

Abstract
The downstream consequences of a single quantitative trait polymorphism can provide important insight into the molecular basis of a trait. However, the molecular consequences of a polymorphism may be complex and only a subset of these may influence the trait of interest. In natural isolates of Saccharomyces cerevisiae, a nonsynonymous polymorphism in cystathione beta-synthase (CYS4) causes a deficiency in both cysteine and glutathione that results in rust-colored colonies and drug-dependent growth defects. Using a single nucleotide allele-replacement, we characterized the effects of this polymorphism on gene expression levels across the genome. To determine whether any of the differentially expressed genes are necessary for the production of rust-colored colonies, we screened the yeast deletion collection for genes that enhance or suppress rust coloration. We found that genes in the sulfur assimilation pathway are required for the production of rust color but not the drug-sensitivity phenotype. Our results show that a single quantitative trait polymorphism can generate a complex set of downstream changes, providing a molecular basis for pleiotropy.
Keywords: QTN, CYS4, pleiotropy, gene expression, hydrogen sulfide
Introduction
The molecular basis of phenotypic variation in natural populations is important to understanding both molecular and phenotypic evolution. Yet, dissecting the molecular basis of a trait is difficult as it involves not only mapping quantitative trait polymorphisms but also understanding how each polymorphism affects a trait by influencing the activity of other genes, pathways and cellular processes involved in the production of a trait. For example, a quantitative trait polymorphism in MKT1 affects high temperature growth (Sinha et al., 2006), sporulation efficiency (Deutschbauer & Davis, 2005), DNA damage sensitivity (Demogines et al., 2008), as well as numerous gene expression levels (Smith & Kruglyak, 2008), but the mechanisms by which the MKT1 polymorphism affects these traits have only begun to be characterized (Lee et al., 2009).
The relationship among molecular, cellular and organismal changes caused by quantitative trait polymorphisms is key to obtaining a general understanding of the molecular basis of a trait. Changes in gene expression provide a rich measurement of molecular variation and have provided valuable insights into the genes and molecular processes related to a trait of interest, e.g. (Cavalieri et al., 2000; de Haan et al., 2002; Fay et al., 2004; Schadt et al., 2003). However, the relationship between gene expression and other phenotypes may often be complex; changes in gene expression that are tightly correlated with a trait may not be involved in the production of the trait. In yeast, changes in the environment cause many genes to change expression even though most are not required to survive or grow in the new environment (Birrell et al., 2002; Giaever et al., 2002). However, environmentally induced changes in gene expression may be different from genetic variation in gene expression segregating in natural populations since the latter is heritable and shaped by natural selection. Thus, while there are numerous examples of changes in gene expression with phenotypic consequences (Carroll et al., 2001; Wray et al., 2003), relatively little is known about the extent to which the effects of a quantitative trait polymorphism are mediated by downstream changes in gene expression.
Identifying the molecular consequences of a single quantitative trait polymorphism is difficult when there are other polymorphisms segregating within the genetic background. In yeast, allele-replacement strains have been used to measure the precise effects of a number of quantitative trait polymorphisms on gene expression levels (Brown et al., 2008; Smith & Kruglyak, 2008; Yvert et al., 2003). Each polymorphism resulted in numerous gene expression changes, but the role that these changes play in generating organismal phenotypes is difficult to know. Indeed, only a subset of the downstream molecular consequences of a quantitative trait polymorphism may mediate its effects. Thus, even when changes in gene expression can be attributed to a single quantitative trait polymorphism, correlative and causative changes must be distinguished to fully understand the molecular basis of a trait.
In yeast, hydrogen sulfide production varies among strains and has a negative impact on wine quality (Jiranek et al., 1995; Spiropoulos & Bisson, 2000). We previously found that a strain isolated from a vineyard in Italy, M22, expresses genes in the sulfur assimilation pathway at high levels and produces high levels of hydrogen sulfide, H2S, a metabolite produced by the sulfur assimilation pathway (Fay et al., 2004). H2S producing strains have been shown to produce dark-brown colonies when grown in the presence of lead acetate (Ono et al., 1991) or on BiGGY medium which uses bismuth as an indicator for sulfide production (Jiranek et al., 1995). Similarly, M22 produces reddish-brown or rust-colored colonies in the presence of copper sulfate. Previous work has shown that a wild-type laboratory strain produces brown-colored colonies when grown on copper containing synthetic complete medium due to copper-sulfur mineral complexes on the cell surface (Yu et al., 1996). These studies support the hypothesis that rust coloration is due to overproduction of hydrogen sulfide leading to the precipitation of hydrogen sulfide and copper ions (Fay et al., 2004). However, the relationship between expression of the sulfur assimilation genes, hydrogen sulfide production and colony color depends on the environment and strain background (Linderholm et al., 2008; Spiropoulos et al., 2000).
To dissect the relationship between changes in gene expression and the production of rust-colored colonies, we conducted a genetic analysis using M22. Based on a cross between M22 and two strains that are white-colored in the presence of copper sulfate, we found rust coloration co-segregates with another M22 phenotype, a drug-dependent delay in growth that occurs across a variety of pharmacological compounds (Kim & Fay, 2007). Quantitative trait mapping, complementation, and a single nucleotide allele-replacement showed that an amino acid polymorphism in cystathione beta-synthase (CYS4), I123N, causes both drug-sensitivity (Kim & Fay, 2007) and rust-colored colonies (Fig. S1). CYS4 encodes the first step in the cysteine biosynthesis pathway, which generates cysteine from homocysteine. (See Fig. 3 for an overview of the pathway.)
Fig. 3.

Genes involved in sulfur assimilation and biosynthesis of cysteine and glutathione. Asterisks and crosses indicate suppressors and enhancers of rust color, respectively. Genes not included in the screen are shown in parenthesis. Genes up-regulated in M22-CYS4::N123I or in M22-CYS4Δ are indicated by a triangle. Dotted lines show ubiquitin mediated negative regulation of Met4 by cysteine and positive regulation of the sulfur assimilation pathway by the Met4 complex.
Abbreviations: APS (adenosine 5′-phosphosulfate), PAPS (3′-phosphoadenosine-5′-phosphosulfate), SAM (S-adenosylmethionine), γ-GluCys (γ-glutamylcysteine), SAH (S-adenosylhomocysteine)
Cysteine regulates degradation of Met4 (Menant et al., 2006), a transcriptional activator of genes in the sulfur assimilation pathway. The sulfur assimilation pathway incorporates sulfur from sulfate into homocysteine via hydrogen sulfide (Thomas & Surdin-Kerjan, 1997). Thus, a deficiency in cysteine provides a mechanism by which the CYS4 polymorphism could cause feedback up-regulation of the sulfur assimilation pathway and high levels of hydrogen sulfide production. If rust coloration is caused by a reaction between copper and hydrogen sulfide ions, rust coloration may be mediated by up-regulation of the sulfur assimilation pathway. However, the mechanism by which rust color is produced could be more complex; both cysteine and glutathione play critical roles in monitoring and regulating the oxidative/reductive state of a cell (Toledano et al., 2007), so deficiencies in cysteine and glutathione could result in alteration of other cellular processes that are required for the production of rust colored colonies.
Here, we describe changes in gene expression caused by a nonsynonymous polymorphism in CYS4 and show that the majority of differentially expressed genes are not required for the production of rust color. We also show that while genes in the sulfur assimilation pathway are required for the rust coloration phenotype, they are not required for the drug-sensitivity phenotype, indicating that the sulfur assimilation pathway plays a role in mediating the pleiotropic effects of the CYS4 polymorphism.
Materials and Methods
Strains and Media
Rich medium [2% (w/v) yeast extract, 1% (w/v) peptone, 2% (w/v) dextrose, with or without 2% (w/v) agar] was supplemented with copper sulfate (1mM) and propargylglycine (320μM PPG) to generate PPG medium for the colony color assays. For chemical complementation, L-cysteine and L-glutathione (Sigma, USA) were dissolved in distilled water, filter-sterilized, and added to YPD medium. M22 is a homothallic diploid isolated from a vineyard in Italy (Fay & Benavides, 2005). An allele replacement strain (M22-CYS4::N123I) was generated by directly transforming M22-CYS4Δ by fusion PCR fragments containing the desired CYS4 allele and by selecting on complete media followed by color selection (Kim & Fay, 2007). Deletion of CYS4, MET10, MET2, and MET17 in M22 was generated using the kanMX deletion cassette and selected on the rich medium supplemented with G418 (Wach et al., 1994).
Expression analysis
Overnight cultures of M22, M22-CYS4Δ, and M22-CYS4::N123I were diluted in fresh YPD to 10% (v/v) and grown for 3 hours at 30°C. Total RNA was isolated using an RNeasy mini kit (QIAGEN, USA), reverse transcribed, labeled with Cy3 or Cy5 fluorescent dyes (Genisphere, USA), and hybridized to epoxy-coated slides (MWG Biotech, USA) spotted with 6388, 70mer oligos (Qiagen-Operon, USA). Arrays were scanned using a ScanArray Express laser scanner (Perkin Elmer, USA). Microarrays were manufactured and hybridizations were completed in the microarray core facility in Washington University’s Genome Center. A dye-swap was performed for one of the three replicates of each strain, where Cy3 instead of Cy5 was used to label the reference RNA, a pool of RNA from all samples. Individual array was normalized using the LOWESS algorithm to control for intensity dependent dye effect (Dudoit et al., 2002). The log transformed median ratio of normalized two channel intensities were used for further analysis. Significant differences in gene expression among strains (M22, M22-CYS4Δ, and M22-CYS4::N123I) were estimated using analysis of variance with the model: yi = μ + Vi + εi, where yi is the ratio of transcripts in strain i compared to the reference pool, μ is the average ratio across all strains, Vi is the effect of strain i on the transcript ratio, and εi is the error. Significant variation in gene expression among the strains was identified using the false discovery rate (FDR) method of Benjamini-Hochberg (Benjamini & Hochberg, 1995). Tukey’s posthoc honest significant difference method implemented in R (http://cran.r-project.org/) was used to identify which of the three pairs of strains were different from one another. Gene expression data have been deposited in Gene Expression Omnibus (GEO, http://www.ncbi.nlm.nih.gov/geo) and are accessible through the GEO Series accession number GSE14102.
Enhancer/suppressor screen
The 4664 homozygous kanMX-deletion strains (BY4743, MATa/α, his3Δ1/his3Δ1, leu2Δ0/leu2Δ0, lys2Δ0/LYS2, MET15/met15Δ0, ura3Δ0/ura3Δ0) were grown on YPD agar plates (384/plate) for 24 hours at 30°C. Colonies were replica-plated to PPG medium using a RoToR robot (Singer Instruments, UK). Photographs were taken after 24 hours growth, images were cropped and red and green channels were saved separately using Photoshop CS2 (Adobe Systems Inc. USA). Spotfinder (Saeed et al., 2003) was used to grid the colonies and measure color intensities. Rust coloration of each strain was measured using the green channel intensities divided by size of the colony. To control for edge effects from the plate, each colony was normalized by either the median of the row or column of the 384 strains in each plate depending on which deviated more from the overall background. Enhancers and suppressors were identified by those strains showing a green intensity of less than 0.80 and greater than 1.20, respectively, from photographs of the strains grown on PPG medium. At these cutoffs, the deletion strains showed slight but noticeably darker or lighter levels of rust color. At a cutoff of 0.60 and 1.30, the strains showed large, easily recognized effects on rust coloration. To assess the significance of deletions with slight effects on rust coloration, a subset of 1,494 strains were phenotyped in replicate. The replicates showed an average standard error of 0.012 and there were no cases where a single green intensity measurement showed a deviation greater than 0.20 from the mean of the five replicates. The probability of observing a single value below 0.80 or above 1.20 from permutations of the error, as measured by each strains deviation from the genotype mean, was 2.7×10-4 and 2.6×10-4, respectively.
Gene ontology analysis
Significantly enriched classes of genes were identified using gene ontology terms and P-values from the hypergeometric distribution followed by Bonferroni correction as implemented in the Saccharomyces Genome Database (http://www.yeastgenome.org/). For the analysis of enhancers and suppressors, named genes in the homozygous deletion collection were used as the background set by which significantly enriched gene ontology terms were identified.
Results
A deficiency in cysteine biosynthesis causes rust coloration in M22
To characterize the molecular mechanisms by which a single amino acid polymorphism within cysteine beta-synthase (Cys4) causes rust coloration. We compared two strains that are identical except for the causative nonsynonymous site within CYS4. We previously showed that the CYS4 amino acid polymorphism causes a deficiency in glutathione, which can be synthesized from cysteine, and that supplementation with either cysteine or glutathione eliminates drug-sensitivity (Kim & Fay, 2007). Similarly, both cysteine and glutathione suppress rust-coloration in M22 and strains lacking CYS4 (Fig. 1). Since glutathione can be converted back into cysteine (Ganguli et al., 2007), these results imply that rust coloration is mediated by a deficiency in intracellular levels of either glutathione or cysteine.
Fig. 1.
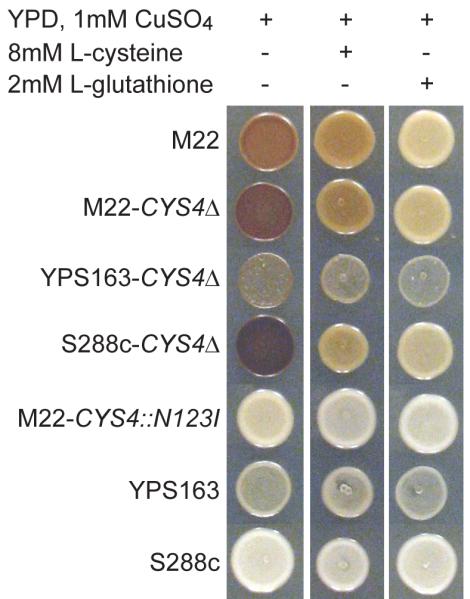
Cysteine and glutathione suppress rust coloration. Deletion of CYS4 in M22, S288c and YPS163 results in rust coloration, similar to M22, on rich medium supplemented with 1mM copper sulfate. The level of rust coloration is reduced in medium supplemented with 8mM L-cysteine or 2mM L-glutathione. Rust coloration is difficult to see in YPS163 which shows reduced growth in the presence of copper sulfate.
Differential gene expression caused by a quantitative trait polymorphism
To characterize the molecular consequences of the CYS4 amino acid polymorphism, we measured genome-wide changes in gene expression levels. We compared expression profiles of M22 (rust-colored) to an allele replacement strain (M22-CYS4::N123I, white-colored). For reference, we also compared the expression profile of the M22 allele-replacement strain to M22 with CYS4 deleted (M22-CYS4Δ, rust-colored). From three replicate comparisons of M22 to M22-CYS4::N123I we identified 24 genes differentially expressed at a false discovery rate (FDR) of 5% and 141 genes at a FDR of 20% (Table S1). Compared to the white-colored allele-replacement, deletion of CYS4 caused a larger set of 45 and 189 changes in gene expression at a 5% and 20% FDR, respectively (Table S1). The average fold change for the 24 and 45 differentially expressed genes was 2.1 and 2.3, respectively. The intersection between the two sets of genes is large; 21 genes intersect at a 5% FDR and 75 genes intersect at a 20% FDR. The larger number of expression differences between M22-CYS4Δ and the allele replacement as well as the large and directionally consistent overlap (71/75) with those expression differences between M22 and the allele replacement is consistent with the M22 allele of CYS4 being a hypomorph, as suggested by previous observations based on glutathione levels (Kim & Fay, 2007).
The 75 genes that differ between the two rust-colored strains and the white allele-replacement strain are significantly enriched for genes involved in sulfur metabolic processes (P = 1.3×10-4) and siderophore-iron transport (P = 0.0058, see Methods). Genes that function in sulfur-assimilation and metabolism of sulfur-containing amino acids, methionine and cysteine are up-regulated in M22 and M22-CYS4Δ (Table 1), consistent with cysteine-dependent feedback regulation by Met4 (Menant et al., 2006). Genes that function in siderophore-iron transport are likely up-regulated in response to glutathione deficiency since glutathione is required for maturation of cytoplasmic iron-sulfur containing proteins and glutathione deficiency causes activation of iron transporters by Aft1 and Aft2 (Rutherford et al., 2005; Sipos et al., 2002).
Table 1.
Rust coloration and drug sensitivity are affected by different set of CYS4 induced genes
| Gene expression relative to M22-CYS4::N123I | Gene deletion phenotype | ||||
|---|---|---|---|---|---|
| Gene class | Gene name | M22 | M22-CYS4Δ | Rust color | Drug sensitivity |
| Sulfate transport | SUL1 | 4.1 | 16.7* | resistant | |
| SUL2 | 1.9 | 3.1* | |||
| OAC1 | 1.4* | 1.1 | |||
| Sulfate assimilation | MET3 | 7.1* | 3.3 | WW | |
| ECM17 | 6.0** | 5.1** | WW | ||
| MET10 | 4.5** | 4.5** | WW | resistant | |
| MET22 | 3.1* | 2.0 | W | ||
| MET16 | 2.4* | 1.9* | W | ||
| Homocysteine biosynthesis | MET17 | 4.6** | 2.5** | R | |
| IRC7 | 1.8** | 1.4** | resistant | ||
| MET2 | 1.6 | 2.1** | |||
| Methionine biosynthesis | MET6 | 5.1* | 2.0 | ||
| S-AdoMet biosynthesis | SAM1 | 1.7* | 1.2 | RR | |
| SAM2 | 1.8* | 2.0* | resistant | ||
| Methionine salvage | ADI1 | 1.7* | 1.0 | ||
| Cysteine biosynthesis | CYS4 | 2.5** | 0.05** | RR | sensitive |
| CYS3 | 1.5* | 1.6* | RR | sensitive | |
| Glutathione biosynthesis | GSH1 | 1.6 | 2.1* | sensitive | |
| Siderophore-iron transport | ARN1 | 1.5** | 3.1** | ||
| SIT1 | 2.9** | 2.7** | resistant | ||
| ENB1 | 2.5* | 2.4* | |||
| FIT2 | 1.6 | 8.5** | resistant | ||
| FIT3 | 1.4 | 7.8** | |||
Genes significant at 5% FDR
genes significant at 20% FDR
Gene expression ratios are the average of three replicates and the mean squared error is 0.13, averaged across genes. Rust coloration and drug sensitivity phenotypes were measured using strains from the homozygous diploid deletion collection. RR indicates a strong enhancer (< 0.6) and WW a strong suppressor (> 1.3), as measured by the intensity of the green channel from photographs. R indicates a moderate enhancer (< 0.8) and W a moderate suppressor (> 1.2). Drug sensitivity was measured by the growth-delay induced by 2mM atenolol supplemented to rich medium. Strains with less than 4 hours of grown-delay are classified as resistant and those with more than 9 hours of growth-delay are classified as sensitive.
Identification of enhancers and suppressors of rust coloration
To identify genes that affect rust coloration, we screened the yeast deletion collection (Winzeler et al., 1999) for enhancers and suppressors of the rust coloration phenotype. Since the deletion collection is white in the presence of copper sulfate, we used propargylglycine (PPG), which inhibits cystathionine gamma-lyase (CYS3) and cysteine biosynthesis, to phenocopy the M22 allele of CYS4. Figure S2 shows that similar to a deficiency in CYS4, PPG causes decreased levels of glutathione and makes it possible to identify deletions that enhance or suppress rust coloration.
Out of 4664 homozygous diploid deletion strains, we identified 120 enhancers and 304 suppressors of rust coloration (Fig. 2, Table S2). Of the 98 enhancers that are named genes, 23 are involved in protein catabolism through endosomal or vacuolar functions, and the most significant gene ontology enrichment is ubiquitin-dependent protein catabolic processes via the multivesicular body pathway (P = 5.8×10-7, Table 2). However, the four strongest enhancers are all genes involved in the production of cysteine. CYS3 and CYS4 generate cysteine from homocysteine, DUG2 generates cysteine by degradation of glutathione (Ganguli et al., 2007), and PDX3 produces pyridoxal 5′-phosphate (Vitamin B6), a prosthetic group required for Cys4 function (McClean et al., 2000). The list of enhancers also includes a number of genes that function in methionine or cysteine biosynthesis (Table 2), and is enriched for genes involved in sulfur amino acid metabolic processes (P = 0.003). The enhancement of rust coloration by deletion of genes involved in cysteine biosynthesis as well as two genes involved in glutathione degradation, DUG2 and DUG3, suggest that rust coloration results from low levels of cysteine rather than glutathione (Fig. 3).
Fig. 2.

Phenotypic distribution of enhancers and suppressors of rust coloration in the homozygous yeast deletion collection. Rust coloration was measured by the average intensity of the green channel from RGB photographs of a strain divided by the median of all strains in the same row or column (See methods for details). Enhancers are shown in red and suppressors in blue. Photographs of selected strains show five replicates per strain illustrating the mean and standard deviation of the rust coloration phenotype.
Table 2.
Functional classification of enhancers and suppressors
| Pathway/function | Subclass | Enhancers | Suppressors |
|---|---|---|---|
| Sulfate-assimilation & sulfur metabolism | Enzyme | SAM1, CYS3/4, DUG2/3, MET17, GLO2 | MET3/10/14/16/22, ECM17 |
| Regulation | BUL1, STP1 | MET28/32 | |
| Siroheme biosynthesis | MET1/8 | ||
| Purine and adenosine biosynthesis | ADE1 | ADE3/5/6 | |
| Transport | MUP1 | ||
| SAM dependent methyl transfer | CHO2 | ||
| Vacuolar, golgi, protein sorting | 23 genes | ||
| Mitochondrial | 221 genes | ||
| Total | 120 genes | 304 genes |
Suppressors of rust coloration include 267 named genes, the majority of which are related to mitochondrial functions (Table 2). Many of the mitochondrial related genes function in the ribosome (29 genes), cytochrome complexes (12 genes), translation (6 genes) and ATP synthase (10 genes). Although there is enrichment for genes involved in cellular respiration (P = 2.0×10-15) and genes that function in the mitochondria (P = 1.1×10-35), there are many genes required for respiration that did not suppress rust coloration. Thus, the suppression of rust color is not simply due to the reduced rate of growth of petite mutants. In addition to mitochondrial related genes, there is also enrichment for genes involved in gene expression (P = 7.2×10-8) and sulfate assimilation (P = 3.0×10-5). All six genes that function in the assimilation of sulfur from sulfate into homocysteine are suppressors of rust coloration (Table 2, Fig. 3). The last step in the sulfur assimilation pathway is the formation of homocysteine from hydrogen sulfide and O-acetyl-L-homoserine by Met17, which is an enhancer of rust coloration. The suppression of rust coloration by deletion of genes that lead to the production of hydrogen sulfide combined with the enhancement of rust coloration by deletion of MET17 indicates that hydrogen sulfide is a key metabolite involved in the production of rust coloration (Fig. 3).
Differentially expressed genes necessary for the production of rust coloration
If genes that are differentially expressed as a result of the CYS4 polymorphism play a role in generating the rust coloration phenotype, then deletion of these genes should either enhance or suppress rust coloration. Seven of the 120 enhancers and eight of the 304 suppressors were differentially expressed between both the rust (M22, M22-CYSΔ) and white-colored (M22-CYS4::N123I) strains. Genes that are up-regulated in M22 should suppress rust coloration when deleted if their expression levels are involved in the production of rust color. Of the eight suppressors, seven are expressed at higher levels in the rust-colored strains and five of these function in the conversion of sulfate into hydrogen sulfide before assimilation into homocysteine (Fig. 3). The other two suppressors are TUF1 and YTA12, required for translation of mitochondrial proteins and assembly of mitochondrial enzyme complexes, respectively. Only one of the enhancers, SWI3, was expressed at lower levels in the two rust-colored strains.
The identification of differentially expressed genes necessary for the production of rust coloration provides insight into the mechanism by which the CYS4 polymorphism results in rust coloration. The up-regulation of both enhancers and suppressors in the same biochemical pathway suggests that hydrogen sulfide is involved in the production of rust coloration. To confirm that hydrogen sulfide production is required for rust coloration in M22, we deleted MET2, MET17 and MET10 in M22. The suppression of rust coloration by MET10 and enhancement of rust coloration by MET2 or MET17 shows that rust coloration in M22 depends on hydrogen sulfide production (Fig. 4). To confirm the requirement for mitochondrial function in M22 and test whether mitochondrial function is required for hydrogen sulfide production, we deleted both TUF1 and YTA12 in M22. Surprisingly, neither deletion suppressed rust coloration. Rust coloration of M22 was also unaffected by deletion of two other genes, MRPL38 and MRPS17, that are required for rust coloration in the deletion collection background.
Fig. 4.

Deletion of MET2 or MET17 enhances rust coloration and deletion of MET10 suppresses rust coloration in M22. The four meiotic progeny of each heterozygous M22 deletion strain were plated on rich medium with 1mM CuSO4 and labels show strain genotypes.
Pleiotropy at the molecular level
In addition to rust coloration, the M22 allele of CYS4 also causes a delay in growth upon exposure to a variety of different pharmacological compounds (Kim & Fay, 2007). While both the rust coloration and drug sensitivity phenotypes are caused by a deficiency in cysteine/glutathione, the molecular basis of these two traits may not be identical. To determine whether any of the differentially expressed genes that function in sulfur assimilation or iron transport affect drug-sensitivity, we measured growth in the presence and absence of atenolol, a beta-adrenergic receptor antagonist that causes a delay in growth dependent on the CYS4 amino acid polymorphism (Kim & Fay, 2007). Four of the genes involved in the production of sulfur-containing amino acids and two of the genes involved in iron transport increase drug resistance when deleted. However, most of the genes in the sulfur assimilation pathway that affect rust coloration do not affect drug-sensitivity (Table 1). In contrast, deletions of genes required for glutathione biosynthesis, CYS3, CYS4 and GSH1, increase drug-sensitivity. Thus, while both rust coloration and drug-sensitivity result from a deficiency in cysteine, they each have a distinct molecular basis since they are enhanced and suppressed by different sets of genes.
Discussion
Dissecting the molecular mechanisms by which a single quantitative trait polymorphism influences a trait of interest provides insight into those biochemical or signaling pathways that underlie a trait. We identified a complex set of downstream changes in gene expression caused by a pleiotropic quantitative trait polymorphism. We then showed that a subset of the differentially expressed genes in the same biochemical pathway is required for the production of rust coloration but not drug-sensitivity. These results provide insight into the molecular basis of pleiotropy.
The molecular basis of rust coloration
The suppression of rust coloration by deletion of genes required for hydrogen sulfide production and enhancement of rust coloration by deletion of genes involved in utilization of hydrogen sulfide implies that rust coloration is mediated by hydrogen sulfide production. Copper-dependent changes in yeast colony color are thought to be due to copper-sulfur mineralization at the cell surface (Ashida et al., 1963; Yu et al., 1996). Consistent with this possibility, electron micrographs show dark granules at the cell surface in M22 but not the white-colored M22 allele replacement strain (Fig. S3). However, hydrogen sulfide production may not be sufficient for the production of rust color. The rust-colored compound may also depend on other cellular perturbations such as a deficiency in glutathione, a potent antioxidant, or altered level of metabolites in the S-adenosylmethionine pathway (Christopher et al., 2002). In the yeast deletion collection background but not M22, rust coloration is dependent on some aspect of mitochondrial function. The large number of suppressors with mitochondrial functions makes it difficult to know what mitochondrial function is required for rust coloration. One possibility is that CuS mineralization occurs in the mitochondrion or requires iron-sulfur containing proteins made in mitochondria (Kispal et al., 1999). However, it is also possible that mitochondrial mutants suppress rust coloration by affecting H2S production, either by reducing the need for glutathione or by affecting the sulfur assimilation genes through mitochondrial retrograde regulation (Liu & Butow, 2006).
In M22, hydrogen sulfide production may be elevated due to differential regulation of the sulfur assimilation pathway. Alternatively, the M22 allele of CYS4 may cause homocysteine utilization to be the rate limiting step in sulfur assimilation. In this scenario, up-regulation of the sulfur assimilation genes would have little or no effect on hydrogen sulfide levels. Consistent with this latter possibility, up-regulation of the sulfur assimilation pathway in another Italian wine strain due to a frameshift in the extracellular amino acid sensor, SSY1 (Brown et al., 2008), does not lead to rust coloration or noticeable levels of hydrogen sulfide production (Kyle Brown, personal communication).
Our enhancer and suppressor screen implies that hundreds of genes with a wide range of biological functions affect rust coloration. In many cases, enhancement and suppression of rust coloration may be mediated by changes in cysteine/glutathione homeostasis and hydrogen sulfide production. Evidence for this possibility can be found in the overlap between enhancers and suppressors of rust coloration and two other screens of the yeast deletion collection. A screen for genes involved in glutathione homeostasis identified 276 genes that excrete high levels of glutathione and one of the largest functionally related groups contains genes involved in the secretory pathway or vacuolar protein sorting (Perrone et al., 2005), similar to enhancers of rust coloration. Of the 276 genes, 96 overlap with enhancers or suppressors of rust coloration. Another screen for genes involved in hydrogen sulfide production identified 89 genes, many of which function in the biosynthesis of sulfur containing amino acids (Linderholm et al., 2008). Of the 89 deletions that cause high levels of sulfite reductase activity, 15 overlap with enhancers of rust coloration. These comparisons imply that some but not all of the enhancers and suppressors of rust coloration affect cysteine/glutathione levels and hydrogen sulfide production in the absence of PPG induced inhibition of cysteine biosynthesis.
The role of gene expression in pleiotropy
We found a large number of genes that are differentially expressed as a consequence of the CYS4 polymorphism. The up-regulation of genes that suppress rust coloration when deleted implies that the differential expression of genes in the sulfur assimilation pathway plays an important role in the production of rust color. However, other CYS4 induced changes in gene expression may have no downstream phenotypic consequences. For example, the up-regulation of genes that enhance drug-sensitivity when deleted, e.g. CYS3 or GSH1, suggests that some expression changes are a response to alterations in cysteine homeostasis but are not responsible for drug-sensitivity. Finally, some CYS4 induced changes in gene expression may only have phenotypic consequences under certain environmental conditions; e.g. rust coloration is only observed in the presence of copper sulfate but up-regulation of the sulfur assimilation genes occurs in both the presence and absence of copper sulfate (Fay et al., 2004). Thus, the differential expression of siderophore iron transporters may also produce a downstream phenotype when measured under the right condition. The up-regulation of SIT1 and FIT2 combined with the higher level of drug-resistance when deleted implies that the differential expression of these two genes may be involved in drug sensitivity.
Implications for phenotypic variation in other yeast isolates
What is the relevance of our results to phenotypic variation in other strains of yeast? While the rust coloration phenotype is rare, progeny of M22 crossed with two white-colored strains show a continuous distribution of rust coloration phenotypes in those strains that inherit the M22 allele of CYS4 (Fig. S4). Furthermore, natural yeast isolates show significant variation in levels of intracellular glutathione (Fig. S5), which affects metabolism of a wide range of pharmacological compounds (Kim & Fay, 2007), and hydrogen sulfide, which affects wine production (Mendes-Ferreira et al., 2002). Thus, our results provide insight into the genes, pathways and processes that affect population variation in cysteine/glutathione metabolism and hydrogen sulfide production. However, we also found evidence for significant effects of genetic background. Deletions of four mitochondrial genes suppress rust coloration in BY4743, the strain used to make the deletion collection, but do not suppress rust coloration in M22. Thus, differences in genetic background may have resulted in missed M22-specific enhancers or suppressors, and presents a significant challenge to obtaining a comprehensive understanding of the molecular basis of cysteine-related phenotypes.
How common are pleiotropic, background dependent quantitative trait nucleotides? A nonsynonymous polymorphism in MKT1 shares many of the characteristics of the CYS4 polymorphism: extensive pleiotropy, background dependent effects and numerous changes in gene expression levels (Demogines et al., 2008; Deutschbauer et al., 2005; Sinha et al., 2006; Smith & Kruglyak, 2008). However, small-effect polymorphisms may have quite different characteristics. For example, quantitative trait polymorphisms with small effects show less pleiotropy than those of large effect (Wagner et al., 2008).
Supplementary Material
Acknowledgements
We thank M. Johnston for sharing yeast deletion collection, B. Cohen for sharing microarrays, L. Riles and J. Dover for technical support, W. Beatty in the Molecular Microbiology Imaging Facility for electron microscopy service, C. Hittinger, M. Dorsett, D. Swain, and B. Engle (All at Washington University) for critical reading and helpful discussions, and K. Brown (Harvard University) for examining phenotypes in another Italian wine strain. This work was supported by a National Institute of Health grant (80669) to JCF.
References
- Ashida J, Higashi N, Kikuchi T. An electronmicroscopic study on copper precipitation by copper-resistant yeast cells. Protoplasma. 1963;57:27–32. [Google Scholar]
- Benjamini Y, Hochberg Y. Controlling the false discovery rate - A practical and powerful approach to multiple testing. J. R. Stat. Soc. B. 1995;57:289–300. [Google Scholar]
- Birrell GW, Brown JA, Wu HI, Giaever G, Chu AM, Davis RW, Brown JM. Transcriptional response of Saccharomyces cerevisiae to DNA-damaging agents does not identify the genes that protect against these agents. P Natl Acad Sci USA. 2002;99:8778–8783. doi: 10.1073/pnas.132275199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown KM, Landry CR, Hartl DL, Cavalieri D. Cascading transcriptional effects of a naturally occurring frameshift mutation in Saccharomyces cerevisiae. Mol Ecol. 2008;17:2985–2997. doi: 10.1111/j.1365-294X.2008.03765.x. [DOI] [PubMed] [Google Scholar]
- Carroll AS, Bishop AC, DeRisi JL, Shokat KM, O’Shea EK. Chemical inhibition of the Pho85 cyclin-dependent kinase reveals a role in the environmental stress response. P Natl Acad Sci USA. 2001;98:12578–12583. doi: 10.1073/pnas.211195798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavalieri D, Townsend JP, Hartl DL. Manifold anomalies in gene expression in a vineyard isolate of Saccharomyces cerevisiae revealed by DNA microarray analysis. P Natl Acad Sci USA. 2000;97:12369–12374. doi: 10.1073/pnas.210395297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christopher SA, Melnyk S, James SJ, Kruger WD. S-adenosylhomocysteine, but not homocysteine, is toxic to yeast lacking cystathionine beta-synthase. Mol Genet Metab. 2002;75:335–343. doi: 10.1016/S1096-7192(02)00003-3. [DOI] [PubMed] [Google Scholar]
- de Haan G, Bystrykh LV, Weersing E, et al. A genetic and genomic analysis identifies a cluster of genes associated with hematopoietic cell turnover. Blood. 2002;100:2056–2062. doi: 10.1182/blood-2002-03-0808. [DOI] [PubMed] [Google Scholar]
- Demogines A, Smith E, Kruglyak L, Alani E. Identification and dissection of a complex DNA repair sensitivity phenotype in Baker’s yeast. PLoS Genet. 2008;4:e1000123. doi: 10.1371/journal.pgen.1000123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deutschbauer AM, Davis RW. Quantitative trait loci mapped to single-nucleotide resolution in yeast. Nat Genet. 2005;37:1333–1340. doi: 10.1038/ng1674. [DOI] [PubMed] [Google Scholar]
- Dudoit S, Yang YH, Callow M, Speed T. Statistical methods for identifying differentially expressed genes in replicated cDNA microarray experiments. Stat. Sin. 2002;12:111–139. [Google Scholar]
- Fay JC, Benavides JA. Evidence for domesticated and wild populations of Saccharomyces cerevisiae. PLoS Genet. 2005;1:66–71. doi: 10.1371/journal.pgen.0010005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fay JC, McCullough HL, Sniegowski PD, Eisen MB. Population genetic variation in gene expression is associated with phenotypic variation in Saccharomyces cerevisiae. Genome Biol. 2004;5:R26. doi: 10.1186/gb-2004-5-4-r26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganguli D, Kumar C, Bachhawat AK. The alternative pathway of glutathione degradation is mediated by a novel protein complex involving three new genes in Saccharomyces cerevisiae. Genetics. 2007;175:1137–1151. doi: 10.1534/genetics.106.066944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giaever G, Chu AM, Ni L, et al. Functional profiling of the Saccharomyces cerevisiae genome. Nature. 2002;418:387–391. doi: 10.1038/nature00935. [DOI] [PubMed] [Google Scholar]
- Jiranek V, Langridge P, Henschke PA. Regulation of hydrogen sulfide liberation in wine-producing Saccharomyces cerevisiae strains by assimilable nitrogen. Appl Environ Microbiol. 1995;61:461–467. doi: 10.1128/aem.61.2.461-467.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim HS, Fay JC. Genetic variation in the cysteine biosynthesis pathway causes sensitivity to pharmacological compounds. P Natl Acad Sci USA. 2007;104:19387–19391. doi: 10.1073/pnas.0708194104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kispal G, Csere P, Prohl C, Lill R. The mitochondrial proteins Atm1p and Nfs1p are essential for biogenesis of cytosolic Fe/S proteins. EMBO J. 1999;18:3981–3989. doi: 10.1093/emboj/18.14.3981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SI, Dudley AM, Drubin D, Silver PA, Krogan NJ, Pe’er D, Koller D. Learning a prior on regulatory potential from eQTL data. PLoS Genet. 2009;5:e1000358. doi: 10.1371/journal.pgen.1000358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linderholm AL, Findleton CL, Kumar G, Hong Y, Bisson LF. Identification of genes affecting hydrogen sulfide formation in Saccharomyces cerevisiae. Appl Environ Microbiol. 2008;74:1418–1427. doi: 10.1128/AEM.01758-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Z, Butow RA. Mitochondrial retrograde signaling. Annu Rev Genet. 2006;40:159–185. doi: 10.1146/annurev.genet.40.110405.090613. [DOI] [PubMed] [Google Scholar]
- McClean KN, Janosík M, Oliveriusová J, Kery V, Kraus JP. Transsulfuration in Saccharomyces cerevisiae is not dependent on heme: purification and characterization of recombinant yeast cystathionine beta-synthase. J. Inorg. Biochem. 2000;81:161–171. doi: 10.1016/s0162-0134(00)00100-8. [DOI] [PubMed] [Google Scholar]
- Menant A, Baudouin-Cornu P, Peyraud C, Tyers M, Thomas D. Determinants of the ubiquitin-mediated degradation of the Met4 transcription factor. J Biol Chem. 2006;281:11744–11754. doi: 10.1074/jbc.M600037200. [DOI] [PubMed] [Google Scholar]
- Mendes-Ferreira A, Mendes-Faia A, Leao C. Survey of hydrogen sulphide production by wine yeasts. J Food Prot. 2002;65:1033–1037. doi: 10.4315/0362-028x-65.6.1033. [DOI] [PubMed] [Google Scholar]
- Ono B, Ishii N, Fujino S, Aoyama I. Role of hydrosulfide ions (HS-) in methylmercury resistance in Saccharomyces cerevisiae. Appl Environ Microbiol. 1991;57:3183–3186. doi: 10.1128/aem.57.11.3183-3186.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perrone GG, Grant CM, Dawes IW. Genetic and environmental factors influencing glutathione homeostasis in Saccharomyces cerevisiae. Mol Biol Cell. 2005;16:218–230. doi: 10.1091/mbc.E04-07-0560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rutherford JC, Ojeda L, Balk J, Muhlenhoff U, Lill R, Winge DR. Activation of the iron regulon by the yeast Aft1/Aft2 transcription factors depends on mitochondrial but not cytosolic iron-sulfur protein biogenesis. J Biol Chem. 2005;280:10135–10140. doi: 10.1074/jbc.M413731200. [DOI] [PubMed] [Google Scholar]
- Saeed AI, Sharov V, White J, et al. TM4: a free, open-source system for microarray data management and analysis. Biotechniques. 2003;34:374–378. doi: 10.2144/03342mt01. [DOI] [PubMed] [Google Scholar]
- Schadt EE, Monks SA, Drake TA, et al. Genetics of gene expression surveyed in maize, mouse and man. Nature. 2003;422:297–302. doi: 10.1038/nature01434. [DOI] [PubMed] [Google Scholar]
- Sinha H, Nicholson BP, Steinmetz LM, McCusker JH. Complex genetic interactions in a quantitative trait locus. PLoS Genet. 2006;2:e13. doi: 10.1371/journal.pgen.0020013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sipos K, Lange H, Fekete Z, Ullmann P, Lill R, Kispal G. Maturation of cytosolic iron-sulfur proteins requires glutathione. J Biol Chem. 2002;277:26944–26949. doi: 10.1074/jbc.M200677200. [DOI] [PubMed] [Google Scholar]
- Smith EN, Kruglyak L. Gene-environmental interaction in yeast gene expression. PLoS Biol. 2008;6:e83. doi: 10.1371/journal.pbio.0060083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spiropoulos A, Bisson LF. MET17 and hydrogen sulfide formation in Saccharomyces cerevisiae. Appl Environ Microbiol. 2000;66:4421–4426. doi: 10.1128/aem.66.10.4421-4426.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas D, Surdin-Kerjan Y. Metabolism of sulfur amino acids in Saccharomyces cerevisiae. Microbiol Mol Biol Rev. 1997;61:503–532. doi: 10.1128/mmbr.61.4.503-532.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toledano MB, Kumar C, Le Moan N, Spector D, Tacnet F. The system biology of thiol redox system in Escherichia coli and yeast: differential functions in oxidative stress, iron metabolism and DNA synthesis. FEBS Lett. 2007;581:3598–3607. doi: 10.1016/j.febslet.2007.07.002. [DOI] [PubMed] [Google Scholar]
- Wach A, Brachat A, Pohlmann R, Philippsen P. New heterologous modules for classical or PCR-based gene disruptions in Saccharomyces cerevisiae. Yeast. 1994;10:1793–1808. doi: 10.1002/yea.320101310. [DOI] [PubMed] [Google Scholar]
- Wagner GP, Kenney-Hunt JP, Pavlicev M, Peck JR, Waxman D, Cheverud JM. Pleiotropic scaling of gene effects and the ‘cost of complexity’. Nature. 2008;452:470–472. doi: 10.1038/nature06756. [DOI] [PubMed] [Google Scholar]
- Winzeler EA, Shoemaker DD, Astromoff A, et al. Functional characterization of the S. cerevisiae genome by gene deletion and parallel analysis. Science. 1999;285:901–906. doi: 10.1126/science.285.5429.901. [DOI] [PubMed] [Google Scholar]
- Wray GA, Hahn MW, Abouheif E, Balhoff JP, Pizer M, Rockman MV, Romano LA. The evolution of transcriptional regulation in eukaryotes. Mol Biol Evol. 2003;20:1377–1419. doi: 10.1093/molbev/msg140. [DOI] [PubMed] [Google Scholar]
- Yu W, Farrell RA, Stillman DJ, Winge DR. Identification of SLF1 as a new copper homeostasis gene involved in copper sulfide mineralization in Saccharomyces cerevisiae. Mol Cell Biol. 1996;16:2464–2472. doi: 10.1128/mcb.16.5.2464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yvert G, Brem RB, Whittle J, et al. Trans-acting regulatory variation in Saccharomyces cerevisiae and the role of transcription factors. Nat Genet. 2003;35:57–64. doi: 10.1038/ng1222. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.