

Abstract
Vesicular stomatitis virus (VSV) induces apoptosis via the mitochondrial pathway. The mitochondrial pathway is regulated by the Bcl-2 family of proteins, which consists of both pro- and antiapoptotic members. To determine the relative importance of the multidomain proapoptotic Bcl-2 family members Bak and Bax, HeLa cells were transfected with Bak and/or Bax small interfering RNA (siRNA) and subsequently infected with recombinant wild-type VSV. Our results showed that Bak is more important than Bax for the induction of apoptosis in this system. Bak is regulated by two antiapoptotic Bcl-2 proteins, Mcl-1, which is rapidly turned over, and Bcl-XL, which is relatively stable. Inhibition of host gene expression by the VSV M protein resulted in the degradation of Mcl-1 but not Bcl-XL. However, inactivation of both Mcl-1 and Bcl-XL was required for cells to undergo apoptosis. While inactivation of Mcl-1 was due to inhibition of its expression, inactivation of Bcl-XL indicates a role for one or more BH3-only Bcl-2 family members. VSV-induced apoptosis was inhibited by transfection with siRNA against Bid, a BH3-only protein that is normally activated by the cleavage of caspase-8, the initiator caspase associated with the death receptor pathway. Similarly, treatment with an inhibitor of caspase-8 inhibited VSV-induced apoptosis. These results indicate a role for cross talk from the death receptor pathway in the activation of the mitochondrial pathway by VSV.
The induction of cell death is a major mechanism by which many viruses cause disease in the tissues they infect (23). In addition, the cytolytic activity of viruses has the potential for therapeutic applications, such as the development of oncolytic viruses for the treatment of cancer (27). Vesicular stomatitis virus (VSV) is well studied as a prototype for negative-strand RNA viruses and is an exceptionally potent inducer of apoptosis in a wide variety of cell types (4, 20, 21). Due to its particularly rapid cytopathic effects, VSV is one of the major viruses being developed as an oncolytic agent (27). VSV is capable of inducing apoptosis by activation of multiple apoptotic pathways. It is important to determine how these pathways are activated and the role that they play in apoptosis induced by VSV in order to understand the virulence and oncolytic activity of the virus, as well as to provide a model to which other viruses can be compared.
Previous work showed that wild-type (wt) VSV induces apoptosis via the mitochondrial (intrinsic) pathway through the initiator caspase caspase-9 (4, 19). This is due in part to the inhibition of host gene expression by the VSV M protein (19). The inhibition of host gene expression by M protein is the mechanism by which VSV inhibits the host antiviral response (2, 31) and leads to induction of apoptosis, similar to that induced by pharmacologic inhibitors of host gene expression (19). Additionally, M protein mutants of VSV that are deficient in the ability to inhibit new host gene expression are effective inducers of apoptosis (12, 13, 19, 20). However, in contrast to wt VSV, induction of apoptosis by M protein mutant virus occurs primarily via the extrinsic pathway through the initiator caspase caspase-8 (12, 13). Infection with M protein mutant VSV results in the expression of proapoptotic genes that are suppressed during infection with wt VSV (12). Therefore, in the case of VSV with wt M protein, the induction of apoptosis is most likely mediated by proteins already present in the host cell. Since it has previously been shown that wt VSV activates the intrinsic pathway, we focused on the Bcl-2 family of proteins to determine the role of Bcl-2 family members in apoptosis induced by wt VSV.
Bcl-2 family proteins function to either suppress or promote mitochondrial outer membrane permeabilization, thereby regulating the release of proapoptotic factors into the cytosol, such as cytochrome c, apoptosis-inducing factor (AIF), and Smac/Diablo (5). Bcl-2 family proteins are subdivided into three groups, depending on the conservation of Bcl-2 homology (BH) domains and function (reviewed in references 8 and 38). The multidomain antiapoptotic Bcl-2 proteins contain BH domains BH1 to BH4 and function to inhibit apoptosis by binding to proapoptotic Bcl-2 family members. Members of this group include Bcl-2, Bcl-XL, Mcl-1, Bcl-w, and BFL-1/A1. The proapoptotic Bcl-2 proteins are comprised of two groups, the multidomain proteins and the BH3-only proteins. Bax and Bak are the two main members of the multidomain group, containing BH domains BH1 to BH3. These proteins are primarily responsible for the permeabilization of the mitochondrial outer membrane, if their activity is not suppressed by antiapoptotic Bcl-2 family members. The BH3-only proteins contain only one Bcl-2 homology domain (BH3) and include Bid, Bad, Bim, Puma, Noxa, and Bik, among others. These proteins function as upstream sensors of signaling pathways and convey to other Bcl-2 family proteins the signals to initiate apoptosis. These death signals can be transmitted from the BH3-only proteins by either binding to antiapoptotic proteins, causing the release of Bak and Bax, or binding to Bak and Bax, thereby causing their activation (6).
The pathways leading to activation of Bak differ from those that activate Bax. Interestingly, only two antiapoptotic Bcl-2 proteins, Mcl-1 and Bcl-XL, have been shown to interact with Bak, while Bax appears to be able to interact with all of the antiapoptotic proteins, with the exception of Mcl-1 (7, 35). BH3-only proteins have strong binding affinities to the antiapoptotic proteins, suggesting that their primary role may be to derepress Bak and Bax by binding and inhibiting the antiapoptotic proteins (36). In addition, BH3-only proteins may play a role in activation of Bak and Bax by binding and inducing an activated conformation (6, 34). For some stimuli, such as the protein kinase inhibitor staurosporine (SSP), the topoisomerase II inhibitor etoposide, and UV radiation, Bak and Bax appear to be redundant, in that the deletion of both is required to render cells resistant to these agents (33). In contrast, Bak and Bax were nonredundant in the induction of apoptosis by Neisseria gonorrhoeae and cisplatin, such that both were required for apoptosis to occur (18).
In the experiments reported here, the silencing of Bak or Bax expression with small interfering RNA (siRNA) showed that Bak is more important than Bax for the induction of apoptosis in HeLa cells infected with wt VSV. Overexpression of both of the antiapoptotic Bcl-2 family proteins known to interact with Bak, Mcl-1 and Bcl-XL, delayed the onset of apoptosis, while depletion of Mcl-1 or Bcl-XL by siRNA transfection prior to infection increased the rate of apoptosis. Furthermore, M protein inhibition of new host gene expression led to the depletion of Mcl-1, enabling the rapid activation of apoptosis. However, inhibition of Bcl-XL was also required for the initiation of apoptosis, indicating a role for one or more BH3-only proteins. Bid, a BH3-only protein that is normally activated by the cleavage of caspase-8, was shown to be important for induction of apoptosis by VSV. Likewise, treatment with an inhibitor of caspase-8 inhibited VSV-induced apoptosis. These results indicate a role for cross talk from the death receptor pathway in the activation of the mitochondrial pathway by VSV.
MATERIALS AND METHODS
Cell lines and viruses.
HeLa cells were cultured in Dulbecco's modified Eagle medium supplemented with 7% fetal bovine serum. The recombinant viruses recombinant wild type (rwt) and rM51R-M were isolated from cDNA clones and grown as previously described (20). Infections were carried out at a multiplicity of infection of 10 PFU per cell. Stably transfected cell lines were generated by transfection of HeLa cells with the h-Mcl-1 pcDNA3 plasmid or the h-Mcl-1-S159A pc3DNA plasmid (gifts from Ulrich Maurer [24]), h-Bcl-XL pcDNA3 (a gift from John Wilkinson), or pcDNA3 plasmid for empty vector (EV) cells, as previously described (19). Stably transfected cells were cultured in Dulbecco's modified Eagle medium supplemented with 7% fetal bovine serum and 200 μg/ml of G418.
RNA interference.
For silencing experiments, HeLa cells were transiently transfected with Bak siRNA (h), Bax siRNA (h), Bcl-XS/L (h), control siRNA-A (Santa Cruz Biotechnology), or On-Target plus duplex J-004501-17 Human Mcl-1 (Dharmacon) using TransIT-siQuest transfection reagent (Mirus Bio Corporation). Briefly, HeLa cells were grown to approximately 75% confluence in 6-well plates, and just before transfection, 1,250 μl of fresh growth medium was added to each well. For each well, a mixture of 250 μl Opti-MEM medium, 2 μl TransIT-siQuest transfection reagent, and 25 nM siRNA was incubated for 20 min before being added to the plate. After 24 h, cells were split into 96-well plates at a density of 8 × 103 for cell viability and caspase-3-like activity assays, 6-well plates for cell lysates to confirm silencing, and 6- or 24-well plates for time-lapse microscopy experiments. At 48 h posttransfection, cells were infected with rwt virus.
Immunoblot analysis.
HeLa cells or siRNA-transfected HeLa cells were grown to approximately 75% confluence in 6-well dishes and infected with recombinant viruses. At the indicated times postinfection, cell lysates were generated and analyzed by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and immunoblotting, as previously described (13). The antibodies used in this study were obtained as follows: Bak and Mcl-1 antibodies were obtained from Santa Cruz Biotechnology, Bax and Bcl-XL antibodies were obtained from Cell Signaling Technologies, and β-actin antibody was obtained from Sigma-Aldrich, Incorporated. Protein band intensities were quantitated by scanning and analysis with Quantity One software (Bio-Rad).
Time-lapse microscopy.
siRNA-transfected HeLa cells (described above) were grown to approximately 50% confluence in 6- or 24-well dishes and infected with rwt virus. Time-lapse microscopy was performed on a Zeiss Axiovert S200, as previously described (13). Fields containing 40 to 80 cells were selected for analysis, and images of the same fields were captured at 15-min intervals for 16 h. The time of entry of each cell into apoptosis was determined by the time of onset of membrane blebbing, which was followed by other morphological changes that are characteristic of apoptosis, as described in detail in reference 20. The data from three separate experiments, including 120 to 200 total cells, were analyzed and evaluated for statistical significance by the log rank test.
Cell viability assay.
Cell viability was assessed by MTT [3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide] assay, according to the manufacturer's instructions (Roche Molecular Biochemicals, Indianapolis, IN). siRNA-transfected cells were seeded in 96-well plates, as described above. Cell viability was evaluated as described previously, and survival was estimated relative to untreated controls (19).
Caspase-3 activity assay.
siRNA-transfected cells were grown to approximately 75% confluence, as described above. Cells were either infected with rwt virus or treated with SSP (1 μg/ml; Cell Signaling Technologies) as a positive control for the activation of the mitochondrial pathway. Duplicate wells were lysed, and caspase-3 activation was determined with a fluorogenic substrate for caspase-3 (DEVD-AFC; R&D Systems, Inc.), as previously described (13). Prior to addition of the fluorogenic substrate, 10 μl was removed and used to determine the protein concentration by Bio-Rad detergent-compatible protein assay (Bio-Rad Laboratories).
Statistical analysis.
A paired Student t test was used to compare the significances of differences between groups at individual time points for experiments containing two experimental groups. Analysis of variance with Dunnett's pairwise comparison with a control was used for experiments containing more than two experimental groups, using SigmaStat software (Systat Software, Inc.). Time-lapse microscopy data were analyzed for statistical significance using the log rank test (http://bioinf.wehi.edu.au/software/russell/logrank/index.html). A P value of <0.05 was considered statistically significant.
RESULTS
VSV induces apoptosis more slowly in HeLa cells lacking Bak than in those lacking Bax.
wt strains of VSV induce apoptosis primarily via the mitochondrial pathway (4, 19). Since the mitochondrial pathway is regulated by the Bcl-2 family of proteins, we determined which Bcl-2 family members play a role in VSV-induced apoptosis. Proapoptotic proteins Bax and Bak in their active forms are the main proteins responsible for the destabilization of the mitochondrial membrane. Bak and Bax have been shown to be redundant in some systems (33), while in other systems, one is more important than the other (18). To determine the relative importance of Bak and Bax in VSV-induced apoptosis, HeLa cells were transfected with siRNA against Bak, Bax, or both or with nontargeting siRNA as a control. At 24, 48, and 72 h posttransfection, cell lysates were prepared and analyzed by SDS-PAGE and immunoblotting. Figure 1A shows a representative immunoblot for the silencing of Bak or Bax, with analysis of actin as a loading control. Levels of Bak and Bax were similarly low at all times posttransfection. Cells were infected with virus at 48 h posttransfection for all subsequent experiments. In the experiments shown in Fig. 1B, cells were infected with an rwt strain of VSV and imaged using time-lapse microscopy. The percentages of cells entering apoptosis were determined as a function of time postinfection by the time of onset of apoptotic membrane blebbing. The graph shows the average results of three experiments. rwt virus rapidly induced apoptosis in the control, nontargeting siRNA-transfected cells, with more than 75% of cells undergoing apoptosis by 16 h postinfection. A similar time course was observed for the Bax siRNA cells. However, the onset of apoptotic membrane blebbing was delayed in both the Bak siRNA cells and the Bak plus Bax siRNA cells, with less than 30% of cells undergoing apoptosis by 16 h postinfection.
FIG. 1.

rwt virus induces apoptosis more slowly in HeLa cells lacking Bak than in those lacking Bax. (A) HeLa cells were transfected with 25 nM Bak, Bax, or nontargeting siRNA as a control. At the indicated times posttransfection, cell lysates were analyzed by immunoblotting with antibodies for Bak, Bax, and actin as a loading control. Cells transfected with Bak or Bax siRNA were analyzed in duplicate. (B) HeLa cells were transfected with 25 nM Bak siRNA, Bax siRNA, and Bak and Bax siRNA, 25 nM nontargeting siRNA as a control for single transfection, and 50 nM nontargeting siRNA as a control for double transfection. At 48 h posttransfection, cells were infected with rwt virus and analyzed by time-lapse microscopy. The cumulative percentage of cells entering apoptosis was determined as the number of cells that underwent apoptotic membrane blebbing and is plotted as a function of time postinfection. The data represent the average of three experiments, which collectively analyzed the time of onset of apoptosis in 120 to 200 total cells. Bak siRNA and Bak plus Bax siRNA data were significantly different from control and Bax siRNA data (P < 0.05) by log rank analysis. In subsequent assays, only one control is shown, as the data are almost identical. (C) Caspase-3-like activity was assayed in HeLa cells transfected for 48 h with the same siRNAs as described above and then infected with rwt virus for the times indicated. Cells were lysed, and caspase-3 activity in cell lysates was measured with a fluorogenic substrate (DEVD-AFC). Data are expressed in arbitrary fluorescence units per microgram of total protein and normalized to the maximum value expressed by HeLa cells incubated with SSP (as a positive control) for 12 h. The data represent the average results ± standard deviations from three experiments. (D) Cell viability was analyzed with an MTT assay. HeLa cells were transfected and infected, as described above. At the indicated times, MTT was added to each sample for 4 h, at which point a solubilization solution was added. Samples were analyzed with an ELISA plate reader. Cell viability was determined as a percentage of a transfected, uninfected control. The data represent the average results ± standard deviations from three experiments (*, P < 0.05 compared to control).
The delay in the induction of apoptosis in Bak siRNA cells was confirmed by an analysis of caspase-3-like activity in cell lysates using a fluorogenic substrate (Fig. 1C) and by analysis of cell viability using the MTT assay (Fig. 1D), which measures metabolically active cells. For the caspase-3 assay, cells treated with SSP were used as a positive control. Bax and Bak siRNA cells both had caspase-3-like activity when treated with SSP, comparable to that of control nontargeting siRNA cells (data not shown). Caspase-3-like activity is presented in Fig. 1C as a percentage of maximal SSP activation. For the MTT assay, data are expressed as a percentage of mock-infected cells (Fig. 1D). Control siRNA cells and Bax siRNA cells had high levels of caspase-3-like activity and reduced viability by 16 h postinfection, while the Bak siRNA cells and Bak plus Bax siRNA cells had significantly less active caspase-3 and higher cell viability than control siRNA cells at 10, 12, and 16 h postinfection (P < 0.05) (Fig. 1C and D). Collectively, the data shown in Fig. 1B to D show that silencing of Bak expression slows the rate of apoptosis, while silencing of Bax expression does not.
Mcl-1 levels are reduced in VSV-infected HeLa cells, while Bcl-XL levels remain unchanged.
In healthy cells, Bak is kept in an inactive state by interaction with antiapoptotic Bcl-2 family members. Only two antiapoptotic family members, Bcl-XL and Mcl-1, have been found to interact directly with Bak. Bcl-XL is a relatively stable protein, with a half-life of over 24 h (1). However, Mcl-1 protein and mRNA have been shown to have a rapid turnover rate in many cell types (1, 26). For example, the half-life of Mcl-1 in cycloheximide-treated HeLa cells is <1 h (1), which we have confirmed (data not shown). VSV inhibits new host gene expression due to the ability of M protein to inhibit host transcription, nuclear-cytoplasmic transport, and translation (23). We hypothesized that Mcl-1 levels would decay rapidly in VSV-infected cells, allowing for the activation of Bak and subsequent apoptosis. To test this hypothesis, HeLa cells were infected with rwt virus, and at different times postinfection, cell lysates were prepared and analyzed for Mcl-1 by SDS-PAGE and immunoblotting (Fig. 2A). There was a visible reduction in the levels of Mcl-1 in rwt virus-infected HeLa cells by 8 h postinfection, with levels being below the limit of detection by 24 h postinfection. The timing of reduction in Mcl-1 levels was consistent with the induction of apoptosis (Fig. 1) and with the inhibition of host gene expression (2). To confirm that the loss of Mcl-1 was due to the shutoff of new host gene expression, HeLa cells were infected with a recombinant M protein mutant virus that is isogenic with rwt virus, except for a point mutation in the M protein, which renders the virus defective in the ability to inhibit host gene expression (rM51R-M virus). Figure 2A shows the quantification of Mcl-1, expressed as a ratio to actin and normalized to mock-infected controls. There was a significant reduction in the levels of Mcl-1 in rwt virus-infected cells by 8 h postinfection, with levels continuing to decline throughout the 24 h time course. In contrast, Mcl-1 levels were increased by 4 h postinfection and were similar to those of mock-infected cells for the remainder of the time course in rM51R-M virus-infected cells. A similar immunoblot analysis of Bcl-XL levels showed that, in contrast to Mcl-1, there were no substantial changes in the level of Bcl-XL in rwt virus-infected cells at any time postinfection compared to the level in mock-infected cells (Fig. 2B). These data support the hypothesis that the loss of Mcl-1 is due to its rapid turnover, combined with the inhibition of host gene expression by wt VSV.
FIG. 2.
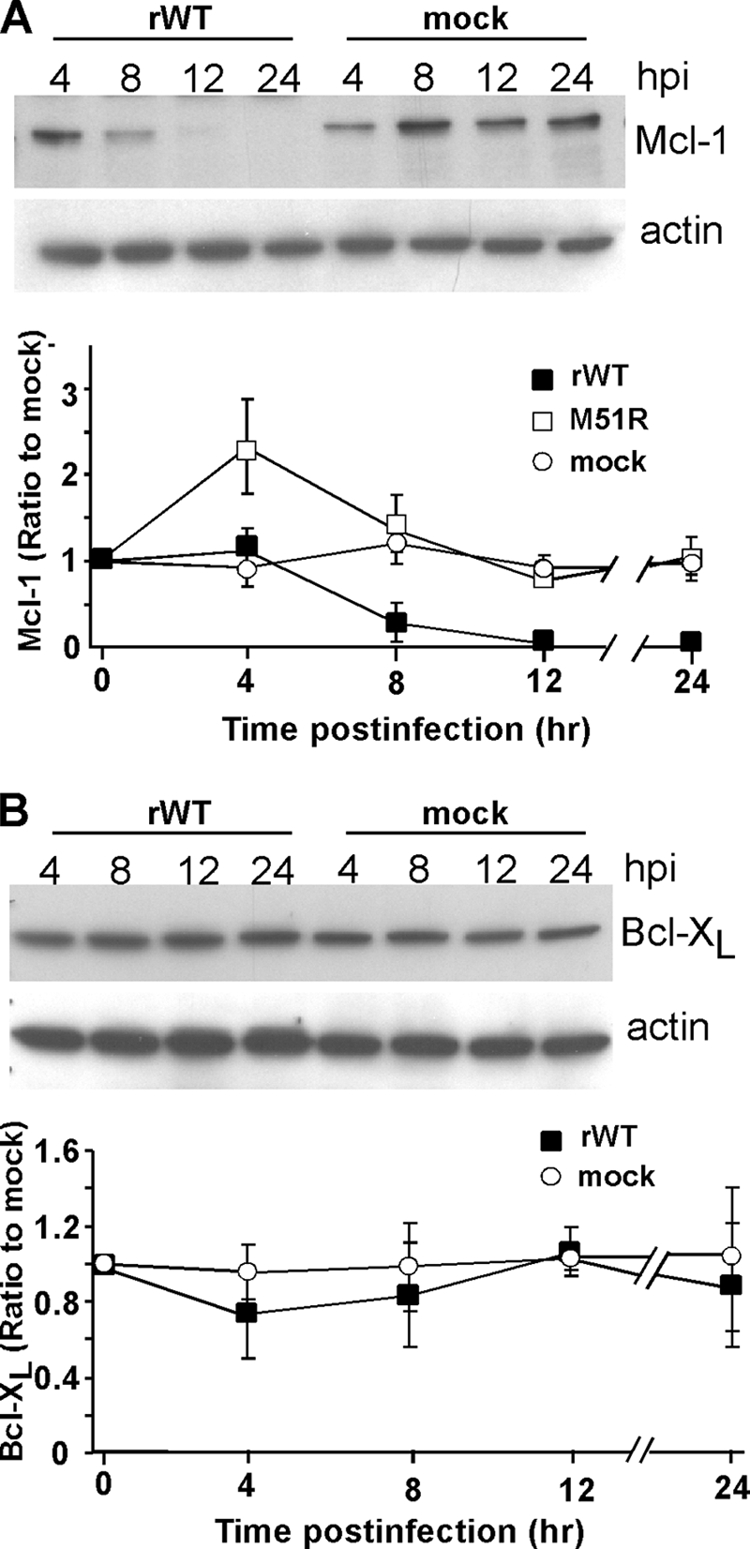
Mcl-1 levels are reduced in rwt virus-infected HeLa cells, while Bcl-XL levels remain unchanged. (A) At the indicated times postinfection with rwt virus, cell lysates were analyzed by immunoblotting with antibodies for Mcl-1. The graph shows the quantitation of Mcl-1 expression normalized to actin expression as percentages of the average ratios in mock-infected cells. The data represent the average results ± standard deviations from three experiments for cells infected with rwt virus or rM51R-M virus or mock-infected cells. (B) Cells were analyzed for Bcl-XL expression, as described for panel A.
Apoptosis is regulated by the levels of both proapoptotic and antiapoptotic Bcl-2 proteins; therefore, we also analyzed the levels of proapoptotic proteins Bak and Bax by immunoblotting (Fig. 3). There were no significant changes in the level of Bak at any time postinfection (Fig. 3A). Levels of Bax began to decline at 12 h postinfection and were significantly decreased by 24 h postinfection (Fig. 3B), likely due to normal turnover following virus-induced inhibition of its synthesis. In contrast, levels of Bak can remain unaffected for long periods of time (>16 h) following inhibition of its synthesis (17). The observation that the levels of Bak did not decline during infection is consistent with the idea that the loss of Mcl-1 contributes to the activation of Bak. Since Bax levels do not have an effect on the rate of apoptosis in rwt virus-infected cells (Fig. 1), the decrease in the level of Bax at later times postinfection is not likely to be important in VSV-induced apoptosis in HeLa cells.
FIG. 3.
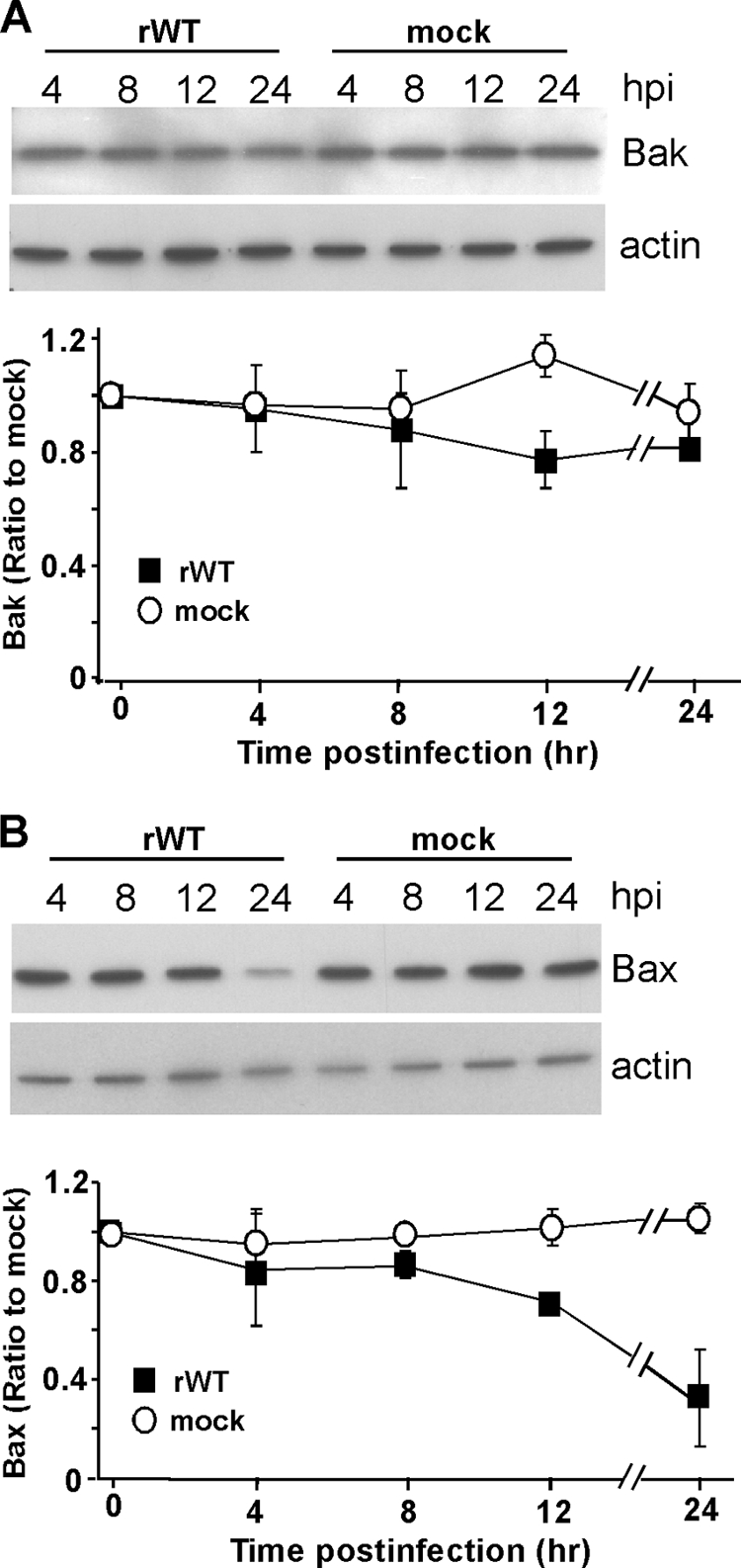
Bak levels in rwt virus-infected HeLa cells are not significantly different from those of mock-infected samples, while Bax levels decrease at later times postinfection. At the indicated times postinfection with rwt virus, cell lysates were generated and analyzed by immunoblotting with antibodies for Bak (A) and Bax (B). Representative gels are shown at the top of panels A and B, the graphs show quantitation of Bak (A) or Bax (B) protein expression normalized to that of actin protein expression, and results are shown as percentages of mock-infected samples. The data represent the average results ± standard deviations from three experiments for rwt virus-infected cells or mock-infected cells. hpi, hours postinfection.
Apoptosis induced by VSV infection is delayed in HeLa cells that overexpress Mcl-1 or Bcl-XL.
To determine whether the rapid decrease in the level of Mcl-1 contributes to the induction of apoptosis in rwt virus-infected cells, HeLa cells that stably overexpress Mcl-1 were generated. Due to the short half-life of Mcl-1, we also generated cell lines that express a mutant Mcl-1, S159A. This is a phosphorylation site mutant, which exhibits enhanced stability and confers increased protection from apoptosis compared to those of wt Mcl-1 (24). Two clonal cell lines were generated for both the wt Mcl-1 and the mutant Mcl-1 and are both shown to indicate that differences are not due to clonal variation. The cell lines overexpress Mcl-1 compared to expression of an EV control cell line, as determined by immunoblotting (Fig. 4A). During infection with rwt virus, the levels of Mcl-1 declined in HeLa-wt Mcl-1 cells, with a time course similar to that of HeLa-mutant Mcl-1 cells, as determined by quantification of immunoblots similar to those shown in Fig. 4A (Fig. 4B). Thus, the greater stability of the mutant Mcl-1 was not sufficient to delay its decline in rwt virus-infected cells. Nonetheless, the levels of Mcl-1 in both HeLa-wt Mcl-1 and HeLa-mutant Mcl-1 cells were higher than those in HeLa-EV control cells throughout the first 12 h postinfection. To determine if the increase in Mcl-1 levels delayed the induction of apoptosis by rwt virus, HeLa-wt Mcl-1, HeLa mutant Mcl-1, and HeLa-EV cells were infected with rwt virus, and cells entering apoptosis were quantitated using time-lapse microscopy (Fig. 4C). HeLa-EV cells entered apoptosis with a time course similar to that of untransfected HeLa cells (Fig. 1). Compared to control cells, there was approximately a 1-h delay in the time it took for 50% of the cells to enter apoptosis for the two wt Mcl-1-overexpressing cell lines and a 2-h delay for the two mutant Mcl-1-overexpressing cell lines. The effects of overexpression of Mcl-1 on virus-induced apoptosis were also determined by assaying caspase-3-like activity. In contrast to the relatively modest change in the time course of the onset of membrane blebbing, the level of caspase-3 activated in HeLa-Mcl-1 cells was dramatically reduced compared to that activated in HeLa-EV cells (Fig. 4D). This difference in the magnitude of the effects of Mcl-1 overexpression may be due to how little caspase-3 activation is required to cause HeLa cells to undergo morphological changes associated with apoptosis. Alternatively, there may be caspase-3-independent mechanisms that lead to the morphological changes assayed as shown in Fig. 4B. From these data, we concluded that Mcl-1 was able to marginally inhibit the onset of the morphological changes accompanying apoptosis in rwt virus-infected HeLa cells, but there was a substantial reduction in the activation of caspase-3-like activity.
FIG. 4.

Apoptosis is delayed in Mcl-1-overexpressing cells infected with rwt virus. Clonal HeLa cell lines that overexpress wt Mcl-1 (Mcl-1-13, Mcl-1-16), a mutant Mcl-1 (S159A-7, S159A-8), or an empty vector control were generated. (A) Cell lysates were analyzed by immunoblotting, using an antibody against Mcl-1. (B) Mcl-1-13, Mcl-1-16, S159A-7, S159A-8, and empty vector cells were infected with rwt virus. The graph shows the quantitation of Mcl-1 expression normalized to that of actin expression as percentages of the average ratios in mock-infected cells. The data represent the average results ± standard deviations from three experiments. (C) Mcl-1-13, Mcl-1-16, S159A-7, S159A-8, and empty vector cells were infected with rwt virus. Cells entering apoptosis were analyzed by time-lapse microscopy, as described in the legend to Fig. 1. The data represent the average results from three experiments, which collectively analyzed the time of onset of apoptosis in 120 to 200 total cells. The data for Mcl-1-13 and Mcl-1-16 cells were significantly different from the data for control empty vector cells, as well as the data for S159A-7 and S159A-8 cells (P < 0.05), by log rank analysis. (D) Mcl-1-overexpressing cells and empty vector cells were infected with rwt virus for the times indicated, and caspase-3-like activity was analyzed using a fluorogenic substrate, as described in the legend to Fig. 1. The data represent the average results ± standard deviations from three experiments.
Since there was a decrease in the rate of apoptosis in cells transfected with Mcl-1, despite the rapid turnover, we hypothesized that overexpression of a more stable antiapoptotic Bcl-2 family member, i.e., Bcl-XL, would have a more dramatic effect on the induction of apoptosis compared to overexpression of Mcl-1. To determine the effects of Bcl-XL overexpression, a HeLa cell line that stably overexpressed Bcl-XL was generated. An immunoblot demonstrating the overexpression of Bcl-XL is shown in Fig. 5A. The induction of apoptosis in HeLa-Bcl-XL cells and HeLa-EV cells infected with rwt virus was quantified by time-lapse microscopy (Fig. 5B) and by an assay measuring caspase-3-like activity (Fig. 5C). rwt virus rapidly induced apoptosis in the control, empty vector cells, with approximately 90% of cells undergoing apoptosis by 16 h postinfection, while only 20% of Bcl-XL-transfected cells underwent apoptosis by the same time point (Fig. 5B). Similar results were obtained for the caspase-3 assay, in which caspase-3-like activity was almost completely abrogated in the Bcl-XL-transfected cells (Fig. 5C). Both assays showed that overexpression of the more stable antiapoptotic protein Bcl-XL caused a dramatic reduction in the rate of induction of apoptosis in rwt virus-infected cells compared to overexpression of Mcl-1.
FIG. 5.

Apoptosis is significantly delayed in Bcl-XL-overexpressing cells infected with rwt virus. HeLa cells that overexpress Bcl-XL (Bcl-XL-3) or an empty vector control were generated. (A) Cell lysates were generated and analyzed by immunoblotting, using an antibody against Bcl-XL. (B) Bcl-XL-3 and empty vector cells were infected with the rwt virus. Cells entering apoptosis were analyzed by time-lapse microscopy, as described in the legend to Fig. 1. The data represent the average results from three experiments, which collectively analyzed the time of onset of apoptosis in 120 to 200 total cells. The data for Bcl-XL-3 cells were significantly different from the data for control empty vector cells (P < 0.05) by log rank analysis. (C) Bcl-XL-3 and empty vector cells were infected with rwt virus for the times indicated, and caspase-3-like activity was analyzed using a fluorogenic substrate, as described in the legend to Fig. 1. The data represent the average results ± standard deviations from three experiments (*, P < 0.05 compared to empty vector control).
Apoptosis induced by VSV infection is accelerated in HeLa cells transfected with Mcl-1 or Bcl-XL siRNA.
The data shown in Fig. 2 to 5 indicate that the rapid induction of apoptosis by rwt virus in HeLa cells is due in part to the depletion of Mcl-1. siRNA silencing experiments were used to determine whether the loss of Mcl-1 or Bcl-XL or both was sufficient to cause cells to undergo apoptosis. Figure 6A shows a representative immunoblot, showing the siRNA silencing of Mcl-1 and Bcl-XL. There was no significant reduction in cell viability of Mcl-1 or Bcl-XL siRNA cells at either 24 or 48 h posttransfection, determined by MTT assay (Fig. 6B). However, the combined silencing of Mcl-1 and Bcl-XL decreased cell viability to approximately 40% of the control at both time points. Thus, silencing of either Mcl-1 or Bcl-XL alone was not sufficient to induce apoptosis, while silencing of both was sufficient to induce cell death in the absence of a viral infection. These results suggest that the loss of Mcl-1 alone in VSV-infected cells is not sufficient to induce apoptosis and that inactivation of Bcl-XL is also required.
FIG. 6.
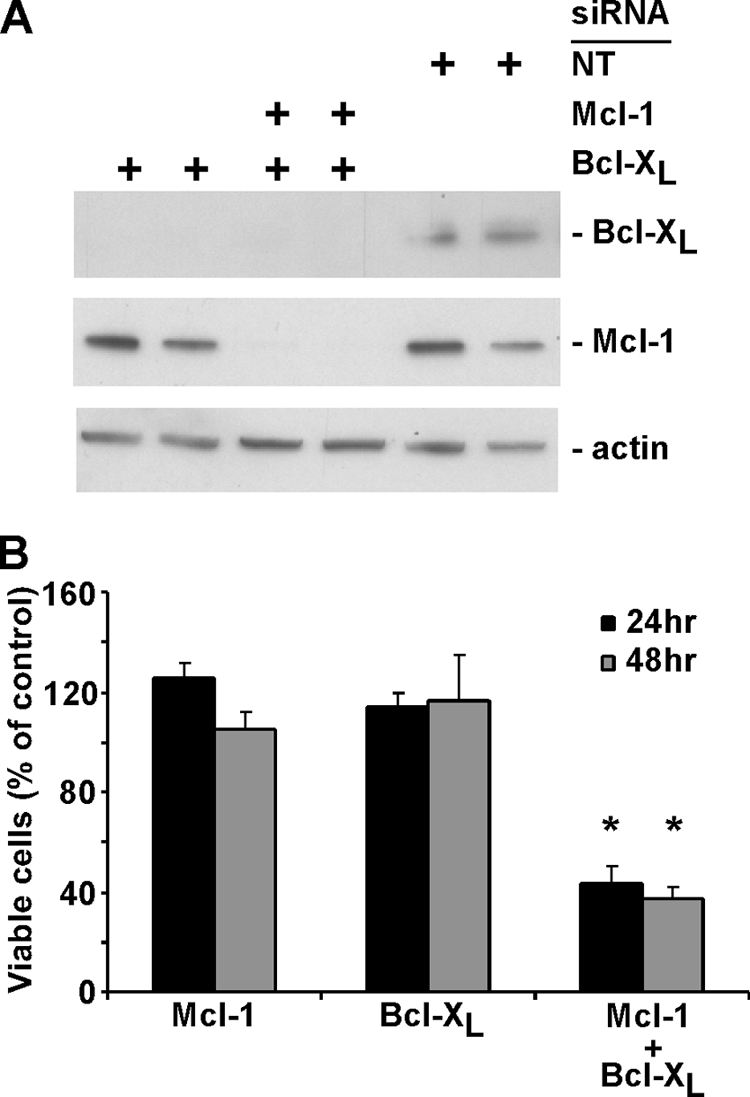
Simultaneous silencing of Bcl-XL and Mcl-1 by siRNA transfection induces cell death. (A) HeLa cells were transfected in duplicate with 25 nM Mcl-1, Mcl-1, or Bcl-XL or with nontargeting (NT) siRNA as a control. At 24 h posttransfection, cell lysates were analyzed by immunoblotting, using antibodies against Mcl-1 and Bcl-XL. (B) HeLa cells were transfected with Mcl-1, Bcl-XL, Mcl-1, or Bcl-XL or with nontargeting siRNA as a control. Cell viability was analyzed at 24 and 48 h posttransfection by MTT assay, as described in the legend to Fig. 1. The data represent the average results and standard deviations from three experiments (*, P < 0.05 compared to single silenced cells).
To test whether silencing of Mcl-1 or Bcl-XL would accelerate virus-induced apoptosis, HeLa cells were transfected with either Bcl-XL, Mcl-1, or nontargeting siRNA for 48 h and then infected with rwt virus. Silencing of Mcl-1 led to a slightly faster induction of apoptosis than silencing in control siRNA cells, as measured by time-lapse microscopy (Fig. 7A). However, silencing of Bcl-XL dramatically increased the rate of virus-induced apoptosis. Similar results were obtained by assaying caspase-3-like activity and cell viability (Fig. 7B and C). There was no significant difference in the amount of virus-induced caspase-3-like activity or reduction in cell viability in Mcl-1 siRNA cells compared to those in control siRNA cells throughout the time course (Fig. 7B). In contrast, virus infection activated significantly more caspase-3 and reduced cell viability to a significantly greater extent in Bcl-XL siRNA cells than in control siRNA cells. These data are consistent with the idea that Mcl-1 is degraded quickly upon infection with VSV; therefore, removal of Mcl-1 prior to infection does not increase the rate of apoptosis. However, inhibition of Bcl-XL requires the activation of one or more BH3-only proteins, so removal of Bcl-XL prior to infection significantly increases the rate of apoptosis, presumably by reducing the time required for inactivation of antiapoptotic proteins by activated BH3-only proteins.
FIG. 7.

Apoptosis is induced significantly more quickly by rwt virus infection in Bcl-XL-silenced cells. (A) HeLa cells were transfected with Mcl-1, Bcl-XL, or nontargeting (control) siRNA for 48 h and then infected with rwt virus. Cells entering apoptosis were analyzed by time-lapse microscopy, as described in the legend to Fig. 1. The data represent the average results from three experiments, which collectively analyzed the time of onset of apoptosis in 120 to 200 total cells. The data for Mcl-1 siRNA cells were significantly different from the data for control siRNA cells, as well as the data for Bcl-XL siRNA cells (P < 0.05), by log rank analysis. (B) HeLa cells were transfected with Mcl-1, Bcl-XL, or nontargeting (control) siRNA for 48 h and then infected with rwt virus for the times indicated, and caspase-3-like activity was analyzed using a fluorogenic substrate, as described in the legend to Fig. 1. The data represent the average results ± standard deviations from three experiments. (C) HeLa cells were transfected with Mcl-1, Bcl-XL, or nontargeting (control) siRNA for 48 h and then infected with rwt virus. Cell viability was analyzed at the indicated times by MTT assay, as described in the legend to Fig. 1. The data represent the average results and standard deviations from three experiments (*, P < 0.05 compared to control).
Apoptosis induced by VSV infection is delayed in HeLa cells transfected with Bid siRNA or treated with caspase-8 inhibitor.
The experiments shown in Fig. 7 raise the question of which BH3-only protein(s) is involved in the inactivation of Bcl-XL. Our previous experiments have shown that VSV infection activates elements of the death receptor pathway as well as the mitochondrial pathway (12, 13, 19). Cross talk between the death receptor pathway and the mitochondrial pathway is mediated primarily by the BH3-only protein Bid, which is cleaved to its active form (truncated Bid or t-Bid) by caspase-8 and caspase-10 (28, 39). We tested the hypothesis that induction of apoptosis by VSV involves activation of Bid by analysis of Bid cleavage by immunoblotting and by silencing Bid expression by transfection with siRNA. HeLa cells were infected with rwt virus, and Bid cleavage was analyzed by immunoblotting (Fig. 8A), using an antibody raised against the N terminus, which is present in full-length Bid but not in t-Bid. Quantification of multiple experiments showed that more than 50% of Bid was cleaved by 8 h postinfection, which generally precedes the onset of the morphological changes associated with the induction of apoptosis (Fig. 1 and 4), and thus is a very early event in the induction of apoptosis by VSV. Also shown in Fig. 8A, the rapid cleavage of full-length Bid was prevented by the treatment of cells with a caspase-8 inhibitor, IETD-fmk (note the difference in time scale), indicating that the disappearance of full-length Bid was due to caspase cleavage and not due to normal turnover following inhibition of its synthesis after VSV infection.
FIG. 8.

Apoptosis is significantly delayed in Bid-silenced cells infected with rwt virus. (A) At the indicated times postinfection with rwt virus, cell lysates were analyzed by immunoblotting with antibodies for Bid. The graph shows the quantitation of Bid expression as percentages of the levels in mock-infected cells. The data represent the average results ± standard deviations from four experiments. Shown on the right are results from HeLa cells incubated in the presence of 100 μM caspase-8 inhibitor (IETD-fmk) for 2 h prior to infection with rwt virus for the indicated times. The inhibitor was present in the medium throughout the time course. (B) HeLa cells were transfected with Bid or nontargeting control siRNA or were mock transfected for 48 h and then analyzed for Bid expression by immunoblotting. Bid siRNA cells or control siRNA cells were infected with rwt virus. Cells entering apoptosis were analyzed by time-lapse microscopy, as described in the legend to Fig. 1. The data represent the average results from three experiments, which collectively analyzed the time of onset of apoptosis in 120 to 200 total cells. The data for Bid siRNA cells were significantly different from the data for control siRNA cells (P < 0.05) by log rank analysis. (C) HeLa cells were transfected with Bid or nontargeting siRNA for 48 h and then infected with rwt virus for the times indicated, and caspase-3-like activity was analyzed using a fluorogenic substrate, as described in the legend to Fig. 1. The data represent the average results ± standard deviations from three experiments. (D) HeLa cells were transfected with Bid or nontargeting siRNA for 48 h and then infected with rwt virus. Cell viability was analyzed at the indicated times by MTT assay, as described in the legend to Fig. 1. The data represent the average results and standard deviations from three experiments (*, P < 0.05 compared to control).
To determine whether silencing of Bid would decrease the rate of VSV-induced apoptosis, HeLa cells were transfected with either Bid or nontargeting siRNA for 48 h and then infected with rwt virus. An immunoblot of transfected cells is shown in Fig. 8B, which indicated that Bid expression was reduced to 24% of that of the control prior to infection. Most of the Bid siRNA cells entered apoptosis more slowly than control siRNA cells, as measured by time-lapse microscopy (Fig. 8B). Similar results were obtained by assaying caspase-3-like activity and cell viability (Fig. 8C and D). These data are consistent with the idea that activation of Bid during VSV infection leads to the inactivation of Bcl-XL and/or the activation of Bak.
Since Bid is cleaved primarily by caspase-8, the experiments shown in Fig. 8 were extended by treating HeLa cells with a caspase-8 inhibitor to determine whether the rate of VSV-induced apoptosis was decreased. HeLa cells were preincubated with either caspase-8 inhibitor (Z-IETD-fmk), pan-caspase inhibitor (Z-VAD-fmk), or no inhibitor for 2 h prior to infection with rwt virus. The inhibitors were present in the medium throughout the time course of the experiment. Cell viability was analyzed at 12, 16, and 24 h postinfection by MTT assay. Similar treatment with IETD-fmk of transfected cells that express M protein in the absence of other viral components did not inhibit apoptosis (19). However, in the case of virus-infected HeLa cells, caspase-8 inhibitor delayed cell death to an extent similar to that of the pan-caspase inhibitor (Fig. 9A). Interestingly, even in the presence of a pan-caspase inhibitor, VSV was able to induce cell death, suggesting that caspase-independent pathways may also be activated. These results, together with those shown in Fig. 8, indicate that elements of the death receptor as well as the mitochondrial pathway are important for VSV-induced apoptosis and that cross talk between the two pathways occurs via caspase-8 cleavage of Bid. This idea was further supported by infecting HeLa-Bcl-XL cells with rwt virus in the presence of caspase-8 inhibitor or pan-caspase inhibitor (Fig. 9B). In the absence of inhibitors, the survival of HeLa-Bcl-XL cells was dramatically prolonged compared to that of control HeLa cells (compare the time scales shown in Fig. 9A and B). The death of HeLa-Bcl-XL cells was delayed slightly in the presence of caspase inhibitors, but the differences in cell viability were not statistically significant at most time points. These data indicate that the contribution of the death receptor pathway to the induction of apoptosis by rwt virus is primarily through cross talk with the mitochondrial pathway.
FIG. 9.
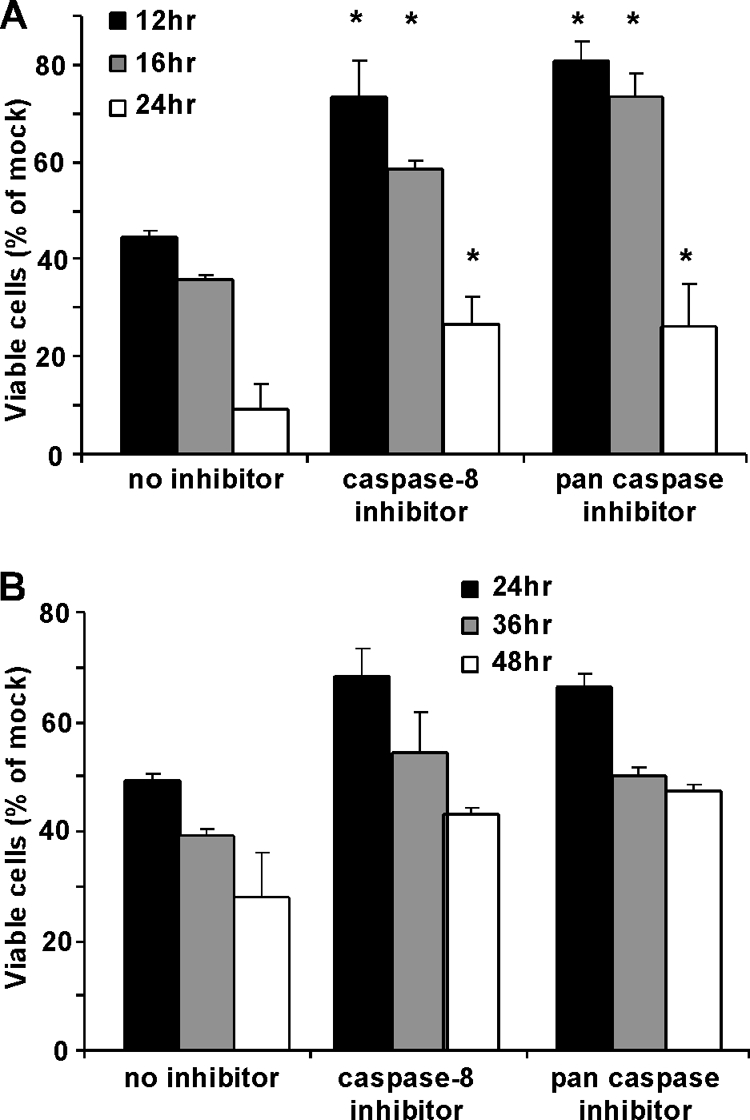
Apoptosis is significantly delayed in HeLa cells infected with rwt virus in the presence of caspase-8 inhibitor. HeLa cells (A) or HeLa cells that stably overexpress Bcl-XL (B) were incubated in the presence of 100 μM caspase-8 inhibitor, 100 μM pan-caspase inhibitor, or no inhibitor for 2 h. Cells were then infected with rwt virus for the indicated times. The inhibitors were present in the medium throughout the time course. Cell viability was analyzed at the indicated times by MTT assay, as described in the legend to Fig. 1. The data represent the average results and standard deviations from three experiments (*, P < 0.05 compared to control).
DISCUSSION
Cell death via the intrinsic pathway depends on the multidomain proapoptotic proteins Bak and Bax (22), which function to permeabilize the outer mitochondrial membrane. In some systems, Bak and Bax have been shown to be functionally redundant. Studies utilizing knockout murine embryonic fibroblasts (MEFs) showed that MEFs deficient in either Bak or Bax alone underwent apoptosis induced by multiple apoptotic stimuli. Conversely, deficiency in both Bak and Bax conferred resistance to these apoptotic stimuli (33). These results indicate that there is functional redundancy for Bak and Bax in MEFs. Subsequently, other groups have reported similar functional overlap in different cell types (9, 15, 33), giving rise to the idea that Bak and Bax are redundant regulators of intrinsic mitochondrial apoptosis and that inactivation of both is necessary to abrogate cell death. The results presented here contrast with this view by showing that Bak plays a more significant role than Bax in the induction of apoptosis in VSV-infected cells (Fig. 1).
The lack of functional redundancy for Bak and Bax is not surprising, given the differences between them. Inactive Bax is a latent monomer in the cytosol (16, 37). Once activated by apoptotic stimuli, Bax undergoes conformational changes, causing altered epitope availability, the formation of homodimers and oligomers, and translocation to the mitochondrial membrane (8). In contrast, Bak resides in a membrane-bound protein complex (14), which suggests that Bak and Bax are activated by different mechanisms. In fact, recent evidence has shown that some apoptotic stimuli, such as SSP, actinomycin D, TRAIL, and overexpression of Puma activate Bak preferentially and that Bak can lead to the release of cytochrome c in the absence of Bax activation (25). Our observation that, in the context of a VSV infection, Bak appears to play a greater role in the induction of apoptosis than Bax is consistent with these more recent results.
Bak and Bax are usually constitutively expressed at relatively constant levels and are regulated posttranslationally mainly by other Bcl-2 proteins. Antiapoptotic proteins such as Bcl-2, Mcl-1, and Bcl-XL keep Bax and Bak from permeabilizing the outer mitochondrial membrane. One mechanism by which they accomplish this is by binding to BH3-only proteins and inhibiting their activity. This is the likely mechanism by which overexpression of Bcl-2 inhibits induction of apoptosis by VSV (10, 19), since Bcl-2 is not known to bind directly to Bak. Since Bax exists as a monomer in the cytosol, there is no repressor bound to Bax in healthy cells (8). However, once apoptotic signals from BH3-only proteins have induced a conformational change in Bax, it translocates to the mitochondrial membrane where Bax can be held in check by Bcl-2, Bcl-B, and Bcl-XL (40). Bak, constitutively present in membranes, has been shown to be kept inactive by interaction with multidomain antiapoptotic Bcl-2 family members Mcl-1 and Bcl-XL in organelle membranes (7, 35). Mcl-1 normally has a rapid turnover rate and is degraded by the ubiquitin-proteosome pathway (1, 26). Therefore, we hypothesized that inhibition of new host gene expression by the VSV M protein induces apoptosis due to the loss of Mcl-1, since the protein cannot be replaced in VSV-infected cells once it has been degraded. This hypothesis was supported by the timing of the reduction of Mcl-1 protein levels during VSV infection, which coincides with the induction of apoptosis (Fig. 2). While the manuscript was in revision, similar results were published, showing degradation of Mcl-1 following VSV infection in other cell types (29). However, depletion of Mcl-1 alone is not sufficient to induce apoptosis (Fig. 6), indicating that inactivation of Bcl-XL is also required for cells to enter apoptosis.
Inactivation of Bcl-XL in VSV-infected cells is likely due to activation of one or more BH3-only proteins. Many BH3-only proteins have been described. However, M protein-mediated shutoff of new host gene expression would likely inhibit the expression of BH3-only proteins that must be newly synthesized, which suggested that BH3-only proteins that are posttranslationally regulated were the most likely candidates. Our previous research has shown that the death receptor pathway is the principal pathway for induction of apoptosis by the M protein mutant VSV rM51R-M (12, 13). This led to our hypothesis that the death receptor pathway might also be activated by VSV with wt M protein and that the BH3-only protein Bid, which is activated by proteolytic cleavage by caspase-8, was involved in Bcl-XL inactivation and Bak activation. This hypothesis was supported by the significant delay in VSV-induced apoptosis in Bid siRNA cells (Fig. 8), as well as in the presence of capase-8 inhibitor (Fig. 9). These results suggest that both the intrinsic and extrinsic pathways are important for apoptosis induced by VSV with wt M protein and that cross talk between the two pathways occurs via caspase-8 cleavage of Bid. The key elements of this pathway are summarized in Fig. 10.
FIG. 10.

Major pathways involving Bcl-2 family members, leading to apoptosis in HeLa cells infected with VSV with wt M protein.
The involvement of the death receptor pathway in the induction of apoptosis by rwt virus contrasts with previous results from our laboratory on the induction of apoptosis by expression of wt M protein in transfected cells in the absence of other viral components (19). Those experiments showed that expression of M protein induced apoptosis through the mitochondrial pathway, similar to pharmacologic inhibitors of host gene expression, and that capsase-8 inhibitor did not inhibit M protein-induced apoptosis. Combining those results with the results presented here suggests that the inhibition of host gene expression by M protein may induce activation of other BH3-only proteins besides Bid, which are independent of caspase-8 and the death receptor pathway (Fig. 10). The presence of such an additional pathway may account for the fact that caspase inhibitors do not completely prevent cell death in HeLa cells (Fig. 9), and other reports indicate that inhibition of caspase-8 does not completely prevent VSV-induced apoptosis (3, 10, 11). The inability of caspase-8 inhibitor to completely prevent cell death could also be due to the activity of other caspases that are coupled to the death receptor pathway, such as caspase-10 (32).
The results presented here provide new insight into why different cell types vary in their sensitivity to induction of apoptosis by VSV with wt versus mutant M protein (12, 13, 19, 20). Cells that depend on Mcl-1 to keep Bak inactive will be particularly sensitive to viruses with wt M protein, which inhibits host gene expression, leading to Mcl-1 degradation. In contrast, some cells are more sensitive to viruses encoding the M51R mutant M protein (13, 20). In these cells, the expression of new proapoptotic gene products is required for the rapid induction of apoptosis, and apoptosis occurs primarily through the death receptor pathway (12, 13). Even though the mitochondrial pathway is activated, induction of apoptosis in these cells is independent of the mitochondrial pathway. In addition, there are cells in which the induction of apoptosis by M protein mutant virus is dependent on the mitochondrial pathway, presumably through cross talk with the death receptor pathway (30). The differences in the requirement for activation of the mitochondrial pathway by M protein mutant VSV is similar to the classification of cell types into type I (mitochondrion independent) or type II (mitochondrion dependent), based on their response to death ligands such as Fas (28). The results presented here, together with previous results, suggest that VSV is a particularly potent inducer of apoptosis because it activates multiple apoptotic pathways, whose expression varies considerably among cell types.
Acknowledgments
We thank Margie McKenzie for performing the Bid immunoblotting and Maryam Ahmed, David Ornelles, and John Wilkinson for helpful advice and comments on the manuscript. We also thank Ulrich Maurer and John Wilkinson for kindly providing the plasmids.
This work was supported by NIH grants R01-AI32983 and R01-AI52304.
Footnotes
Published ahead of print on 8 July 2009.
REFERENCES
- 1.Adams, K. W., and G. M. Cooper. 2007. Rapid turnover of Mcl-1 couples translation to cell survival and apoptosis. J. Biol. Chem. 282:6192-6200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ahmed, M., M. O. McKenzie, S. Puckett, M. Hojnacki, L. Poliquin, and D. S. Lyles. 2003. Ability of the matrix protein of vesicular stomatitis virus to suppress beta interferon gene expression is genetically correlated with the inhibition of host RNA and protein synthesis. J. Virol. 77:4646-4657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Balachandran, S., M. Porosnicu, and G. N. Barber. 2001. Oncolytic activity of vesicular stomatitis virus is effective against tumors exhibiting aberrant p53, Ras, or myc function and involves the induction of apoptosis. J. Virol. 75:3474-3479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Balachandran, S., P. C. Roberts, T. Kipperman, K. N. Bhalla, R. W. Compans, D. R. Archer, and G. N. Barber. 2000. Alpha/beta interferons potentiate virus-induced apoptosis through activation of the FADD/caspase-8 death signaling pathway. J. Virol. 74:1513-1523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bernardi, P., L. Scorrano, R. Colonna, V. Petronilli, and F. Di Lisa. 1999. Mitochondria and cell death. Mechanistic aspects and methodological issues. Eur. J. Biochem. 264:687-701. [DOI] [PubMed] [Google Scholar]
- 6.Bouillet, P., and A. Strasser. 2002. BH3-only proteins—evolutionarily conserved proapoptotic Bcl-2 family members essential for initiating programmed cell death. J. Cell Sci. 115:1567-1574. [DOI] [PubMed] [Google Scholar]
- 7.Cuconati, A., C. Mukherjee, D. Perez, and E. White. 2003. DNA damage response and MCL-1 destruction initiate apoptosis in adenovirus-infected cells. Genes Dev. 17:2922-2932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Danial, N. N., and S. J. Korsmeyer. 2004. Cell death: critical control points. Cell 116:205-219. [DOI] [PubMed] [Google Scholar]
- 9.Degenhardt, K., R. Sundararajan, T. Lindsten, C. Thompson, and E. White. 2002. Bax and Bak independently promote cytochrome c release from mitochondria. J. Biol. Chem. 277:14127-14134. [DOI] [PubMed] [Google Scholar]
- 10.Desforges, M., G. Despars, S. Berard, M. Gosselin, M. O. McKenzie, D. S. Lyles, P. J. Talbot, and L. Poliquin. 2002. Matrix protein mutations contribute to inefficient induction of apoptosis leading to persistent infection of human neural cells by vesicular stomatitis virus. Virology 295:63-73. [DOI] [PubMed] [Google Scholar]
- 11.Gadaleta, P., X. Perfetti, S. Mersich, and F. Coulombie. 2005. Early activation of the mitochondrial apoptotic pathway in vesicular stomatitis virus-infected cells. Virus Res. 109:65-69. [DOI] [PubMed] [Google Scholar]
- 12.Gaddy, D. F., and D. S. Lyles. 2007. Oncolytic vesicular stomatitis virus induces apoptosis via signaling through PKR, Fas, and Daxx. J. Virol. 81:2792-2804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gaddy, D. F., and D. S. Lyles. 2005. Vesicular stomatitis viruses expressing wild-type or mutant M proteins activate apoptosis through distinct pathways. J. Virol. 79:4170-4179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Griffiths, G. J., L. Dubrez, C. P. Morgan, N. A. Jones, J. Whitehouse, B. M. Corfe, C. Dive, and J. A. Hickman. 1999. Cell damage-induced conformational changes of the pro-apoptotic protein Bak in vivo precede the onset of apoptosis. J. Cell Biol. 144:903-914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hahn, P., T. Lindsten, A. Lyubarsky, G. S. Ying, E. N. Pugh, Jr., C. B. Thompson, and J. L. Dunaief. 2004. Deficiency of Bax and Bak protects photoreceptors from light damage in vivo. Cell Death Differ. 11:1192-1197. [DOI] [PubMed] [Google Scholar]
- 16.Hsu, Y. T., K. G. Wolter, and R. J. Youle. 1997. Cytosol-to-membrane redistribution of Bax and Bcl-X(L) during apoptosis. Proc. Natl. Acad. Sci. USA 94:3668-3672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jackson, S., C. Harwood, M. Thomas, L. Banks, and A. Storey. 2000. Role of Bak in UV-induced apoptosis in skin cancer and abrogation by HPV E6 proteins. Genes Dev. 14:3065-3073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kepp, O., K. Rajalingam, S. Kimmig, and T. Rudel. 2007. Bak and Bax are non-redundant during infection- and DNA damage-induced apoptosis. EMBO J. 26:825-834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kopecky, S. A., and D. S. Lyles. 2003. Contrasting effects of matrix protein on apoptosis in HeLa and BHK cells infected with vesicular stomatitis virus are due to inhibition of host gene expression. J. Virol. 77:4658-4669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kopecky, S. A., M. C. Willingham, and D. S. Lyles. 2001. Matrix protein and another viral component contribute to induction of apoptosis in cells infected with vesicular stomatitis virus. J. Virol. 75:12169-12181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Koyama, A. H. 1995. Induction of apoptotic DNA fragmentation by the infection of vesicular stomatitis virus. Virus Res. 37:285-290. [DOI] [PubMed] [Google Scholar]
- 22.Lindsten, T., A. J. Ross, A. King, W. X. Zong, J. C. Rathmell, H. A. Shiels, E. Ulrich, K. G. Waymire, P. Mahar, K. Frauwirth, Y. Chen, M. Wei, V. M. Eng, D. M. Adelman, M. C. Simon, A. Ma, J. A. Golden, G. Evan, S. J. Korsmeyer, G. R. MacGregor, and C. B. Thompson. 2000. The combined functions of proapoptotic Bcl-2 family members bak and bax are essential for normal development of multiple tissues. Mol. Cell 6:1389-1399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lyles, D. S. 2000. Cytopathogenesis and inhibition of host gene expression by RNA viruses. Microbiol. Mol. Biol. Rev. 64:709-724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Maurer, U., C. Charvet, A. S. Wagman, E. Dejardin, and D. R. Green. 2006. Glycogen synthase kinase-3 regulates mitochondrial outer membrane permeabilization and apoptosis by destabilization of MCL-1. Mol. Cell 21:749-760. [DOI] [PubMed] [Google Scholar]
- 25.Neise, D., V. Graupner, B. F. Gillissen, P. T. Daniel, K. Schulze-Osthoff, R. U. Janicke, and F. Essmann. 2008. Activation of the mitochondrial death pathway is commonly mediated by a preferential engagement of Bak. Oncogene 27:1387-1396. [DOI] [PubMed] [Google Scholar]
- 26.Nijhawan, D., M. Fang, E. Traer, Q. Zhong, W. Gao, F. Du, and X. Wang. 2003. Elimination of Mcl-1 is required for the initiation of apoptosis following ultraviolet irradiation. Genes Dev. 17:1475-1486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Parato, K. A., D. Senger, P. A. Forsyth, and J. C. Bell. 2005. Recent progress in the battle between oncolytic viruses and tumours. Nat. Rev. Cancer 5:965-976. [DOI] [PubMed] [Google Scholar]
- 28.Scaffidi, C., S. Fulda, A. Srinivasan, C. Friesen, F. Li, K. J. Tomaselli, K. M. Debatin, P. H. Krammer, and M. E. Peter. 1998. Two CD95 (APO-1/Fas) signaling pathways. EMBO J. 17:1675-1687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Schache, P., E. Gurlevik, N. Struver, N. Woller, N. Malek, L. Zender, M. Manns, T. Wirth, F. Kuhnel, and S. Kubicka. 2009. VSV virotherapy improves chemotherapy by triggering apoptosis due to proteasomal degradation of Mcl-1. Gene Ther. 16:849-861. [DOI] [PubMed] [Google Scholar]
- 30.Sharif-Askari, E., P. Nakhaei, S. Oliere, V. Tumilasci, E. Hernandez, P. Wilkinson, R. Lin, J. Bell, and J. Hiscott. 2007. Bax-dependent mitochondrial membrane permeabilization enhances IRF3-mediated innate immune response during VSV infection. Virology 365:20-33. [DOI] [PubMed] [Google Scholar]
- 31.Stojdl, D. F., B. D. Lichty, B. R. tenOever, J. M. Paterson, A. T. Power, S. Knowles, R. Marius, J. Reynard, L. Poliquin, H. Atkins, E. G. Brown, R. K. Durbin, J. E. Durbin, J. Hiscott, and J. C. Bell. 2003. VSV strains with defects in their ability to shutdown innate immunity are potent systemic anti-cancer agents. Cancer Cell 4:263-275. [DOI] [PubMed] [Google Scholar]
- 32.Wang, J., H. J. Chun, W. Wong, D. M. Spencer, and M. J. Lenardo. 2001. Caspase-10 is an initiator caspase in death receptor signaling. Proc. Natl. Acad. Sci. USA 98:13884-13888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wei, M. C., W.-X. Zong, E. H. Y. Cheng, T. Lindsten, V. Panoutsakopoulou, A. J. Ross, K. A. Roth, G. R. MacGregor, C. B. Thompson, and S. J. Korsmeyer. 2001. Proapoptotic BAX and BAK: a requisite gateway to mitochondrial dysfunction and death. Science 292:727-730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Willis, S. N., and J. M. Adams. 2005. Life in the balance: how BH3-only proteins induce apoptosis. Curr. Opin. Cell Biol. 17:617-625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Willis, S. N., L. Chen, G. Dewson, A. Wei, E. Naik, J. I. Fletcher, J. M. Adams, and D. C. S. Huang. 2005. Proapoptotic Bak is sequestered by Mcl-1 and Bcl-xL, but not Bcl-2, until displaced by BH3-only proteins. Genes Dev. 19:1294-1305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Willis, S. N., J. I. Fletcher, T. Kaufmann, M. F. van Delft, L. Chen, P. E. Czabotar, H. Ierino, E. F. Lee, W. D. Fairlie, P. Bouillet, A. Strasser, R. M. Kluck, J. M. Adams, and D. C. S. Huang. 2007. Apoptosis initiated when BH3 ligands engage multiple Bcl-2 homologs, not Bax or Bak. Science 315:856-859. [DOI] [PubMed] [Google Scholar]
- 37.Wolter, K. G., Y. T. Hsu, C. L. Smith, A. Nechushtan, X. G. Xi, and R. J. Youle. 1997. Movement of Bax from the cytosol to mitochondria during apoptosis. J. Cell Biol. 139:1281-1292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Youle, R. J., and A. Strasser. 2008. The BCL-2 protein family: opposing activities that mediate cell death. Nat. Rev. Mol. Cell Biol. 9:47-59. [DOI] [PubMed] [Google Scholar]
- 39.Zamzami, N., C. El Hamel, C. Maisse, C. Brenner, C. Munoz-Pinedo, A. S. Belzacq, P. Costantini, H. Vieira, M. Loeffler, G. Molle, and G. Kroemer. 2000. Bid acts on the permeability transition pore complex to induce apoptosis. Oncogene 19:6342-6350. [DOI] [PubMed] [Google Scholar]
- 40.Zhai, D., C. Jin, Z. Huang, A. C. Satterthwait, and J. C. Reed. 2008. Differential regulation of Bax and Bak by anti-apoptotic Bcl-2 family proteins Bcl-B and Mcl-1. J. Biol. Chem. 283:9580-9586. [DOI] [PMC free article] [PubMed] [Google Scholar]