

Abstract
The Inhibitor of Growth (ING) tumor suppressors are implicated in oncogenesis, control of DNA damage repair, cellular senescence and apoptosis. All members of the ING family contain unique amino-terminal regions and a carboxy-terminal plant homeodomain (PHD) finger. While the amino-terminal domains associate with a number of protein effectors including distinct components of histone deacetylase (HDAC) and histone acetyltransferase (HAT) complexes, the PHD finger binds strongly and specifically to histone H3 trimethylated at lysine 4 (H3K4me3). In this review we describe the molecular mechanism of H3K4me3 recognition by the ING1-5 PHD fingers, analyze the determinants of the histone specificity and compare the biological activities and structures within subsets of PHD fingers.
The atomic-resolution structures of the ING PHD fingers in complex with a H3K4me3 peptide reveal that the histone tail is bound in a large and deep binding site encompassing nearly one-third of the protein surface. An extensive network of intermolecular hydrogen bonds, hydrophobic and cation-π contacts, and complementary surface interactions coordinate the first six residues of the H3K4me3 peptide. The trimethylated Lys4 occupies an elongated groove, formed by the highly conserved aromatic and hydrophobic residues of the PHD finger, whereas the adjacent groove accommodates Arg2. The two grooves are connected by a narrow channel, the small size of which defines the PHD finger’s specificity, excluding interactions with other modified histone peptides. Binding of the ING PHD fingers to H3K4me3 plays a critical role in regulating chromatin acetylation. The ING proteins function as tethering molecules that physically link the HDAC and HAT enzymatic complexes to chromatin. In this review we also highlight progress recently made in understanding the molecular basis underlying biological and tumorigenic activities of the ING tumor suppressors.
Keywords: Plant homeodomain, PHD finger, ING tumor suppressor, histone, H3K4me3, structure
OVERVIEW OF THE DOMAIN STRUCTURE OF ING1-5
The first member of the Inhibitor of Growth family, ING1 was discovered over a decade ago in a screen designed to isolate candidate tumor suppressor genes [1]. The other four ING family members (ING2-5), along with their multiple isoforms and splice variants, were identified through homology searches [2-5]. Since their discovery, the ING proteins have been implicated in a number of vital cellular events including p53 signaling [4-10], control of cell growth and proliferation [2], apoptosis [11-13], DNA damage repair [14, 15], and chromatin remodeling [16-21]. Interestingly, all ING genes with the exception of ING3 are found near the ends of chromosomes, and the function and expression of ING5 could be affected by telomere erosion [22].
Overall, the ING proteins show a relatively high degree of sequence conservation, with identities ranging from 59% (ING2) to 27% (ING3) in comparison to ING1, and share similar domain architecture. The amino-terminal region of ING1-5 functions as a protein-binding module that targets distinct nuclear components and chromatin-remodeling complexes. Thus, ING1 and ING2 associate with the Sin3A/HDAC1/2 repressor complex through their SAP30-interacting domain (SAID) [16, 17], whereas the N-terminal domains of ING3, ING4 and ING5 bind the EPC1/2, Jade1 and BRPF1 subunits of the hNuA4/Tip60, HBO1 and MOZ/MORF HAT complexes, respectively [19]. Additionally, the N-terminal region of ING1 is directly involved in the interactions with other effectors, including PCNA and Lamin A [12, 23, 24]. The Lamin A-binding sequence is conserved among ING1-5, suggesting that the association with nuclear lamina is a common feature of this family of proteins. A large insertion in the N-terminal region distinguishes ING3. Downstream of the protein-interacting region, all ING members contain a nuclear localization signal (NLS) that plays an essential role in transporting these proteins to the nucleus. The NLS is followed by the carboxy-terminal plant homeodomain (PHD) finger, and a short polybasic region (PBR) found only in ING1 and ING2, which binds phosphoinositides [8]. The PHD fingers of ING1-5 have recently been shown to recognize histone H3 trimethylated at lysine 4 (H3K4me3) [20, 25].
STRUCTURES OF THE ING1-5 PHD FINGERS
The PHD module is the most conserved region within the ING family showing sequence homology greater than 78% (Fig. 1). To date ten atomic-resolution structures of the ING1-5 PHD fingers have been determined by NMR spectroscopic and X-ray crystallographic techniques with the atomic coordinates deposited to Protein Data Bank (PDB) [references [21, 25-29] and unpublished data]. Five crystal structures of the ING PHD fingers have been solved in complex with the histone H3K4me3 peptide [PDB codes 2QIC (ING1), 2G6Q (ING2), 2PNX (ING4), 2VNF (ING4), and 3C6W (ING5)], whereas all structures of the ligand-free states were obtained exclusively in solution by NMR spectroscopy [PDB codes 1WES (ING2), 1X4I (ING3), 1WEU (ING4), 1WEN (ING4), and 2K1J (ING4)]. This fact suggests that the ING PHD fingers contain flexible regions that may preclude crystallization of this domain in the absence of the ligand. Indeed, the PHD finger fold consists of three loops, L1 (residues 219-221 of ING1), L2 (residues 226-234) and L3 (residues 242-253), stabilized by two zinc-binding clusters. The zinc ions are coordinated in a cross-brace arrangement by a Cys4-His-Cys3 consensus sequence (Fig. 2A). In this review, the numbering of residues used in both the sequence alignment and structural analysis refers to the ING1 PHD finger unless specifically noted. The core of each ING PHD finger is composed of a central double-stranded anti-parallel beta sheet (β3, residues 222-225 and β4, residues 235-237), which is surrounded by two small beta strands at the N-terminus (β1, residues 211-212 and β2, residues 217-218), and two short C-terminal alpha helices (α1, residues 239-241 and α2, residues 254-257) (Figs. 1 and 2A).
Fig. (1).
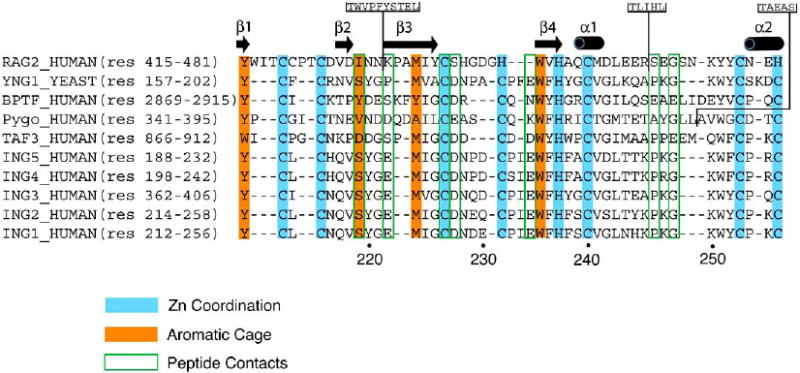
Alignment of amino acid sequences of PHD fingers known to bind H3K4me2/3. A structure-based sequence alignment (Expresso) [64] reveals the conserved Cys and His residues required for the coordination of two zinc ions, residues that make up the aromatic cage, and residues that form specific hydrogen bond contacts to the H3K4me3 peptide. Insertions in the RAG2 and Pygo sequences are indicated. The amino acid numbering below the alignment corresponds to the ING1 protein sequence. The secondary structure elements of the ING1 PHD finger were determined with DSSP [65].
Fig. (2).
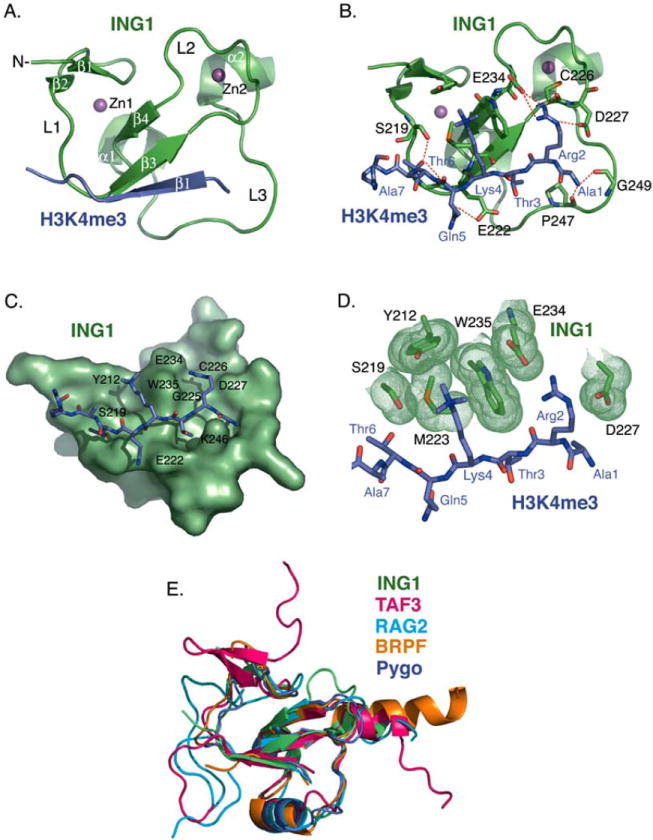
Structural analysis of the ING1 PHD finger. (A) A ribbon diagram of the human ING1 PHD finger (green) in complex with the H3K4me3 peptide (purple). The secondary structure elements were generated by DSSP [65]. The zinc ions are depicted in magenta. (B) Coordination of the histone peptide by the ING1 PHD finger. Specific intermolecular hydrogen bonds are indicated by a red dotted line. The intermolecular beta-strand hydrogen bond contacts are omitted for clarity. (C) The H3K4me3 binding site. The PHD finger is shown as solid surface. The histone peptide is depicted as a stick model. (D) A close up view of the ING1 aromatic cage. Residues of the ING1 PHD finger involved in the coordination of H3K4me3 and H3R2 are shown in green. (E) Superimposed structures of the PHD domains of ING1 (green), TAF3 (pink), RAG2 (light blue), BPTF (gold), and Pygo (purple). The structures were generated using Pymol and superimposed using Swiss-PDB Viewer [66].
THE ING1-5 PHD FINGERS BIND H3K4me3
The specificity of the ING1-5 PHD fingers toward H3K4me3 was initially determined by western blot analysis and NMR, while precise binding affinities were measured using tryptophan fluorescence [20, 25]. The PHD finger of ING2 binds the histone H3K4me3 tail with a Kd of 1.5 μM (Table 1). Removal of each methyl group attached to Lys4 results in a 10-fold decrease of binding affinity (Table 1). Recognition of H3K4me3 appears to be very specific, as no interaction with unmodified histone tails or histone peptides methylated at other lysine residues is detected [25]. Furthermore, the PHD fingers of all other human ING and yeast YNG proteins associate with the histone H3K4me3 peptide in the 1-10 μM range, indicating that recognition of this epigenetic mark is a general function of the ING/YNG protein family.
Table 1.
Binding Affinities of the Wild Type ING1-5 PHD Fingers for the Modified Histone H3 Tail Peptides
| Protein | Peptide | Kd ± s.d. (μM) | Method | Reference |
|---|---|---|---|---|
| ING1 | H3K4me3 | 3.3 ± 1.6 | Fluorescence | Pena, P. et al., 2006 |
| H3K4me2 | 17.3 ± 3.9 | Pena, P. et al., 2008 | ||
| H3K4me1 | 419 ± 71 | |||
| ING2 | H3K4me3 | 1.5 ± 1 | Fluorescence | Pena, P. et al., 2006 |
| H3K4me2 | 15 ± 4 | |||
| H3K4me1 | 208 ± 80 | |||
| ING3 | H3K4me3 | 6.9 ± 1.1 | Fluorescence | Pena, P. et al., 2006 |
| ING4 | H3K4me3 (12-mer) | 7.9 ± 2 | Fluorescence | Pena, P. et al., 2006 |
| H3K4me2 | 16.5 ± 1.1 | Hung, T. et al., 2009 | ||
| H3K4me1 | 159 ± 20 | |||
| H3K4me0 | 205 ± 28 | |||
| H3K4me3 (15-mer) | 4.0 ± 0.7 | NMR | Palacios, A. et al., 2006 | |
| H3K4me2 | 1.8 ± 1 | |||
| H3K4me1 | 1.6 ± 0.8 | |||
| H3K4me0 | 370 ± 20 | |||
| H3K4me3 (10-mer) | 3.0 ± 0.6 | NMR | Palacios, A. et al., 2008 | |
| H3K4me2 | 9.2 ± 1.4 | |||
| H3K4me1 | 34 ± 4 | |||
| H3K4me0 | 274 ± 6 | |||
| H3R2me2/K4me3 | 19.2 ± 1.7 | |||
| H3K4me3 (10-mer) | 6.4 ± 0.8 | ITC | Palacios, A. et al., 2008 | |
| H3K4me2 | 9.7 ± 0.6 | |||
| H3K4me1 | 45.8 ± 2.5 | |||
| H3K4me0 | 474 ± 67 | |||
| ING5 | H3K4me3 | 2.4 ± 1.0 | Fluorescence | Pena, P. et al., 2006 |
| H3K4me2 | 16 ± 1.2 | Champagne, K.S. et al., 2008 | ||
| H3K4me1 | 222 ± 17 | |||
| H3K4me0 | 261 ± 34 | |||
The H3K4me3-binding mechanism was defined through analyzing the crystal structures of the ING PHD fingers in complex with the histone tail (Fig. 2A and B). Upon the interaction, the H3K4me3 peptide forms a third anti-parallel beta strand that pairs with the existing double-stranded β-sheet of the PHD finger (Fig. 2A). Interestingly, the structures of the complexes appear to be more similar to each other than to the structures of the apo-states. For example, a root mean squared deviation (rmsd) between the H3K4me3-bound PHD fingers of ING4 (2PNX) and ING5 (3C6W) is 0.3 Å, whereas an rmsd between the ING4 complex (2PNX) and the apo-state of ING4 (1WEU) is almost five times as large (1.8 Å).
H3K4me3 lies in a deep and extensive binding site that covers nearly one-third of the PHD finger surface (Fig. 2C). The binding pocket consists of two large grooves connected by a narrow channel. The side chain of trimethylated Lys4 fits snugly into a hydrophobic pocket formed by Y212, S219, M223 and W235, whereas Arg2 occupies the adjacent groove and is surrounded by G225, C226, D227 and E234. The aromatic ring of W235 protrudes orthogonally to the protein’s surface, separating Arg2 and Lys4 of the peptide, and together with F238, I224, E222, and K246 creates a narrow channel where Thr3 is bound. The small size of the channel most likely defines the specificity of the PHD finger for methylated H3K4 versus methylated H3K9, H3K27, H3K36 and H4K20 sequences, which contain a bulky Arg or Val in place of a small threonine residue at the position -1 in respect to the methylated lysine.
Numerous intermolecular hydrogen bonds stabilize the ING PHD finger-H3K4me3 peptide complex. Arg2, Lys4 and Thr6 of the peptide make characteristic backbone β-sheet contacts with the G225, M223 and G221 residues of the PHD finger. Additionally, the first six residues of the histone tail form specific hydrogen bonds in the complex (Fig. 2B). The N-terminal amino group of Ala1 is coordinated by the carbonyl groups of P247 and G249. Contacts with the carboxyl groups of D227 and E234, and carbonyls of C226 and G225 restrain the guanidino moiety of Arg2. The hydroxyl group of Thr6 interacts with G221 and the hydroxyl group of S219.
Besides the dense scaffold of hydrogen bonds, complementary surface interactions contribute substantially to the binding. The side chain of each peptide residue fits precisely into the distinct binding pocket (Fig. 2C). The well-defined grooves for Arg2 and Lys4 are apparent. The methyl groups of Ala1 and Thr3 are packed in deep hydrophobic cavities formed by the core resides W251 and I224. The side chain of Thr6 is positioned in the site framed by S219, Y220, and G221. Thus, H3K4me3 is coordinated by the PHD finger through an extensive network of hydrogen bonding and complementary surface interactions. These contacts, which involve the first six N-terminal residues of the histone H3 tail, are responsible for the PHD finger specificity and unique recognition of the ARTK(me3)QT peptide sequence.
TRIMETYLATED LYS4 IS ENCLOSED BY AN AROMATIC CAGE
The trimethylated Lys4 of the histone H3K4me3 peptide lies in a groove composed of two aromatic residues, Y212 and W235, a hydrophobic M223 residue, and S219 of the ING1 PHD finger (Fig. 2D). Both the aromatic and hydrophobic residues are required for the histone binding since substitution of either residue abolishes this interaction. The aromatic side chains of Y212 and W235 are positioned perpendicular to each other and to the protein surface. They form a semi-aromatic cage around the tri-methylammonium group of Lys4 and make cation-π, hydrophobic and van der Waals contacts with this group. Similar coordination of a methylated lysine residue by an aromatic cage is seen in other histone-binding modules, including chromodomains, MBT and Tudor [30-36]. Although each of these modules contains three to four aromatic residues in the cage instead of two as the ING1-5 PHD fingers, the mechanism of methyllysine recognition appears to be well conserved. Methylation of a lysine residue is known to enhance its interaction with aromatic residues, particularly Trp, due to the increased hydrophobic contacts involving the aromatic side chain and the lipophilic methyl groups and ε-CH2 of Lys [37]. Requirement for the precise coordination of methylated lysine may be the driving force behind a remarkably high conservation of Trp in the aromatic cage of such structurally unrelated modules as ING PHD fingers, chromodomains and MBT and Tudor domains.
STRUCTURAL DIFFERENCES WITHIN THE ING FAMILY OF PHD FINGERS
Comparison of the crystal structure of the ING1 PHD finger bound to the H3K4me3 peptide with that of ING2, ING4 and ING5 reveals that the overall fold of these modules and the histone-binding mechanism are highly conserved, with ING1 (PDB code 2QIC) being most closely related to ING2 (PDB code 2G6Q, rmsd of 0.3 Å over Cα atoms), followed by ING4 (2PNX, rmsd of 0.6 Å), and ING5 (3C6W, rmsd of 0.7 Å). The H3K4me3 peptide occupies analogous binding sites in all structures with only small differences seen in the coordination pattern. For example, the solvent-exposed K249 residue makes unique hydrogen bonds to Ala1 and Thr3 of the peptide in the ING2 PHD-H3K4me3 complex (2G6Q). On the other hand, the direct interaction between Arg2 and G228, observed in the ING1 PHD-H3K4me3 complex (2QIL), is absent in the ING2 complex.
Currently there is no three dimensional structure of the ING3 PHD finger bound to the histone peptide deposited in PDB, however the apo- form of ING3 (1X4I) superimposes well with the ligand-free states of other ING PHD fingers [rmsd = 1.3 Å between ING3 (1X4I) and ING2 (1WES), and rmsd = 2.0 Å between ING3 (1X4I) and ING4 (1WEU)]. In the ING4 PHD-H3K4me3 complex (2PNX), the side chains of Arg2 and Gln5 of the peptide are displaced by 4.5 Å and 2 Å, respectively, in comparison to how these residues are bound by the ING2 PHD finger. While both the ING1 and ING2 PHD fingers coordinate Arg2 through a glutamate side chain (E234 in ING1, and E237 in ING2), this interaction is not observed in the ING4 and ING5 complexes. In addition, the ING4 PHD-H3K4me3 complex has fewer hydrogen bond contacts than the other ING complexes, possibly accounting for a slight decrease in binding affinity of the ING4 PHD finger (Table 1).
The crystal structure of a longer construct of the ING4 PHD finger in complex with the H3K4me3 peptide (PDB code 2VNF) demonstrates that the C-terminal Arg8, Lys9 and Ser10 residues of the histone peptide could also contribute to the binding [28]. In this structure the H3K4me3 peptide is kinked at Ala7, allowing Arg8 and Lys9 to make a cation-π interaction with W206 of the protein, while Ser10 forms hydrogen bonds to S205 and E195. Both crystal structures of the H3K4me3-bound ING4 PHD finger (2PNX and 2VNF) are similar to the structure of the ligand-free protein (1WEU, rmsds of 2.0 Å and 1.8 Å, respectively), indicating that binding to the histone peptide causes no significant conformational change in the PHD finger. It has been suggested that the extent of methylation at Lys4 in a 15-residue histone H3 peptide does not alter the binding affinity of the ING4 PHD finger [26]. Conversely, two separate reports have demonstrated that as in the case of other ING PHD modules, affinity of ING4 for the 10- and 12-residue H3 tails increases concomitantly with the number of methyl groups attached to Lys4 [21, 28] (Table 1).
OTHER KNOWN PHD FINGERS
According to the SMART database there are 132 Homo sapiens, 17 Saccharomyces cerevisiae, 49 Drosophila melanogaster, 33 Caenorhabditis elegans, and 217 Arabidopsis thaliana proteins that contain PHD fingers. This zinc-coordinating motif shows topological similarities to the RING [38] and FYVE [39, 40] domains and is commonly found in nuclear proteins that regulate transcription and chromatin remodeling. Although the PHD module was identified fifteen years ago [41], its biological role became clear only recently. In 2004 it was demonstrated that the PHD finger of transcriptional cofactor p300 localizes to nucleosomes and has a function related to chromatin binding [42], whereas the ACF1 PHD fingers were found to associate with the central domain of core histones [43]. Two years later, the PHD fingers of ING2 and BPTF were shown to recognize posttranslational modifications on histone tails, particularly H3K4me3 [20, 25, 44, 45]. Since then it has been demonstrated that many other PHD fingers bind H3K4me3, including all members of the ING family of tumor suppressors, their yeast homologues YNG1, YNG2 and PHO23, and the PHD fingers of RAG2, TAF3 and Pygopus (Pygo) [46-54].
Recent studies suggest that the large family of PHD fingers can be divided into at least four subsets based on the specificity of this domain toward post-translational histone modifications. Besides the H3K4me3-recognizing PHD fingers, a distinct subset comprising the PHD modules of BHC80, AIRE and the cysteine-rich domain of DNMT3L binds unmodified H3, or H3K4me0 [55-57], while two other subsets are specific for H3K9me3 and H3K36me3 [51, 58-60].
A structural comparison of the ING1 PHD finger with other known H3K4me3-binding proteins shows that it is most closely related to the PHD domain of TAF3, a subunit of the basal transcription factor TFIID (an rmsd = 2.1 Å between ING1 (PDB code 2QIC) and TAF3 (PDB code 2K17)) (Fig. 2E). The TAF3 PHD finger has an additional double-stranded anti-parallel beta sheet upstream of the beta strand corresponding to β1 of ING1, and a longer α2 helix. Unlike ING1 and TAF3, the PHD fingers of BPTF (PDB code 2FUU), Pygo (PDB code 2DX8), and RAG2 (PDB code 2V89) contain a third alpha helix in loop 3. A large insertion in loop 1 is also seen in RAG2, while BPTF has the longest C-terminal α-helix.
The histone-binding activity is conserved among this subset of PHD fingers. Like the PHD domains of ING1-5, the BPTF, Pygo and RAG2 PHD fingers recognize H3K4me2/3 peptides with low μM affinities [25, 44, 49, 50, 53] (Tables 1 and 2). The PHD finger of TAF3 binds about ten-fold more tightly to H3K4me3 than other PHD fingers, exhibiting a Kd of 160-310 nM [52, 54] (Table 2). While asymmetric di-methylation of H3R2 inhibits binding of the TAF3 PHD finger to H3K4me3, acetylation of H3K9 and H3K14 enhances it [52, 54]. Interestingly, all of these PHD fingers prefer trimethylated H3K4 peptides over dimethylated, monomethylated or unmodified, except for Pygo, which binds equally to H3K4me2 and H3K4me3 [53]. The high selectivity of Pygo for both tri- and dimethylated lysine is due to D352 that forms one of the walls in the aromatic cage of the PHD finger. The carboxylic side chain of D352 makes an additional hydrogen bond with the di-methylammonium proton of Lys4, consequently increasing the binding affinity toward H3K4me2 up to the level of the interaction with H3K4me3.
Table 2.
Binding Affinities of other Known H3K4me2/3-Recognizing PHD Fingers
| Protein | Peptide | Kd ± s.d. (μM) | Method | Reference |
|---|---|---|---|---|
| BPTF | H3K4me3 | 2.7 | ITC | Li, H. et al., 2006 |
| H3K4me2 | 5.0 | |||
| Pygo | H3K4me3 | 2.5 ± 0.2 | ITC | Fiedler, M. et al., 2008 |
| H3K4me2 | 2.4 ± 0.4 | |||
| H3K4me1 | 9.0 ± 0.4 | |||
| H3K4me0 | no binding | |||
| H3R2me2a/K4me2 | 2.1 ± 0.1 | |||
| RAG2 | H3K4me3 | 4 | Fluorescence | Matthews, A.G.W. et al., 2007 |
| H3K4me2 | 60 | |||
| H3K4me1 | 120 | |||
| H3K4me0 | 500 | |||
| H3K4me3 | 33.8 ± 1.9 | Fluorescence | Ramon-Maiques, S. et al.,2007 | |
| H3K4me2 | 173.2 ± 25.1 | |||
| H3R2me2s/K4me3 | 25.1 ± 3.5 | |||
| H3R2me2a/K4me3 | 34.6 ± 5.0 | |||
| TAF3 | H3K4me3 | 0.16 | Fluorescence | Vermeulen, M. et al., 2007 |
| H3K4me2 | 1.7 | |||
| H3K4me1 | >200 | |||
| H3K4me0 | >200 | |||
| H3K4me3 | 0.31 ± 0.09 | Fluorescence | van Ingen, H., 2008 | |
| BHC80 | H3K4me2 | no binding | ITC | Lan, F. et al., 2007 |
| H3K4me1 | 459 105 | |||
| H3K4me0 | 33 6.4 | |||
| DMNT3L | H3K4me3 | >500 | Fluorescence | Ooi, S.K.T. et al., 2007 |
| H3K4me2 | >500 | |||
| H3K4me1 | 36.5 ± 3.5 | |||
| H3K4me0 | 2.1 ± 0.5 | |||
| AIRE | H3K4me2 | >500 | Fluorescence | Org, T. et al., 2008 |
| H3K4me1 | 21.4 ± 5.9 | |||
| H3K4me0 | 4.7 ± 0.8 | |||
| H3K4me2 | 714 ± 90 | |||
| H3K4me1 | 55.6 ± 1.2 | ITC | Org, T. et al., 2008 | |
| H3K4me0 | 6.5 ± 0.2 | |||
Selection for the lower methylated state of lysine seems to often correlate with the presence of an acidic residue in the aromatic cage. Indeed, the engineered PHD finger of BPTF, in which a tyrosine in the aromatic cage is replaced with glutamate, shows a preference for dimethylated over trimethylated H3K4, and has an increased affinity for H3K4me1 [35]. On the other hand, the TAF3 PHD finger also contains an acidic residue, D877, in the aromatic cage, nevertheless it selects for the tri-methylammonium group over di-methylammonium, exhibiting a ~5-fold higher affinity for H3K4me3 [54]. As in the case of ING1-5, Pygo and TAF3, the semi-aromatic cage of the RAG2 PHD finger contains two aromatic residues, but this cage is entirely aromatic in BPTF, consisting of four Tyr/Trp residues. Unlike TAF3, the binding affinity of the RAG2 PHD finger for the histone tail is slightly increased in the presence of a double modification, H3R2me2/K4me3 [50]. However, the Pygo, BPTF and ING2 PHD fingers appear to be insensitive to methylation of H3R2 [50, 52, 53] (Table 2). Most likely, replacement of the carboxylic D230 and E237 residues, which tightly coordinate H3R2 in the ING2 PHD complex, with a tyrosine residue (Y445) in the PHD finger of RAG2 results in a more favorable interaction with methylated arginine.
A newly identified subset of PHD fingers, including those of AIRE and BHC80 and a PHD-like cysteine-rich domain of DNMT3L, binds to the unmodified histone H3 tail (H3K4me0) [55-57]. The specificity of BHC80 is determined through the recognition of Arg2, Lys4 and Arg8 residues of the histone tail [55]. The side chain of M502 of the BHC80 PHD finger is placed between Arg2 and Lys4 of the peptide, while D489 inserts between Lys4 and Arg8 and makes an electrostatic bridge. The amino group of Lys4 is further restrained by contacting the side chain of H487 and forming a hydrogen bond to the carbonyl group of E488. Mono-methylation of Lys4 causes a 15-fold decrease in affinity of BHC80 PHD, and no binding is detected to the histone tail carrying the dimethylated mark (Table 2). It was suggested that a second or a third methyl group would clash with the side chains of D489 and H486 and/or the carbonyl group of E488, preventing the interaction with the histone tail [55].
Close comparison of AIRE, BHC80 and DNMT3L reveals that all three proteins contain an aspartic acid in the same position as D489 of BHC80, indicating that the H3K4me0-binding mechanism is conserved within this subset of PHD fingers. Interestingly, the AIRE PHD finger and the DNMT3L PHD-like module are both unable to bind histone peptides di- or trimethylated at Lys4, and are insensitive to other modifications such as methylation of H3K9 [56, 57] (Table 2). Thus, the substrate specificity of the AIRE and BHC80 PHD fingers and the PHD-like domain of DNMT3L is driven by the formation of a hydrogen bond to the epsilon amino group of Lys4 and by steric exclusion of bulky methyl groups, rather than the formation of an aromatic/hydrophobic cage, which provides essential cation-π and hydrophobic interactions in the PHD-H3K4me3 complexes.
The PHD fingers of SMCX, ICBP90 and Lid2 have recently been shown to recognize the histone H3K9me3 mark [58-60]. Like H3K4me3, H3K9me3 is important for regulation of gene expression, however this modification is enriched in transcriptionally inactive regions of the genome, or heterochromatin, whereas H3K4me3 is usually associated with euchromatin. SMCX is an enzyme that demethylates H3K4me3, producing di- and monomethylated species, but not H3K4me0, and is a target gene for X-linked mental retardation (XLMR) [61]. The A388P mutation of SMCX found in XLMR patients reduces binding of its N-terminal PHD1 finger to H3K9me3 by 60% [58]. The ICBP90 protein targets heterochromatic regions in interphase nuclei via its PHD and SET- and RING-associated (SRA) domains that interact with H3K9me3 [59]. ICBP90 is required for higher order chromatin organization and may be involved in herterochromatin maintenance.
The S. pombe JmjC-domain protein Lid2 is a tri-methyl H3K4 demethylase responsible for H3K4 hypomethylation, and is also necessary for the coordination of H3K4 and H3K9 methylation in heterochromatin [60]. Lid2 contains three PHD domains, with the second PHD2 finger shown to bind H3K9me3. Apparently, Lid2 is involved in two opposing activities, i.e. it cooperates with Clr4 to demethylate H3K4 and methylate H3K9 in heterochromatin. On the other hand, Lid2 functions as a structural link between Set1 and Lsd1 to ensure the H3K4 hypermethylation and H3K9 hypomethylation of active genes in euchromatin. This example nicely illustrates how the methylation patterns of the H3K4 and H3K9 can be tightly cross-regulated [60]. Currently, no three-dimensional structure of any PHD finger bound to H3K9me3 is available. It will be interesting to learn how the coordination of this epigenetic mark differs from that of H3K4me3 and H3K4me0. The structural information will also be critical for better understanding of how key histone modifications regulate the diverse functions of PHD finger-containing proteins.
BIOLOGICAL ROLES OF PHD FINGER-CONTAINING PROTEINS
Expression analyses of multiple tumor types show that the ING1-5 genes are often down-regulated, up-regulated, or mutated in human malignancies including melanoma, leukemia, brain, breast and gastrointestinal cancers [62]. These facts suggest a correlation between the oncogenic activities of the ING1-5 tumor suppressors and their mechanistic roles in the regulation of gene transcription, DNA repair and recombination, chromatin remodeling, and apoptosis [2-14, 16-21]. The human ING proteins can be divided into three groups based on their association with three distinct enzymatic complexes. The first group is composed of ING1 and ING2 which associate primarily with the Sin3A/HDAC1/2 repressive histone deacetylase complexes [16, 17, 19]. ING3 is a component of the NuA4/Tip60 HAT complex and is required for acetylation of chromatin substrates including the N-terminal tails of histones H4 and H2A [18, 19]. ING4 and ING5 are subunits of HAT complexes containing MOZ (monocytic leukemia zinc finger protein)/MORF (Moz-related factor) and HBO1 [19].
ING1 mediates the cellular response to genotoxic stresses and is critical for maintaining the genomic stability of the cell. Depending on the severity of the DNA damage, ING1 either promotes cell cycle arrest followed by DNA damage repair, or stimulates apoptosis if the damage cannot be repaired. While the PHD finger of ING1 binds H3K4me3, its N-terminal SAID domain associates with the Sin3A/HDAC1/2 repressor complexes. Human Sin3A/HDAC1/2 complexes deacetylate nucleosomes near Sin3-regulated promoters causing silencing of gene expression. These multisubunit complexes are comprised of a catalytic core, and 8 to 10 tightly associated subunit proteins including SAP30 and SAP18. The SAP30 subunit interacts with a number of effectors including ING1 [16, 17]. The ING1 SAID domain-SAP30 interaction may bridge or stabilize the Sin3A/HDAC1/2 complexes at the nucleosomes for subsequent deacetylation of acetylated lysine residues of histone tails, leading to transcriptional repression. Accordingly, ING1 links chromatin remodeling and gene expression with its DNA repair, apoptotic and tumor suppressor functions.
ING2 is also a component of the Sin3A/HDAC1/2 repressive complexes that negatively regulates cell proliferation through modulation of p53 acetylation [62]. The PHD domain of ING2 binds H3K4me3, directing HDAC1/2 complexes to actively transcribed genes, which may be essential for rapid suppression of gene transcription [20, 25]. The carboxy-terminal polybasic region of ING2 is able to interact with nuclear phosphoinositides, such as phosphatidylinositol 5-phosphate [PtdIns(5)P] [8]. It was demonstrated that ING2-mediated activation of p53 and p53-dependent apoptosis requires binding of PtdIns(5)P. Thus, the multiple functionality of the C-terminus of ING2 integrates various nuclear signaling pathways, including chromatin remodeling, DNA damage repair, cell death and tumor development and progression.
ING3 has been identified as a native subunit of the human NuA4/Tip60 HAT complex [18, 19]. This twelve-component acetyltransferase complex is responsible for acetylation of the N-terminal histone H4 and H2A tails. In addition to Tip60, which contains the catalytic MYST domain, the hNuA4 complex includes Eaf6, the enhancer of polycomb homology domain 1 (EPC1), and the ING3 subunit among others. ING3 plays a key role in regulating the enzymatic activity of NuA4/Tip60. It bridges the HAT complex to chromatin through concurrently associating with the EPC1 subunit and H3K4me3 through its N-terminal region and the C-terminal PHD finger, respectively. Similarly to ING1 and ING2, the ING3 tumor suppressor is also thought to cooperate with p53 in transcription regulation, cell cycle control and apoptosis [18, 19].
ING4 is a stable subunit of the HBO1 HAT complex and is necessary for acetyltransferase activity on free histones and chromatin [19]. Although the PHD finger of ING4 is not essential for HBO1 acetylation of histone H4, its recognition of H3K4me3 promotes acetylation of histone H3 [21]. It has been suggested that the ING4 PHD finger serves to sense the methylation state of H3K4 for re-directing the HBO1 HAT activity from H4 toward H3. Furthermore, the ability of ING4 to bind H3K4me3 can regulate gene expression in response to DNA damage. Mediating crosstalk between H3K4 methylation and H3 acetylation, ING4 may be responsible for transducing critical tumor suppressor signals [21].
ING5 was previously shown to co-purify with both the MOZ/MORF and HBO1 HAT complexes which acetylate histone H3 at Lys14, and histone H4 at Lys5, Lys8, and Lys12, respectively [19]. The MOZ/MORF complex is composed of the MOZ/MORF catalytic subunit, and the ING5, BRPF, and hEaf6 effector subunits. HAT assays demonstrate that the presence of full-length ING5 and BRPF results in preferential acetylation of methylated H3K4 peptides [27]. This suggests that ING5 acts as an adapter molecule which tethers and/or stabilizes the MOZ/MORF HAT complex at chromatin via concomitant binding to methylated H3K4 and BRPF.
In addition to the ING family, a number of other PHD finger-containing proteins have been identified that bind H3K4me3 and link this epigenetic mark to diverse biological outcomes. For example, TAF3 acts as a transcriptional co-activator, anchoring transcription factor TFIID in a PHD finger-dependent manner to gene promoters, which could stimulate subsequent rounds of transcription [52]. Furthermore, the crosstalk between histone modifications and functioning of TFIID (dimethylation of H3R2 selectively inhibits TFIID/TAF3 association with H3K4me3, while H3K9 and H3K14 acetylation augments it) may play an important role in regulation of RNA polymerase II-mediated transcription [52]. The BPTF PHD finger stabilizes the ATP-dependent chromatin remodeling complex NURF at the chromatin, resulting in NURF-catalyzed nucleosome sliding and activation of HOX genes [44]. Decoding the methylation state of histone H3 by Pygo is critical for controlling Wnt-induced transcription [53]. To target histones efficiently the Pygo PHD finger associates with the HD1 domain of BCL9. Binding of the Pygo/BCL9 complex to dimethylated H3R2 could expose this mark for demethylation prior to the recruitment of SET1 which converts H3K4me2 into H3K4me3. This would allow a full transcriptional activation of TCF target genes during Wnt signaling [53]. Recognition of H3K4me3 by the RAG2 PHD finger is essential for the function of the RAG1/2 V(D)J recombinase, which recognizes and cleaves the recombination signal sequences [63]. The W453R mutation in RAG2 is found in patients with a severe combined immunodeficiency disease known as Omenn’s syndrome. W453 is one of the ‘aromatic cage’ residues required for the coordination of the tri-methylammonium group of Lys4. Substitution of this residue disrupts the integrity of the aromatic cage, abolishing the RAG2 interaction with H3K4me3 [49, 50]. These examples highlight the pivotal role of recognition of the H3K4me epigenetic mark by protein effectors in cell physiology and prevention of pathologic states.
Acknowledgments
This work is supported by the NIH grants CA113472 and GM071424, Cancer League of Colorado and the University of Colorado Cancer Center. K.S.C. is an NIH NRSA postdoctoral Fellow (F32 GM083462).
References
- 1.Garkavtsev I, Kazarov A, Gudkov A, Riabowol K. Nat Genet. 1996;14(4):415–420. doi: 10.1038/ng1296-415. [DOI] [PubMed] [Google Scholar]
- 2.Nagashima M, Shiseki M, Miura K, Hagiwara K, Linke SP, Pedeux R, Wang XW, Yokota J, Riabowol K, Harris CC. Proc Natl Acad Sci USA. 2001;98(17):9671–9676. doi: 10.1073/pnas.161151798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gunduz M, Ouchida M, Fukushima K, Ito S, Jitsumori Y, Nakashima T, Nagai N, Nishizaki K, Shimizu K. Oncogene. 2002;21(28):4462–4470. doi: 10.1038/sj.onc.1205540. [DOI] [PubMed] [Google Scholar]
- 4.Nagashima M, Shiseki M, Pedeux RM, Okamura S, Kitahama-Shiseki M, Miura K, Yokota J, Harris CC. Oncogene. 2003;22(3):343–350. doi: 10.1038/sj.onc.1206115. [DOI] [PubMed] [Google Scholar]
- 5.Shiseki M, Nagashima M, Pedeux RM, Kitahama-Shiseki M, Miura K, Okamura S, Onogi H, Higashimoto Y, Appella E, Yokota J, Harris CC. Cancer Res. 2003;63(10):2373–2378. [PubMed] [Google Scholar]
- 6.Garkavtsev I, Grigorian IA, Ossovskaya VS, Chernov MV, Chumakov PM, Gudkov AV. Nature. 1998;391(6664):295–298. doi: 10.1038/34675. [DOI] [PubMed] [Google Scholar]
- 7.Zeremski M, Hill JE, Kwek SS, Grigorian IA, Gurova KV, Garkavtsev IV, Diatchenko L, Koonin EV, Gudkov AV. J Biol Chem. 1999;274(45):32172–32181. doi: 10.1074/jbc.274.45.32172. [DOI] [PubMed] [Google Scholar]
- 8.Gozani O, Karuman P, Jones DR, Ivanov D, Cha J, Lugovskoy AA, Baird CL, Zhu H, Field SJ, Lessnick SL, Villasenor J, Mehrotra B, Chen J, Rao VR, Brugge JS, Ferguson CG, Payrastre B, Myszka DG, Cantley LC, Wagner G, Divecha N, Prestwich GD, Yuan J. Cell. 2003;114(1):99–111. doi: 10.1016/s0092-8674(03)00480-x. [DOI] [PubMed] [Google Scholar]
- 9.Pedeux R, Sengupta S, Shen JC, Demidov ON, Saito S, Onogi H, Kumamoto K, Wincovitch S, Garfield SH, McMenamin M, Nagashima M, Grossman SR, Appella E, Harris CC. Mol Cell Biol. 2005;25(15):6639–6648. doi: 10.1128/MCB.25.15.6639-6648.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Abad M, Menendez C, Fuchtbauer A, Serrano M, Fuchtbauer EM, Palmero I. J Biol Chem. 2007;282(42):31060–31067. doi: 10.1074/jbc.M701639200. [DOI] [PubMed] [Google Scholar]
- 11.Helbing CC, Veillette C, Riabowol K, Johnston RN, Garkavtsev I. Cancer Res. 1997;57(7):1255–1258. [PubMed] [Google Scholar]
- 12.Scott M, Boisvert FM, Vieyra D, Johnston RN, Bazett-Jones DP, Riabowol K. Nucleic Acids Res. 2001;29(10):2052–2058. doi: 10.1093/nar/29.10.2052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wang Y, Li G. J Biol Chem. 2006;281(17):11887–11893. doi: 10.1074/jbc.M511309200. [DOI] [PubMed] [Google Scholar]
- 14.Cheung KJ, Jr, Mitchell D, Lin, Li G. Cancer Res. 2001;61(13):4974–4977. [PubMed] [Google Scholar]
- 15.Kichina JV, Zeremski M, Aris L, Gurova KV, Walker E, Franks R, Nikitin AY, Kiyokawa H, Gudkov AV. Oncogene. 2006;25(6):857–866. doi: 10.1038/sj.onc.1209118. [DOI] [PubMed] [Google Scholar]
- 16.Skowyra D, Zeremski M, Neznanov N, Li M, Choi Y, Uesugi M, Hauser CA, Gu W, Gudkov AV, Qin J. J Biol Chem. 2001;276(12):8734–8739. doi: 10.1074/jbc.M007664200. [DOI] [PubMed] [Google Scholar]
- 17.Kuzmichev A, Zhang Y, Erdjument-Bromage H, Tempst P, Reinberg D. Mol Cell Biol. 2002;22(3):835–848. doi: 10.1128/MCB.22.3.835-848.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Doyon Y, Selleck W, Lane WS, Tan S, Cote J. Mol Cell Biol. 2004;24(5):1884–1896. doi: 10.1128/MCB.24.5.1884-1896.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Doyon Y, Cayrou C, Ullah M, Landry AJ, Cote V, Selleck W, Lane WS, Tan S, Yang XJ, Cote J. Mol Cell. 2006;21(1):51–64. doi: 10.1016/j.molcel.2005.12.007. [DOI] [PubMed] [Google Scholar]
- 20.Shi X, Hong T, Walter KL, Ewalt M, Michishita E, Hung T, Carney D, Pena P, Lan F, Kaadige MR, Lacoste N, Cayrou C, Davrazou F, Saha A, Cairns BR, Ayer DE, Kutateladze TG, Shi Y, Cote J, Chua KF, Gozani O. Nature. 2006;442(7098):96–99. doi: 10.1038/nature04835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hung T, Binda O, Champagne KS, Kuo AJ, Johnson K, Chang H, Simon M, Kutateladze TG, Gozani O. Mol Cell. 2009 doi: 10.1016/j.molcel.2008.12.016. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.He GH, Helbing CC, Wagner MJ, Sensen CW, Riabowol K. Mol Biol Evol. 2005;22(1):104–116. doi: 10.1093/molbev/msh256. [DOI] [PubMed] [Google Scholar]
- 23.Han X, Feng X, Rattner JB, Smith H, Bose P, Suzuki K, Soliman MA, Scott MS, Burke BE, Riabowol K. Nat Cell Biol. 2008;10(11):1333–1340. doi: 10.1038/ncb1792. [DOI] [PubMed] [Google Scholar]
- 24.Gordon PM, Soliman MA, Bose P, Trinh Q, Sensen CW, Riabowol K. BMC Genom. 2008;9:426. doi: 10.1186/1471-2164-9-426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Peña PV, Davrazou F, Shi X, Walter KL, Verkhusha VV, Gozani O, Zhao R, Kutateladze TG. Nature. 2006;442(7098):100–103. doi: 10.1038/nature04814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Palacios A, Garcia P, Padro D, Lopez-Hernandez E, Martin I, Blanco FJ. FEBS Lett. 2006;580(30):6903–6908. doi: 10.1016/j.febslet.2006.11.055. [DOI] [PubMed] [Google Scholar]
- 27.Champagne KS, Saksouk N, Pena PV, Johnson K, Ullah M, Yang XJ, Cote J, Kutateladze TG. Proteins. 2008;72(4):1371–1376. doi: 10.1002/prot.22140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Palacios A, Munoz IG, Pantoja-Uceda D, Marcaida MJ, Torres D, Martin-Garcia JM, Luque I, Montoya G, Blanco FJ. J Biol Chem. 2008;283(23):15956–15964. doi: 10.1074/jbc.M710020200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Peña PV, Hom RA, Hung T, Lin H, Kuo AJ, Wong RP, Subach OM, Champagne KS, Zhao R, Verkhusha VV, Li G, Gozani O, Kutateladze TG. J Mol Biol. 2008;380:303–312. doi: 10.1016/j.jmb.2008.04.061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jacobs SA, Khorasanizadeh S. Science. 2002;295(5562):2080–2083. doi: 10.1126/science.1069473. [DOI] [PubMed] [Google Scholar]
- 31.Nielsen PR, Nietlispach D, Mott HR, Callaghan J, Bannister A, Kouzarides T, Murzin AG, Murzina NV, Laue ED. Nature. 2002;416(6876):103–107. doi: 10.1038/nature722. [DOI] [PubMed] [Google Scholar]
- 32.Fischle W, Wang Y, Jacobs SA, Kim Y, Allis CD, Khorasanizadeh S. Genes Dev. 2003;17(15):1870–1881. doi: 10.1101/gad.1110503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Huang Y, Fang J, Bedford MT, Zhang Y, Xu RM. Science. 2006;312(5774):748–751. doi: 10.1126/science.1125162. [DOI] [PubMed] [Google Scholar]
- 34.Botuyan MV, Lee J, Ward IM, Kim JE, Thompson JR, Chen J, Mer G. Cell. 2006;127(7):1361–1373. doi: 10.1016/j.cell.2006.10.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Li H, Fischle W, Wang W, Duncan EM, Liang L, Murakami-Ishibe S, Allis CD, Patel DJ. Mol Cell. 2007;28(4):677–691. doi: 10.1016/j.molcel.2007.10.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Min J, Allali-Hassani A, Nady N, Qi C, Ouyang H, Liu Y, MacKenzie F, Vedadi M, Arrowsmith CH. Nat Struct Mol Biol. 2007;14(12):1229–1230. doi: 10.1038/nsmb1340. [DOI] [PubMed] [Google Scholar]
- 37.Hughes RM, Wiggins KR, Khorasanizadeh S, Waters ML. Proc Natl Acad Sci U S A. 2007;104(27):11184–11188. doi: 10.1073/pnas.0610850104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Barlow PN, Luisi B, Milner A, Elliott M, Everett R. J Mol Biol. 1994;237(2):201–211. doi: 10.1006/jmbi.1994.1222. [DOI] [PubMed] [Google Scholar]
- 39.Misra S, Hurley JH. Cell. 1999;97(5):657–666. doi: 10.1016/s0092-8674(00)80776-x. [DOI] [PubMed] [Google Scholar]
- 40.Kutateladze TG, Overduin M. Science. 2001;291:1793–1796. doi: 10.1126/science.291.5509.1793. [DOI] [PubMed] [Google Scholar]
- 41.Schindler U, Beckmann H, Cashmore AR, Schindler TF. Plant J. 1993;4(1):137–150. doi: 10.1046/j.1365-313x.1993.04010137.x. [DOI] [PubMed] [Google Scholar]
- 42.Ragvin A, Valvatne H, Erdal S, Arskog V, Tufteland KR, Breen K, ØYan AM, Eberharter A, Gibson TJ, Becker PB, Aasland R. J Mol Biol. 2004;337(4):773–788. doi: 10.1016/j.jmb.2004.01.051. [DOI] [PubMed] [Google Scholar]
- 43.Eberharter A, Vetter I, Ferreira R, Becker PB. EMBO J. 2004;23(20):4029–4039. doi: 10.1038/sj.emboj.7600382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Li H, Ilin S, Wang W, Duncan EM, Wysocka J, Allis CD, Patel DJ. Nature. 2006;442(7098):91–95. doi: 10.1038/nature04802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wysocka J, Swigut T, Xiao H, Milne TA, Kwon SY, Landry J, Kauer M, Tackett AJ, Chait BT, Badenhorst P, Wu C, Allis CD. Nature. 2006;442(7098):86–90. doi: 10.1038/nature04815. [DOI] [PubMed] [Google Scholar]
- 46.Martin DG, Baetz K, Shi X, Walter KL, MacDonald VE, Wlodarski MJ, Gozani O, Hieter P, Howe L. Mol Cell Biol. 2006;26(21):7871–7879. doi: 10.1128/MCB.00573-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Taverna SD, Ilin S, Rogers RS, Tanny JC, Lavender H, Li H, Baker L, Boyle J, Blair LP, Chait BT, Patel DJ, Aitchison JD, Tackett AJ, Allis CD. Mol Cell. 2006;24(5):785–796. doi: 10.1016/j.molcel.2006.10.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Liu Y, Subrahmanyam R, Chakraborty T, Sen R, Desiderio S. Immunity. 2007;27(4):561–571. doi: 10.1016/j.immuni.2007.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Matthews AG, Kuo AJ, Ramon-Maiques S, Han S, Champagne KS, Ivanov D, Gallardo M, Carney D, Cheung P, Ciccone DN, Walter KL, Utz PJ, Shi Y, Kutateladze TG, Yang W, Gozani O, Oettinger MA. Nature. 2007;450(7172):1106–1110. doi: 10.1038/nature06431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ramon-Maiques S, Kuo AJ, Carney D, Matthews AG, Oettinger MA, Gozani O, Yang W. Proc Natl Acad Sci U S A. 2007;104(48):18993–18998. doi: 10.1073/pnas.0709170104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Shi X, Kachirskaia I, Walter KL, Kuo JH, Lake A, Davrazou F, Chan SM, Martin DG, Fingerman IM, Briggs SD, Howe L, Utz PJ, Kutateladze TG, Lugovskoy AA, Bedford MT, Gozani O. J Biol Chem. 2007;282(4):2450–2455. doi: 10.1074/jbc.C600286200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Vermeulen M, Mulder KW, Denissov S, Pijnappel WW, van Schaik FM, Varier RA, Baltissen MP, Stunnenberg HG, Mann M, Timmers HT. Cell. 2007;131(1):58–69. doi: 10.1016/j.cell.2007.08.016. [DOI] [PubMed] [Google Scholar]
- 53.Fiedler M, Sanchez-Barrena MJ, Nekrasov M, Mieszczanek J, Rybin V, Muller J, Evans P, Bienz M. Mol Cell. 2008;30(4):507–518. doi: 10.1016/j.molcel.2008.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.van Ingen H, van Schaik FM, Wienk H, Ballering J, Rehmann H, Dechesne AC, Kruijzer JA, Liskamp RM, Timmers HT, Boelens R. Structure. 2008;16(8):1245–1256. doi: 10.1016/j.str.2008.04.015. [DOI] [PubMed] [Google Scholar]
- 55.Lan F, Collins RE, De Cegli R, Alpatov R, Horton JR, Shi X, Gozani O, Cheng X, Shi Y. Nature. 2007;448(7154):718–722. doi: 10.1038/nature06034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ooi SK, Qiu C, Bernstein E, Li K, Jia D, Yang Z, Erdjument-Bromage H, Tempst P, Lin SP, Allis CD, Cheng X, Bestor TH. Nature. 2007;448(7154):714–717. doi: 10.1038/nature05987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Org T, Chignola F, Hetenyi C, Gaetani M, Rebane A, Liiv I, Maran U, Mollica L, Bottomley MJ, Musco G, Peterson P. EMBO Rep. 2008;9(4):370–376. doi: 10.1038/embor.2008.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Iwase S, Lan F, Bayliss P, de la Torre-Ubieta L, Huarte M, Qi HH, Whetstine JR, Bonni A, Roberts TM, Shi Y. Cell. 2007;128(6):1077–1088. doi: 10.1016/j.cell.2007.02.017. [DOI] [PubMed] [Google Scholar]
- 59.Karagianni P, Amazit L, Qin J, Wong J. Mol Cell Biol. 2008;28(2):705–717. doi: 10.1128/MCB.01598-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Li F, Huarte M, Zaratiegui M, Vaughn MW, Shi Y, Martienssen R, Cande WZ. Cell. 2008;135(2):272–283. doi: 10.1016/j.cell.2008.08.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Jensen LR, Amende M, Gurok U, Moser B, Gimmel V, Tzschach A, Janecke AR, Tariverdian G, Chelly J, Fryns JP, Van Esch H, Kleefstra T, Hamel B, Moraine C, Gecz J, Turner G, Reinhardt R, Kalscheuer VM, Ropers HH, Lenzner S. Am J Hum Genet. 2005;76(2):227–236. doi: 10.1086/427563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Campos EI, Chin MY, Kuo WH, Li G. Cell Mol Life Sci. 2004;61(1920):2597–2613. doi: 10.1007/s00018-004-4199-4. [DOI] [PubMed] [Google Scholar]
- 63.Gellert M. Annu Rev Biochem. 2002;71:101–132. doi: 10.1146/annurev.biochem.71.090501.150203. [DOI] [PubMed] [Google Scholar]
- 64.Armougom F, Moretti S, Poirot O, Audic S, Dumas P, Schaeli B, Keduas V, Notredame C. Nucleic Acids Res. 2006;34(Web Server issue):W604–608. doi: 10.1093/nar/gkl092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kabsch W, Sander C. Biopolymers. 1983;22(12):2577–2637. doi: 10.1002/bip.360221211. [DOI] [PubMed] [Google Scholar]
- 66.Guex N, Peitsch MC. Electrophoresis. 1997;18(15):2714–2723. doi: 10.1002/elps.1150181505. [DOI] [PubMed] [Google Scholar]