

Abstract
Lanthanide(III) complexes of macrocycles 1,4,7,10-tetrakis(2-hydroxyethyl)-1,4,7,10-tetraazacyclododecane (THED) and (1S,4S,7S,10S)-1,4,7,10-tetrakis(2-hydroxypropyl)-1,4,7,10-tetraazacyclododecane (S-THP) were studied as chemical exchange saturation transfer (CEST) agents for magnetic resonance imaging (MRI) applications. The four hyperfine-shifted alcohol protons of these Ln(III) complexes gave rise to a single 1H resonance in wet d3-acetonitrile that was separated from the bulk water resonance (Δω) by 8 ppm (Ce), 2 ppm (Nd), 7 ppm (Eu) or 17 ppm (Yb). A CEST peak corresponding to the alcohol protons was observed for all Ln(THED)3+ or Ln(S-THP)3+ complexes except Nd(III) at low water concentrations (< 1%). In 100% aqueous buffered solutions, the CEST hydroxyl peak is observed for the Eu(III), Ce(III) and Yb(III) complexes over a range of pH values. The optimal pH range for the CEST effect of each complex is related to the pKa of the hydroxyl/water ligands of the complex. Optimum pH values for the CEST effect from alcohol proton exchange are pH = 6.0 for Ce(S-THP)3+, pH = 4.5 for Eu(THED)3+, and pH= 3.0 for Yb(S-THP)3+.
Introduction
MRI contrast agents that function through chemical exchange saturation transfer (CEST)1,2 contain mobile protons that are in slow exchange on the NMR time scale with bulk water. CEST experiments use a presaturation pulse on the resonance frequency of protons at the exchangeable site to saturate spins in exchange with the protons of the bulk water solvent. To avoid direct saturation of the bulk water signal, paramagnetic lanthanide(III) complexes (PARACEST agents) are used to shift the resonances of the exchangeable sites away from that of bulk water2. The net CEST effect is a decrease in the proton signal of bulk water upon applying a pulse at the resonance frequency of the contrast agent, producing a negative contrast image3. Despite the successful development of lanthanide PARACEST agents,2,4,5 there are relatively few examples of ligand donor groups with exchangeable protons. Reported donors include NH groups of amides or amines, or OH groups of metal bound water.2,6 In an attempt to rectify this, we reported the CEST properties of a Eu(III) complex (Eu(CNPHC)3+, Chart 1) which possesses five magnetically inequivalent proton exchangeable sites.7 However, this complex showed CEST spectra for the alcohol proton exchange with water protons in wet acetonitrile, but not in pure water. Recently, we communicated the CEST properties of Eu(S-THP)3+ in pure water at near neutral pH and showed that the CEST spectrum was sensitive to both outer and innersphere ligand binding.8 Here we show that judicious choice of lanthanide ion and pH leads to a new class of PARACEST agent with hydroxyl proton exchangeable groups that are active in water.
Chart 1.

There are several unique features of macrocyclic complexes with pendent alcohol groups that make them good candidates for PARACEST agents. First, both THED and S-THP complexes of the Ln(III) are kinetically resistant to dissociation.9-11 Ln(III) complexes of S-THP are especially inert with half-lives for dissociation on the order of 100 days in aqueous solution at 37 °C. Second, direct excitation luminescence studies of Eu(THED)3+ and Eu(S-THP)3+ (Chart 1) show that these complexes are nine-coordinate with an octadentate macrocycle and a single water ligand completing the coordination sphere in aqueous solutions at neutral pH.12 At slightly basic pH, either an alcohol pendent groups or a water ligand ionizes with a pKa value of 7.5 and 7.7 for Eu(THED)3+ and Eu(S-THP)3+, respectively. As we show here, the near neutral ionization of the alcohol groups leads to pH dependent CEST properties under physiologically relevant conditions. In addition, Eu(THED)3+ and analogous complexes bind strongly to anionic ligands of biological importance.8,13 This solution chemistry is expected to influence CEST properties of the THED and S-THP complexes and may be useful in the development of responsive MRI contrast agents. A fourth property of the lanthanide cyclen based complexes that affects CEST spectra is the existence of multiple stereoisomers in solution. The stereoisomerism displayed by Ln(III) complexes of cyclen derivatives with pendent groups involves two elements of chirality. One element arises from pendent group orientation (clockwise or counterclockwise) and the other from the two conformations of the cyclen ring. This gives rise to four stereoisomers which exist as two enantiomeric pairs14-18. For amide pendent group macrocycles, two diastereomers are generally observed in solution by NMR spectroscopy15,19,20 and by Eu(III) excitation luminescence spectroscopy21. These two diastereomers differ by the orientation of their pendent arms to give rise to either a square antiprismic geometry (SA) or a twisted square antiprismic geometry (TSA). By contrast, lanthanide(III) complexes of THED show a single diastereomeric form in solution at room temperature as shown by NMR9,11,12,14 and luminescence spectroscopy.12 The presence of just one diastereomeric form is potentially advantageous for PARACEST experiments because there is a single set of proton resonances rather than two sets of mobile proton resonances that may be resolved in complexes that have large hyperfine shifts. Lanthanide(III) complexes of S-THP also show a single diastereomeric form in solution, the TSAP (twisted square antiprism) form11,18,22.
Here we present a study of the CEST properties of several lanthanide(III) complexes of THED and S-THP. Different lanthanide(III) ions were chosen in order to: 1) vary Δω, the chemical shift difference between bulk water and the alcohol protons, to allow for larger proton transfer rate constants, and to 2) vary the alcohol proton exchange rate constants by modulation of the pKa of the alcohol pendent groups. This work shows that lanthanide(III) complexes with alcohol donor groups constitute a new class of PARACEST agent that are highly modulated by solution pH.
Experimental Section
Materials
Acetonitrile was dried over molecular sieves. Cyclen (1,4,7,10-tetraazacyclododecane) was purchased from Strem Chemicals. Eu(CF3SO3)3, Yb(CF3SO3)3, Nd(CF3SO3)3, ethanol anhydrous and ethylene oxide were purchased from Aldrich and Ce(CF3SO3)3 was purchased from TCI America. THED, S-THP, [Eu(THED)](CF3SO3)3, [Eu(S-THP)](CF3SO3)3 and [Yb(S-THP)](CF3SO3)3 were prepared as reported previously.23-25 MES buffer was used at pH 5.5 to 6.5 and HEPES buffer was used at pH 6.5 to 7.5.
[Yb(THED)](CF3SO3)3. A solution of THED (0.293g, 0.84mmol) and Yb(CF3SO3)3 (0.541g, 0.87mmol) was heated under reflux in dry ethanol (20 mL) under argon for 2h. The solution was cooled and the solvent was removed under vacuum. The resulting solid was dissolved in a minimum amount of ethanol, 2-propanol was added until the solution turned cloudy and the solution was placed in the freezer overnight. The mixture was filtered, and the white precipitate was collected and dried under vacuum. Yield : 76 %. 1H NMR (500 MHz, CD3CN) δ = 55.4, 12.3, 7.9, -8.4, -10.6, -17.5, -18.8, -35.7 (each resonance is a s, 32 H total, cyclen -CH2-, NCH2CH2OH and NCH2CH2OH), 18.5 (s, 4H, -OH); ESI m/e {Yb(THED)-2H+} 516.4 (9%), 517.4 (40%), 518.3 (62%), 519.4 (55%), 520.4 (100%), 521.3 (22%), 522.4 (38%); {[Yb(THED)](CF3SO3)-H+} 666.0 (6%), 667.0 (43%), 668.0 (73%), 669.0 (63%), 670.0 (100%), 671.0 (20%), 672.0 (46%), 673.0 (10%); {[Yb(THED)](CF3SO3)2+} 815.6 (9%), 816.5 (35%), 817.7 (50%), 818.7 (73%), 819.7(100%), 820.9 (11%), 821.7 (34%); {[Yb(THED)](CF3SO3)3+Na+} 983.8 (33%), 984.8 (80%), 985.8 (81%), 986.8 (100%), 987.7 (39%), 988.7 (59%), 989.7 (20%).
[Nd(S-THP)](CF3SO3)3. A procedure similar to that for preparation of [Yb(THED)](CF3SO3)3 was used to give the product in 75% yield. 1H NMR (500 MHz, CD3CN) δ 9.8, 9.3, 8.4, 8.1, 4.6, 2.6, -0.43 (each resonance is a s, 28H total, cyclen -CH2-, NCH2CH2OH and NCH2CH(CH3)OH), 2.4 (s, 12H, NCH2CH(CH3)OH), 4.3 (s, 4H, -OH); ESI m/e {Nd(S-THP)-2H+} 544.2 (97%), 545.3 (60%), 546.2 (100%), 547.3 (45%), 548.3 (71%), 549.3 (22%), 550.3 (22%); {[Nd(S-THP)](CF3SO3)-H+} 694.1 (94%), 695.1 (71%), 696.1(100%), 697.1(58%), 698.1(83%), 699.1(20%), 700.1(25%); {[Nd(S-THP)](CF3SO3)2+} 843.7 (23%), 844.7 (32%), 845.7 (37%), 846.7 (20%), 847.7 (19%), 848.7 (8%),849.7 (8%). {[Nd(S-THP)](CF3SO3)3+Na+} 1010.7 (93%), 1011.8 (63%), 1012.8 (100%), 1013.8 (57%), 1014.8 (79%), 1015.7 (36%), 1016.8 (40%).
[Ce(S-THP)](CF3SO3)3. A procedure similar to that for preparation of [Yb(THED)](CF3SO3)3 was used to give the product in 72% yield. 1H NMR (500 MHz, CD3CN) δ 9.36, 8.66, 6.49, 5.94, 2.30, 1.47, -6.8 (each resonance is a s, 28H total, cyclen -CH2-, NCH2CH2OH and NCH2CH(CH3)OH), 3.19 (s, 12H, NCH2CH(CH3)OH), 10.2 (s, 4H, -OH); ESI m/e {Ce(S-THP)-2H+} 542.1 (100%), 543.2 (27%), 544.1 (15%), 545.2 (4%); {[Ce(S-THP)](CF3SO3)-H+} 692.0 (100%), 693.0 (25%), 693.9 (20%), 694.9 (5%); {[Ce(S-THP)](CF3SO3)2+} 840.7 (100%), 841.5 (24%), 842.7 (19%), 843.6 (6%).
NMR experiments
Two-Dimensional EXSY experiments were recorded on an Varian Inova-500 spectrometer at room temperature using conventional NOESY 90°-t1-90°-τmix-90°-acq phase sensitive pulse sequence with 2048 t2 and 1024 t1 data points and a mixing time of 5 ms26. All CEST experiments were acquired on a Varian Inova-500 Spectrometer at room temperature, B1 = 800 Hz with an irradiation time of 3 seconds as the optimal irradiation time (Figure S1). Lanthanide(III) complex concentrations for CEST, 1H NMR experiments and EXSY were 10 mM unless otherwise noted. CEST spectra were fit to modified Bloch equations with three exchangeable sites to obtain rate constants for exchange by using Matlab.27 The longitudinal relaxation time (T1) of bulk water protons was measured by inversion recovery method to give T1 values of 1.3 s (Yb(S-THP)3+) or 1.4 s (Ce(S-THP)3+) used in the calculations.
Results
1H NMR studies
To assign the chemical shifts of the alcohol protons, the 1H NMR spectra of the various lanthanide(III) complexes were recorded in acetonitrile and in water. The symmetrical structure of the Ln(THED)3+ and Ln(S-THP)3+ complexes in d3-acetonitrile (Ln = Ce, Nd for S-THP); (Ln = Eu, Yb for THED) gives rise to a simple 1H NMR spectrum consisting of nine distinct proton resonances of equal intensity. Four resonances arise from the protons in the macrocycle backbone, four resonances arise from the protons on the ethylene portion or methyl group of the pendent arms and one resonance arises from the hydroxyl protons (Figure 1). The simplicity of these 1H NMR spectra is consistent with a single enantiomeric pair in solution.10,14 As shown previously, Ln(THED)3+ complexes form a pair of enantiomers that are either in the form of a twisted square antiprism or a square antiprism.14 Ln(S-THP)3+ complexes have an additional element of chirality in the pendent propanol group that gives a single diastereomer in solution,11 the twisted square antiprism.22
Figure 1.
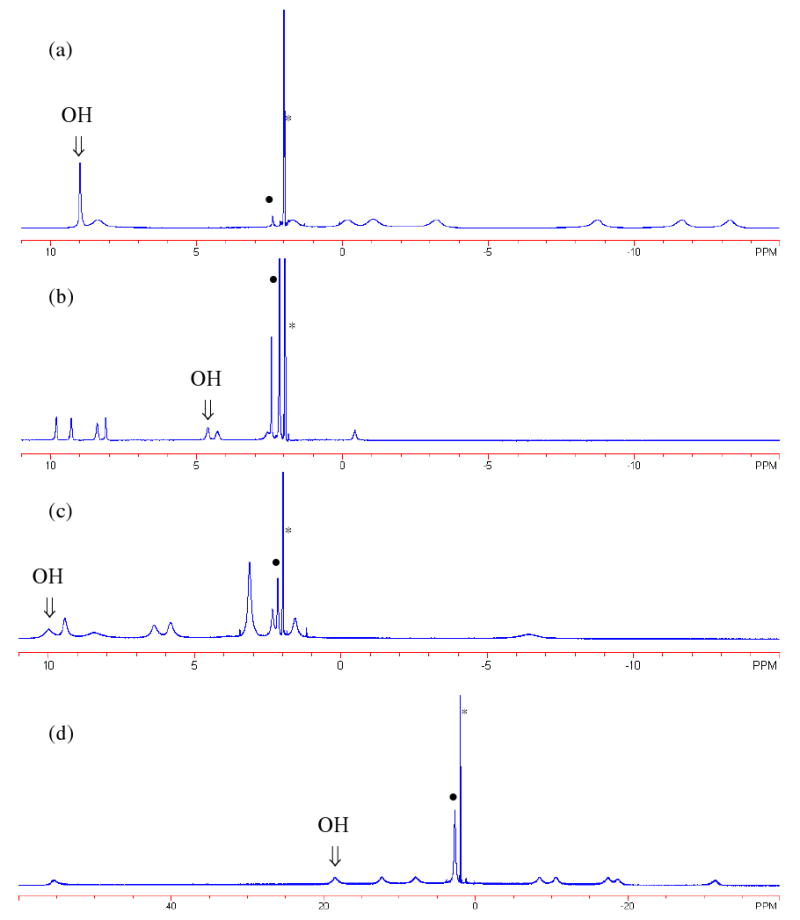
1H NMR spectra of (a) Eu(THED)3+, (b) Nd(S-THP)3+, (c) Ce(S-THP)3+, and (d) Yb(THED)3+ in d3-acetonitrile (* solvent; (●) H2O).
For Eu(THED)3+, the chemical shifts of the proton resonances (-14 to 6 ppm) correspond closely to those previously reported for the related complex, Eu(S-THP)3+, including the most downfield resonance at ∼9 ppm that is assigned to the alcohol protons7 (Figure 1a). The chemical shift difference (Δω) between the hydroxyl proton and bulk water of Eu(THED)3+ is modest, at about 6.5 ppm. Similar to Eu(S-THP)3+, this resonance broadens and shifts toward the bulk water peak as H2O is added to the acetonitrile solution (Figure S2a) as a result of an increase in the rate constant for chemical exchange between the two sites.7 The assignment of this resonance as arising from the alcohol protons is supported by replacement of these exchangeable protons with deuterium by addition of D2O, followed by removal of the solvent and addition of fresh d3-acetonitrile. This leads to multiple resonances at 9 ppm due to the formation of partially deuterated isomers, all of which give rise to alcohol proton resonances that differ slightly from the all protonated form (Figure S2b). Finally, the EXSY and CEST spectra also support the alcohol proton resonance assignment (see below). Two other proton resonances were assigned. The proton resonance for Eu(THED)3+ at ca. 8 ppm for which there is no corresponding resonance for Eu(S-THP)3+, is assigned as one set of protons in the CH2 group α to the alcohol group. This peak shifts by approximately 3 ppm upon addition of water although, consistent with its assignment, it is not in exchange with the bulk water protons as shown by EXSY and CEST experiments. This shows that the environment of these protons is more highly influenced by the concentration of water in the sample than the other non-alcohol protons in the macrocycle due to their close proximity to the alcohol protons. For Eu(THED)3+, there is an additional minor resonance observed at ∼2.4 ppm in solutions containing very dry acetonitrile that integrates to two protons. As water was added to the sample, this resonance broadened and shifted to the resonance characteristic of the bulk water resonance at 4.1 ppm. We assign this resonance to the bound water of Eu(THED)3+. The related complex Eu(S-THP)3+ has a bound water resonance at 3.0 ppm.7
The Nd(S-THP)3+ complex has 1H NMR resonances dispersed from approximately -0.2 ppm to 10 ppm with eight resonances of equal intensity and the protons of the methyl groups located at ∼2 ppm in d3-acetonitrile (Figure 1b). Several proton resonances shift upon addition of water or D2O (Figure S3), leading to merging of the resonances at 9.2 and 8.5 ppm, and shifting of the peak at -0.2 ppm to 1.8 ppm. The resonance at 4.5 ppm disappears upon addition of H2O, supportive of its assignment as the alcohol protons. The chemical shift (Δω) between the hydroxyl protons and bulk water of the Nd(III) complex is only 2 ppm in wet d3-acetonitrile, the smallest of all of the lanthanide complexes studied here.
The Ce(S-THP)3+ complex has 1H NMR resonances in d3-acetonitrile dispersed from approximately -7 ppm to 10 ppm with eight resonances of equal intensity and with the resonance for the protons of methyl groups at 3 ppm (Figure 1c). The disappearance of the most downfield resonance at 10 ppm upon addition of H2O and the reduced intensity of the resonance upon addition of D2O support the assignment of this resonance as the alcohol protons (Figure S4). The CEST spectra also support this assignment (see below). The chemical shift difference (Δω) between the hydroxyl proton and bulk water of the Ce(III) complex is about 6 ppm, similar to that of Eu(S-THP)3+ and Eu(THED)3+.
The Yb(THED)3+ complex (Figure 1d) has 1H NMR resonances that are highly dispersed from 60 ppm to -40 ppm with nine resonances of equal intensity in d3-acetonitrile. Addition of 1% H2O to the Yb(THED)3+ solution led to the disappearance of the resonance at 19 ppm, supportive of its assignment as hydroxyl protons (Figure S5a). The chemical shift of these hydroxyl protons is close to that of the related Yb(S-THP)3+ complex reported previously22. An EXSY spectrum of this complex also confirms the assignment of the alcohol protons because they are in exchange with water protons (see below). The chemical shift difference (Δω) between the hydroxyl proton and bulk water of Yb(THED)3+ is approximately 17 ppm, the largest of all complexes here.
Two dimensional proton exchange spectroscopy (EXSY) confirms the structural fluxionality of both of Yb(THED)3+ and Eu(THED)3+ (figure S6). The cross-peaks in the EXSY spectrum of Yb(THED)3+ show that four pairs of protons of the macrocyclic ligand undergo mutual chemical exchange. This represents the pairwise exchange of protons as the two enantiomers interconvert. For example, there are two sets of protons of the macrocycle backbone that interconvert and two sets of protons on the ethylene of the pendent group that interconvert. In addition, the cross peaks between the resonance at ∼20 ppm and ∼4 ppm for Yb(THED)3+ demonstrate that there is proton exchange between the hydroxyl peak and bulk water peak. The five cross peaks observed in the EXSY spectrum of Eu(THED)3+ show a similar result.
CEST spectra
CEST spectra were taken under several different conditions for the four lanthanide complexes. CEST spectra show a plot of Mz/Mo% for the percent reduction of the intensity of the bulk water peak as a function of the frequency of irradiation with a presaturation pulse. Note that all of the CEST spectra shown here have the bulk water peak set at 0.0 ppm. CEST spectra were taken in d3-acetonitrile with added water, conditions similar to those of the 1H NMR spectroscopy studies for assignment of resonances. In addition, 1H NMR and CEST spectra were taken in water at different pH values.
All complexes with the exception of Nd(S-THP)3+ showed CEST spectra in acetonitrile (<1% water) with a peak attributed to exchange of alcohol protons (Figs S7-S8). However, the appearance of the CEST spectra changed dramatically upon addition of more water to the acetonitrile solutions. For example, the CEST spectrum of Eu(THED)3+ observed in 1% or 5% H2O had a single CEST peak for the alcohol protons at 10 ppm. The efficiency of the complex as measured by Mz/Mo% of the hydroxyl proton peak decreased ∼10% at the higher water concentration. Similarly, a CEST spectrum is observed for Yb(THED)3+ dissolved in 100% d3-acetonitrile with trace water (Figure S5). When 1% H2O was added to the sample the alcohol CEST peak disappears, corresponding to the disappearance of the hydroxyl peak in the 1H NMR spectrum of Yb(THED)3+. A comparison of the CEST spectra of Yb(THED)3+, Eu(THED)3+ and Ce(S-THP)3+ in acetonitrile with 1% H2O is shown in Figure S8 along with the spectrum of Eu(CNPHC)3+. It is important to note that the pH was not controlled in any of the CEST NMR spectroscopy experiments in water/acetonitrile mixtures.
Under conditions of controlled pH, THED and S-THP complexes of Eu(III), Ce(III) and Yb(III) show CEST peaks for alcohol OH groups in 100% aqueous solutions, but the magnitude of these peaks is highly pH dependent. For Eu(THED)3+ and Ce(S-THP)3+ complexes, there is a pH range for the observation of the alcohol CEST peak with an optimum at slightly acidic conditions (Figure 2). For Eu(THED)3+, saturation transfer from alcohol protons to water is observed between pH 3.5 to 6.5 with an optimum at approximately pH 4.5 (Figure 2 and S9). For Ce(S-THP)3+, the CEST alcohol shoulder is observed between pH 4 to 8 with an optimum at about pH 6. This shows that Ce(S-THP)3+ is the most effective CEST agent at near neutral pH. Variation of the concentration of the Ce(III) complex shows that the CEST effect can be observed down to 1.00 mM concentrations (Figure 3) at pH 6.5 to give the enhancement of 5% Mz/Mo28 for on peak irradiation (5.0 ppm) compared to off peak irradiation (-5.0 ppm) that has been cited as a benchmark for detection of CEST-MRI image contrast.28 For Yb(S-THP)3+, the alcohol CEST peak is only observed at very low pH (Figure 4a). Effective CEST is observed for the Yb(III) complex between pH 6 to 3.0 (Figure 4b) with an optimal pH of 3.0 of the conditions that were studied.
Figure 2.

CEST effect for 10.0 mM Ce(S-THP)3+ (●) and Eu(THED)3+(x025CB;) at 6 ppm in water with no buffer at 296 K.
Figure 3.

CEST spectra of Ce(S-THP)3+ at ((○) 10 mM; (●) 5.0 mM; (X) 2.5 mM; (Δ) 1.0 mM; (▲) 0.50 mM at pH=6.5 with 20.0 mM MES buffer, 100 mM NaCl at 296 K.
Figure 4.

(a) CEST spectra of 10mM Yb(S-THP)3+ at pH 3.1; (b) The CEST effect of 5mM Yb(S-THP)3+ at 20 ppm as a function of pH.
CEST spectra for Ce(S-THP)3+ and Yb(S-THP)3+ were fit to modified Bloch equations with three exchangeable sites to obtain rate constants for exchange according to the procedure published by Woessner.27 Of special interest is the comparison of rate constants for these complexes as a function of pH. For Ce(S-THP)3+, the rate constants increase slightly as a function of pH from k = 230 s-1 at pH 4.0 to 450 s-1 at pH 7.0. For Yb(S-THP)3+, the rate constant for proton exchange decreased with pH from k= 370 s-1 at pH 3.1 to 52 s-1 at pH 6.2. Eu(THED)3+ CEST spectra could not be readily fit because of the close proximity of the CEST alcohol peak to bulk water.
High resolution 1H NMR spectra of the Ce(III) and Eu(III) complexes are consistent with an increase in the rate constant for alcohol proton exchange with increasing pH (Figure S10). At low pH values, (4.5 for Ce(S-THP)3+, and 4.0 for Eu(THED)3+), resonances for the alcohol protons of the complexes are observed. Raising the pH gives rise to broadening of the alcohol resonance into the baseline, consistent with more rapid exchange of the alcohol protons with bulk water. The 1H NMR alcohol proton resonance of the Yb(S-THP)3+ could not be observed in water under similar conditions at acidic pH (3.0) or neutral pH.
Discussion
PARACEST agents with alcohol groups
Our studies show that lanthanide(III) complexes with alcohol groups are CEST agents in water. Although the chemical shift separation between alcohol protons and water is modest for the Eu(III) and Ce(III) complexes studied here, the slow exchange regime was attained by variation of pH to retard the rate of proton exchange as discussed further below. The small chemical shift dispersion of the Eu(III) macrocyclic complexes with alcohol groups compared to analogous complexes with different pendent arms (Eu(DOTA) or Eu(TCMC), Chart 1) is related to the ligand field factor D and is affected by the distance and orientation of the protons relative to the lanthanide metal ion.29 Alcohols are poor donor groups for lanthanides, produce weak ligand fields and induce relatively small hyperfine chemical shifts.7 The acetate groups of DOTA and derivatives are better donors and thus produce stronger ligand fields and larger hyperfine chemical shifts, including a large (Δω) chemical shift different between the protons of bound and bulk water. Interestingly, Eu(CNPHC)3+, which has a single amide group, has a larger Δω (∼18 ppm) than Eu(THED)3+ which has all alcohol pendent groups.7 A large Δω is beneficial as it may lead to increased efficiency of the CEST agent by accommodating larger rate constants for proton exchange between CEST agent and bulk water and by minimizing the direct saturation of the water resonance.2
In order to increase Δω, complexes of THED and S-THP with Yb(III) were prepared. Yb(III) induces a large chemical shift dispersion in neighboring proton resonances, largely of pseudocontact shift origin.30,31 Yb(III) complexes of THED and S-THP have highly hyperfine-shifted ligand proton resonances and the largest Δω of the complexes studied here. However, addition of 1% water to an acetonitrile solution of Yb(THED)3+ led to disappearance of the hydroxyl proton resonances, and a loss of the CEST alcohol peak. Similarly, addition of water to an acetonitrile solution of Eu(THED)3+ led to broadening and, at high water concentrations, near disappearance of the alcohol proton CEST peak. However, CEST spectra are observable in 100% water if the pH is controlled. Under these conditions at approximately pH 5.2, the Eu(THED)3+ CEST spectrum exhibits a broad shoulder that partially overlaps with the bulk water, similar to that observed for the Ce(III) complex at pH 6.5 (Figure 3). The CEST spectrum of Yb(S-THP)3+ exhibits a well-separated CEST peak at 20 ppm at pH 3.1 (Figure 4). Thus, there is a unique pH-dependence for the CEST spectra of each of these three complexes with the pH of CEST optimal efficiency in the order Ce > Eu > Yb. Notably, the pKa values of the alcohol/water ligands of the complexes9 decrease in the same order.
The rate constants of exchangeable ligand protons on CEST agents are often dependent on the pH of the solution.6,32-34 For amide complexes, there is an increase in the CEST efficiency of the complexes with increased pH from neutral to basic pH as the rate constant for proton exchange increases due to base catalysis, followed by a decrease in the CEST peak as exchange rates exceed slow exchange conditions on the 1H NMR time scale.6 The pH optimum for Ln(III) complexes with mobile NH protons in amide groups ranges from pH 7.5 to 8.56,28,34 and rate constants for proton exchange are in the same range as those observed here for alcohol protons.28 Analogously, the weakly acidic alcohol groups of the Ln(III) complexes of THED and S-THP are anticipated to give rise to pH-dependent CEST properties. This is indeed observed for all complexes studied here. However, the pH dependence of the alcohol CEST effect is more dramatic than that of amide protons because the hydroxyl groups of the pendent alcohols bind directly to the lanthanide(III) so that there is a marked ligand pKa difference across the Ln(III) series.
The pKa values of Ln(III) complexes of S-THP vary by two units across the lanthanide series, from a pKa value of 8.4 for La(S-THP)3+ to 6.4 for Lu(S-THP)3+.9,11 This pKa value tracks the decrease in ionic radius and increasing Lewis acidity of Ln(III) ions across the lanthanide series. In contrast, there is little variation with the addition of the methyl group as a substituent (pKa of 7.5 for Eu(THED)3+ and 7.7 for Eu(S-THP)3+). This ionization is attributed either to an alcohol ligand or to a water ligand of the lanthanide(III) complex9,11,12 or perhaps a combination of the two involving a bound water with hydrogen bonding to the alcohol groups. The variation in ligand ionization values should produce different pH-dependent CEST spectra for each complex. If proton exchange is base-catalyzed, the CEST pH optimum is predicted to increase for Ln(III) ion complexes that are less acidic in comparison to Ln(III) complexes that are stronger acids. Consistent with this postulate, Ce(S-THP)3+ as the least acidic Ln(III) complex has a higher pH optimum for the CEST alcohol peak than does the Eu(III) complex, and the Eu(III) complex pH optimum is higher than the Yb(III) complex (Figures 2, 4).
For Ce(S-THP)3+, the rate constant for alcohol proton exchange increases only modestly with an increase in pH. An increase in rate constant with increasing pH is consistent with base-catalyzed alcohol proton exchange, but it is not clear why the pH dependence of the rate constant is not more pronounced. An increase in proton exchange rate constant will enhance the CEST effect so long as the system remains in the slow exchange regime.2,35,36 As the rate constant exceeds the slow exchange limit, the alcohol peak broadens and merges with the bulk water peak. For base catalyzed exchange, the lower pKa of Eu(THED)3+ would lead to more efficient CEST at low pH values compared to the Ce(III) complex.
The alcohol CEST effect decreases from pH 3.0 to 7.0 for Yb(S-THP)3+, but in contrast to the Eu(III) and Ce(III) complexes, the rate constant for proton exchange decreases with increasing pH. This suggests that there is a distinctly different mechanism for proton-catalyzed exchange for the Yb(III) complex, perhaps driven by the smaller size of this late Ln(III) ion. Data shows that the CEST effect and proton exchange rate constant are largest under conditions where the neutral Yb(S-THP)3+ complex would predominate in solution, rather than the ionized complex.9 In addition, the increase in the CEST peak from pH 5 to 3 suggests that acid catalyzed exchange may play a role in the pH dependence of CEST for the Yb(III) complex.
Conclusions
This work highlights the importance of the choice of lanthanide(III) ion in PARACEST agents with alcohol donor groups because both Δω and the pKa values of the alcohol/water complex protons vary across the lanthanide series and both affect the CEST spectrum. The combination of moderate alcohol/water ligand ionization values for early Ln(III) ion complexes of S-THP and moderate chemical shift dispersion of Ce(III) makes Ce(S-THP)3+ the first PARACEST agent to exhibit a CEST spectrum for alcohol proton exchange at pH 7.0 in the absence of other coordinating ligands that influence CEST.8 The pH modulation of the CEST spectrum suggests that Ln(III) complexes of THED or S-THP have rich potential as pH responsive agents. Future work will focus on modulating the pH optimum of the CEST spectrum by tuning the macrocyclic complex pKa.12 This work shows that alcohols are alternative ligating groups for lanthanide(III) complexes for the preparation of PARACEST agents.
Supplementary Material
Acknowledgments
We thank the National Institutes of Health for support of this work (EB-04609). We thank Dean Sherry (University of Texas, Dallas and University of Texas Southwestern Medical Center) and Mark Woods (Portland State University) for the CEST spectrum fitting program and for suggestions on fitting of CEST spectra.
References
- 1.Ward KM, Aletras AH, Balaban RS. J Magn Reson. 2000;143:79–87. doi: 10.1006/jmre.1999.1956. [DOI] [PubMed] [Google Scholar]
- 2.Woods M, Donald EWC, Sherry AD. Chem Soc Rev. 2006;35:500–511. doi: 10.1039/b509907m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Baguet E, Roby C. J Magn Reson. 1997;128:149–160. doi: 10.1006/jmre.1997.1230. [DOI] [PubMed] [Google Scholar]
- 4.Aime S, Crich SG, Gianolio E, Giovenzana GB, Tei L, Terreno E. Coord Chem Rev. 2006;250:1562–1579. [Google Scholar]
- 5.Yoo B, Raam MS, Rosenblum RM, Pagel MD. Contrast Med & Molec Imag. 2007;2:189–198. doi: 10.1002/cmmi.145. [DOI] [PubMed] [Google Scholar]
- 6.Zhang S, Michaudet L, Burgess S, Sherry AD. Angew Chem, Int Ed. 2002;41:1919–1921. [PubMed] [Google Scholar]
- 7.Woods M, Woessner DE, Zhao PY, Pasha A, Yang MY, Huang CH, Vasalitiy O, Morrow JR, Sherry AD. J Am Chem Soc. 2006;128:10155–10162. doi: 10.1021/ja061498t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Huang CH, Morrow JR. J Am Chem Soc. 2009;131:4206–4207. doi: 10.1021/ja900290z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chin KOA, Morrow JR. Inorg Chem. 1994;33:5036–5041. [Google Scholar]
- 10.Morrow JR, Chin KOA. Inorg Chem. 1993;32:3357–3361. [Google Scholar]
- 11.Chin KOA, Morrow JR, Lake CH, Churchill MR. Inorg Chem. 1994;33:656–664. [Google Scholar]
- 12.Chappell LL, Voss DA, Horrocks WD, Morrow JR. Inorg Chem. 1998;37:3989–3998. doi: 10.1021/ic980191v. [DOI] [PubMed] [Google Scholar]
- 13.Supkowski RM, Horrocks WD., Jr Inorg Chem. 1999;38:5616–5619. doi: 10.1021/ic990597n. [DOI] [PubMed] [Google Scholar]
- 14.Pittet PA, Früh D, Tissières V, Bünzli JCG. Dalton Trans. 1997;5:895–900. [Google Scholar]
- 15.Parker D, Dickins RS, Puschmann H, Crossland C, Howard JAK. Chem Rev. 2002;102:1977–2010. doi: 10.1021/cr010452+. [DOI] [PubMed] [Google Scholar]
- 16.Aime S, Botta M, Ermondi G. Inorg Chem. 1992;31:4291–4299. [Google Scholar]
- 17.Marques MPM, Geraldes CFGC, Sherry AD, Merbach AE, Powell H, Pubanz D, Aime S, Botta M. J Alloys Comp. 1995;225:303–307. [Google Scholar]
- 18.Corsi DM, Vander Elst L, Muller RN, van Bekkum H, Peters JA. Chem Eur J. 2001;7:1383–1389. doi: 10.1002/1521-3765(20010401)7:7<1383::aid-chem1383>3.0.co;2-5. [DOI] [PubMed] [Google Scholar]
- 19.Zhang S, Kovacs Z, Burgess S, Aime S, Terreno E, Sherry AD. Chem Eur J. 2001;7:288–296. doi: 10.1002/1521-3765(20010105)7:1<288::aid-chem288>3.0.co;2-6. [DOI] [PubMed] [Google Scholar]
- 20.Dunand FA, Aime S, Merbach AE. J Am Chem Soc. 2000;122:1506–1512. [Google Scholar]
- 21.Muller G, Kean SD, Parker D, Riehl JP. J Phys Chem A. 2002;106:12349–12355. [Google Scholar]
- 22.Lelli M, Pintacuda G, Cuzzola A, Di Bari L. Chiralit. 2005;17:201–211. doi: 10.1002/chir.20151. [DOI] [PubMed] [Google Scholar]
- 23.Morrow JR, Chin KOA. Inorg Chem. 1993;32:3357–3361. [Google Scholar]
- 24.Buøen S, Dale J, Groth P, Krane J. J Chem Soc, Chem Commu. 1982:1172–1174. [Google Scholar]
- 25.Baker BF, Khalili H, Wei N, Morrow JR. J Am Chem Soc. 1997;119:8749–8755. [Google Scholar]
- 26.Jacques V, Desreux JF. Inorg Chem. 1994;33:4048–4053. [Google Scholar]
- 27.Woessner DE, Zhang S, Merritt ME, Sherry AD. Magn Res Med. 2005;53:790–799. doi: 10.1002/mrm.20408. [DOI] [PubMed] [Google Scholar]
- 28.Terreno EP, Castelli DDP, Cravotto GP, Milone LP, Aime SP. Invest Radiol. 2004;39:235–243. doi: 10.1097/01.rli.0000116607.26372.d0. [DOI] [PubMed] [Google Scholar]
- 29.Pintacuda G, John M, Su XC, Otting G. Acc Chem Res. 2007;40:206–212. doi: 10.1021/ar050087z. [DOI] [PubMed] [Google Scholar]
- 30.Geraldes CFGC, Luchinat C. In: Metal Ions in Biological Systems. Sigel A, Sigel H, editors. Vol. 40. Marcel Dekker; New York, NY: 2003. pp. 513–588. [PubMed] [Google Scholar]
- 31.Piguet C, Geraldes CFGC. Handbook on the Physics and Chemistry of Rare Earth. 2003;33:353–463. [Google Scholar]
- 32.Woods M, Zhang S, Von Howard E, Sherry AD. Chem Eur J. 2003;9:4634–4640. doi: 10.1002/chem.200305159. [DOI] [PubMed] [Google Scholar]
- 33.Zhang S, Kovacs Z, Burgess S, Aime S, Terreno E, Sherry AD. Chem Eur J. 2001;7:288–296. doi: 10.1002/1521-3765(20010105)7:1<288::aid-chem288>3.0.co;2-6. [DOI] [PubMed] [Google Scholar]
- 34.Aime S, Barge A, Castelli DD, Fedeli F, Mortillaro A, Nielsen FU, Terreno E. Magn Reson Med. 2002;47:639–648. doi: 10.1002/mrm.10106. [DOI] [PubMed] [Google Scholar]
- 35.Zhang S, Merritt M, Woessner DE, Lenkinski RE, Sherry AD. Acc Chem Res. 2003;36:783–790. doi: 10.1021/ar020228m. [DOI] [PubMed] [Google Scholar]
- 36.Sherry AD, Woods M. In: Molecular and Cellular MR Imaging. Modo MMJ, Bulte JW, editors. CRC Press; Boca Raton, FL: 2007. pp. 101–122. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.