is localized at VE-cadherin-containing cell-cell contacts 28-31, it is conceivable that PTP1B localized at adherens junction may form multiple complex with VEGFR2 and VE-cadherin to downregulate VEGF signaling, thereby limiting angiogenic program. Density-enhanced phosphatase-1 (DEP1)/CD148 indirectly associates with VE-cadherin/VEGFR2 complex, which in turn promotes contact-dependent inhibition of VEGFR2 phosphorylation, ERK activation and EC proliferation 32. Thus, PTP1B-mediated inhibition of VEGFR2-ERK-EC proliferation may be at least in part through promoting cell-cell contacts, thereby contributing to DEP-1-mediated contact-dependent inhibition of VEGFR2 signaling in ECs. Additionally, vascular endothelial PTP (VE-PTP) 33 and SHP-2 34 bind to VE-cadherin and β-catenin, respectively, which are involved in VE-cadherin-mediated barrier function in confluent ECs. These suggest that PTP1B and other PTPs which localize at cell-cell contacts may collaborate to maintain low levels of tyrosine phosphorylation, and thus stabilizing junctional integrity, which may contribute to contact-dependent inhibition of VEGF signaling and EC proliferation.
PTPs have a reactive Cys residue in the enzyme active site (e.g., Cys-215 in human PTP1B) which is deprotonated at neutral pH and is susceptible to the reversible oxidative inactivation by agonist-induced ROS 35. We and others demonstrated that ROS are involved in VEGFR2 autophosphorylation 36-38 as well as loss of cell-cell adhesions through increasing tyrosine phosphorylation of VE-cadherin and/or other junctional proteins 39-41. Whether VEGF-induced ROS are involved in oxidative inactivation of PTP1B in ECs is currently under investigation.
General PTP inhibitors promote VEGFR2 activation, blood flow recovery or angiogenesis after hindlimb ischemia 5, 6, suggesting PTPs as potential therapeutic targets to promote neovascularization. Using mouse hindlimb ischemia model, we demonstrate that PTP1B expression is increased at lectin positive ECs and skeletal myocyte in a time-dependent manner with a peak at 7days after hindlimb ischemia, which is associated with an increase in PTP1B activity. This staining pattern is consistent with the previous report for the induction of VEGFR2 in ischemic skeletal muscle 42. Together with our in vitro data, ischemia-induced upregulation of PTP1B may represent an important compensatory mechanism that blunts over-activation of angiogenic signaling in vivo at least in part by inhibiting tyrosine phosphorylation of VEGFR2 and VE-cadherin. Our results are consistent with previous reports that PTP1B inhibits PDGF- or FGF-induced proliferation of cultured vascular smooth muscle, while PTP1B expression and neointima formation are increased in vascular injury model 43, 44. This was interpreted as counter-regulatory function of PTP1B in neointima formation in response to vascular injury. In contrast, SHP-2 expression was not changed after ischemia and SHP-2 siRNA had no effects on VEGFR2 autophosphorylation in cultured ECs, suggesting specific involvement of PTP1B in angiogenic responses in vitro and in vivo. Sugano et al. 17 reported that SHP-1 protein is increased in a rat hindlimb ischemia model, which prevents TNFα-induced negative inhibitory effects on VEGF signaling. We also found that SHP-1 protein is increased after ischemia, but the extent of its increase is much less than that of PTP1B. Previous reports show that PTP1B inhibitor enhances VEGF-mediated angiogenesis using mouse Matrigel model 45; which cannot eliminate the non-specific effects. Since our ischemia hindlimb data implicates but does not conclude that PTP1B may play a role in VEGF-induced angiogenesis in vivo, further investigation will be required using PTP1B-/- mice in future study. Given that PTP1B-/- mice prevent type 2 diabetes and obesty 8, 9 in which ischemia-induced angiogenesis is impaired, inhibiting PTP1B may be important therapeutic strategy to promote new vessel formation in cardiovascular diseases.
In summary, we demonstrate that PTP1B functions as negative regulator of VEGF signaling by dephosphorylating VEGFR2 via binding to the receptor as well as by stabilizing adherens junctions via inhibiting tyrosine phosphorylation of VE-cadherin that mediates contact-dependent inhibition of VEGFR2 signaling. These mechanisms may contribute to specific inhibition of ERK-EC proliferation pathway. Induction of PTP1B by hindlimb ischemia may represent essential counter-regulatory mechanism that blunts widespread, uncontrolled activities of VEGFR2 and other RTKs during angiogenesis in vivo. These findings should provide novel insight into PTP1B as a potential therapeutic target to promote neovascularization in ischemic cardiovascular diseases.
Supplementary Material
Acknowledgments
Sources of Funding: This research was supported by NIH R01 HL077524 (to M.U.-F.), AHA Grant-In-Aid 0555308B (to M.U.-F.) and 0755805Z (to M.U.-F.), NIH grants DH 43396 (to B.J.G.) and HL70187 (to T.F.), Ministry of Education, Culture, Sports, Science Grant-in Aid (to Y.N.), and Technology of the Japanese government (No. 17688006)(to Y.N.), and Japan Heart Foundation / Bayer Yakuhin Research Grant Abroad (to H.I).
Footnotes
Disclosures: None
References
- 1.Matsumoto T, Claesson-Welsh L. VEGF receptor signal transduction. Sci STKE. 2001:RE21. doi: 10.1126/stke.2001.112.re21. [DOI] [PubMed] [Google Scholar]
- 2.Takahashi T, Yamaguchi S, Chida K, Shibuya M. A single autophosphorylation site on KDR/Flk-1 is essential for VEGF-A-dependent activation of PLC-gamma and DNA synthesis in vascular endothelial cells. Embo J. 2001;20:2768–2778. doi: 10.1093/emboj/20.11.2768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lamalice L, Houle F, Jourdan G, Huot J. Phosphorylation of tyrosine 1214 on VEGFR2 is required for VEGF-induced activation of Cdc42 upstream of SAPK2/p38. Oncogene. 2004;23:434–445. doi: 10.1038/sj.onc.1207034. [DOI] [PubMed] [Google Scholar]
- 4.Wallez Y, Vilgrain I, Huber P. Angiogenesis: the VE-cadherin switch. Trends Cardiovasc Med. 2006;16:55–59. doi: 10.1016/j.tcm.2005.11.008. [DOI] [PubMed] [Google Scholar]
- 5.Sugano M, Tsuchida K, Makino N. A protein tyrosine phosphatase inhibitor accelerates angiogenesis in a rat model of hindlimb ischemia. J Cardiovasc Pharmacol. 2004;44:460–465. doi: 10.1097/01.fjc.0000143275.45289.0a. [DOI] [PubMed] [Google Scholar]
- 6.Carr AN, Davis MG, Eby-Wilkens E, Howard BW, Towne BA, Dufresne TE, Peters KG. Tyrosine phosphatase inhibition augments collateral blood flow in a rat model of peripheral vascular disease. American journal of physiology. 2004;287:H268–276. doi: 10.1152/ajpheart.00007.2004. [DOI] [PubMed] [Google Scholar]
- 7.Tonks NK. PTP1B: from the sidelines to the front lines! FEBS Lett. 2003;546:140–148. doi: 10.1016/s0014-5793(03)00603-3. [DOI] [PubMed] [Google Scholar]
- 8.Elchebly M, Payette P, Michaliszyn E, Cromlish W, Collins S, Loy AL, Normandin D, Cheng A, Himms-Hagen J, Chan CC, Ramachandran C, Gresser MJ, Tremblay ML, Kennedy BP. Increased insulin sensitivity and obesity resistance in mice lacking the protein tyrosine phosphatase-1B gene. Science. 1999;283:1544–1548. doi: 10.1126/science.283.5407.1544. [DOI] [PubMed] [Google Scholar]
- 9.Klaman LD, Boss O, Peroni OD, Kim JK, Martino JL, Zabolotny JM, Moghal N, Lubkin M, Kim YB, Sharpe AH, Stricker-Krongrad A, Shulman GI, Neel BG, Kahn BB. Increased energy expenditure, decreased adiposity, and tissue-specific insulin sensitivity in protein-tyrosine phosphatase 1B-deficient mice. Mol Cell Biol. 2000;20:5479–5489. doi: 10.1128/mcb.20.15.5479-5489.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Flint AJ, Tiganis T, Barford D, Tonks NK. Development of “substrate-trapping” mutants to identify physiological substrates of protein tyrosine phosphatases. Proc Natl Acad Sci U S A. 1997;94:1680–1685. doi: 10.1073/pnas.94.5.1680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Goldstein BJ, Ahmad F, Ding W, Li PM, Zhang WR. Regulation of the insulin signalling pathway by cellular protein-tyrosine phosphatases. Mol Cell Biochem. 1998;182:91–99. [PubMed] [Google Scholar]
- 12.Sheth P, Seth A, Atkinson KJ, Gheyi T, Kale G, Giorgianni F, Desiderio DM, Li C, Naren A, Rao R. Acetaldehyde dissociates the PTP1B-E-cadherin-beta-catenin complex in Caco-2 cell monolayers by a phosphorylation-dependent mechanism. Biochem J. 2007;402:291–300. doi: 10.1042/BJ20060665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Balsamo J, Arregui C, Leung T, Lilien J. The nonreceptor protein tyrosine phosphatase PTP1B binds to the cytoplasmic domain of N-cadherin and regulates the cadherin-actin linkage. J Cell Biol. 1998;143:523–532. doi: 10.1083/jcb.143.2.523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yamaoka-Tojo M, Ushio-Fukai M, Hilenski L, Dikalov SI, Chen YE, Tojo T, Fukai T, Fujimoto M, Patrushev NA, Wang N, Kontos CD, Bloom GS, Alexander RW. IQGAP1, a novel vascular endothelial growth factor receptor binding protein, is involved in reactive oxygen species-dependent endothelial migration and proliferation. Circ Res. 2004;95:276–283. doi: 10.1161/01.RES.0000136522.58649.60. [DOI] [PubMed] [Google Scholar]
- 15.Hernandez MV, Sala MG, Balsamo J, Lilien J, Arregui CO. ER-bound PTP1B is targeted to newly forming cell-matrix adhesions. J Cell Sci. 2006;119:1233–1243. doi: 10.1242/jcs.02846. [DOI] [PubMed] [Google Scholar]
- 16.Frangioni JV, Beahm PH, Shifrin V, Jost CA, Neel BG. The nontransmembrane tyrosine phosphatase PTP-1B localizes to the endoplasmic reticulum via its 35 amino acid C-terminal sequence. Cell. 1992;68:545–560. doi: 10.1016/0092-8674(92)90190-n. [DOI] [PubMed] [Google Scholar]
- 17.Sugano M, Tsuchida K, Maeda T, Makino N. SiRNA targeting SHP-1 accelerates angiogenesis in a rat model of hindlimb ischemia. Atherosclerosis. 2007;191:33–39. doi: 10.1016/j.atherosclerosis.2006.04.021. [DOI] [PubMed] [Google Scholar]
- 18.Mitola S, Brenchio B, Piccinini M, Tertoolen L, Zammataro L, Breier G, Rinaudo MT, den Hertog J, Arese M, Bussolino F. Type I collagen limits VEGFR-2 signaling by a SHP2 protein-tyrosine phosphatase-dependent mechanism 1. Circ Res. 2006;98:45–54. doi: 10.1161/01.RES.0000199355.32422.7b. [DOI] [PubMed] [Google Scholar]
- 19.Huang L, Sankar S, Lin C, Kontos CD, Schroff AD, Cha EH, Feng SM, Li SF, Yu Z, Van Etten RL, Blanar MA, Peters KG. HCPTPA, a protein tyrosine phosphatase that regulates vascular endothelial growth factor receptor-mediated signal transduction and biological activity. The Journal of biological chemistry. 1999;274:38183–38188. doi: 10.1074/jbc.274.53.38183. [DOI] [PubMed] [Google Scholar]
- 20.Liu F, Hill DE, Chernoff J. Direct binding of the proline-rich region of protein tyrosine phosphatase 1B to the Src homology 3 domain of p130(Cas) The Journal of biological chemistry. 1996;271:31290–31295. doi: 10.1074/jbc.271.49.31290. [DOI] [PubMed] [Google Scholar]
- 21.Liu F, Chernoff J. Protein tyrosine phosphatase 1B interacts with and is tyrosine phosphorylated by the epidermal growth factor receptor. Biochem J. 1997;327:139–145. doi: 10.1042/bj3270139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Milarski KL, Zhu G, Pearl CG, McNamara DJ, Dobrusin EM, MacLean D, Thieme-Sefler A, Zhang ZY, Sawyer T, Decker SJ, et al. Sequence specificity in recognition of the epidermal growth factor receptor by protein tyrosine phosphatase 1B. The Journal of biological chemistry. 1993;268:23634–23639. [PubMed] [Google Scholar]
- 23.Holmqvist K, Cross MJ, Rolny C, Hagerkvist R, Rahimi N, Matsumoto T, Claesson-Welsh L, Welsh M. The adaptor protein shb binds to tyrosine 1175 in vascular endothelial growth factor (VEGF) receptor-2 and regulates VEGF-dependent cellular migration. The Journal of biological chemistry. 2004;279:22267–22275. doi: 10.1074/jbc.M312729200. [DOI] [PubMed] [Google Scholar]
- 24.Dayanir V, Meyer RD, Lashkari K, Rahimi N. Identification of tyrosine residues in vascular endothelial growth factor receptor-2/FLK-1 involved in activation of phosphatidylinositol 3-kinase and cell proliferation. The Journal of biological chemistry. 2001;276:17686–17692. doi: 10.1074/jbc.M009128200. [DOI] [PubMed] [Google Scholar]
- 25.Vercauteren M, Remy E, Devaux C, Dautreaux B, Henry JP, Bauer F, Mulder P, Hooft van Huijsduijnen R, Bombrun A, Thuillez C, Richard V. Improvement of peripheral endothelial dysfunction by protein tyrosine phosphatase inhibitors in heart failure. Circulation. 2006;114:2498–2507. doi: 10.1161/CIRCULATIONAHA.106.630129. [DOI] [PubMed] [Google Scholar]
- 26.Sallee JL, Wittchen ES, Burridge K. Regulation of cell adhesion by protein-tyrosine phosphatases: II. Cell-cell adhesion. The Journal of biological chemistry. 2006;281:16189–16192. doi: 10.1074/jbc.R600003200. [DOI] [PubMed] [Google Scholar]
- 27.Chen WL, Harris DL, Joyce NC. Effects of SOV-induced phosphatase inhibition and expression of protein tyrosine phosphatases in rat corneal endothelial cells. Exp Eye Res. 2005;81:570–580. doi: 10.1016/j.exer.2005.03.015. [DOI] [PubMed] [Google Scholar]
- 28.Lampugnani MG, Corada M, Andriopoulou P, Esser S, Risau W, Dejana E. Cell confluence regulates tyrosine phosphorylation of adherens junction components in endothelial cells. J Cell Sci. 1997;110:2065–2077. doi: 10.1242/jcs.110.17.2065. [DOI] [PubMed] [Google Scholar]
- 29.Carmeliet P, Lampugnani MG, Moons L, Breviario F, Compernolle V, Bono F, Balconi G, Spagnuolo R, Oostuyse B, Dewerchin M, Zanetti A, Angellilo A, Mattot V, Nuyens D, Lutgens E, Clotman F, de Ruiter MC, Gittenberger-de Groot A, Poelmann R, Lupu F, Herbert JM, Collen D, Dejana E. Targeted deficiency or cytosolic truncation of the VE-cadherin gene in mice impairs VEGF-mediated endothelial survival and angiogenesis. Cell. 1999;98:147–157. doi: 10.1016/s0092-8674(00)81010-7. [DOI] [PubMed] [Google Scholar]
- 30.Shay-Salit A, Shushy M, Wolfovitz E, Yahav H, Breviario F, Dejana E, Resnick N. VEGF receptor 2 and the adherens junction as a mechanical transducer in vascular endothelial cells. Proc Natl Acad Sci U S A. 2002;99:9462–9467. doi: 10.1073/pnas.142224299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zanetti A, Lampugnani MG, Balconi G, Breviario F, Corada M, Lanfrancone L, Dejana E. Vascular endothelial growth factor induces SHC association with vascular endothelial cadherin: a potential feedback mechanism to control vascular endothelial growth factor receptor-2 signaling. Arteriosclerosis, thrombosis, and vascular biology. 2002;22:617–622. doi: 10.1161/01.atv.0000012268.84961.ad. [DOI] [PubMed] [Google Scholar]
- 32.Grazia Lampugnani M, Zanetti A, Corada M, Takahashi T, Balconi G, Breviario F, Orsenigo F, Cattelino A, Kemler R, Daniel TO, Dejana E. Contact inhibition of VEGF-induced proliferation requires vascular endothelial cadherin, beta-catenin, and the phosphatase DEP-1/CD148. J Cell Biol. 2003;161:793–804. doi: 10.1083/jcb.200209019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nawroth R, Poell G, Ranft A, Kloep S, Samulowitz U, Fachinger G, Golding M, Shima DT, Deutsch U, Vestweber D. VE-PTP and VE-cadherin ectodomains interact to facilitate regulation of phosphorylation and cell contacts. Embo J. 2002;21:4885–4895. doi: 10.1093/emboj/cdf497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ukropec JA, Hollinger MK, Salva SM, Woolkalis MJ. SHP2 association with VE-cadherin complexes in human endothelial cells is regulated by thrombin. The Journal of biological chemistry. 2000;275:5983–5986. doi: 10.1074/jbc.275.8.5983. [DOI] [PubMed] [Google Scholar]
- 35.Tonks NK. Redox redux: revisiting PTPs and the control of cell signaling. Cell. 2005;121:667–670. doi: 10.1016/j.cell.2005.05.016. [DOI] [PubMed] [Google Scholar]
- 36.Ushio-Fukai M, Tang Y, Fukai T, Dikalov S, Ma Y, Fujimoto M, Quinn MT, Pagano PJ, Johnson C, Alexander RW. Novel role of gp91phox-containing NAD(P)H oxidase in vascular endothelial growth factor-induced signaling and angiogenesis. Circ Res. 2002;91:1160–1167. doi: 10.1161/01.res.0000046227.65158.f8. [DOI] [PubMed] [Google Scholar]
- 37.Colavitti R, Pani G, Bedogni B, Anzevino R, Borrello S, Waltenberger J, Galeotti T. Reactive oxygen species as downstream mediators of angiogenic signaling by vascular endothelial growth factor receptor-2/KDR. The Journal of biological chemistry. 2002;277:3101–3108. doi: 10.1074/jbc.M107711200. [DOI] [PubMed] [Google Scholar]
- 38.Ushio-Fukai M. VEGF signaling through NADPH oxidase-derived ROS. Antioxid Redox Signal. 2007;9:731–739. doi: 10.1089/ars.2007.1556. [DOI] [PubMed] [Google Scholar]
- 39.van Wetering S, van Buul JD, Quik S, Mul FP, Anthony EC, ten Klooster JP, Collard JG, Hordijk PL. Reactive oxygen species mediate Rac-induced loss of cell-cell adhesion in primary human endothelial cells. J Cell Sci. 2002;115:1837–1846. doi: 10.1242/jcs.115.9.1837. [DOI] [PubMed] [Google Scholar]
- 40.Lin MT, Yen ML, Lin CY, Kuo ML. Inhibition of vascular endothelial growth factor-induced angiogenesis by resveratrol through interruption of Src-dependent vascular endothelial cadherin tyrosine phosphorylation. Mol Pharmacol. 2003;64:1029–1036. doi: 10.1124/mol.64.5.1029. [DOI] [PubMed] [Google Scholar]
- 41.Yamaoka-Tojo M, Tojo T, Kim HW, Hilenski L, Patrushev NA, Zhang L, Fukai T, Ushio-Fukai M. IQGAP1 mediates VE-cadherin-based cell-cell contacts and VEGF signaling at adherence junctions linked to angiogenesis. Arteriosclerosis, thrombosis, and vascular biology. 2006;26:1991–1997. doi: 10.1161/01.ATV.0000231524.14873.e7. [DOI] [PubMed] [Google Scholar]
- 42.Germani A, Di Carlo A, Mangoni A, Straino S, Giacinti C, Turrini P, Biglioli P, Capogrossi MC. Vascular endothelial growth factor modulates skeletal myoblast function. Am J Pathol. 2003;163:1417–1428. doi: 10.1016/S0002-9440(10)63499-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chang Y, Zhuang D, Zhang C, Hassid A. Increase of PTP levels in vascular injury and in cultured aortic smooth muscle cells treated with specific growth factors. American journal of physiology. 2004;287:H2201–2208. doi: 10.1152/ajpheart.00520.2004. [DOI] [PubMed] [Google Scholar]
- 44.Chang Y, Ceacareanu B, Zhuang D, Zhang C, Pu Q, Ceacareanu AC, Hassid A. Counter-regulatory function of protein tyrosine phosphatase 1B in platelet-derived growth factor- or fibroblast growth factor-induced motility and proliferation of cultured smooth muscle cells and in neointima formation. Arteriosclerosis, thrombosis, and vascular biology. 2006;26:501–507. doi: 10.1161/01.ATV.0000201070.71787.b8. [DOI] [PubMed] [Google Scholar]
- 45.Soeda S, Shimada T, Koyanagi S, Yokomatsu T, Murano T, Shibuya S, Shimeno H. An attempt to promote neo-vascularization by employing a newly synthesized inhibitor of protein tyrosine phosphatase. FEBS Lett. 2002;524:54–58. doi: 10.1016/s0014-5793(02)03002-8. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.