

Abstract
Trichloroethylene (TCE) is a well-known carcinogen in rodents and concerns exist regarding its potential carcinogenicity in humans. Oxidative metabolites of TCE, such as dichloroacetic acid (DCA) and trichloroacetic acid (TCA), are thought to be hepatotoxic and carcinogenic in mice. The reactive products of glutathione conjugation, such as S-(1,2-dichlorovinyl)-L-cysteine (DCVC), and S-(1,2-dichlorovinyl) glutathione (DCVG), are associated with renal toxicity in rats. Recently, we developed a new analytical method for simultaneous assessment of these TCE metabolites in small-volume biological samples. Since important gaps remain in our understanding of the pharmacokinetics of TCE and its metabolites, we studied a time-course of DCA, TCA, DCVG and DCVG formation and elimination after a single oral dose of 2100 mg/kg TCE in male B6C3F1 mice. Based on systemic concentration-time data, we constructed multi-compartment models to explore the kinetic properties of the formation and disposition of TCE metabolites, as well as the source of DCA formation. We conclude that TCE-oxide is the most likely source of DCA. According to the best-fit model, bioavailability of oral TCE was ~74%, and the half-life and clearance of each metabolite in the mouse were as follows: DCA: 0.6 hr, 0.081 ml/hr; TCA: 12 hr, 3.80 ml/hr; DCVG: 1.4 hr, 16.8 ml/hr; DCVC: 1.2 hr, 176 ml/hr. In B6C3F1 mice, oxidative metabolites are formed in much greater quantities (~3600 fold difference) than glutathione-conjugative metabolites. In addition, DCA is produced to a very limited extent relative to TCA, while most of DCVG is converted into DCVC. These pharmacokinetic studies provide insight into the kinetic properties of four key biomarkers of TCE toxicity in the mouse, representing novel information that can be used in risk assessment.
Keywords: Trichloroethylene; metabolism; pharmacokinetics; trichloroacetic acid; dichloroacetic acid; S-(1, 2-dichlorovinyl)glutathione; S-(1, 2-dichlorovinyl)-L-cysteine
INTRODUCTION
Trichloroethylene (TCE) is a widely-used industrial chemical and also is applied as a dry cleaning agent (ATSDR, 1997). Due to high production volume, broad applications, and chemical mobility, TCE is one of the most common environmental contaminants found in soil, ground and surface water. Acute exposure to TCE (at doses in excess of 250 ppm) causes behavioral effects consistent with the impairment of the central nervous system (Kishi et al., 1993), while long-term exposures are thought to cause hepatotoxicity (Bull, 2000), nephrotoxicity (Lash et al., 2000b), and neurotoxicity (Barton and Clewell, III, 2000). Adverse effects on the reproductive, developmental and immune systems have also been reported but remain controversial (Pastino et al., 2000). TCE is classified as reasonably anticipated to be a human carcinogen by the US National Toxicology Program and as Group 2A: Probably carcinogenic in humans by the International Agency for Research on Cancer based on limited evidence of carcinogenicity from studies in humans and sufficient evidence of carcinogenicity from studies in experimental animals. Significant gaps remain in our knowledge of TCE toxicity and its risk assessment, for instance, formation and disposition of DCA and glutathione conjugates (National Research Council, 2006).
The metabolism of TCE is complex, and it is well-accepted that TCE metabolites are responsible for the wide spectrum of toxicity and organ-specific effects associated with this chemical (Lash et al., 2000a). There are two primary metabolic pathways (Figure 1): oxidation via microsomal mixed-function oxidase (MOF) such as cytochrome P450 (CYP), and conjugation with glutathione (GSH) by glutathione-S-transferases (GSTs) (Lash et al., 2000a). Of the various metabolites, oxidative ones [e.g. dichloroacetic acid (DCA), trichloroacetic acid (TCA), and chloral hydrate (CH)] have been suggested to contribute to liver toxicity and carcinogenicity in rodents; while S-(1,2-dichlorovinyl)-L-cysteine (DCVC) and S-(1,2-dichlorovinyl) glutathione (DCVG) are considered penultimate metabolites for kidney toxicity (Bull, 2000; Bruning and Bolt, 2000; Lash et al., 2000a; Chiu et al., 2006a). Although TCE metabolism has been regarded to be qualitatively similar across species (ATSDR, 1997), the mode of action of TCE and dose-response are known to differ both within and among species (Lash et al., 2000a).
Figure 1. Schematic representation of the metabolic pathways of trichloroethylene.
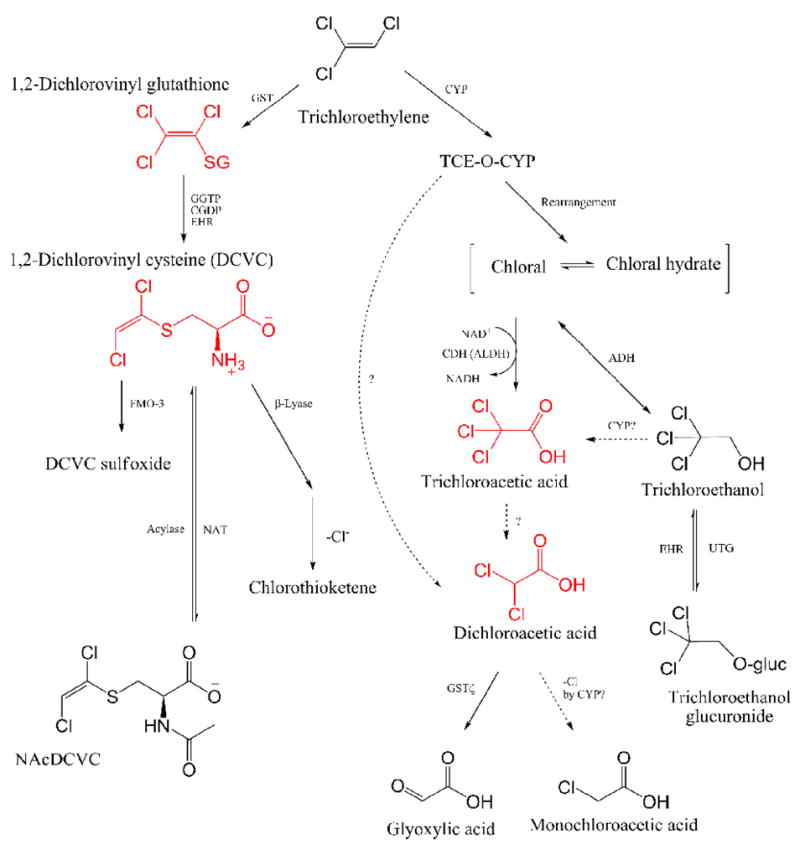
NAcDCVC, N-acetyl-DCVC; GST, glutathione S-transferase; GGTP, γ-glutamyltransferase; CGDP, cysteinyl-glycine dipeptidase; EHR, enterohepatic recirculation; FMO3, flavin monooxigenase-3; NAT, N-acetyltransferase; CYP, Cytochrom P450 enzyme; TCE-O-CYP, oxygenated TCE-CYP transition state complex; ADH, alcohol dehydrogenase; ALDH, aldehyde dehydrogenase; CDH, chloral dehydrogenase; UTG, uridine diphosphate-glucuronosyltransferase [Adaped from (Lash et al., 2000a; Schultz et al., 2002; National Research Council, 2006; Bronley-DeLancey et al., 2006; Chiu et al., 2006a)].
Many studies have contributed to our understanding of TCE metabolism and pharmacokinetics; however, several important issues remain to be addressed. For example, while DCA is thought to be a product of TCE in biological systems (Merdink et al., 1998; Bull, 2000), the mechanism of DCA formation is still being debated (Merdink et al., 2008). Several groups suggested that DCA may be a product of dechlorination of TCA; however, this theory is plagued by unreliable measurements of DCA in vivo (Ketcha et al., 1996; Merdink et al., 1998). It has also been suggested that DCA could be formed either from TCE-oxide, an intermediate chemical complex with CYP by chlorine-shift (Guengerich, 1986; Cai and Guengerich, 1999), or from TCA by reductive dehalogenation (Merdink et al., 2000). Only some groups successfully detected DCA in vivo after dosing with TCE, and there are few reports of reliable measurements of GSH-conjugates (Lash et al., 2000a; Lash et al., 2006). In addition, several physiologically-based pharmacokinetic (PBPK) models were constructed to detail TCE metabolism (Clewell, III et al., 2000; Fisher, 2000; Bois, 2000; US AF-EPA, 2004), but even the most up-to-date ones may need to be re-evaluated due to paucity of data on DCA and GSH-conjugates (National Research Council, 2006).
A new analytical method was developed for simultaneous assessment of TCA, DCA, DCVG and DCVC in small-volume samples of mouse serum (Kim et al., submitted). We applied this new methodology to measure the time profile of key oxidative and conjugative TCE metabolites after a single oral dose of TCE in B6C3F1 mice. Using these data, we constructed compartmental pharmacokinetic models to describe formation and disposition of TCA, DCA, DCVG and DCVC. With this approach, we report the pharmacokinetic properties (formation and disposition) of these TCE metabolites and evaluate the source of DCA formation.
MATERIALS AND METHODS
Chemicals
HPLC-grade acetonitrile, HPLC-grade water, HPLC-grade methanol, spectrophotometric grade diethyl ether, ammonium hydroxide (28%), trichloroacetic acid (TCA, 100%), ACS grade sulfuric acid (98%), ACS grade acetic acid (100%) and 2-(2-methoxyethoxy)ethanol (2-MEE, 99%) were purchased from Fisher Scientific Company (Pittsburgh, PA). Dichloroacetic acid (DCA, 99%), difluoroacetic acid (DFA, 98%), trifluoroacetic acid (TFA, 99.5%), and ammonium formate salt (99.9%) were purchased from Sigma (St. Louis, MO). DCVC, [13C5,15N]DCVC, DCVG and [13C4,15N]DCVG were synthesized by modification of a published method (McKinney et al., 1959), details to be published elsewhere.
Animal treatments
Male mice (B6C3F1, aged 14–16 weeks) were obtained from the Jackson Laboratory (Bar Harbor, ME) and housed in polycarbonate cages on Sani-Chips irradiated hardwood bedding (P.J. Murphy Forest Products Corp., Montville, NJ). Animals were fed NTP-2000 wafer diet (Zeigler Brothers, Inc., Gardners, PA) and water ad libitum, and maintained on a 12 h light-dark cycle. Mice received a single dose (2100 mg/kg) of TCE diluted with corn oil (10 ml/kg) by gavage. The dose selected is expected to be carcinogenic based on the report of significant increases in the incidence of hepatocellular carcinomas and adenomas in male and female B6C3F1 mice treated with TCE (1000 mg/kg, gavage) for 2 years (National Toxicology Program, 1990). Animals (three per group) were sacrificed 0.5, 1, 2, 6, 8, 12 and 24 hrs after dosing under nembutal (100 mg/kg, i.p., Abott Laboratories, Chicago, IL) anesthesia and blood was collected from the posterior vena cava. Serum was prepared by centrifugation using 1.1-ml Z-gel tubes (Sarstedt AG & Co., Germany) at 16,000 × g for 15 min, and stored at −80 °C until assayed. The animal studies were conducted under a protocol approved by the Institutional Animal Care and Use Committee at the University of North Carolina at Chapel Hill.
Sample preparation of DCA, TCA, DCVG and DCVC in serum
Concentrations of DCA, TCA, DCVG and DCVC in serum were determined according to the method of Kim et al. (submitted). Briefly, aqueous mixture of internal standards (5 μl; DFA and TFA, 500 nmol/ml each; [13C5,15N]DCVC and [13C4,15N]DCVG, 2.5 nmol/ml each) was spiked with a serum specimen (50 μl) diluted with water (100 μl). Serum proteins were removed by filter centrifugation (Microcon YM-10, Danvers, MA) at 14,000 × g for 30 min at 25 °C. Subsequently, 2 ml of diethyl ether was added to extract haloacetic acids after acidifying the sample with 100 μl of 3% (v/v) sulfuric acid. The ether layer was transferred to another vial, reduced under N2 and transferred to 300-μl glass vial insert containing 5 μl of water for solvent transfer before dryness. The residue was reconstituted in 100 μl of mobile phase: 68.6% ACN, 29.4% 40 mM ammonium formate (pH 9.1) and 2% 2-MEE. The aqueous fraction remaining ether extraction process above was neutralized with 5 μl of 28% NH4OH before another extraction of DCVG and DCVC through a solid phase extraction (SPE) cartridge (StrataTM X-AW, 30 mg in 96-well plate; Phenomenex, Torrance, CA). After conditioning with 300 μl of methanol, followed by equilibration with 300 μl of water, the samples (~ 300 μl) were loaded, washed with 300 μl of water, and eluted with 250 μl of basic methanol (pH adjusted at 10.8). Light vacuum (up to 50 mmHg) was applied to expedite washing and elution. The final eluant was collected into 300 μl glass vial inserts and dried in a Speed Vac Concentrator (Fisher Scientific) before being reconstituted with 20 μl of 80:20 water/methanol containing 0.1% acetic acid.
Determination of TCE metabolites using HPLC-MS/MS
Determination of DCA and TCA was performed by HPLC-ESI-MS/MS with a Finnigan Surveyor autosampler and pump coupled to a Finnigan TSQ Quantum triple-quadrupole mass spectrometer (Thermo Finnigan, San Jose, CA). The source was operated in the negative electrospray mode. The mass analyzer was operated in the single reaction monitoring (SRM) mode: DCA (m/z 127→83), DFA (m/z 95→51), TCA m/z 161→117), and TFA (m/z 113→69). Sample injection volume was 15 μl. A Luna amino column (150 × 2.0 mm, 3 μm; Phenomenex) was used in an isocratic mode (200 μl/min) with a mobile phase as described above. During the first three minutes the effluent flow was diverted to waste. MS settings were as follows: electrospray voltage (3 kV), capillary temperature (275 °C), sheath and auxiliary gas pressures (nitrogen; 40 and 10 arbitrary units), collision energy (12 V), tube lens offset (SRM table reference), Q2 collision gas pressure (0.5 mTorr). Quantification was based on peak areas relative to the fluoride analogues. On the other hand, determination of DCVC and DCVG was performed by HPLC-ESI-MS/MS with an Aquity UPLC® system (Waters, Milford, MA) coupled to a TSQ Quantum Ultra triple quadrupole mass analyzer (Thermo Finnigan) using a heat-assisted electrospray ionization (HESI) source in positive ion mode. Sample injection volume was 20 μl. A YMC ODS-AQ analytic column (150 × 2 mm, 3 μm; Waters) was operated with a linear gradient of 20% methanol 0.1% acetic acid for 0.5 min, then to 52% methanol 0.1% acetic acid in 7.5 min, and subsequently to 20% methanol 0.1% acetic acid at a flow rate of 200 μl/min. During the 15 min run, the effluent for the first 2.5 min was diverted to waste to prevent salts from entering the MS. Analytes were detected in multiple reaction monitoring mode, monitoring the transition of the m/z 216 → 127 and 199 for DCVC, m/z 222 → 129 for [13C5,15N]DCVC, m/z 402 → 273 for DCVG and m/z 407 → 278 for [13C4,15N]DCVG. MS settings were as follows: electrospray voltage (3.5 kV), capillary temperature (250 °C), sheath and auxiliary gas pressures (nitrogen; 30 and 35 arbitrary units), collision energy (14–20 V), tube lens offset (SRM table reference), Q2 collision gas pressure (1.0 mTorr). The MS and electrospay ionization parameters were optimized by direct infusion of standards using the Xcalibur software (Thermo Finnigan).
Quantification was based on peak areas relative to the stable isotope-labeled internal standards and the calibration curve was constructed in each batch. In order to address possible nonlinearity or heteroscedacity in calibration, log-log transformed data were used in construction of calibration curves, where the logged peak area ratio was regressed on logged concentration of each TCE metabolite in serum (Georgita et al., 2008; Srinivas, 2008). Limits of detection were determined by a signal-to-noise ratio of 3:1 based on the 15 μl injection volume. Measurements less than limit of detection (LOD) were removed before analysis. Limits of detection were determined by a signal-to-noise ratio of 3:1 based on the 15 μl injection volume. Measurements less than limit of detection (LOD) were removed before analysis. The overall assay precision for each analyte was estimated as coefficient of variation (CV, %) from three sets of calibration curve samples and positive controls which were measured on different time points. The CV was estimated as , where is the estimated error variance obtained from a mixed-effects model, where log-transformed levels of each analytes were regressed on a fixed effect (nominal levels of QC samples) and three random effects (batch, assay date, residual error) by implementing SAS PROC MIXED (Kim et al., 2006; Rappaport and Kupper, 2008).
Non-compartmental analysis
Non-compartmental analysis was performed to provide descriptive kinetic parameters (i.e., area under the curve [AUC], Cmax and Tmax) for TCE metabolites. The AUC was estimated with the linear trapezoidal method and extrapolation through infinite time according to the terminal elimination rate constant, which was recovered by log-linear regression of the concentration-time profile. Regression analysis utilized the inverse of the metabolite concentration as a weighting factor. Cmax and Tmax were determined by inspection of the concentration-time data.
Description of the pharmacokinetic model (multi-compartment model)
We assumed first-order kinetics for the rate of absorption, distribution, metabolism and disposition of TCE and its metabolites. Multi-compartment models were constructed to describe the PK profiles of TCE metabolites (Figure 2). The model structure reflected the known metabolic pathways of TCE (Lash et al., 2000a; Chiu et al., 2006b), and was composed of a two-compartment model (for parent compound) and four sub-models for TCE metabolites (TCA, DCA, DCVG and DCVC). Depending on the putative source of DCA, the sub-models for TCA and DCA were classified as models A, B, C and D, among which models A, B and C were one compartment models for each metabolite, while model D was a two-compartment model. A best-fit model was selected by comparison of the fitting status using Akaike’s Information Criteria (AIC) and χ2–test of log-likelihood ratio between models (Burnham and Anderson, 2002; Rappaport and Kupper, 2008).
Figure 2. Pharmacokinetic model for trichloroethylene and its metabolites.
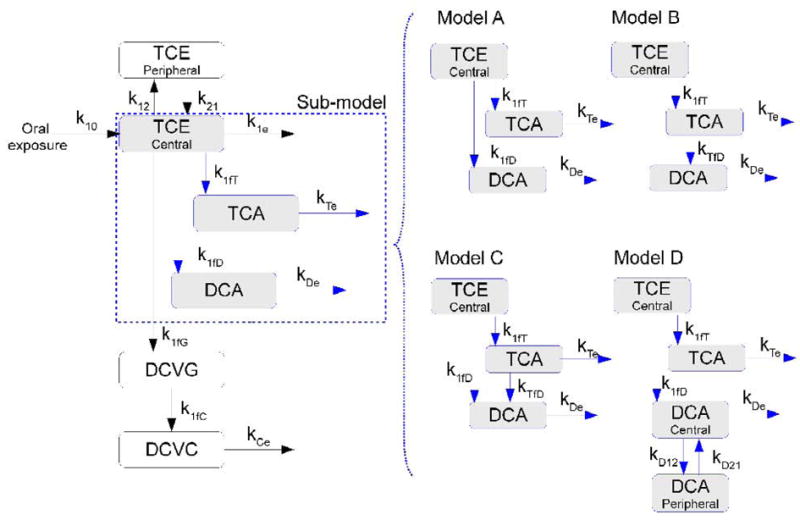
Left panel shows a basic structure and model A, while right panel shows variations of the model depending on the putative source of DCA formation: model B (DCA from TCA), model C (DCA from TCE and TCA), and model A (DCA from TCE). Abbreviations: k10 – TCE absorption; k12 – TCE disposition from central compartment (blood) to peripheral compartment; k21 – reabsorption from peripheral compartment to blood; k1e – TCE elimination at blood; k1fT – TCE → TCA formation; kTe –TCA elimination; k1fD – TCE→DCA formation; kTfD – TCA → DCA formation; kDe –DCA elimination; k1fG – TCE → DCVG formation; k1fC – DCVG → DCVC formation; kCe –DCVC elimination; kD12 – DCA distribution from central to peripheral compartment; kD21 – DCA distribution from peripheral to central compartment.
Model optimization
First, we fit the two-compartment model to previously-published TCE concentration-time data in blood from B6C3F1 mice. The TCE dose (2000 mg/kg) and vehicle (corn oil) were compatible with the present experiment (Abbas and Fisher, 1997). Kinetic parameters for TCE absorption and disposition were obtained and used in the multi-compartment models for TCE metabolites. Parameter estimation was performed using all concentration-time data for the four metabolites concurrently after subtracting background (pre-dose) concentrations of TCA and DCA. The direction set algorithm, with tolerance set at 1×10−5, was used to optimize the parameters, which was fortified by Brent’s method (Brent, 1972) to avoid a local minimum problem. The initial values in optimization procedures were obtained from the preliminary analyses for the individual metabolites such as non-compartment analysis or one-compartment models using WinNonlin (Pharsight, Carry, NC) and SAS PROC NLIN (SAS, Cary, NC). Model optimization and parameter estimation were carried out using AcslX (AEgis, Huntsville, AL) without restrictions (bound statements) except in Model B.
Uncertainly analysis on parameter estimates
Parameter uncertainty associated with the selected model/submodels was assessed with Monte Carlo simulation analyses (Clewell et al., 1999). Briefly, it was assumed that each of the pharmacokinetic parameters in the model would be normally distributed in a “population” of mice. The “population distribution” of each parameter was defined with a “population mean” (the best-fit parameter estimate) and a “population variance” (obtained from the uncertainty with which each parameter was recovered by the nonlinear least-squares regression routine). This hypothetical “population” of animals was randomly sampled 500 times (i.e., to provide 500 independent sets of parameters), allowing estimation of the relative standard deviation (RSD) of all parameter estimates in the model.
Sensitivity analysis
Since each model incorporated somewhat different kinetic parameters for DCA formation and disposition, sensitivity analysis was performed to gauge the quantitative importance of each parameter. Briefly, the degree to which predicted DCA concentrations were altered by a 1% change in each parameter value was assessed using normalized coefficient of sensitivity (NSC). NSC were calculated using the forward difference method: , where m is the response variable (i.e., concentration of DCA in serum), Δm the change in the response variable, p the value of the parameter estimates, and Δp is the change in the parameter value (Evans and Andersen, 2000).
RESULTS
Time-course of TCE metabolism
The concentration-time profiles (0.5 to 24 hrs) for the four TCE metabolites, TCA, DCA, DCVG and DCVC, were determined in serum of B6C3F1 mice gavaged with 2100 mg/kg TCE in corn oil (Table 1). The nominal LOD of each metabolite was as follows: 0.01 nmol/ml (DCA), 0.4 nmol/ml (TCA) and 0.001 nmol/ml (DCVG and DCVC). Estimates of CVs representing measurement error were: DCA, 9.1%; TCA, 12.3%; DCVG, 9.3%; and DCVC, 14.7%. For the subsequent pharmacokinetic analysis the dose of TCE was estimated at 581.4 μmol (male B6C3F1 mice with mean body weight – 35.7 g).
Table 1.
Time profiles of TCE metabolites in B6C3F1 mice after dosing 2100 mg/kg TCE with corn oil vehicle by gavage.
| Time, hr | TCA, nmol/ml | DCA, nmol/ml | DCVG, pmol/ml | DCVC, pmol/ml |
|---|---|---|---|---|
| 0 | 2.15 ± 0.85 | 0.055 ± 0.017 | ND | ND |
| 0.5 | 139 ± 11.6 | 0.143 ± 0.012 | 38.9 ± 16.2 | ND |
| 1 | 178 ± 30.6 | 0.132 ± 0.005 | 32.2 ± 3.71 | 1.05 ± 0.48 |
| 2 | 256 ± 29.7 | 0.122 ± 0.014 | 37.3 ± 14.8 | 2.43 ± 0.87 |
| 6 | 339 ± 7.76 | 0.135 ± 0.014 | 28.0 ± 9.42 | 0.80 ± 0.49 |
| 8 | 390 ± 19.6 | 0.112 ± 0.025 | 19.0 ± 1.50 | 2.06 ± 1.32 |
| 12 | 324 ± 30.6 | 0.127 ± 0.030 | 1.43a | 0.71b ± 0.62 |
| 24 | 214 ± 18.9 | 0.096 ± 0.034 | ND | ND |
ND, not detected or lower than limit of detection. Each cell represents mean ± standard deviation of three replicates except for
single measurement;
average of two measurements.
Non-compartmental analysis of TCE metabolism
The parameter estimates from non-compartmental analysis are shown in Table 2. Total AUC of oxidative metabolites (DCA and TCA) was about 40,000-fold higher than that of GSH-conjugates (DCVC and DCVG). The AUC of TCA was about 4,000-fold higher than that of DCA, while the AUC of DCVG was about 7-fold higher than that of DCVC. According to Tmax, DCA and DCVG peaked rapidly (Tmax, about 0.5 hr or less), while TCA and DCVC accumulated more slowly (Tmax = 8 hr).
Table 2.
Parameter estimates from non-compartmental analysis.
| Parameters | TCEa | TCAb | DCAb | DCVGb | DCVCb |
|---|---|---|---|---|---|
| Cmax, μmol/l | 1,647 | 388 | 0.088 | 0.0389 | 0.0028 |
| Tmax, hr | 0.43 | 8 | 0.5 | 0.5 | 8 |
| T1/2, hr | 3.32 | 7.92 | 7.41 | 3.00 | 4.15 |
| AUClast, μmol/l ·hr | 5,213 | 6,855 | 1.52 | 0.281 | 0.0212 |
| AUC∞, μmol/l ·hr | 5,260 | 12,612 | 3.02 | 0.283 | 0.0416 |
| AUMC∞, μmol/l ·hr2 | 26,273 | 372,721 | 108 | 1.25 | 0.751 |
| MRT∞, hr | 5.00 | 29.6 | 35.2 | 4.41 | 18.1 |
Cmax, peak concentration (please note that background concentrations of TCA and DCA in mouse serum were subtracted from each time point prior to pharmacokinetic modeling); Tmax, peak time; T1/2, terminal half life; AUClast, area under the serum concentration-time curve between 0 and last observation; AUC∞, area under the serum concentration-time curve between 0 and ∞; AUMC, area under first-moment curve; MRT, mean residence time.
Experimental dose of 2000 mg/kg TCE in 0.0275 kg B6C3F1 mice in average (corresponding to 419 μmoles of TCE) (Abbas and Fisher, 1997).
Experimental dose of 2,140 mg/kg TCE onto 0.0357 kg B6C3F1 mice in average (corresponding to 581 μmoles of TCE) in the present study.
Disposition of TCE in a two-compartment model
The concentration-time profile for TCE in serum of B6C3F1 mice (2000 mg/kg gavage) was reported previously (Abbas and Fisher, 1997). Figure 3 shows these data together with predictions from a physiologically-based pharmacokinetic model [dashed line, (Abbas and Fisher, 1997)] and the present two-compartment model. According to the two-compartment model, the half life (T1/2) and oral clearance were estimated at 1.77 hr and 0.074 L/hr, respectively.
Figure 3. Disposition profile of TCE in blood.
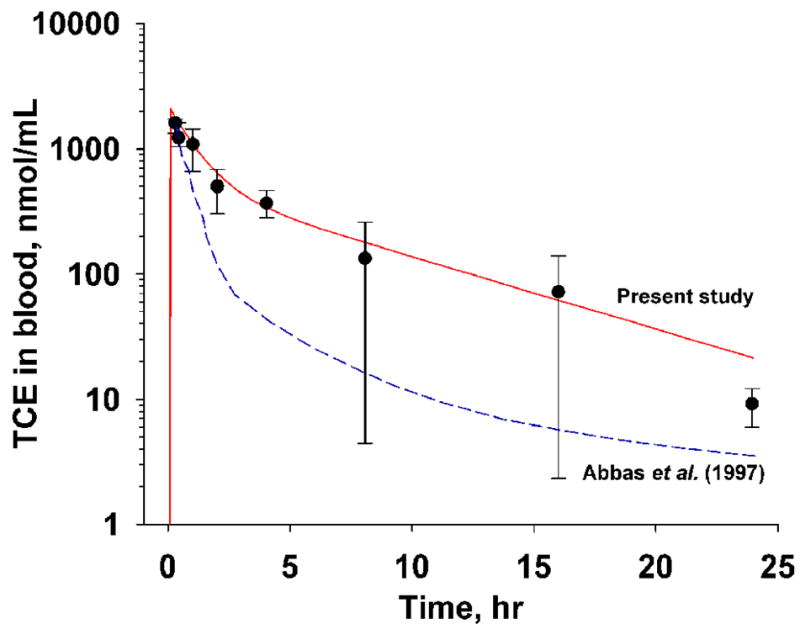
Measurements (means ± SD, n =4) were adapted from (Abbas and Fisher, 1997) where B6C3F1 mice were dosed with 2000 kg/mg TCE in corn oil vehicle by gavage. Solid line, model prediction of the two-compartment in the present study. Dotted line, PBPK prediction (Abbas and Fisher, 1997).
Pharmacokinetic modeling of TCE metabolism
Once we obtained the pharmacokinetic parameters for TCE, we fixed these values and fit the multi-compartment model to TCE metabolite concentration-time data after scaling the body weight and the actual dose for the animals in this study. Four multi-compartment models were constructed based on the previous mechanistic studies of TCE metabolism (see Methods). While there is little controversy regarding the metabolic fate of DCVG and DCVC, our main attention was focused on capturing various proposed mechanisms of DCA formation (Figure 2). By evaluating the fit of each model to the experimental data the following conclusions can be made. In graphical analysis, model A (TCE→DCA) showed a relatively poor fit to data at the late time points, while model B (TCE→TCA→DCA) did not fit the highest concentrations of DCA in the first 2 hrs after dosing (Figure 4A and B). Indeed, model A provided a better description of the data than model B, which had the poorest fit of all 4 models (Table 2).
Figure 4. Model optimization.
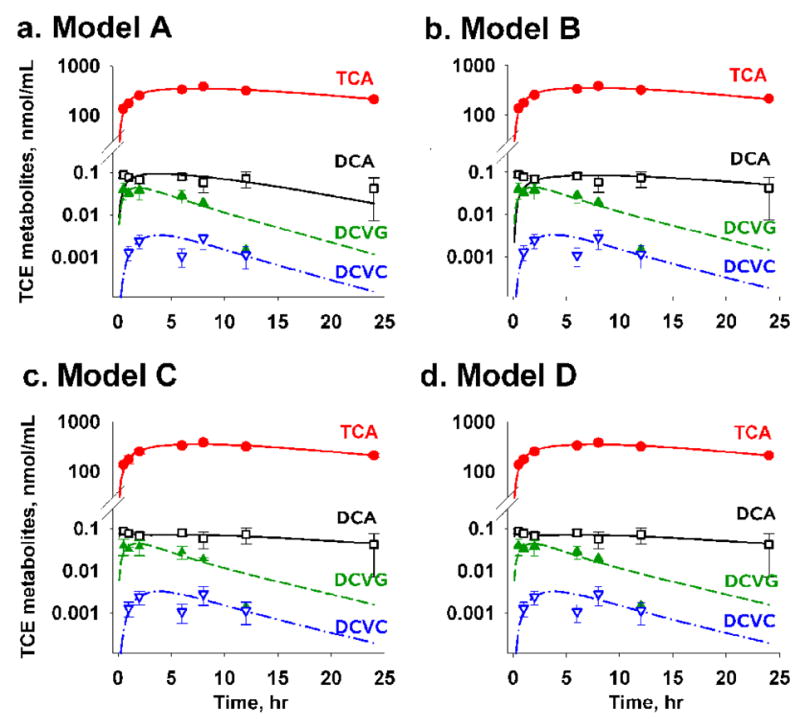
Model fitness status showed that DCA measurements were not supported by model B, in which the source of DCA formation was TCA only. (A) Model A for TCE → DCA, (B) Model B for TCA → DCA, (C) Model C for TCE+TCA → DCA, and (D) Model D for TCE → DCA (two-compartment). Filled circle, symbol and error bars represent mean of three replicates (animals) and standard deviation of each TCE metabolite at a given time point.
Models C (TCE+TCA→DCA) and D (TCE→DCA with 2-compartments) exhibited a very good fit to the experimental data (Figures 4C and D). Model C showed a significantly better fit [ ] than model A; however, model D was superior to other models based on AIC and likelihood ratio (LR) tests (Table 3). Both models C and D captured the bi-exponential trends of DCA elimination well; however, model D provided a significantly better fit status based on a substantial difference of AIC (ΔAIC = 6.6) and LR test statistics (model C vs D: p < 0.005).
Table 3.
Model comparison statistics.
| Model | AIC | (−)2LL | No. Parameter | Comparison, χ2 | p-value | |
|---|---|---|---|---|---|---|
| Model A | TCE → DCA | −65.5 | −87.6 | 11 | - | - |
| Model B | TCA → DCA | −62.0 | −84.0 | 11 | - | - |
| Model C | TCE+TCA → DCA | −77.5 | −99.6 | 12 | A vs. C, 12.0 | A vs. C, p <0.001 |
| Model D | TCE → DCA (2) | −82.1 | −108.0 | 13 | C vs. D, 8.4 | C vs. D, p < 0.005 |
AIC – Akaike Information Criteria, (−)2LL – 2Log-likelihood function, χ2 – test statistic (likelihood ratio), DCA(2) – two compartmental model of DCA.
Table 4 shows the pharmacokinetic parameters of the best-fit model (model D). Parameter estimates for model C are provided in Supplemental Table 1. Each TCE metabolite appeared to follow first-order kinetics for formation and disposition, and one-compartment model was enough for TCA, DCVG and DCVC, while two-compartment model was needed for DCA. It should be noted that while the measurements of DCVG at 12 hr after dosing represent a potential outlier, inclusion or exclusion of this data point in the modeling did not affect the outcome (data not shown).
Table 4.
Kinetic parameter estimates of the best-fit model (model D).
| Parameter | Notes | Final Value | Clearance, L/hr | Half-life, hr |
|---|---|---|---|---|
| k01 | TCE (GI) → TCE (blood) | 104 | - | - |
| k1e | TCE (blood) elimination | 0.393 | 0.0742 | 1.77 |
| k12 | TCE (blood) → TCE (peripheral) | 0.384 | - | - |
| k21 | TCE (peripheral) → TCE (blood) | 0.33 | - | - |
| V1 | Volume of distribution for TCE | 0.189 | - | - |
| k1fT | TCE → TCA formation | 0.0303 | - | - |
| kTe | TCA elimination | 0.0578 | 0.00381 | 12.0 |
| VmT | Volume of distribution for TCA | 0.0660 | - | - |
| k1fD | TCE → DCA formation | 1.46×10−7 | - | - |
| kD12 | DCA: central → peripheral | 10.9 | - | - |
| kD21 | DCA: peripheral → central | 0.500 | - | - |
| kDe | DCA elimination | 1.16 | 8.07×10−5 | 0.598 |
| VmD | Volume of distribution for DCA | 6.96×10−5 | - | - |
| k1fG | TCE → DCVG formation | 4.16×10−6 | - | - |
| kGfC | DCVG → DCVC formation | 0.490 | 0.0168 | 1.41 |
| VmG | Volume of distribution for DCVG | 0.0342 | - | - |
| kCe | DCVC elimination | 0.600 | 0.176 | 1.16 |
| VmC | Volume of distribution for DCVC | 0.293 | - | - |
Clearance is the product between elimination constant and volume of distribution of TCE or each metabolite, reflecting systemic clearance. Units of the reaction constants and volume of distributions are hr−1 and liter, respectively.
Based on Tmax and Cmax values in model D, we concluded that DCA peaks earlier than TCA, while DCVG elimination is followed by that of DCVC (Figures 4D and 5). DCA showed a very short half-life (T1/2 = 0.598 hr) relative to TCA (T1/2 = 12.0 hr). The half-lives of DCVG (T1/2 = 1.41 hr) and DCVC (T1/2 = 1.16 hr) were comparable. Furthermore, the time-profile of each analyte was simulated to obtain the AUC∞ values: 5,824 μmol/l ·hr (TCE), 10,905 μmol/l ·hr (TCA), 2.48 μmol/l ·hr (DCA), 0.341 μmol/l ·hr (DCVG), 0.0325 μmol/l ·hr (DCVC), respectively. Then, we estimated total clearance after oral dosing [(CLt)oral] using an equation (Boroujerd, 2001), , where F is systemic bioavailability (the fraction of the dose absorbed into the systemic circulation), and D is the dose. Rearranging this equation, (CLt)oral/F was estimated 0.0998 l/hr, and the estimated CLt was 0.0742 (Table 4), the bioavailability of TCE orally dosed was estimated at 74.4%.
Figure 5. Assessment of parameter uncertainty in model D using Monte Carlo analysis.
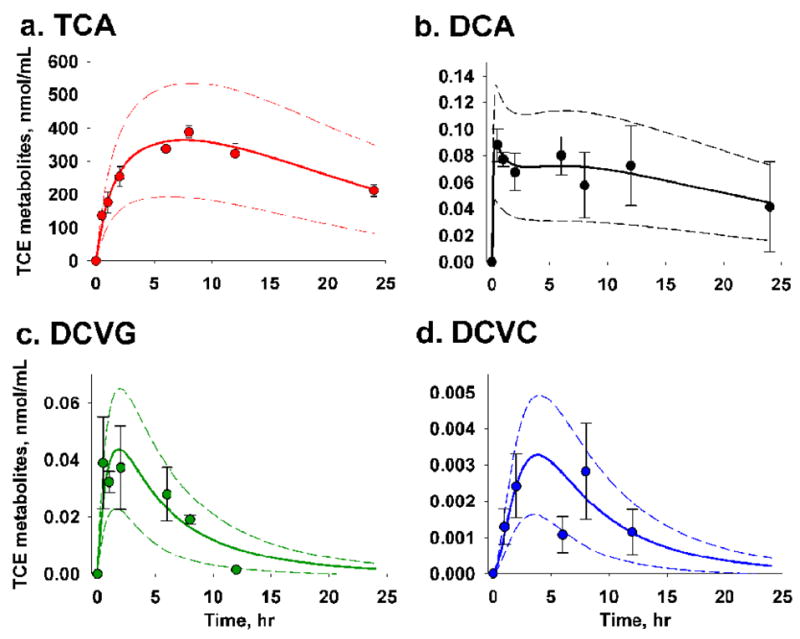
Symbol and error bar represent mean of three replicates (animals) and standard deviation of each TCE metabolite at a given time point. The middle, upper and lower boundary lines depict the mean and three-fold standard deviation from 500 random sampling given 10% relative standard deviation in all parameter estimates in the model.
We also estimated the amount of a metabolite eliminated ( ) at infinity using an equation (Boroujerd, 2001), , where CLM is clearance of the metabolite M (Table 4) and AUCM∞ is AUC of a metabolite at infinity. Interestingly, while the elimination value for TCA were relatively high (41.5 μmol), that for DCA was much lower (0.2 nmol), even less than for DCVG (5.73 nmol) and DCVC (5.72 nmol).
Parameter uncertainty in model D was assessed using the Monte Carlo analysis (Clewell et al., 1999). By evaluating the probability distribution of values of model inputs we conclude that the variation in our measurements of TCE metabolites fits well within the three-fold standard deviation of the model prediction (Figure 5).
Sensitivity analysis
Four parameters are crucial for predicting serum DCA concentrations in model D: TCE-to-DCA formation (k1fD), DCA elimination (kDe), DCA distribution from central to peripheral compartment (kD12) and DCA reverse distribution from peripheral to central compartments (kD21). Sensitivity analysis of these parameters relative to the whole model was carried out to gauge the extent to which the model prediction of DCA disposition may be altered by the changes in these parameters (Figure 6). Change in k1fD affects DCA levels uniformly as time increased, while the effect of kDe on serum DCA was negatively correlated with time. The change in kD12 and kD21 was inversely related to DCA concentrations; this relationship subsided as time increased. Increases in kD12 reduced DCA concentrations more rapidly within the first 1 hr, while increases in kD21 increased DCA concentrations until 5 hr (Figure 6A).
Figure 6. Sensitivity analysis of the DCA-related rate constants in pharmacokinetic models.
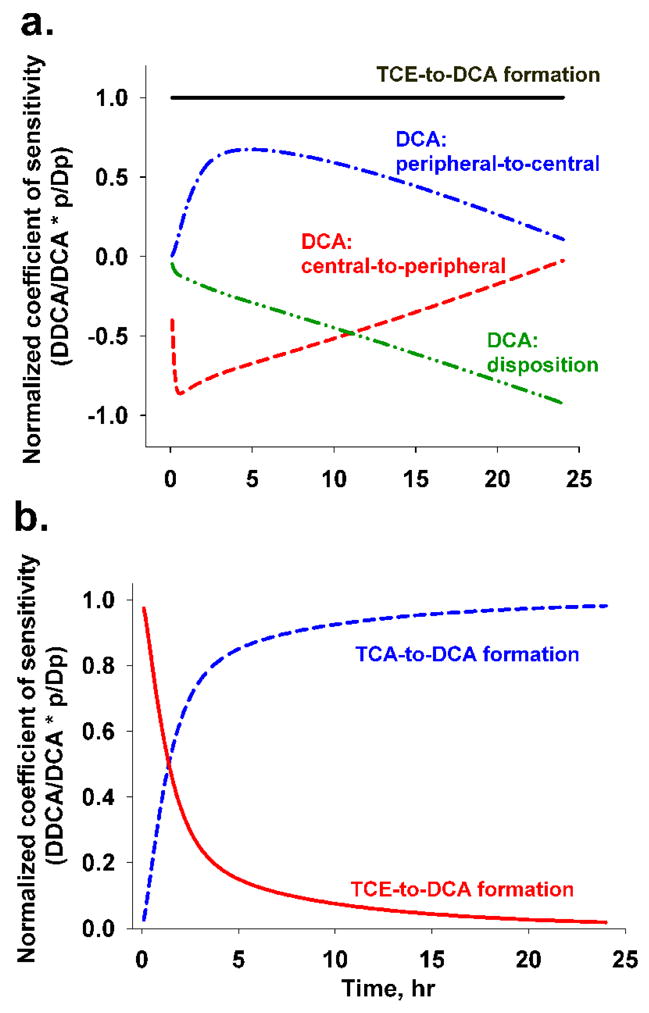
(A) Normalized coefficients of sensitivity for DCA-related kinetic constants in model D: solid line for TCE→DCA formation (k1fD), line-dot-line for DCA distribution from peripheral to central compartment (kD21), dashed line for DCA distribution from central to peripheral compartment (kD12), line-dot-dot-line for DCA elimination (kDe). (B) Normalized coefficients of sensitivity for DCA-related kinetic constants in model C: dashed line for TCA→DCA formation (kTfD), solid line for TCE→DCA formation (k1fD).
Since model C was the second-best model, and provided comparable model predictions, we performed a sensitivity analysis to assess the contribution of the two sources of DCA formation (K1fD and KTfD). TCE-to-DCA formation (K1fD) was the governing factor to generate DCA for the first 1.5 hrs showing peak concentrations of DCA in serum, whereas TCA-to-DCA formation (KTfD) likely contributed to the remaining DCA in serum afterwards (Fig. 6b).
DISCUSSION
Despite the fact that a large body of knowledge on adverse health effects of TCE in both rodents and humans exists, significant gaps in understanding its mode of action for organ toxicity and carcinogenicity remain. Not surprisingly, the regulatory agencies (e.g., US EPA, Occupational Safety and Health Administration, and California EPA) and a broad scientific community show continuing interest in research on TCE toxicity. The putative adverse health effects of TCE are thought to arise from metabolism of TCE, which proceeds along several paths, as illustrated in Figure 1. Of the various metabolites, sulfoxidated metabolites DCVC and DCVG are widely believed to be contributing to kidney toxicity; however, deficiencies in the available analytical techniques led to challenges in estimating pharmacokinetic properties in the TCE metabolism through GSH conjugation (National Research Council, 2006; Lash et al., 2006). Conversely, TCA and DCA have been reported as liver tumorigens in mice; however, the key events in the hypothesized mode of action are not well understood in rodents and their relevance to human risk assessment is still uncertain (Caldwell et al., 2008). Furthermore, the source of DCA formation, and the amounts formed in vivo in susceptible species remain controversial (Bull, 2000; Merdink et al., 2000; National Research Council, 2006). Our study performed pharmacokinetic modeling of TCE metabolites of biological relevance: DCA, TCA, DCVG and DCVC. We evaluated the time course of TCE metabolic pathways concurrently by utilizing a newly-developed analytical method. The sensitivity and accuracy of this technique afforded the possibility of addressing some of the important lingering issues in TCE metabolism research.
Since it is known that TCE disposition in rodents follows a bi-exponential function, in the present study we selected a classical two-compartment modeling approach (Figure 2), similar to that used previously (D’Souza et al., 1985). The model-predicted disposition of TCE exhibited good fit to the experimental data (Abbas and Fisher, 1997), and was superior to that from the PBPK modeling (Figure 3). Rapid oral absorption of TCE (Withey et al., 1983) was also confirmed in our model, which could be explained by passive diffusion and non-varying factor in pharmacokinetic studies. Considering this, the half-life for TCE in mouse blood (1.8 hr) in the present study was comparable to that in rat blood (2 hr) (D’Souza et al., 1985).
The multi-compartment models constructed in our study reflected various hypothetical sub-models for DCA formation (Figure 2). According to model selection criteria, model B (TCA only → DCA) was the least appropriate since it did not fit the data for the first 1 – 2 hr associated with peak concentrations of DCA (Table 3). In contrast, models A, C and D, all of which presumed TCE to be the source for DCA formation, described the early peak concentrations of DCA well. Of these models, model D was chosen as a best-fit model based on AIC and LR tests. Interestingly, the second best-fit model (model C) provided the same model predictions as in model D. While model D has a single source of DCA formation (i.e., TCE) plus additional compartment for back-distribution of DCA, model C has two sources of DCA formation, directly from TCE and via TCA.
The putative mechanism of DCA formation from TCE is thought to involve TCE-oxide, followed by hydrolysis and Cl-shift (Miller and Guengerich, 1982; Cai and Guengerich, 2000). The possibility of biotransformation of TCA into DCA also was proposed, in part based on the evidence of formation of dichloroacetate radicals from TCA (Merdink et al., 2000). While many studies suggested that DCA is formed in high amounts from TCA, problems with analytical methodologies may have led to considerable overestimation of DCA formation (Ketcha et al., 1996). Indeed, in a recent study which used chloral hydrate as an in vivo treatment, DCA concentrations were reported at less than LOD or LOQ (Merdink et al., 2008). This result supports our hypothesis that the pathways through chloral hydrate and TCA might not be a source for DCA formation. While the experimental evidence on biotransformation pathways for DCA remains limited, our modeling results suggest that: (i) it is unlikely that DCA is formed only from TCA (model B); (ii) most of the DCA is likely to be formed from one of the other TCE-derived intermediates (model D, see below) whereby repartitioning between blood and tissues takes place; and (iii) it is also likely that some fraction of DCA may be a product of TCA biotransformation (Merdink et al., 2000) especially 2 hrs after TCE dose (see model C and Figure 6B).
A two-compartment model (D) for DCA disposition is likely since the DCA concentration-time data conformed to a bi-exponential function (Figure 5B). Our observations concur with a recent study of DCA metabolism in B6C3F1 mice which received a high dose of DCA as an intravenous bolus (20 mg/kg, about 3 μmoles of DCA) (Schultz et al., 2002). Using the data from the latter study we found that a two-compartment model was superior to one-compartment model (ΔAIC = 17.5) in describing DCA disposition (data not shown). While the exact mechanism of the bi-exponential disposition of DCA is yet to be elucidated, several hypotheses have been proposed: involvement of another enzyme (e.g. dechlorination by CYP in Figure 1) (Lash et al., 2000a), enterohepatic re-circulation of TCE and metabolites with possible dechlorination of TCA to DCA by gut microflora (Moghaddam et al., 1996), or mechanism-based inactivation of GSTζ after long-term exposure to DCA (Schultz et al., 2002).
While our data show that ~10% of retained TCE was biotransformed into TCA, DCA was formed in very limited amounts in B6C3F1 mice. In addition, it has been suggested that DCA has an extremely rapid disposition (Merdink et al., 1998; National Research Council, 2006), which is supported by the results of the two-compartment modeling in the present study. DCA itself is a liver carcinogen in mice and in rats (Herren-Freund et al., 1987; Bull et al., 1990). Exposure of male B6C3F1 mice to DCA in drinking water for up to 100 weeks resulted in a dose-dependent statistically significant increase in incidence of hepatocellular carcinoma at doses greater than 1000 mg/L, even though liver tumor multiplicity was significantly increased at doses as low as 50 mg/L (Deangelo et al., 1999). In view of our time-course observations for DCA formation following administration of a single large (2.1 g/l) dose of TCE, it is unlikely that DCA would ever reach comparable concentrations when animals are exposed to TCE. It should be noted, however, that degradation of DCA in liver cytosol is less efficient in humans than in rodents (Lipscomb et al., 1995), and further studies are needed to assess pharmacokinetics of TCA and DCA in species other than mice, as well as in tissues.
Another important issue is the bioavailability of TCE following an oral dose. According to the literature (Withey et al., 1983), type of vehicle can alter the bioavailability of TCE. For instance, use of corn oil as vehicle in rats resulted in 15-fold lower blood TCE concentrations relative to that given in water. Based on the fact that mice absorbed TCE in greater amounts and faster than rats (Prout et al., 1985; Fisher et al., 1991), 74% of bioavailability of TCE estimated in this study suggests that while the corn oil effect cannot be ruled out, it may not be as pronounced as in the rat.
In this study, the amounts of major oxidative and conjugative metabolites were assessed from the same animal and thus allow for comparison of relative proportion between the two pathways of TCE metabolism. We estimate that oxidative metabolites (TCA and DCA) were formed in the amounts at least 3600-fold higher than conjugative (DCVG and DCVC) ones. Furthermore, we observed that most of DCVG is metabolized further to DCVC. The relative advantage of the oxidative metabolism pathway in the mouse fits well with the historic observations suggesting that GSH conjugation is the minor pathway in mice, but not in rats (Prout et al., 1985; Goeptar et al., 1995; Bull, 2000; Lash et al., 2006). In humans, TCE metabolism appears to be substantially less efficient than in rodents (Lash et al., 2000a) and while DCVG has been detected in blood of human volunteers exposed to TCE vapors [(Lash et al., 1999), up to 100 ppm for 4 hrs], little information is available to compare the efficiency of oxidative and conjugative pathways in man. Thus, further studies are needed to address between- and within-species differences in TCE metabolism to provide quantitative data necessary for human risk assessment.
One additional limitation of this study is its focus on a small number of oxidative metabolites of TCE. While information on TCA and DCA kinetics provides for reasonable estimation of TCE metabolism through cytochrome P450-dependent pathway, assessment of trichloroethanol and chloral hydrate would also be desirable. Similarly, this work focused on only two key liver-derived glutathione conjugates rather than terminal nephrotoxic metabolites (e.g., DCVC sulfoxide, chlorothioketene, etc) or ultimately excreted metabolites (i.e., NAcDCVC). While additional improvements to our analytical approach are being considered to extend the number of metabolites that can be detected simultaneously, the difficulties of small-volume sample extraction and sensitivity of detection are formidable challenges. Finally, an extension of the time-course and assessment of the lower doses of TCE is needed to increase relevance of the data to human risk assessment. In order to monitor TCA and TCEOH, we might need a longer monitoring window, which is also needed to confirm the fate of DCA.
In conclusion, this study provides several important clues that may be useful for TCE risk assessment. First, we show the utility of the analytical method capable of assessing both oxidative and conjugative metabolites of TCE in a single small-volume sample. Second, using the time-course data obtained on the same animals we built a pharmacokinetic model which fits the data well and provides the kinetic parameters for TCA, DCA, DCVG and DCVC in the male B6C3F1 mice. Third, the model provides novel mechanistic justification for the pathway of biotransformation of TCE into DCA in the mouse. While not without limitations (i.e., limited number of metabolites assessed, high dose of TCE, etc.), this study provides an important foundation for further improvements of the analytical methodology, enables additional studies, and provides novel data and kinetic properties that may be used for re-assessment of existing models of TCE used in risk assessment.
Supplementary Material
Acknowledgments
The authors are grateful to Dr. Jeannie Padowski at UNC for assistance with modeling. This work was supported by NIEHS (P42 ES005948).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abbas R, Fisher JW. A physiologically based pharmacokinetic model for trichloroethylene and its metabolites, chloral hydrate, trichloroacetate, dichloroacetate, trichloroethanol, and trichloroethanol glucuronide in B6C3F1 mice. Toxicol Appl Pharmacol. 1997;147:15–30. doi: 10.1006/taap.1997.8190. [DOI] [PubMed] [Google Scholar]
- ATSDR. Agency for Toxic Substances and Disease Registry. U.S. Department of Health and Human Services; Washington, DC: 1997. Toxicological profile for trichloroethylene. [PubMed] [Google Scholar]
- Barton HA, Clewell HJ., III Evaluating noncancer effects of trichloroethylene: dosimetry, mode of action, and risk assessment. Environ Health Perspect. 2000;108(Suppl 2):323–334. doi: 10.1289/ehp.00108s2323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bois FY. Statistical analysis of Clewell et al. PBPK model of trichloroethylene kinetics. Environ Health Perspect. 2000;108(Suppl 2):307–316. doi: 10.1289/ehp.00108s2307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boroujerd M. Pharmacokinetics: Principles and Applications. McGraw-Hill/Appleton & Lange; New York: 2001. [Google Scholar]
- Brent RP. Algorithms for minimization without derivatives. Prentice-Hall; Englewood Cliffs, N.J.: 1972. [Google Scholar]
- Bronley-DeLancey A, McMillan DC, McMillan JM, Jollow DJ, Mohr LC, Hoel DG. Application of cryopreserved human hepatocytes in trichloroethylene risk assessment: relative disposition of chloral hydrate to trichloroacetate and trichloroethanol. Environ Health Perspect. 2006;114:1237–1242. doi: 10.1289/ehp.9047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruning T, Bolt HM. Renal toxicity and carcinogenicity of trichloroethylene: key results, mechanisms, and controversies. Crit Rev Toxicol. 2000;30:253–285. doi: 10.1080/10408440091159202. [DOI] [PubMed] [Google Scholar]
- Bull RJ. Mode of action of liver tumor induction by trichloroethylene and its metabolites, trichloroacetate and dichloroacetate. Environ Health Perspect. 2000;108(Suppl 2):241–259. doi: 10.1289/ehp.00108s2241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bull RJ, Sanchez IM, Nelson MA, Larson JL, Lansing AJ. Liver tumor induction in B6C3F1 mice by dichloroacetate and trichloroacetate. Toxicology. 1990;63:341–359. doi: 10.1016/0300-483x(90)90195-m. [DOI] [PubMed] [Google Scholar]
- Burnham KP, Anderson DR. Model selection and multimodel inference: a practical information-theoretic approach. Springer; New York: 2002. [Google Scholar]
- Cai H, Guengerich FP. Mechanism of Aqueous Decomposition of Trichloroethylene Oxide. J Am Chem Soc. 1999;121:11656–11663. [Google Scholar]
- Cai H, Guengerich FP. Acylation of protein lysines by trichloroethylene oxide. Chem Res Toxicol. 2000;13:327–335. doi: 10.1021/tx000003p. [DOI] [PubMed] [Google Scholar]
- Caldwell JC, Keshava N, Evans MV. Difficulty of mode of action determination for trichloroethylene: An example of complex interactions of metabolites and other chemical exposures. Environ Mol Mutagen. 2008;49:142–154. doi: 10.1002/em.20350. [DOI] [PubMed] [Google Scholar]
- Chiu WA, Caldwell JC, Keshava N, Scott CS. Key scientific issues in the health risk assessment of trichloroethylene. Environ Health Perspect. 2006a;114:1445–1449. doi: 10.1289/ehp.8690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiu WA, Okino MS, Lipscomb JC, Evans MV. Issues in the pharmacokinetics of trichloroethylene and its metabolites. Environ Health Perspect. 2006b;114:1450–1456. doi: 10.1289/ehp.8691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clewell HJ, Gearhart JM, Gentry PR, Covington TR, VanLandingham CB, Crump KS, Shipp AM. Evaluation of the uncertainty in an oral reference dose for methylmercury due to interindividual variability in pharmacokinetics. Risk Anal. 1999;19:547–558. doi: 10.1023/a:1007017116171. [DOI] [PubMed] [Google Scholar]
- Clewell HJ, III, Gentry PR, Covington TR, Gearhart JM. Development of a physiologically based pharmacokinetic model of trichloroethylene and its metabolites for use in risk assessment. Environ Health Perspect. 2000;108(Suppl 2):283–305. doi: 10.1289/ehp.00108s2283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Souza RW, Bruckner JV, Feldman S. Oral and intravenous trichloroethylene pharmacokinetics in the rat. J Toxicol Environ Health. 1985;15:587–601. doi: 10.1080/15287398509530688. [DOI] [PubMed] [Google Scholar]
- Deangelo AB, George MH, House DE. Hepatocarcinogenicity in the male B6C3F1 mouse following a lifetime exposure to dichloroacetic acid in the drinking water: dose-response determination and modes of action. J Toxicol Environ Health A. 1999;58:485–507. doi: 10.1080/009841099157115. [DOI] [PubMed] [Google Scholar]
- Evans MV, Andersen ME. Sensitivity analysis of a physiological model for 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD): assessing the impact of specific model parameters on sequestration in liver and fat in the rat. Toxicol Sci. 2000;54:71–80. doi: 10.1093/toxsci/54.1.71. [DOI] [PubMed] [Google Scholar]
- Fisher JW. Physiologically based pharmacokinetic models for trichloroethylene and its oxidative metabolites. Environ Health Perspect. 2000;108(Suppl 2):265–273. doi: 10.1289/ehp.00108s2265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisher JW, Gargas ML, Allen BC, Andersen ME. Physiologically based pharmacokinetic modeling with trichloroethylene and its metabolite, trichloroacetic acid, in the rat and mouse. Toxicol Appl Pharmacol. 1991;109:183–195. doi: 10.1016/0041-008x(91)90167-d. [DOI] [PubMed] [Google Scholar]
- Georgita C, Albu F, David V, Medvedovici A. Nonlinear calibrations on the assay of dilitiazem and two of its metabolites from plasma samples by means of liquid chromatography and ESI/MS(2) detection: application to a bioequivalence study. Biomed Chromatogr. 2008;22:289–297. doi: 10.1002/bmc.927. [DOI] [PubMed] [Google Scholar]
- Goeptar AR, Commandeur JN, Van Ommen B, van Bladeren PJ, Vermeulen NP. Metabolism and kinetics of trichloroethylene in relation to toxicity and carcinogenicity. Relevance of the mercapturic acid pathway. Chem Res Toxicol. 1995;8:3–21. doi: 10.1021/tx00043a001. [DOI] [PubMed] [Google Scholar]
- Guengerich FP. Covalent binding to apoprotein is a major fate of heme in a variety of reactions in which cytochrome P-450 is destroyed. Biochem Biophys Res Commun. 1986;138:193–198. doi: 10.1016/0006-291x(86)90265-2. [DOI] [PubMed] [Google Scholar]
- Herren-Freund SL, Pereira MA, Khoury MD, Olson G. The carcinogenicity of trichloroethylene and its metabolites, trichloroacetic acid and dichloroacetic acid, in mouse liver. Toxicol Appl Pharmacol. 1987;90:183–189. doi: 10.1016/0041-008x(87)90325-5. [DOI] [PubMed] [Google Scholar]
- Ketcha MM, Stevens DK, Warren DA, Bishop CT, Brashear WT. Conversion of trichloroacetic acid to dichloroacetic acid in biological samples. J Anal Toxicol. 1996;20:236–241. doi: 10.1093/jat/20.4.236. [DOI] [PubMed] [Google Scholar]
- Kim S, Vermeulen R, Waidyanatha S, Johnson BA, Lan Q, Rothman N, Smith MT, Zhang L, Li G, Shen M, Yin S, Rappaport SM. Using urinary biomarkers to elucidate dose-related patterns of human benzene metabolism. Carcinogenesis. 2006;27:772–781. doi: 10.1093/carcin/bgi297. [DOI] [PubMed] [Google Scholar]
- Kishi R, Harabuchi I, Ikeda T, Katakura Y, Miyake H. Acute effects of trichloroethylene on blood concentrations and performance decrements in rats and their relevance to humans. Br J Ind Med. 1993;50:470–480. doi: 10.1136/oem.50.5.470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lash LH, Fisher JW, Lipscomb JC, Parker JC. Metabolism of trichloroethylene. Environ Health Perspect. 2000a;108(Suppl 2):177–200. doi: 10.1289/ehp.00108s2177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lash LH, Parker JC, Scott CS. Modes of action of trichloroethylene for kidney tumorigenesis. Environ Health Perspect. 2000b;108(Suppl 2):225–240. doi: 10.1289/ehp.00108s2225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lash LH, Putt DA, Brashear WT, Abbas R, Parker JC, Fisher JW. Identification of S-(1,2-dichlorovinyl)glutathione in the blood of human volunteers exposed to trichloroethylene. J Toxicol Environ Health A. 1999;56:1–21. doi: 10.1080/009841099158204. [DOI] [PubMed] [Google Scholar]
- Lash LH, Putt DA, Parker JC. Metabolism and tissue distribution of orally administered trichloroethylene in male and female rats: identification of glutathione- and cytochrome P-450-derived metabolites in liver, kidney, blood, and urine. J Toxicol Environ Health A. 2006;69:1285–1309. doi: 10.1080/15287390500360133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipscomb JC, Mahle DA, Brashear WT, Barton HA. Dichloroacetic acid: metabolism in cytosol. Drug Metab Dispos. 1995;23:1202–1205. [PubMed] [Google Scholar]
- McKinney LL, Picken JC, Weakley FB, Eldridge AC, Campbell RE, Cowan JC, Biester HE. Possible toxic factor of trichloroethylene-extracted soybean oil mea. J Am Chem Soc. 1959;81:909–915. [Google Scholar]
- Merdink JL, Bull RJ, Schultz IR. Trapping and identification of the dichloroacetate radical from the reductive dehalogenation of trichloroacetate by mouse and rat liver microsomes. Free Radic Biol Med. 2000;29:125–130. doi: 10.1016/s0891-5849(00)00330-0. [DOI] [PubMed] [Google Scholar]
- Merdink JL, Gonzalez-Leon A, Bull RJ, Schultz IR. The extent of dichloroacetate formation from trichloroethylene, chloral hydrate, trichloroacetate, and trichloroethanol in B6C3F1 mice. Toxicol Sci. 1998;45:33–41. doi: 10.1006/toxs.1998.2500. [DOI] [PubMed] [Google Scholar]
- Merdink JL, Robison LM, Stevens DK, Hu M, Parker JC, Bull RJ. Kinetics of chloral hydrate and its metabolites in male human volunteers. Toxicology. 2008;245:130–140. doi: 10.1016/j.tox.2007.12.018. [DOI] [PubMed] [Google Scholar]
- Miller RE, Guengerich FP. Oxidation of trichloroethylene by liver microsomal cytochrome P-450: evidence for chlorine migration in a transition state not involving trichloroethylene oxide. Biochemistry. 1982;21:1090–1097. doi: 10.1021/bi00534a041. [DOI] [PubMed] [Google Scholar]
- Moghaddam AP, Abbas R, Fisher JW, Stavrou S, Lipscomb JC. Formation of dichloroacetic acid by rat and mouse gut microflora, an in vitro study. Biochem Biophys Res Commun. 1996;228:639–645. doi: 10.1006/bbrc.1996.1709. [DOI] [PubMed] [Google Scholar]
- National Research Council. Assessing the human health risks of trichloroethylene: Key scientific issues. The National Academies Press; Washington, D.C: 2006. [Google Scholar]
- National Toxicology Program. Carcinogenesis Studies of Trichloroethylene (Without Epichlorohydrin) (CAS No. 79-01-6) in F344/N Rats and B6C3F1 Mice (Gavage Studies) Natl Toxicol Program Tech Rep Ser. 1990;243:1–174. [PubMed] [Google Scholar]
- Pastino GM, Yap WY, Carroquino M. Human variability and susceptibility to trichloroethylene. Environ Health Perspect. 2000;108(Suppl 2):201–214. doi: 10.1289/ehp.00108s2201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prout MS, Provan WM, Green T. Species differences in response to trichloroethylene. I. Pharmacokinetics in rats and mice. Toxicol Appl Pharmacol. 1985;79:389–400. doi: 10.1016/0041-008x(85)90137-1. [DOI] [PubMed] [Google Scholar]
- Rappaport SM, Kupper LL. Quantitative exposure assessment. Rappaport, SM; El Cerrito, CA, U.S.A: 2008. [Google Scholar]
- Schultz IR, Merdink JL, Gonzalez-Leon A, Bull RJ. Dichloroacetate toxicokinetics and disruption of tyrosine catabolism in B6C3F1 mice: dose-response relationships and age as a modifying factor. Toxicology. 2002;173:229–247. doi: 10.1016/s0300-483x(02)00034-3. [DOI] [PubMed] [Google Scholar]
- Srinivas NR. Applicability of nonlinear calibration regression for quantitative determination of parent and metabolite(s) in bioequivalence assessment. Biomed Chromatogr. 2008;22:1315–1317. doi: 10.1002/bmc.1077. [DOI] [PubMed] [Google Scholar]
- US AF-EPA, 2004. Development of a Physiologically-Based Pharmacokinetic Model of Trichloroethylene and Its Metabolites for Use in Risk Assessment. Prepared for U.S. Air Force by U.S. Air Force-U.S. Environmental Protection Agency Trichloroethylene Physiogically-Based Pharmacokinetic Model Workgroup
- Withey JR, Collins BT, Collins PG. Effect of vehicle on the pharmacokinetics and uptake of four halogenated hydrocarbons from the gastrointestinal tract of the rat. J Appl Toxicol. 1983;3:249–253. doi: 10.1002/jat.2550030506. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.