

Abstract
We describe a new stochastic search algorithm for linear regression models called the bounded mode stochastic search (BMSS). We make use of BMSS to perform variable selection and classification as well as to construct sparse dependency networks. Furthermore, we show how to determine genetic networks from genomewide data that involve any combination of continuous and discrete variables. We illustrate our methodology with several real-world data sets.
Keywords: Bayesian regression analysis, Dependency networks, Gene expression, Stochastic search, Variable selection
1. INTRODUCTION
Nowadays, the identification of biological pathways from genomewide studies is the focus of a considerable research effort. The overarching goal is to use gene expression, genotype, and clinical and physiological information to create a network of interactions that could potentially be representative for underlying biological processes. One pertinent example we will focus on in this paper is the determination of genetic networks associated with lymph node positivity status (LNPos) in human breast cancer. Our data have been previously analyzed in Pittman and others (2004) and Hans and others (2007). It comprises 100 low-risk (node-negative) samples and 48 high-risk (high node-positive) samples. The data set records the expression levels of 4512 genes, estimated tumor size (in centimeters) and estrogen receptor status (binary variable determined by protein assays).
The numerous approaches to learning networks developed so far are quite diverse and, for this reason, are complementary to each other. Most of these techniques have been successful in unraveling various parts of the complex biology that induced the patterns of covariation represented in the observed data. The biggest challenge comes from the large number of biological entities that need to be represented in a network. The links (edges) between these entities (vertices) are determined from a relatively small number of available samples. Inducing sparsity in the resulting network is key both for statistical reasons (a small sample size can support a reduced number of edges) and for biological reasons (only a small set of regulatory factors are expected to influence a given entity).
Two coexpressed genes are likely to be involved in the same biological pathways, hence an association network in which missing edges correspond with genes having low absolute correlation of their expression levels could reveal groups of genes sharing the same functions (Butte and others, 2000), (Steuer and others, 2003). Shortest path analysis in such networks can uncover other genes that do not have the same expression pattern but are involved in the same biological pathway (Zhou and others, 2002). Another important type of networks for expression data are represented by Gaussian graphical models (Dobra and others, 2004), (Schafer and Strimmer, 2005), (Li and Gui, 2006), (Castelo and Roverato, 2006), (Wille and Bühlmann, 2006). The observed variables are assumed to follow a multivariate normal distribution. The edges in this network correspond with nonzero elements of the inverse of the covariance matrix. The biological relevance of paths in Gaussian graphical models networks is studied in Jones and West (2005). Other types of networks are derived from Bayesian networks whose graphical representation is a directed acyclic graph (Segal and others, 2003), (Yu and others, 2004), (Friedman, 2004).
A related question that appears in genomewide studies is the identification of a reduced set of molecular and clinical factors that are related to a certain phenotype of interest. This is known as the variable selection problem and can be solved based on univariate rankings that individually measure the dependency between each candidate factor and the response – see, for example, Golub and others (1999), Nguyen and Rocke (2002), Dudoit and others (2002), Tusher and others (2001). Other approaches consider regression that involve combinations of factors, which lead to a huge increase in the number of candidate models that need to be examined. The stepwise methods of Furnival and Wilson (1974) can only be used for very small data sets due to their inability to escape local modes created by complex patterns of collinear predictors. A significant step forward were Markov chain Monte Carlo (MCMC) algorithms that explore the models space by sampling from the joint posterior distribution of the candidate models and regression parameters – see, for example, George and McCulloch (1993), George and McCulloch (1997), Green (1995), Raftery and others (1997), Nott and Green (2004). Excellent review papers about Bayesian variable selection for Gaussian linear regression models are Carlin and Chib (1995), Chipman and others (2001), and Clyde and George (2004). Lee and others (2003) make use of MCMC techniques in the context of probit regression to develop cancer classifiers based on expression data. Theoretical considerations related to the choice of priors for regression parameters are discussed in Fernández and others (2003) and Liang and others (2008).
MCMC methods can have a slow convergence rate due to the high model uncertainty resulting from the small number of available samples. Yeung and others (2005) recognize this problem and develop a multiclass classification method by introducing a stochastic search algorithm called the iterative Bayesian model averaging. While this method performs very well in the context of gene selection in microarray studies, it is still based on a univariate ordering of the candidate predictors. Hans and others (2007) make another step forward and propose the shotgun stochastic search (SSS) algorithm that is capable of quickly moving toward high-probable models while evaluating and recording complete neighborhoods around the current most promising models.
The aim of this paper was to combine stochastic search methods for linear regressions and the identification of biological networks in a coherent and comprehensive methodology. We introduce a new stochastic search algorithm called the bounded mode stochastic search (BMSS). We make use of this algorithm to learn dependency networks (Heckerman and others, 2000) that further allow us to infer sparse networks of interactions. We allow for the presence of any combination of continuous and binary variables and, as such, we are no longer restricted to the multivariate normal assumption required by the Gaussian graphical models. Moreover, the edges we identify are indicative of complex nonlinear relationships and generalize correlation-based networks.
The structure of this paper is as follows. In Section 2, we describe the BMSS algorithm. In Section 3 we discuss dependency networks, and in Section 4 we show how to infer genetic networks. In Section 5, we make some concluding remarks. Our proposed methodology is illustrated throughout with the lymph node status data.
2. A STOCHASTIC SEARCH ALGORITHM FOR SMALL SUBSETS REGRESSIONS
We assume that a response variable Y = X1 is associated with the first component of a random vector X = (X1,…,Xp), while the remaining components are the candidate explanatory covariates. Let V = {1,2,…,p}. Denote by D the n × p data matrix, where the rows correspond with samples and the i-th column corresponds with variable Xi. For A⊂V, denote by DA the submatrix of D formed with the columns with indices in A.
A regression model for Y given a subset XA = (Xi)i∈A, A⊂V\{1} of the remaining variables is denoted by [1|A]. We follow the prior specification for regression parameters described in Appendix A for normal linear regression (Y continuous) and logistic regression (Y binary). If the marginal likelihood p(D|[1|A]) of the regression [1|A] can be calculated exactly or approximated numerically, the posterior probability of [1|A] is readily available up to a normalizing constant:
where p([1|A]) is the prior probability of model [1|A].
Genomewide data sets are characterized by a very large p/n ratio. As such, we are interested in regressions that contain a number of predictors much smaller than p. There are 2 ways to focus on these small subset regressions. The first approach involves choosing a prior on the candidate regressions space that down weights richer regressions (Chipman, 1996), (Kohn and others, 2001), (Scott and Berger, 2006). While such priors encourage sparsity and seem to work reasonably well (Hans and others, 2007), we found that in practice it is not straightforward to calibrate them to completely avoid evaluating the marginal likelihood of models with many predictors. Such calculations are prone to lead to numerical difficulties especially when there are no formulas available for the corresponding high-dimensional integrals. This is the case of logistic regressions whose marginal likelihoods are estimated using the Laplace approximation – see (A.6) in the Appendix A.
The second approach involves reducing the space of candidate models to 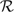 pmax – the set of regressions with at most pmax predictors. This implies that only
pmax – the set of regressions with at most pmax predictors. This implies that only 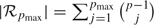 regressions need to be considered which represents a significant reduction compared to 2p − 1 – the total number of regressions for Y. There is no substantive need to further penalize for model complexity and we assume throughout that the models in pmax are apriori equally likely, i.e.,
regressions need to be considered which represents a significant reduction compared to 2p − 1 – the total number of regressions for Y. There is no substantive need to further penalize for model complexity and we assume throughout that the models in pmax are apriori equally likely, i.e.,
| (2.1) |
Since the size of pmax precludes its exhaustive enumeration, we need to make use of stochastic techniques to determine high posterior probability regressions in pmax. One such procedure is called the MCMC model composition algorithm (MC3) and was introduced by Madigan and York (1995). MC3 moves around pmax by sampling from {p([1|A]|D):[1|A]∈pmax}. As such, the probability of identifying the highest posterior probability regression in pmax could be almost 0 if |pmax| is large and n is small, which means that MC3 could be inefficient in finding models with large posterior probability.
Hans and others (2007) recognized this issue and proposed the SSS algorithm that aggressively moves toward regions with high posterior probability in pmax by evaluating the entire neighborhood of the current regression instead of only one random neighbor. Hans and others (2007) empirically show that SSS finds models with high probability faster than MC3. This is largely true if one does not need to make too many changes to the current model to reach the highest posterior probability regression in pmax or other models with comparable posterior probability. If a significant number of changes are required, fully exploring the neighborhoods of all the models at each iteration could be extremely inefficient. In this case, MC3 might end up reaching higher posterior probability regressions after visiting fewer models than SSS. Each iteration of SSS is computationally more expensive than an iteration of MC3 since the entire neighborhood of each regression needs to be visited and recorded. For this reason, SSS does not stay at the same model for 2 consecutive iterations as MC3 does. While SSS can significantly benefit from cluster computing that allows a simultaneous examination of subsets of neighbors, it still moves around pmax by selecting the regression whose neighborhood will be studied at the next iteration from the neighbors of the current regression [1|Ak]. This constitutes a limitation of SSS because it is very likely that other models from the list ℒ could lead to the highest posterior probability regressions faster than a regression from nbdpmax([1|Ak]).
Berger and Molina (2005) pointed out that the model whose neighbor(s) could be visited at the next iteration should be selected from the list of models identified so far with probabilities proportional with the posterior model probabilities. This leads to more aggressive moves in the models space.
We develop a novel stochastic search algorithm which we call the BMSS. Our method combines MC3, SSS, and some of the ideas of Berger and Molina (2005) in 2 different stages. In the first stage, we attempt to advance in the space of models fast by exploring only one model at each iteration. Once higher posterior probability models have been reached, we proceed to exhaustively explore their neighborhoods at the second stage to make sure we do not miss any relevant models that are close to the models already identified. There is no benefit in exploring the same model twice at the second stage, hence we keep track of the models explored at the previous iterations.
We record in a list 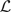 the highest posterior probability regressions determined by BMSS. This list is sequentially updated by adding and deleting regressions at each iteration of BMSS. We define
the highest posterior probability regressions determined by BMSS. This list is sequentially updated by adding and deleting regressions at each iteration of BMSS. We define
where c∈(0,1) and [1|A*] = argmax[1|A′]∈p([1|A′]|D} is the regression in with the highest posterior probability. According to Kass and Raftery (1995), a choice of c in one of the intervals (0,0.01], (0.01,0.1], (0.1,1/3.2], (1/3.2,1] means that the models in \(c) have, respectively, decisive, strong, substantial or “not worth more than a bare mention” evidence against them with respect to [1|A*].
We further introduce the set (c,m) that consists of the top m highest posterior probability models in (c). This reduced set of models is needed because (c) might still contain a large number of models for certain values of c especially if there are many models having almost the same posterior probability.
We define the neighborhood of a regression [1|A] as (Hans and others, 2007):
The 3 subsets of neighbors are obtained by including an additional predictor Xj in regression [1|A], by substituting a predictor Xj that is currently in [1|A] with another predictor Xj′ that does not belong to [1|A] and by deleting a predictor Xj from [1|A]:
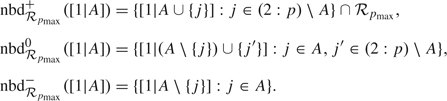 |
The regression neighborhoods are defined so that any regression in pmax can be connected with any other regression in pmax through a sequence of regressions in pmax such that any 2 consecutive regressions in this sequence are neighbors. We remark that the size of the neighborhoods of the regressions in pmax is not constant. If the regression [1|A] contains the maximum number of predictors (i.e., |A| = pmax), no other variable can be added to the model (i.e.,  ). As such, the neighborhoods would be too constrained if we would not allow the substitution of a variable currently in the model with some variable currently outside the model.
). As such, the neighborhoods would be too constrained if we would not allow the substitution of a variable currently in the model with some variable currently outside the model.
We are now ready to give a description of our stochastic search method:
- procedure BMSS(pmax,c,m,kmax1,kmax2)
- ▸ Start at a random regression [1|A1]∈pmax. Set = {[1|A1]}.
- ▸ Stage 1. For k = 1,2,…,kmax1 do:
- Uniformly draw a regression from nbdpmax([1|Ak])\, where [1|Ak] is the current model. Calculate the posterior probability and include in .
- Prune the lowest posterior probability models from so that = (c,m).
- Sample a regression [1|Ak + 1] from with probability proportional with {p([1|A]|D):[1|A]∈}.
- □
- ▸ Mark all the models in as unexplored.
- ▸ Stage 2. For k = 1,…,kmax2 do:
- LetU⊂ the subset of unexplored models. If U = ∅, STOP.
- Sample a model fromU with probability proportional with {p([1|A]|D):[1|A]∈U}. Mark as explored.
- For every regression in nbdpmax([1|])\, calculate its posterior probability, include it in and mark it as unexplored.
- Prune the lowest posterior probability models from so that = (c,m).
□
At the second stage, BMSS might end if no unexplored models are found in before completing kmax2 iterations. Setting m to a larger value (e.g., m = 500 or m = 1000) leads to very good results as the highest probability models identified are captured in the output of BMSS. The value of the parameter c determines the elimination of regressions with a small Bayes factor with respect to the highest posterior probability regression in ℒ. Particular choices of c are interpreted as we described before according to the criteria from Kass and Raftery (1995). The first stage of the algorithm should be run for a larger number of iterations (e.g., kmax1 = 100000) since only one regression has to be evaluated at each iteration. The second stage of BMSS should be run for a reduced number of iterations (e.g., kmax2 = 100) since the entire neighborhood of a regression has to be evaluated and recorded. It is recommended that BMSS should be restarted several times to make sure that it outputs the same regressions. If this does not happen, BMSS needs to be run for an increased number of iterations kmax1 and kmax2.
2.1. Example: lymph node status data
We used BMSS with pmax = 6, c = 0.25, m = 1000, kmax1 = 100000, kmax2 = 100 to identify high posterior probability logistic regressions associated with LNPos involving the 4514 candidate predictors in the lymph node status data. The choice c = 0.25 means that regressions having decisive evidence against them with respect to the highest posterior probability regression identified are discarded from the final list of regressions reported by BMSS. BMSS returns 11 regressions in which 17 genes appear. These regressions together with their marginal likelihoods are shown in the 2 leftmost columns of Table 1. The selected genes and their corresponding posterior inclusion probabilities are: SFRS17A (1.0), W26659 (1.0), RGS3 (1.0), ATP6V1F (1.0), GEM (0.385), WSB1 (0.346), PJA2 (0.209), SDHC (0.207), XPO1 (0.134), TOMM40 (0.132), ARF6 (0.116), CD19 (0.102), DPY19L4 (0.089), UBE2A (0.081), HSPE1 (0.075), RAD21 (0.065), KEAP1 (0.060).
Table 1.
Comparison of the effectiveness of BMSS, SSS and MC3. The 3 rightmost columns show which regressions have been identified by each algorithm
| Model | Log-posterior | BMSS | SSS | MC3 |
| SFRS17A, GEM, RGS3, SDHC, W26659, ATP6V1F | − 42.33 | Yes | Yes | No |
| SFRS17A, TOMM40, RGS3, PJA2, W26659, ATP6V1F | − 42.78 | Yes | No | No |
| SFRS17A, RGS3, W26659, ATP6V1F, WSB1, CD19 | − 43.03 | Yes | No | No |
| SFRS17A, DPY19L4, RGS3, W26659, ATP6V1F, WSB1 | − 43.17 | Yes | No | Yes |
| SFRS17A, RGS3, W26659, ATP6V1F, WSB1, UBE2A | − 43.27 | Yes | Yes | No |
| SFRS17A, RGS3, PJA2, W26659, ATP6V1F, XPO1 | − 43.31 | Yes | Yes | Yes |
| SFRS17A, RGS3, HSPE1, W26659, ATP6V1F, WSB1 | − 43.34 | Yes | Yes | No |
| SFRS17A, GEM, RGS3, W26659, ATP6V1F, RAD21 | − 43.49 | Yes | Yes | Yes |
| SFRS17A, GEM, ARF6, KEAP1, W26659, ATP6V1F | − 43.57 | Yes | Yes | No |
| SFRS17A, GEM, RGS3, ARF6, W26659, ATP6V1F | − 43.62 | Yes | Yes | No |
| SFRS17A, GEM, RGS3, W26659, ATP6V1F, XPO1 | − 43.63 | Yes | Yes | Yes |
We compare the effectiveness of BMSS in identifying high posterior probability regressions in 6 with respect to SSS and MC3. With the settings above, BMSS performs 2805400 marginal likelihood evaluations. We run SSS and MC3 until they completed the same number of evaluations. It turns out that SSS and MC3 do not find any regression in 6 with a larger posterior probability than the regressions reported by BMSS. In fact, the 11 regressions found by BMSS are the top 11 regressions identified by all 3 algorithms. However, as shown in Table 1, SSS finds only 8 of these 11 regressions, while MC3 finds only 4 of these models. This indicates that BMSS explores 6 more effectively than SSS and MC3.
We assess the convergence of BMSS by running 5 separate instances of the procedure with various random seeds. Figure 1 shows the largest posterior probability of a regression in the list kept by BMSS plotted against the number of marginal likelihood evaluations performed across consecutive iterations. BMSS always calculates one marginal likelihood at each iteration of its first stage. In this particular example, BMSS computes the marginal likelihood of 27054 regressions at every iteration of its second stage. We see that both stages are needed to obtain the highest posterior probability regressions. BMSS outputs the same 11 regressions in each of the 5 instances, which means we could be fairly confident that we have found the top models in 6. We remark that BMSS finds the top models after evaluating only 2805400 regressions – a small number compared to 1.173×1019, the number of regressions in 6.
Fig. 1.

Convergence of BMSS for the lymph node status data. The vertical dotted line indicates the transition of BMSS from stage 1 to stage 2, that occurs after evaluating kmax1 = 100000 models. Each of the 5 solid lines corresponds with an instance of BMSS.1
What is the relevance of these 11 regressions with respect to LNPos? The model-averaged prediction probabilities lead to 141 (95.27%) samples correctly predicted with a Brier score of 9.03 having a standard error of 0.80. Here, we use the generalized version of the Brier score given in Yeung and others (2005). We perform leave-one-out cross-validation prediction by recomputing the model posterior probabilities as well as the prediction probabilities after sequentially leaving out each of the 148 observations. We obtain 132 (89.19%) samples correctly predicted with a Brier score of 13.5 having a standard error of 1.47.
By comparison, Hans and others (2007) show their prediction results based on the top 10 logistic regression models they identify. These models involve 18 genes out of which 4 (RGS3, ATP6V1F, GEM, and WSB1) are also present in our list of 17 genes. Their fitted prediction probabilities correctly predict 135 samples (91.22%), while their leave-one-out cross-validation predictive performance has a sensitivity of 79.2% and a specificity of 76%. Our leave-one-out predictive performance has a sensitivity of 85.4% and a specificity of 93% which indicates that we have discovered combinations of genes with better predictive power.
2.2. Selecting pmax
The choice of the maximum number of predictors pmax defines the search space for BMSS. Increasing pmax leads to richer sets of candidate regressions and potentially to more complex combinations of predictors that are ultimately identified. Selecting a sensible value for pmax is therefore key to our methodology. This determination should involve a careful consideration of model fit, model complexity, and the inclusion of explanatory variables that are expected to be relevant based on expert knowledge about the data.
Here we show how to choose pmax for the lymph node status data. We run separate instances of BMSS with pmax∈{1,2,…,15} while keeping the other parameters at their values from Section 2.1. In each case, we recorded the number of regressions identified in pmax, the number of variables that appear in these regressions as well as 5-fold cross-validation prediction results (Brier score, sensitivity, and specificity) (see Table 2).
Table 2.
Regressions selected by BMSS for various values of pmax in the lymph node status data. The standard error associated with the 5-fold cross-validation Brier score is given in parentheses. The number of variables that appear in the top regressions is given in column Variables, while the number of top regressions is given in column Regressions. The last column shows whether tumor size was present in the top regressions
| pmax | Variables | Regressions | Brier score | Sensitivity (%) | Specificity (%) | Tumor size |
| 1 | 1 | 1 | 26.13 (0.47) | 39.6 | 90 | No |
| 2 | 6 | 5 | 26.24 (1.43) | 54.2 | 86 | No |
| 3 | 15 | 11 | 25.8 (1.83) | 62.5 | 87 | No |
| 4 | 12 | 8 | 20.4 (1.51) | 75 | 88 | No |
| 5 | 11 | 7 | 15 (1.52) | 79.2 | 93 | No |
| 6 | 17 | 11 | 13.5 (1.47) | 85.4 | 93 | No |
| 7 | 20 | 11 | 10.1 (1) | 89.6 | 96 | No |
| 8 | 53 | 51 | 9 (0.74) | 85.4 | 98 | No |
| 9 | 68 | 69 | 7.61 (0.85) | 91.2 | 97 | No |
| 10 | 166 | 173 | 6.04 (0.48) | 91.2 | 99 | No |
| 11 | 60 | 69 | 5.69 (0.64) | 97.9 | 99 | Yes |
| 12 | 391 | 1000 | 4.58 (0.51) | 95.8 | 98 | Yes |
| 13 | 341 | 1000 | 4.17 (0.56) | 97.9 | 100 | Yes |
| 14 | 465 | 1000 | 3.18 (0.36) | 99 | 100 | Yes |
| 15 | 460 | 1000 | 3.55 (0.4) | 97.9 | 100 | Yes |
We see that the prediction results constantly improve as pmax increases until they reach a plateau starting with pmax = 11. For pmax > 11, the Brier score has a marginal decrease with respect to pmax = 11, while the sensitivity and specificity remain about the same. The model-averaged fitted prediction probabilities for pmax = 11 lead to 148 (100%) samples correctly predicted with a Brier score of 3.02 having a standard error of 0.25 (Figure 2). The choice pmax = 11 is also related to the presence of tumor size in the highest posterior probability regressions identified by BMSS. We would certainly expect that the size of a tumor should be relevant for lympth node positivity status. Table 2 shows that tumor size is not present in the top regressions for pmax < 11 and it is always present if pmax ≥ 11. Moreover, the number of variables in the top regressions seems to increase by an order of magnitude if pmax > 11 which could mean that the corresponding combinations of predictors are too complex to be relevant. As such, pmax = 11 is an appropriate choice for the lymph node status data.
Fig. 2.

Prediction results corresponding with pmax = 11 for the lymph node status data. The solid circles represent low-risk patients, while the open circles represent high-risk patients. The vertical lines are 80% confidence intervals.
3. LEARNING AND INFERENCE FOR DEPENDENCY NETWORKS
We denote by X − j = XV\{j}, for j = 1,…,p. A dependency network (Heckerman and others, 2000) is a collection of conditional distributions or regressions of each variable given the rest:
Each of these local probability distributions can be modeled and learned independently of the others. We can make use of BMSS to determine a set j = {[j|Alj]:l = 1,…,|j|} of high posterior probability regressions of Xj given X − j. It follows that
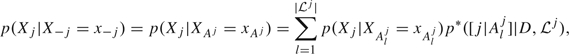 |
(3.1) |
where Aj = ∪l = 1|j|Alj are the indices of all the regressors that appear in at least one regression in j. The weight of each regression in the mixture (3.1) is given by its posterior probability normalized within j:
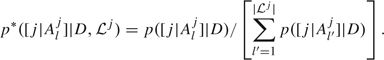 |
(3.2) |
If the number p of observed variables is extremely large and pmax is small, it is likely that the size of each Aj will also be much smaller than p − 1. Equation (3.1) implies that Xj is conditionally independent of XV\({j}∪Aj) given XAj. Hence the dependency network 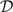 is sparse and embeds conditional independence constraints that creates a parsimonious structure among the observed covariates. This structure reflects the uncertainty of a particular choice of regressions associated with each variable through Bayesian model averaging (Kass and Raftery, 1995). The parameters pmax, c and m of BMSS control the size as well as the number of models in the lists j.
is sparse and embeds conditional independence constraints that creates a parsimonious structure among the observed covariates. This structure reflects the uncertainty of a particular choice of regressions associated with each variable through Bayesian model averaging (Kass and Raftery, 1995). The parameters pmax, c and m of BMSS control the size as well as the number of models in the lists j.
BMSS can be used to perform an initial variable selection with respect to a response variable of interest. The variables selected appear in the highest posterior probability regressions associated with the response. We performed variable selection for the lymph node data and determined 60 explanatory variables that are predictive of LNPos (see Section 4.1). We subsequently constructed dependency networks on this restricted set of 61 variables. However, the variable selection step is not required in our framework. Our inference approach can be used on data sets involving an arbitrarily large number of variables provided that enough computational resources are available. From a theoretical perspective, it is possible to construct dependency networks involving all the 4515 variables in the lymph node status data, but such an experiment was beyond our computing capabilities. There is always a good chance of missing relevant associations by performing any kind of prior variable selection, hence the selection step should be avoided if the required computing effort is not too daunting.
Once has been fully determined, we can sample from using an ordered Gibbs sampling algorithm (Geman and Geman, 1984). Assume that the current state of the chain is x(t) = (x1(t),…,xp(t)). For each j = 1,…,p, sample
| (3.3) |
which gives the next state of the chain x(t + 1). In (3.3), p(Xj|X − j = x − j) is given in (3.1). Sampling from p(Xj|X − j = x − j) is performed as follows:
(i) sample a regression [j|Alj] from
j with probabilities proportional with p*([j|Alj]|D,j) given in (3.2).(ii) sample a set of regression coefficients βj corresponding with [j|Alj] from their posterior distributions. If Xj is continuous, we sample βj from the normal inverse-Gamma posteriors (A.2) and (A.3). If Xj is binary, we sample βj from the joint posterior (A.5) using the Metropolis–Hastings algorithm described in the Appendix.
(iii) sample from p(Xj|X − j = x − j,[j|Alj],βj) = p(Xj|XAlj = xAlj,βj). If Xj is continuous, we sample from (A.1). If Xj is binary, we sample from (A.4).
We remark that, given enough samples, we should have j1∈Aj2 if and only if j2∈Aj1 for any j1≠j2. This means that Xj1 appears in the conditional of Xj2 and vice versa. Bayesian model averaging is key in this context because it eliminates the need to make an explicit decision relative to the choice of covariates that appear in each conditional distribution. As such, the order in which we sample from the local probability distributions should be irrelevant. We emphasize that the symmetry of the sets Aj does not have to be explicitly enforced.
The most important question relates to the existence of a joint probability distribution p(XV) associated with the local probability distributions . Given a positivity condition usually satisfied in practice, a dependency network uniquely identifies a joint distribution p(XV) up to a normalizing constant (Besag, 1974). If p(XV) exists, it is unique and is called consistent. If p(XV) does not exist, is called inconsistent (Heckerman and others, 2000). Hobert and Casella (1998) study the more general case when is inconsistent but still determines an improper joint distribution. Arnold and others (2001) provide a comprehensive discussion related to conditionally specified distributions. Related results are presented in Gelman and Speed (1993) and Besag and Kooperberg (1995), among others.
The ordered Gibbs sampling algorithm can be used to sample from p(XV) if is consistent (Heckerman and others, 2000). Unfortunately the output from the Gibbs sampler does not offer any indication whether is indeed consistent (Hobert and Casella, 1998). We make use of the samples generated from only to estimate relevant quantities of interest, such as bivariate dependency measures. These samples reflect the structure of and do not necessarily come from a proper joint distribution p(XV).
4. GENETIC NETWORKS
We show how construct a network associated with a vector of continuous and discrete random variables XV. After learning a dependency network as described in Section 3, we use the ordered Gibbs sampler to generate a random sample  from . This random sample embeds the structural constraints implied by . We identify 2 different types of networks as follows:
from . This random sample embeds the structural constraints implied by . We identify 2 different types of networks as follows:
(a) Association networks. Estimate the pairwise associations d(Xj1,Xj2), j1,j2∈V based on . Here d(,) denotes Kendall's tau, Spearman's rho, or the correlation coefficient. We prefer using Kendall's tau or Spearman's rho because they measure the concordance between 2 random variables (Nelsen, 1999). On the other hand, the correlation coefficient reflects only linear dependence. The edges in the resulting association network connect pairs of variables whose pairwise associations are different from 0.
(b) Liquid association networks. Li (2002) introduced the concept of liquid association to quantify the dynamics of the association between to random variables Xj1 and Xj2 given a third random variable Y. We denote this measure with d(Xj1,Xj2|Y). The liquid association is especially relevant for pairs of random variables with a low absolute value of their pairwise association d(Xj1,Xj2). Such pairs will not be captured in an association network. However, the association between Xj1 and Xj2 could vary significantly as a function of Y. If Y is continuous, the liquid association between Xj1 and Xj2 given Y is defined as the expected value of the derivative of dY = y(Xj1,Xj2) with respect to Y, i.e.,
| (4.1) |
where dY = y(Xj1,Xj2) is the measure of association d(,) between Xj1 and Xj2 evaluated for the samples Y = y. Li (2002) proved that d(Xj1,Xj2|Y) = E[Xj1Xj2Y] if Xj1, Xj2 and Y are normal random variables with zero mean and unit variance, while d(,) is the correlation coefficient, that is, d(Xj1,Xj2) = E[Xj1Xj2]. If Y∈{0,1} is a binary random variable, we define the liquid association between Xj1 and Xj2 given Y as the absolute value of the change between the association of Xj1 and Xj2 for the samples with Y = 1 versus the samples with Y = 0:
| (4.2) |
As suggested in Li (2002), a permutation test can be used to assess statistical significance in (4.1) and (4.2). We generate random permutations Y* of the observed values of Y and compute the corresponding liquid association score. The p-value is given by the number of permutations that lead to a score higher than the observed score divided by the total number of permutations. The liquid association can be constructed with respect to Kendall's tau, Spearman's rho, or the correlation coefficient.
Estimating the strength of pairwise interactions based on instead of the observed samples D leads to a significant decrease in the number of edges of the resulting networks. Inducing sparsity in the network structure is the key to identify the most relevant associations by shrinking most pairwise dependencies to 0. The inherent correlation that exists between the corresponding test statistics is significantly decreased, thus the use of false discovery rate techniques to decide which edges are present in the network becomes less problematic and avoids the serious multiple testing issues discussed in Efron (2007) and Shi and others (2008).
The genetic networks (a) and (b) complement each other with respect to their biological significance. For example, 2 genes that are directly connected in an association network are likely to be functionally related since their expression levels are either positively or negatively associated (Butte and others, 2000), (Steuer and others, 2003). On the other hand, an edge between the same 2 genes in a liquid association network means that the relationship between them is likely to be influenced by the gene or phenotype with respect to which the network was constructed. These genes might be strongly coexpressed in one experimental condition, while their expression levels could be unrelated in some other condition. The identification of functionally related genes based on liquid association is discussed in Lee (2004) and Li and others (2007).
4.1. Example: lymph node status data
We learn a dependency network involving LNPos and the 60 regressors (59 genes and tumor size) present in the top 69 highest posterior probability models in 11. We employed BMSS with pmax = 11, c = 0.001, m = 1000, kmax1 = 10000, kmax2 = 100 and 5 search replicates to learn the highest posterior probability regressions with at most 11 regressors for each of the 61 variables. We simulate 25000 samples from this dependency network with a burn-in of 2500 samples and a gap of 100 between 2 consecutive saved samples. We use the resulting 250 samples to estimate Kendall's tau for any pair of the 61 covariates. There are 326 pairs of variables having a value of Kendall's tau different than 0 at a false discovery rate of 1%.
Since it is generally believed that highly connected genes (hubs) play a central role in the underlying biological processes, we express the topology of the resulting association network by the degree of each vertex, that is, the number of direct neighbors. We sort the regressors in decreasing order with respect to their degrees in this association network. We also sort them in decreasing order with respect to their posterior inclusion probabilities with respect to LNPos. The 2 sets of ranks define the axes of Figure 3. Therefore, this plot gives a 2-dimensional representation of the relevance of each explanatory variable with respect to LNPos. We see that tumor size ranks high with respect to its predictive relevance but has a smaller number of association with the 59 genes. The most interesting regressors have high ranks on both scales. Three of them seem to stand out: CLDN5, LAT, and WSB1. Another relevant gene is RGS3: it still has a high connectivity in the association network and it is the only gene with a posterior inclusion probability of 1 for each pmax∈{1,2,…,15} (Table 2).
Fig. 3.

Revelance of the 60 variables that appear in the association network for LNPos.
We use the simulated data from the dependency network to identify the pairwise interactions between regressors that are dependent on LNpos. Figure 4 shows the 84 pairs whose p-value was below 0.05. We see that LAT and WSB1 are connected in this liquid association network. Moreover, WSB1 is a neighbor of tumor size. The other 2 neighbors of tumor size are ANKHD1-EIF4EBP3 and DMBT1. Our principled examination of the 2 genetic networks leads us to the 6 genes from Table 3. Five of them are upregulated and one is downregulated.
Fig. 4.

Liquid association network for LNPos. Each edge corresponds with a pair of genes whose association differs for low-risk and high-risk samples. This graph was produced with Cytoscape (http://www.cytoscape.org/).
Table 3.
Relevant genes for LNPos as determined from predictive regression models, the association network and the liquid association network
| Gene | Description | Expression |
| RGS3 | Regulator of G-protein signalling 3 | Up |
| LAT | Linker for activation of T cells | Up |
| WSB1 | WD Repeat and SOCS box-containing 1 | Up |
| CLDN5 | Transmembrane protein deleted in velocardiofacial syndrome | Up |
| DMBT1 | Deleted in malignant brain tumors 1 | Up |
| ANKHD1-EIF4EBP3 | Readthrough transcript | Down |
This set of genes seems to be involved in various processes related to cancer. For example, RGS3 is upregulated in p53-mutated breast cancer tumors (Ooe and others, 2007). LAT plays key roles in cellular defense response, the immune response and the inflammatory response processes. Archange and others (2008) showed that overexpression of WSB1 controls apoptosis and cell proliferation in pancreatic cancer cells xenografts. CLDN5 is associated with schizophrenia (Ishiguro and others, 2008) and human cardiomyopathy (Mays and others, 2008). It is also differentially expressed in human lung squamous cell carcinomas and adenocarcinomas (Paschoud and others, 2007). DMBT1 could play a role in the prevention of inflammation and if it is impaired could lead to Crohn's disease (Renner and others, 2007). Moreover, DMBT1 might be involved in the suppression of mammary tumors in both mice and women (Blackburn and others, 2007). The ANKHD1-EIF4EBP3 mRNA is a co-transcribed product of the neighboring ANKHD1 and EIF4EBP3 genes. ANKHD1 is overexpressed in acute leukemias (Traina and others, 2006), while EIF4EBP3 is involved in the negative regulation of translational initiation.
5. DISCUSSION
The methodology we developed is relevant in 2 different albeit related areas. First of all, we proposed a stochastic search algorithm called BMSS for linear regression models that explores the space of candidate models more efficiently than other related model determination methods. We showed how BMSS performs for normal linear regressions and logistic regressions for several high-dimensional data sets. The classifiers constructed through Bayesian model averaging from the set of regressions identified by BMSS hold their performance for out-of-sample prediction. We do not discuss our choice of priors for regression parameters because we believe that this is only one choice among many other choices available in the literature. Our priors work well for the applications described in this paper, but we would not be surprised to see that other priors perform even better. We stress that our main contribution is the BMSS algorithm. Our procedure can be employed with any other choice of prior parameters as long as the marginal likelihood of each regression model can be explicitly computed or at least accurately approximated in a reasonable amount of time. For example, regression models for discrete variables with more than 2 categories can also be determined using BMSS.
Second, we proposed using BMSS to learn dependency networks that further determine sparse genetic networks. The advantages of our approach are as follows: (i) the edges of the network are no longer restricted to linear associations; (ii) any combination of continuous and discrete variables can be accommodated in a coherent manner; (iii) there is no need to assume a multivariate normal model for expression data sets as required by the Gaussian graphical models; (iv) the approach scales to high-dimensional data sets since the regression models associated with each variable can be learned independently – possibly on many computing nodes if a cluster of computers is available; (v) sparsity constraints can be specified in a straightforward manner. Model uncertainty is explicitly taken into account when sampling from the dependency network since we model the conditional distribution of each variable as a mixture of regressions.
Complete source code, data, and sample input files are available for download from http://www.stat.washington.edu/adobra/software/bmss/.
FUNDING
This work was supported in part by the National Institutes of Health [R01 HL092071].
Acknowledgments
The author thanks Chris Hans who provided the lymph node status data. The author also thanks the Associate Editor and two anonymous reviewers for their comments that significantly improved the quality of this paper. Conflict of Interest: None declared.
APPENDIX A
A.1. Bayesian inference for normal regression
Let Y = X1 be a continuous response variable and X − 1 = (X2,…,Xp) be the vector of explanatory variables. Denote by D1 the first column of the n×p data matrix D, by D(2:p) the columns 2,…,p of D. To keep the notation simple, we assume that all the explanatory variables are present in the regression [1|(2:p)] with coefficients β = (β2,…,βp). We center and scale the observed covariates such that their sample means are 0 and their sample standard deviations are 1. We assume
| (A.1) |
The prior for σ2 is p(σ2) = IG((p + 2)/2,1/2) and, conditional on σ2, the regression coefficients have independent priors p(βj) = N(0,σ2), j = 2,…,p. Dobra and others (2004) show that the corresponding posterior distributions are
| (A.2) |
| (A.3) |
where M = Ip − 1 + D(2:p)TD(2:p). The marginal likelihood of [1|(2:p)] therefore given by
Bayesian inference for logistic regression
Let Y = X1 be a binary response variable. We denote by Di the i-th row of the data matrix D and define Di,p + 1 = 1, for i = 1,…,n. The coefficients of the regression [1|(2:p)] are β = (β2,…,βp) and βp + 1 – the intercept term. We center and scale the explanatory variables X − 1 such that their sample means are 0 and their sample standard deviations are 1. We assume that
| (A.4) |
with g(β,βp + 1,x) = (1 + exp( − xTβ − βp + 1)) − 1 and that the regression coefficients have independent priors p(βj) = N(0,1), j = 2,…,p + 1. The posterior distribution of β is therefore given by
| (A.5) |
where
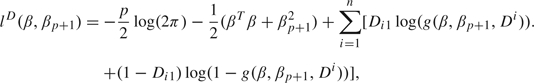 |
and p(D|[1|(2:p)]) = ∫ℜp + 1exp(lD(β,βp + 1))∏j = 2p + 1dβj is the marginal likelihood. The Laplace approximation (Tierney and Kadane, 1986) to p(D|[1|(2:p)]) is
| (A.6) |
where 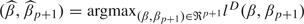 is the posterior mode and HD is the p×p Hessian matrix associated with lD. The gradient of lD(β,βp + 1) is hD(β,βp + 1) = (hjD(β,βp + 1))1 ≤ j ≤ p where hjD(β,βp + 1) = − βj + 1 + ∑i = 1n(Di1 − g(β,βp + 1,Di))Di,j + 1, j = 1,…,p. It follows that the entries of HD(β,βp + 1) are
is the posterior mode and HD is the p×p Hessian matrix associated with lD. The gradient of lD(β,βp + 1) is hD(β,βp + 1) = (hjD(β,βp + 1))1 ≤ j ≤ p where hjD(β,βp + 1) = − βj + 1 + ∑i = 1n(Di1 − g(β,βp + 1,Di))Di,j + 1, j = 1,…,p. It follows that the entries of HD(β,βp + 1) are
where δjk = 1 if j = k and δjk = 0 if j≠k.
The posterior mode is determined using the Newton–Raphson algorithm that produces a sequence (β0,βp + 1) = 0,(β1,βp + 11),…,(βk,βp + 1k),… such that
Sampling from the posterior distribution p(β,βp + 1|D) is done with the Metropolis–Hastings algorithm. At iteration k, generate 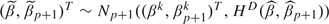 . Set with probability
. Set with probability
Otherwise set (βk + 1,βp + 1k + 1) = (βk,βp + 1k).
References
- Archange C, Nowak J, Garcia S, Moutardier V, Calvo EL, Dagorn J, Iovanna J. The WSB1 gene is involved in pancreatic cancer progression. PLoS ONE. 2008;25:e2475. doi: 10.1371/journal.pone.0002475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnold BC, Castillo E, Sarabia JM. Conditionally specified distributions: an introduction. Statistical Science. 2001;16:249–274. [Google Scholar]
- Berger JO, Molina G. Posterior model probabilities via path-based pairwise priors. Statistica Neerlandica. 2005;59:3–15. [Google Scholar]
- Besag J. Spatial interaction and the statistical analysis of lattice systems (with discussion) Journal of Royal Statistical Society, Series A. 1974;36:192–236. [Google Scholar]
- Besag J, Kooperberg C. On conditional and intrinsic autoregressions. Biometrika. 1995;82:733–746. [Google Scholar]
- Blackburn A, Hill L, Roberts A, Wang J, Aud D, Jung J, Nikolcheva T, Allard J, Peltz G, Otis CN, and others Genetic mapping in mice identifies DMBT1 as a candidate modifier of mammary tumors and breast cancer risk. American Journal of Pathology. 2007;170:2030–2041. doi: 10.2353/ajpath.2007.060512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butte AJ, Tamayo P, Slonim D, Golub TR, Kohane IS. Discovering functional relationships between RNA expression and chemotherapeutic susceptibility using relevance networks. Proceedings of the National Academy of Sciences. 2000;97:12182–12186. doi: 10.1073/pnas.220392197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlin BP, Chib S. Bayesian Model Choice via Markov Chain Monte Carlo. Journal of the Royal Statistical Society, Series B. 1995;57:473–484. [Google Scholar]
- Castelo R, Roverato A. A robust procedure for Gaussian graphical model search from microarray data with p larger than n. Journal of Machine Learning Reasearch. 2006;7:2621–2650. [Google Scholar]
- Chipman H. Bayesian variable selection with related predictors. Canadian Journal of Statistics. 1996;24:17–36. [Google Scholar]
- Chipman H, George EI, McCullogh RE. The practical implementation of Bayesian model selection (with discussion) In: Lahiri P, editor. Model Selection. Beachwood: IMS; 2001. pp. 66–134. [Google Scholar]
- Clyde M, George EI. Model uncertainty. Statistical Science. 2004;19:81–94. [Google Scholar]
- Dobra A, Hans C, Jones B, Nevins JR, Yao G, West M. Sparse graphical models for exploring gene expression data. Journal of Multivariate Analysis. 2004;90:196–212. [Google Scholar]
- Dudoit S, Fridlyand J, Speed TP. Comparison of discrimination methods for the classification of tumors using gene expression data. Journal of the American Statistical Association. 2002;97:77–87. [Google Scholar]
- Efron B. Correlation and large-scale simultaneous significance testing. Journal of the American Statistical Association. 2007;102:93–103. [Google Scholar]
- Fernández C, Ley E, Steel MF. Benchmark priors for Bayesian model averaging. Journal of Econometrics. 2003;75:317–343. [Google Scholar]
- Friedman N. Inferring cellular networks using probabilistic graphical models. Science. 2004;30:799–805. doi: 10.1126/science.1094068. [DOI] [PubMed] [Google Scholar]
- Furnival GM, Wilson RW. Regression by leaps and bounds. Technometrics. 1974;16:499–511. [Google Scholar]
- Gelman A, Speed TP. Characterizing a joint probability distribution by conditionals. Journal of Royal Statistical Society, Series B. 1993;55:185–188. [Google Scholar]
- Geman S, Geman D. Stochastic relaxation, Gibbs distributions and the Bayesian restoration of images. IEEE Transactions of Pattern Analysis and Machine Intelligence. 1984;6:721–742. doi: 10.1109/tpami.1984.4767596. [DOI] [PubMed] [Google Scholar]
- George EI, McCulloch RE. Variable Selection via Gibbs Sampling. Journal of the American Statistical Association. 1993;88:881–889. [Google Scholar]
- George EI, McCulloch RE. Approaches for Bayesian Variable Selection. Statistica Sinica. 1997;7:339–373. [Google Scholar]
- Golub TR, Slonim DK, Tamayo P, Huard C, Gaasenbeek M, Mesirov JP, Coller H, Loh ML, Downing JR, Caligiuri MA, and others Molecular classification of cancer: class discovery and class prediction by gene expression monitoring. Science. 1999;286:531–537. doi: 10.1126/science.286.5439.531. [DOI] [PubMed] [Google Scholar]
- Green PJ. Reversible jump Markov Chain Monte Carlo computation and Bayesian model determination. Biometrika. 1995;82:711–732. [Google Scholar]
- Hans C, Dobra A, West M. Shotgun stochastic search for “Large p” regression. Journal of the American Statistical Association. 2007;102:507–516. [Google Scholar]
- Heckerman D, Chickering DM, Meek C, Rounthwaite R, Kadie C. Dependency networks for inference, collaborative filtering and data visualization. Journal of Machine Learning Research. 2000;1:1–48. [Google Scholar]
- Hobert JP, Casella G. Functional compatibility, Markov chains, and Gibbs sampling with improper posteriors. Journal of Computational and Graphical Statistics. 1998;7:42–60. [Google Scholar]
- Ishiguro H, Imai K, Koga M, Horiuchi Y, Inada T, Iwata N, Ozaki N, Ujike H, Itokawa H, Kunugi M, and others Replication study for associations between polymorphisms in the CLDN5 and DGCR2 genes in the 22q11 deletion syndrome region and schizophrenia. Psychiatric Genetics. 2008;18:255–256. doi: 10.1097/YPG.0b013e328306c7dc. [DOI] [PubMed] [Google Scholar]
- Jones B, West M. Covariance decomposition in undirected Gaussian graphical models. Biometrika. 2005;92:779–786. [Google Scholar]
- Kass R, Raftery AE. Bayes factors. Journal of American Statistical Association. 1995;90:773–95. [Google Scholar]
- Kohn R, Smith M, Chan D. Nonparametric regression using linear combinations of basis functions. Statistics and Computing. 2001;11:313–322. [Google Scholar]
- Lee KE, Sha N, Dougherty ER, Vanucci M, Mallick BK. Gene selection: a Bayesian variable selection approach. Bioinformatics. 2003;19:90–97. doi: 10.1093/bioinformatics/19.1.90. [DOI] [PubMed] [Google Scholar]
- Li H, Gui J. Gradient directed regularization for sparse Gaussian concentration graphs, with application to inference of genetic networks. Biostatistics. 2006;2:302–317. doi: 10.1093/biostatistics/kxj008. [DOI] [PubMed] [Google Scholar]
- Li K-C. Genome-wide coexpression dynamics: theory and application. Proceedings of the National Academy of Sciences. 2002;99:16875–16880. doi: 10.1073/pnas.252466999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li K-C, Liu C-T, Sun W, Yuan S, Yu T. A system for enhancing genome-wide coexpression dynamics study. Proceedings of the National Academy of Sciences. 2004;101:15561–15566. doi: 10.1073/pnas.0402962101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li K-C, Palotie A, Yuan S, Bronnikov D, Chen D, Wei X, Wa CO, Saarela J, Peltonen L. Finding disease candidate genes by liquid association. Genome Biology. 2007;8:R205. doi: 10.1186/gb-2007-8-10-r205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang F, Paulo R, Molina G, Clyde M, Berger JO. Mixtures of g-priors for Bayesian Variable Selection. Journal of the American Statistical Association. 2008;103:410–423. [Google Scholar]
- Madigan D, York J. Bayesian graphical models for discrete data. International Statistical Review. 1995;63:215–232. [Google Scholar]
- Mays TA, Binkley PF, Lesinski A, Doshi AA, Quaile MP, Margulies KB, Janssen P, Rafael-Fortney JA. Claudin-5 levels are reduced in human end-stage cardiomyopathy. Journal of Molecular and Cell Cardiology. 2008;81:81–87. doi: 10.1016/j.yjmcc.2008.04.005. [DOI] [PubMed] [Google Scholar]
- Nelsen RB. An Introduction to Copulas. Volume 139 of Lecture Notes in Statistics. New York: Springer-Verlag; 1999. [Google Scholar]
- Nguyen DV, Rocke DM. Tumor classification by partial least squares using microarray gene expression data. Bioinformatics. 2002;18:39–50. doi: 10.1093/bioinformatics/18.1.39. [DOI] [PubMed] [Google Scholar]
- Nott D, Green P. Bayesian variable selection and the Swendsen-Wang algorithm. Journal of Computational and Graphical Statistics. 2004;13:1–17. [Google Scholar]
- Ooe A, Kato K, Noguchi S. Possible involvement of CCT5, RGS3, and YKT6 genes up-regulated in p53-mutated tumors in resistance to docetaxel in human breast cancers. Breast Cancer Research and Treatment. 2007;101:305–315. doi: 10.1007/s10549-006-9293-x. [DOI] [PubMed] [Google Scholar]
- Paschoud S, Bongiovanni M, Pache JC, Citi S. Claudin-1 and claudin-5 expression patterns differentiate lung squamous cell carcinomas from adenocarcinomas. Modern Pathology. 2007;20:947–954. doi: 10.1038/modpathol.3800835. [DOI] [PubMed] [Google Scholar]
- Pittman J, Huang E, Dressman H, Horng CF, Cheng SH, Tsou MH, Chen CM, Bild A, Iversen ES, Huang AT. Integrated modeling of clinical and gene expression information for personalized prediction of disease outcomes. Proceedings of the National Academy of Sciences. 2004;101:8431–8436. doi: 10.1073/pnas.0401736101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raftery AE, Madigan D, Hoeting J. Bayesian model averaging for linear regression models. Journal of the American Statistical Association. 1997;92:1197–1208. [Google Scholar]
- Renner M, Bergmann G, Krebs I, End C, Lyer S, Hilberg F, Helmke B, Gassler N, Autschbach F, Bikker F, and others DMBT1 confers mucosal protection in vivo and a deletion variant is associated with Crohn's disease. Gastroenterology. 2007;133:1499–1509. doi: 10.1053/j.gastro.2007.08.007. [DOI] [PubMed] [Google Scholar]
- Schafer J, Strimmer K. An empirical Bayes approach to inferring large-scale gene association networks. Bioinformatics. 2005;21:754–764. doi: 10.1093/bioinformatics/bti062. [DOI] [PubMed] [Google Scholar]
- Scott JG, Berger JO. An exploration of aspects of Bayesian multiple testing. Journal of Statistical Planning and Inference. 2006;136:2144–2162. [Google Scholar]
- Segal E, Shapira M, Regev A, Pe'er D, Botstein D, Koller D, Friedman N. Module networks: identifying regulatory modules and their condition-specific regulators from gene expression data. Nature Genetics. 2003;34:166–176. doi: 10.1038/ng1165. [DOI] [PubMed] [Google Scholar]
- Shi J, Levinson DF, Whittemore AS. Significance levels for studies with correlated test statistics. Biotstatistics. 2008;9:458–466. doi: 10.1093/biostatistics/kxm047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steuer R, Kurths J, Fiehn O, Weckwerth W. Observing and interpreting correlation in metabolomic networks. Bioinformatics. 2003;19:1019–1026. doi: 10.1093/bioinformatics/btg120. [DOI] [PubMed] [Google Scholar]
- Tierney L, Kadane J. Accurate approximations for posterior moments and marginal densities. Journal of American Statistical Association. 1986;81:82–86. [Google Scholar]
- Traina F, Favaro PM, Medina SS, Duarte AS, Winnischofer SM, Costa FF, Saad ST. ANKHD1, ankyrin repeat and KH domain containing 1, is overexpressed in acute leukemias and is associated with SHP2 in K562 cells. Biochimicia et Biophysica Acta. 2006;1762:828–834. doi: 10.1016/j.bbadis.2006.07.010. [DOI] [PubMed] [Google Scholar]
- Tusher VG, Tibshirani R, Chu G. Significance analysis of microarrays applied to the ionizing radiation response. Proceedings of the National Academy of Sciences. 2001;98:5116–5121. doi: 10.1073/pnas.091062498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wille A, Bühlmann P. Low-order conditional independence graphs for inferring genetic networks. Statistical Applications in Genetics and Molecular Biology. 2006 doi: 10.2202/1544-6115.1170. 5, article 1. [DOI] [PubMed] [Google Scholar]
- Yeung K, Bumgarner R, Raftery A. Bayesian model averaging: development of an improved multi-class, gene selection and classification tool for microarray data. Bioinformatics. 2005;21:2394–2402. doi: 10.1093/bioinformatics/bti319. [DOI] [PubMed] [Google Scholar]
- Yu J, Smith VA, Wang PP, Hartemink AJ, Jarvis ED. Advances in Bayesian network inference for generating causal networks from observational biological data. Bioinformatics. 2004;20:3594–3603. doi: 10.1093/bioinformatics/bth448. [DOI] [PubMed] [Google Scholar]
- Zhou X, Kao M-CJ, Wong WH. Transitive functional annotation by shortest-path analysis of gene expression data. Proceedings of the National Academy of Sciences. 2002;99:12783–12788. doi: 10.1073/pnas.192159399. [DOI] [PMC free article] [PubMed] [Google Scholar]