

Abstract
Merkel cell carcinoma (MCC) is a rare but aggressive human skin cancer that typically affects elderly and immunosuppressed individuals, a feature suggestive of an infectious origin. We studied MCC samples by digital transcriptome subtraction and detected a fusion transcript between a previously undescribed virus T antigen and a human receptor tyrosine phosphatase. Further investigation led to identification and sequence analysis of the 5387-base-pair genome of a previously unknown polyomavirus that we call Merkel cell polyomavirus (MCV or MCPyV). MCV sequences were detected in 8 of 10 (80%) MCC tumors but only 5 of 59 (8%) control tissues from various body sites and 4 of 25 (16%) control skin tissues. In six of eight MCV-positive MCCs, viral DNA was integrated within the tumor genome in a clonal pattern, suggesting that MCV infection and integration preceded clonal expansion of the tumor cells. Thus, MCV may be a contributing factor in the pathogenesis of MCC.
Polyomaviruses have been suspected as potential etiologic agents in human cancer since the discovery of murine polyoma virus (MuPyV) by Gross in 1953 (1). However, although polyomavirus infections can produce tumors in animal models, there is no conclusive evidence that they play a role in human cancers (2). These small double-stranded DNA viruses [∼5200 base pairs (bp)] encode a variably spliced oncoprotein, the tumor (T) antigen (3, 4), and are divided into three genetically distinct groups: (i) avian polyomaviruses, (ii) mammalian viruses related to MuPyV, and (iii) mammalian polyomaviruses related to simian virus 40 (SV40) (5). All four known human polyomaviruses [BK virus (BKV), JCV, KIV, and WUV (6, 7)] belong to the SV40 subgroup. In animals, integration of polyomavirus DNA into the host genome often precedes tumor formation (8).
Merkel cell carcinoma (MCC) is a neuroectodermal tumor arising from mechanoreceptor Merkel cells (Fig. 1A). MCC is rare, but its incidence has tripled over the past 2 decades in the United States to 1500 cases per year (9). It is one of the most aggressive forms of skin cancer; about 50% of advanced MCC patients live 9 months or less. Gene expression profiling studies indicate that MCC may comprise two or more clinically similar diseases with distinct etiologies (10). Like Kaposi’s sarcoma (KS), MCC occurs more frequently than expected among immunosuppressed transplant and AIDS patients (11). These similarities to KS, an immune-related tumor caused by KS-associated herpesvirus (12), raise the possibility that MCC may also have an infectious origin.
Fig. 1.
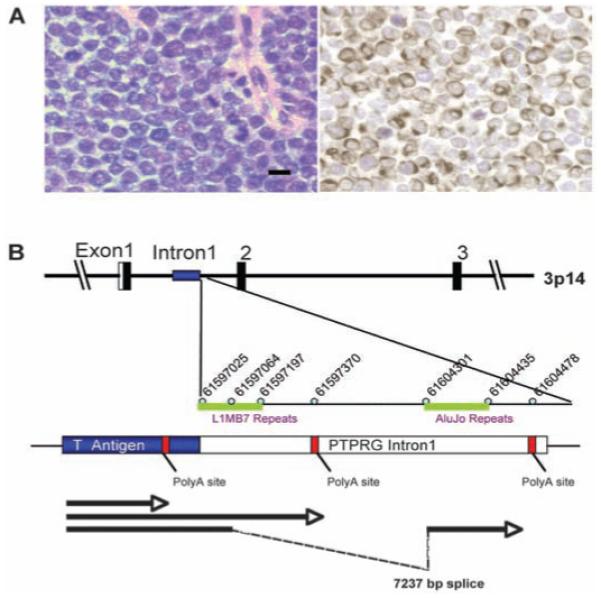
(A) MCC is an aggressive skin cancer derived from Merkel mechanoreceptor cells that expresses neuroendocrine and perinuclear cytokeratin 20 markers, distinguishing it from other small round cell tumors (MCC349, left, hematoxylin and eosin; right, cytokeratin 20 staining, 40×. Scale bar represents 10 μm). (B) Discovery of Merkel cell polyomavirus transcripts in (MCC). 3′-RACE mapping of an MCC fusion transcript between the MCV T antigen and human PTPRG. A cDNA corresponding to a polyomavirus-like T antigen transcript was found by DTS analysis of MCC. This T antigen cDNA was extended by 3′-RACE to map three mRNA sequences (arrows), one of which terminates at a viral polyadenylation site and two of which extend into flanking human sequence and terminate in intron 1 of the human PTPRG gene on chromosome 3p14, indicative of viral DNA integration into the tumor cell genome. The two viral-human chimeric transcripts were generated by read-through of a weak polyadenylation signal in the viral T antigen gene. Identical RACE products were also sequenced from a lymph node metastasis of this primary tumor.
To search for viral sequences in MCC, we used digital transcriptome subtraction (DTS), a methodology we developed that can identify foreign transcripts by using human high-throughput cDNA sequencing data (13). We generated two cDNA libraries from a total of four anonymized MCC tumors. One library was prepared with the use of mRNA from a single tumor (MCC347), and the other was prepared with mRNA pooled from three tumors (MCC337, 343, and 346) to increase the likelihood of detecting rare viral sequences (table S1).
From these two libraries, we respectively pyro-sequenced 216,599 and 179,135 cDNA sequences (∼150 to 200 bp). These 395,734 cDNA sequences were trimmed with LUCY stringency equivalent to PHRED scores of 20 or higher (14). Copolymers of adenine or thymidine [poly(A) and poly (T), respectively], dust (low-complexity), human repeat, and primer adaptor sequences were then removed, leaving 382,747 sequences to form a high-fidelity (HiFi) data set. Of these, 380,352 (99.4%) aligned to human RefSeq RNA, mitochondrial, assembled chromosomes, or immunoglobulin sequences in National Center for Biotechnology Information (NCBI) databases. Of the remaining 2395 HiFi candidate sequences, one transcript (DTS1) from MCC347 cDNA aligned with high homology to African green monkey (AGM) lymphotropic polyomavirus (LPyV) and to human BK polyomavirus T antigen sequences. A second DTS transcript (DTS2) had no homology to deposited polyomavirus sequences but was subsequently identified by aligning HiFi candidates to the full-length viral genome (see below). These two sequences define a previously unknown human polyomavirus that we call Merkel cell polyomavirus (MCV or MCPyV) because of its close association with MCC.
Rapid amplification of cDNA ends (3′-RACE) extended DTS1 to three different cDNAs (Fig. 1B): One transcript terminated at a poly(A) site in the T antigen sequence, and two cDNAs read through this weak poly(A) site to form different length fusions with intron 1 of the human receptor tyrosine phosphatase type G gene (PTPRG) (GenBank:18860897) on chromosome 3p14.2. Viral integration at this site was confirmed by sequencing DNA polymerase chain reaction (PCR) products with the use of a viral primer and a human PTPRG primer. The same three RACE products were independently cloned from MCC348, a lymph node metastasis from the MCC347 primary tumor, indicating that this tumor was seeded from a single tumor cell already positive for the T antigen-PTPRG fusion transcript.
By viral genome walking, we sequenced the complete closed circular genome of MCV (5387 bp, prototype) from tumor MCC350. A second genome, MCV339 (5201 bp), was then sequenced by using MCV-specific primers. The sequences of MCV350 and MCV339 have GenBank accession numbers EU375803 and EU375804, respectively. Both viruses encode sequences with high homology to polyomavirus T antigen, VP1, VP2/3, and replication origin sequences (Fig. 2A). MCV has an early gene expression region [196 to 3080 nucleotides (nt)] containing the T antigen locus with large T and small T open reading frames and a late gene region containing VP1 and VP2/3 open reading frames between 3156 and 5118 nt. The T antigen locus has features conserved with other polyomavirus T antigens, including cr1, DnaJ, pRB1-binding Leu-X-Cys-X-Glu (LXCXE) motif, origin-binding, and helicase/adenosine triphosphatase (ATPase) domains. Mutations in the C terminus of MCV350 and 339 large T open reading frames are predicted to truncate large T protein but are unlikely to affect small T antigen protein expression. The replication origin is highly conserved with that of other polyomaviruses and includes features such as a poly(T) tract and conserved T antigen binding boxes (fig. S1). MCV has highest homology to viruses belonging to the MuPyV subgroup and is most closely related to AGM LPyV (Fig. 2B) (15). It is more distantly related to known human polyomaviruses and SV40. The principal differences between MCV350 and MCV339 are a 191-bp (1994 to 2184 nt) deletion in the MCV339 T antigen gene and a 5-bp (5216 to 5220 nt) insertion in the MCV339 late promoter. Excluding these sites, only 41 (0.8%) nucleotides differ between MCV350 and 339.
Fig. 2.
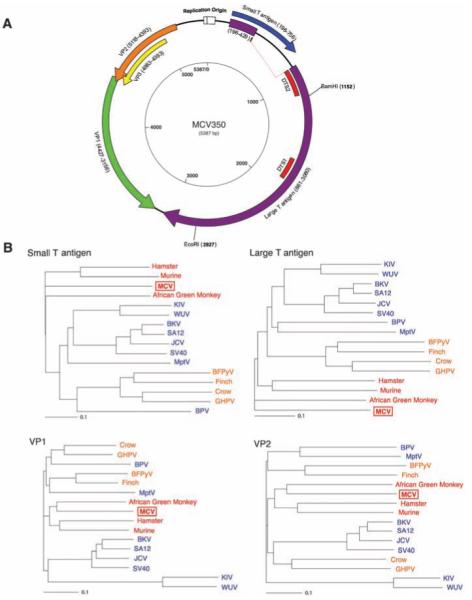
(A) Schematic of MCV genome. Genome walking was used to clone the full MCV genome from tumor MCC350. The genome encodes typical features of a polyomavirus, including large T (purple) and small T (blue) open reading frames. Also shown are predicted VP1 (green) and overlapping VP2 (orange) and VP3 (yellow) genes. DTS1 and DTS2 (red) represent cDNA fragments originally identified by DTS screening. The former was used to identify MCV, and the latter is a spliced transcript with no homology to known polyomavirus sequences. (B) Neighbor-joining trees for putative MCV large T, small T, VP1, and VP2 proteins. The four known human polyomaviruses (BKV, JCV, KIV, and WUV) cluster together in the SV40 subgroup (blue), whereas MCV is most closely related to MuPyV subgroup viruses (red). Both subgroups are distinct from the avian polyomavirus subgroup (orange). Scale bars indicate an evolutionary distance of 0.1 amino acid substitutions per position in the sequence.
To investigate the association between MCV infection and MCC, we compared tumors from 10 MCC patients to two tissue control groups. The first control group was composed of unselected tissues from various body sites (including nine skin samples) from 59 patients without MCC (table S2). These samples were taken consecutively on a single surgical day and tested for MCV positivity with two PCR primer sets in the T antigen locus (LT1 and LT3) and one in the VP1 gene (VP1). These primers do not amplify cloned human BKV or JCV genomic DNA or SV40 genome from COS-7 cells. A second control group composed of skin and skin tumor samples from 25 immunocompetent and immunosuppressed patients without MCC were tested with LT1 and VP1 primers (table S2). Samples were randomized and tested in a blinded fashion. Southern blotting of PCR products was performed to increase sensitivity (fig. S2).
Of the 10 MCC tumors from different patients, 8 (80%) were positive for MCV sequences by PCR (Table 1 and table S1). Seven tumors showed robust amplification, and one tumor was positive only after PCR-Southern hybridization. MCC348 (metastasis from MCC347) and MCC338 (infiltrating tumor from MCC339) were also positive. Two tumors, MCC343 and 346, remained negative after testing with 13 PCR primer pairs spanning the MCV genome. None of the 59 control tissues, including nine skin samples, was positive by PCR alone, but five gastrointestinal tract tissues tested weakly positive after PCR-Southern hybridization (8%, P < 0.0001, table S2). Viral T antigen sequences were recovered from three of these samples, confirming low copy number infection. Similarly, only 4 of 25 (16%, P = 0.0007, table S2) additional skin and non-MCC skin tumor samples from immunocompetent and immunosuppressed patients tested positive for MCV sequences (Table 2 and table S2).
Table 1.
PCR for MCV DNA in MCC tissues. A plus symbol indicates that the sample was strongly positive by ethidium bromide staining only with one or more primers. A minus symbol indicates that the tissue was negative for all primers. Entries with both plus and minus symbols indicate that the sample was negative by ethidium bromide staining but positive after Southern hybridization of PCR products
| MCC cases (n = 10) | ||
|---|---|---|
| Patient | Tissue ID | MCV positivity |
| 1 | MCC337 | -/+ |
| 2† | MCC338 | + |
| 2 | MCC339 | + |
| 3 | MCC343 | - |
| 4 | MCC344 | + |
| 5 | MCC345 | + |
| 6 | MCC346 | - |
| 7 | MCC347 | + |
| 7‡ | MCC348 | + |
| 8 | MCC349 | + |
| 9 | MCC350 | + |
| 10 | MCC352 | + |
| Total (%) | 8/10 (80) | |
MCC338 was from an infiltrating tumor in skin tissue adjacent to MCC339 tumor.
MCC348 taken from a metastatic lymph node from MCC347.
Table 2.
PCR for MCV DNA in comparison control tissues (n = 84). For detailed description of tissues and tissue sites, see table S2. MCV positivities marked with plus and minus symbols together are as in Table 1. For the various body site tissues, there were 59 samples; for the skin and skin tumor tissues, the sample size was 25 (table S2)
| MCV positivity | ||
|---|---|---|
| Various body site tissues | ||
| Total MCV negative (%) | 54/59 (92) | |
| Total MCV positive (%) | 5/59 (8) | |
| Appendix control 1 | -/+ | |
| Appendix control 2 | -/+ | |
| Gall bladder | -/+ | |
| Bowel | -/+ | |
| Hemorrhoid | -/+ | |
| Skin and skin tumor tissues | ||
| Total MCV negative (%) | 21/25 (84) | |
| Total MCV positive (%) | 4/25 (16) | |
| Skin | -/+ | |
| KS skin tumor 1 | -/+ | |
| KS skin tumor 2 | -/+ | |
| KS skin tumor 3 | -/+ |
To determine whether MCV DNA was integrated into the tumor genome, we examined MCC samples by direct Southern blotting without PCR amplification. When MCV DNA in MCC tumor is digested by single-cutter restriction endonucleases, such as EcoRI or BamHI, and probed with viral sequence, four possible patterns are predicted to occur: (i) if the viral DNA exists as freely replicating circular episomes, then a ∼5.4 kilobase (kb) band will be present (integrated-concatenated virus will also generate a ∼5.4 kb band); (ii) if MCV DNA integrates polyclonally, as might occur during secondary infection of the tumor if MCV is a passenger virus, then diffuse hybridization from different band sizes is expected; (iii) if MCV DNA integrates at one or a few chromosomal sites, then the tumors will have identical or near-identical non-5.4-kb banding patterns; or (iv) if MCV DNA integrates at different chromosomal sites before clonal expansion of the tumor cells, then distinct bands of different sizes will be present (monoclonal viral integration).
Eight of 11 MCC DNA samples (including MCC348 metastasis from MCC347) digested with either BamHI or EcoRI showed robust MCV hybridization, and these corresponded to the same tumors positive by PCR analysis with multiple primers (Fig. 3A and fig. S3). Monoclonal viral integration (pattern iv) was evident with one or both enzymes in six tumors: MCC339, 345, 347, 348, 349, and 352 (solid arrowheads). EcoRI digestion of MCC339, for example, produced two distinct 7.5- and 12.2-kb bands that would arise only if MCV is integrated at a single site in the majority of tumor cells. MCC344 and 350 bands have episomal or integrated-concatemeric bands (open arrowhead, pattern i). MCC352 has a monoclonal integration pattern (solid arrowheads, pattern iv) on BamHI digestion as well as an intense 5.4-kb band (open arrowhead), consistent with an integrated concatemer. All three tumors negative by PCR with ethidium bromide staining (MCC337, 343, and 346) were also negative by direct Southern blotting.
Fig. 3.
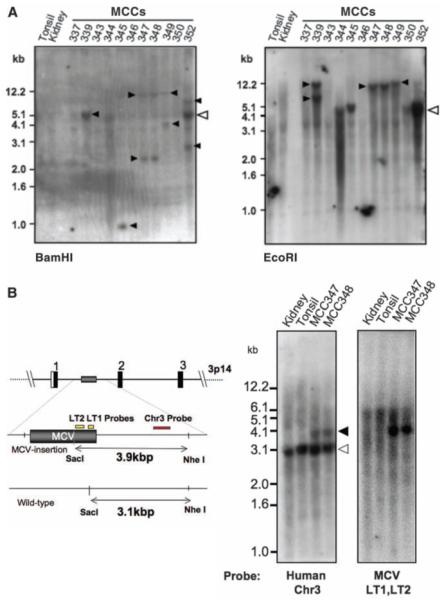
Clonal MCV integration in MCC tumors detected by direct Southern hybridization. (A) DNA digested with BamHI (left) or EcoRI (right) and Southern-blotted with MCV DNA probes reveals different banding patterns in each tumor, including >5.4-kb bands. Open arrowhead shows the expected position for MCV episomal or concatenated-integrated genome (5.4 kb) with corresponding bands present in tumors MCC344 and 350. Tumors MCC339, 345, 347, 348, and 349 have different band sizes and doublet bands (solid arrowheads), consistent with genomic monoclonal integration. MCC352 has a prominent 5.4-kb band as well as higher and lower molecular weight monoclonal integration bands (BamHI), consistent with an integrated concatemer. Tumors MCC337, 343, and 346 have no MCV DNA detected by Southern blotting [bands at 1.5 kb (kidney) and 1.2 kb (MCC346) are artifacts]. (B) Viral and cellular monoclonality in MCC347 and 348. Tumor MCC347 and its metastasis MCC348 were digested with SacI and NheI and Southern-blotted with unique human flanking sequence probe [Chr3 (red), left] or viral probes [LT1 and LT2 (yellow), right]. The wild-type human allele is present in all samples at 3.1 kb (left). The MCC tumors, however, have an additional 3.9-kb allelic band formed by MCV DNA insertion into chromosome 3p14. Hybridization with probes for MCV T antigen sequence (yellow, right) generates an identical band.
The Southern blot banding patterns (Fig. 3A) were identical for MCC347 and its metastasis, MCC348, in line with 3′-RACE results (Fig. 1B) and confirming that MCC348 arose as a metastatic clone of MCC347. Because the genomic integration site (the PTPRG locus on chromosome 3p14) is mapped for these tumors, we performed Southern blotting with flanking human sequence probes to examine cellular monoclonal integration. NheI-SacI digestion of MCC347 and 348 is predicted to generate a 3.1-kb fragment from the wild-type allele and a 3.9-kb fragment from the allele containing the integrated MCV DNA. Hybridization with a flanking human PTPRG sequence probe revealed that the 3.9-kb allele was present in MCC347 and 348 DNA but not in control tissue DNA (Fig. 3B). As predicted, the same fragment hybridized to a MCV T antigen sequence probe, consistent with both cellular and viral monoclonality in this tumor. These results provide evidence that MCV infection and genome integration occurred in this tumor before clonal expansion of tumor cells. MCV in MCC may have some parallels to high-risk human papillomavirus (HPV), which causes cervical cancer mainly after viral episome disruption and integration into the cervical epithelial cell genome (16).
If MCV plays a causal role in tumorigenesis, it could conceivably do so by several mechanisms, including T antigen expression, insertional mutagenesis, or both. Our DTS results show tumor expression of MCV T antigen, which has conserved DnaJ (4), pocket protein-binding LXCXE (17), and pp2A-binding (18, 19) domains previously shown to play roles in polyomavirus-induced cell transformation. Mutational disruption of the PTPRG gene, which is suspected to be a tumor suppressor (20), could also play a role in MCC, although our Southern blot data suggest that MCV integration occurs at various genomic sites in different MCC tumors.
Our study validates the utility of DTS for the discovery of cryptic human viruses, but it has also revealed some limitations of the approach. Of the four tumors we sampled, only one (MCC347) was infected at high copy number. MCV transcripts in this tumor were present at 10 transcripts per million or about 5 transcripts per tumor cell. In future searches for other directly transforming tumor viruses (21), DTS should be used on multiple highly uniform samples sequenced to a depth of 200,000 transcripts or greater. Because DTS is quantitative, it is less likely to be useful in its current form for discovery of low-abundance viruses in autoimmune disorders or other chronic infectious diseases. Discovery of MCV by DTS nonetheless shows that DTS and related approaches (22) are promising methods to identify previously unknown human tumor viruses.
Supplementary Material
Acknowledgments
We thank the National Cancer Institute-supported Cooperative Human Tissue Network for tissues used in this study, M. Aquafondata for tissue staining, P. S. Schnable for sharing cDNA data sets used in DTS pilot testing, O. Gjoerup and R. D. Wood for helpful comments, and J. Zawinul for help with the manuscript.
Supported in part by funds from NIH R33CA120726 and the Pennsylvania Department of Health. The Pennsylvania Department of Health specifically disclaims responsibility for any analyses, interpretations, or conclusions.
References and Notes
- 1.Gross L. Proc. Soc. Exp. Biol. Med. 1953;83:414. doi: 10.3181/00379727-83-20376. [DOI] [PubMed] [Google Scholar]
- 2.Poulin DL, DeCaprio JA. J. Clin. Oncol. 2006;24:4356. doi: 10.1200/JCO.2005.03.7101. [DOI] [PubMed] [Google Scholar]
- 3.Dilworth SM. Nat. Rev. Cancer. 2002;2:951. doi: 10.1038/nrc946. [DOI] [PubMed] [Google Scholar]
- 4.Pipas JM. J. Virol. 1992;66:3979. doi: 10.1128/jvi.66.7.3979-3985.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Crandall KA, Perez-Losada M, Christensen RG, McClellan DA, Viscidi RP. Adv. Exp. Med. Biol. 2006;577:46. doi: 10.1007/0-387-32957-9_3. [DOI] [PubMed] [Google Scholar]
- 6.Allander T, et al. J. Virol. 2007;81:4130. doi: 10.1128/JVI.00028-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gaynor AM, et al. PLoS Pathog. 2007;3:e64. doi: 10.1371/journal.ppat.0030064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hollanderova D, Raslova H, Blangy D, Forstova J, Berebbi M. Int. J. Oncol. 2003;23:333. [PubMed] [Google Scholar]
- 9.Lemos B, Nghiem P. J. Investig. Dermatol. 2007;127:2100. doi: 10.1038/sj.jid.5700925. [DOI] [PubMed] [Google Scholar]
- 10.Van Gele M, et al. Oncogene. 2004;23:2732. doi: 10.1038/sj.onc.1207421. [DOI] [PubMed] [Google Scholar]
- 11.Engels EA, Frisch M, Goedert JJ, Biggar RJ, Miller RW. Lancet. 2002;359:497. doi: 10.1016/S0140-6736(02)07668-7. [DOI] [PubMed] [Google Scholar]
- 12.Chang Y, et al. Science. 1994;266:1865. doi: 10.1126/science.7997879. [DOI] [PubMed] [Google Scholar]
- 13.Feng H, et al. J. Virol. 2007;81:11332. doi: 10.1128/JVI.00875-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chou HH, Holmes MH. Bioinformatics. 2001;17:1093. doi: 10.1093/bioinformatics/17.12.1093. [DOI] [PubMed] [Google Scholar]
- 15.Pawlita M, Clad A, zur Hausen H. Virology. 1985;143:196. doi: 10.1016/0042-6822(85)90108-4. [DOI] [PubMed] [Google Scholar]
- 16.Durst M, Gissmann L, Ikenberg H, zur Hausen H. Proc. Natl. Acad. Sci. U.S.A. 1983;80:3812. doi: 10.1073/pnas.80.12.3812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.DeCaprio JA, et al. Cell. 1988;54:275. doi: 10.1016/0092-8674(88)90559-4. [DOI] [PubMed] [Google Scholar]
- 18.Pallas DC, et al. Cell. 1990;60:167. doi: 10.1016/0092-8674(90)90726-u. [DOI] [PubMed] [Google Scholar]
- 19.Walter G, Ruediger R, Slaughter C, Mumby M. Proc. Natl. Acad. Sci. U.S.A. 1990;87:2521. doi: 10.1073/pnas.87.7.2521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pitterle DM, Jolicoeur EM, Bepler G. In Vivo (Athens) 1998;12:643. [PubMed] [Google Scholar]
- 21.Parsonnet J. In: Microbes and Malignancy. Parsonnet J, editor. Oxford Univ. Press; New York: 1999. pp. 3–18. [Google Scholar]
- 22.Xu Y, et al. Genomics. 2003;81:329. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.