

Abstract
The zinc-dependent enzyme LpxC catalyzes the deacetylation of UDP-3-O-acyl-GlcNAc, the first committed step of lipid A biosynthesis. Lipid A is an essential component of the outer membranes of most Gram-negative bacteria, including Escherichia coli, Salmonella enterica and Pseudomonas aeruginosa, making LpxC an attractive target for antibiotic design. The inhibition of LpxC by a novel N-aroyl-L-threonine hydroxamic acid (CHIR-090) from a recent patent application (International Patent WO 2004/062601 A2 to Chiron and the University of Washington) is reported here. CHIR-090 possesses remarkable antibiotic activity against both E. coli and P. aeruginosa, comparable to that of ciprofloxacin. The biological activity of CHIR-090 is explained by its inhibition of diverse LpxC orthologs at low nM concentrations, including that of Aquifex aeolicus, for which structural information is available. The inhibition of A. aeolicus LpxC by CHIR-090 occurs in two steps. The first step is rapid and reversible, with a Ki of 1.0 - 1.7 nM, depending on the method of assay. The second step involves the conversion of the EI complex with a half-life of about a minute to a tightly bound form. The second step is functionally irreversible but does not result in the covalent modification of the enzyme, as judged by electrospray-ionization mass spectrometry. CHIR-090 is the first example of a slow, tight-binding inhibitor for LpxC, and may be the prototype for a new generation of LpxC inhibitors with therapeutic applicability.
The emergence of multi-drug resistant bacterial pathogens is a growing public health concern (1). Human and animal pathogens are developing resistance to every major class of commercial antibiotic, both natural and synthetic. New antibiotics directed against previously unexploited bacterial targets are urgently needed (2-4).
Zinc-dependent hydrolases are a well-studied class of proteins, many of which have set successful precedents for mechanism-based inhibitor design (5-7). Several bacterial metallo-amidases have been identified as potential antibiotic targets (7). Among them is LpxC, a zinc-dependent, cytoplasmic deacetylase involved in the biosynthesis of the lipid A component of lipopolysaccharide (Scheme 1) (8-11).
Scheme 1. Role of the deacetylase LpxC in the biosynthesis of lipid A.
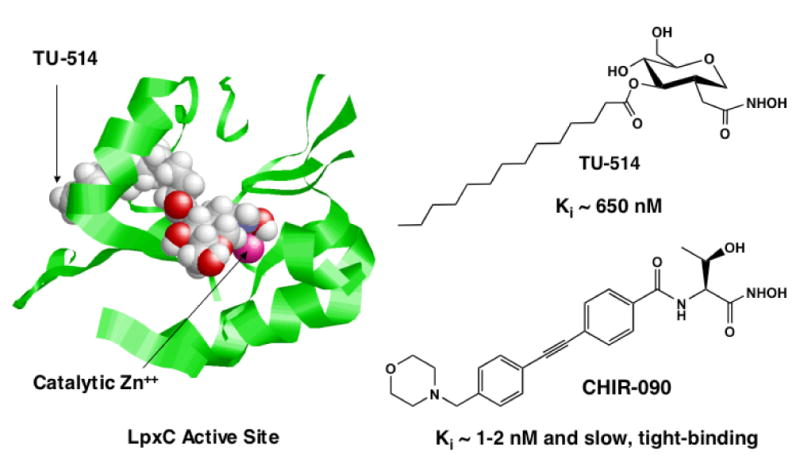
LpxA and LpxC are cytoplasmic enzymes. The enzymology and genetics of the lipid A pathway are best characterized in E. coli (15).
LpxC removes the acetate group from the nitrogen atom at the glucosamine 2 position of UDP-3-O-acyl-N-acetylglucosamine (Scheme 1) (12, 13). This reaction is the first committed step of lipid A biosynthesis (14) and is essential for bacterial growth (12, 13). LpxC is encoded by a single-copy gene, present in almost all Gram-negative bacteria (15), including Pseudomonas aeruginosa (16) and Escherichia coli (17). LpxC does not display sequence homology to other deacetylases or eucaryotic proteins, suggesting it can be targeted selectively with minimal risk of toxicity.
Despite its unique substrate specificity and sequence, LpxC does share some mechanistic features with other important metallo-amidases. As in thermolysin (18), angiotensin-converting enzyme (5, 19), the matrix metallo-proteinases (6), and peptide N-deformylase (20), a single transition metal ion is required for LpxC catalytic activity (9) and a glutamate side chain in the LpxC active site is thought to activate the Zn2+-bound water (21). Selective chelation of the LpxC active site Zn2+ ion by certain small molecules containing hydroxamic acid groups results in inhibition (Fig. 1) (8, 10, 11, 22, 23). The phenyloxazoline hydroxamic acid, L-161,240 (Fig. 1), is a competitive inhibitor of E. coli LpxC (Ki ∼ 50 nM) and displays a minimal inhibitory concentration (MIC) of 1-3 μg/mL against wild-type E. coli (8, 10). However, L-161, 240 is not active as an antibiotic against P. aeruginosa and other clinically important pathogens. A different series of sulfonamide-containing hydroxamates, such as BB 78485 (Fig. 1), possesses a slightly broader antibacterial spectrum, but the best of these compounds is still not effective enough to be developed as a therapeutic agent (11). Although the available pharmacologic (8, 11) and genetic work (13, 24, 25) validates LpxC as an excellent target, inhibitors with low nM Kis and potent antibiotic activity against diverse Gram-negative pathogens have not been reported.
Figure 1. Structures and LpxC inhibitory activities of selected hydroxamic acids.
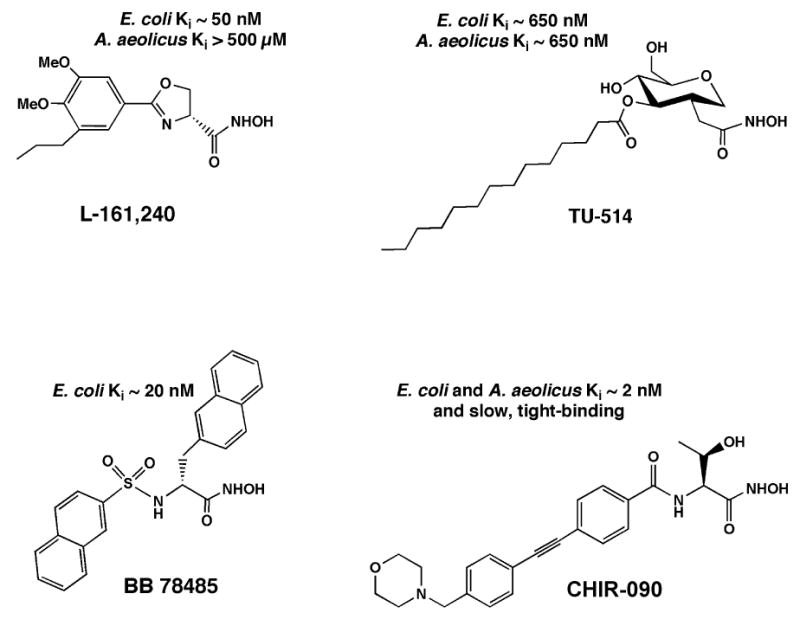
CHIR-090 is from International Patent WO 2004/062601 A2 (31). The slow, tight-binding inhibitory activity of CHIR-090 against E. coli and A. aeolicus LpxC was established in the present study. The structures of L-161,240, TU-514 and BB 78485, as well as their estimated Kis versus LpxC of E. coli and A. aeolicus, are from the literature (8, 10, 11).
Substrate-analog LpxC inhibitors, like TU-514 (10, 26), which contain a hydroxamic acid moiety (Fig. 1), are less potent than the above compounds. However, they effectively inhibit diverse LpxC orthologs in vitro, ranging from E. coli to Aquifex aeolicus LpxC. The latter displays only 32 % sequence identity to E. coli LpxC (10, 27, 28) and is completely resistant to L-161,240 (10). TU-514 possesses little or no antibacterial activity (10), because it probably cannot penetrate the Gram-negative cell envelope. However, the NMR solution structure of Aquifex LpxC in complex with TU-514 (Fig. 2) was recently solved (28, 29). In this structure (29) the hydroxamic acid group of the inhibitor is positioned to bind to the active site Zn2+ ion (Fig. 2), and the enzyme contains an unusual hydrophobic passage that captures the acyl chain of TU-514 (Fig. 2) (29). These features are presumed to contribute most of the binding energy for the natural substrate, UDP-3-O-acyl-N-acetylglucosamine (Scheme 1). The active site also contains a basic and a hydrophobic patch (29), which might provide additional binding energy. The x-ray structure of the zinc-inhibited form of Aquifex LpxC in the absence of TU-514 was reported simultaneously with the NMR structure (30). It revealed the same overall protein fold, fatty acid binding tunnel and zinc ligands.
Figure 2. Refined NMR structure of the TU-514/AaLpxC complex.
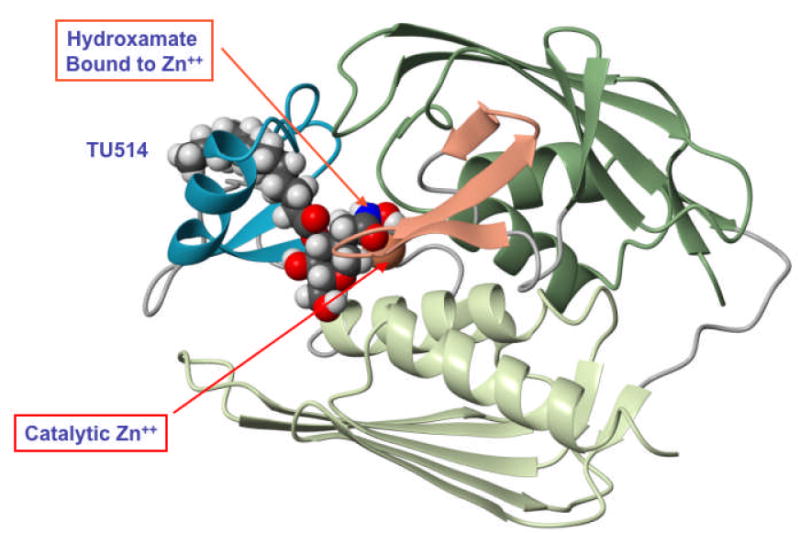
This ribbon diagram is based on the recent work of Coggins et al. (29).
We have now characterized CHIR-090, a member of a new series of LpxC-specific inhibitors that was recently disclosed in a joint Chiron/University of Washington patent application (International Patent WO 2004/062601 A2) (31). We demonstrate that the N-aroyl-L-threonine hydroxamic acid, designated as compound 15 (CHIR-090) (Fig. 1) in the patent application (31), displays extraordinary antibacterial activity against both E. coli and P. aeruginosa. Binding studies1 had revealed that CHIR-090 displayed high affinity for the active sites of both E. coli and P. aeruginosa LpxC, since CHIR-090 displaced a variety of 19F-labeled inhibitors (22, 31) from both orthologs, as judged by NMR assays. We now show that the inhibition of A. aeolicus and E. coli LpxC by CHIR-090 occurs with low nM potency and is also time-dependent. Our data support a two-step inhibition mechanism, which is competitive with low nM potency in the first stage and irreversible in the second. Although CHIR-090 inactivates A. aeolicus LpxC completely in the second stage, there is no evidence of a covalent modification, as judged by mass spectrometry. The time-dependent inhibition of LpxC by CHIR-090, which was not reported in the patent application (31), may contribute to its potent antibiotic activity. CHIR-090 is the first example of a slow, tight-binding LpxC inhibitor. It may be possible to improve the potency of CHIR-090 even further, given that it was synthesized prior to recent structural studies of LpxC.
Materials and Methods
Buffers and Reagents
[α-32P]-UTP was purchased from NEN Dupont. PEI-cellulose TLC plates were purchased from EMD Chemicals, Gibbstown, NJ. Bovine serum albumin (BSA) and sodium phosphate were obtained from Sigma-Aldrich, St. Louis, MO. Dimethylsulfoxide (DMSO) was purchased from Mallinckrodt, Phillipsburg, NJ. Tube-O-Dialyzer dialysis tubes were purchased from GenoTech, St. Louis, MO. Filter paper (# 42) was purchased from Whatman, Florham Park, NJ. All other reagents were of high commercial grade. The LpxC inhibitor CHIR-090 was synthesized at the University of Washington, Seattle, and an initial sample was provided from that synthetic lot. Later samples were prepared on a 200 mg scale at the Duke University Chemical Biology facility, according to the published procedure (31).
Activity assays
A. aeolicus LpxC-catalyzed deacetylation was measured with the substrate [α-32P]-UDP-3-O-(R-3-hydroxymyristoyl)-N-acetylglucosamine, prepared enzymatically as previously described (9, 10). The specific radioactivity of the substrate varied from 200,000 to 4,000,000 cpm/nmol. Assays were performed in 1 mg/mL BSA and 25 mM sodium phosphate buffer, pH 7.4. The concentration of non-radioactive carrier substrate was 5 μM, unless otherwise indicated. Reactions were performed at 30 °C in a heat block and initiated with the addition of E. coli (Ec) LpxC (0.1 nM) or A. aeolicus (Aa) LpxC (0.7 - 0.9 nM). Final inhibitor concentrations in the assay were varied as indicated. Inhibitor solution stocks were stored in 100 % DMSO at −20 °C. The final concentration of DMSO maintained in the assay was 10 %. Assays were performed using a combination of thin layer chromatography and PhosphorImager analysis, as previously described (9, 10, 21).
Antibacterial disc tests
Liquid cultures of each strain (32) (E. coli R477, E. coli R477 [LpxC G17S] or P. aeruginosa PAO1) were grown from single colonies in LB broth (33) at 37 °C to A600 ∼ 0.2. A lawn of cells of each strain was spread onto LB-agar plates with sterile cotton swabs. Sterile filter-paper discs (Whatman # 42) were placed on top of the bacterial lawn, and then 20 μg of compound was spotted onto each disc (2 μl of 10 mg/mL stocks). DMSO (100 %) was also spotted onto a separate disc as a control. Plates were then incubated overnight at 37 °C. A halo of killing was clearly visible on the next day around discs that had been spotted with active compounds. The diameter of each halo was measured for each compound and strain.
Initial screening of CHIR-090 for time-dependent inhibition
CHIR-090 was preincubated at 0.02 μg/mL (46 nM) with 3.3 nM of AaLpxC in 1 mg/mL BSA and 25 mM sodium phosphate buffer, pH 7.4, for 1 min at 30 °C. AaLpxC was also preincubated by itself with DMSO under the same conditions as a control. The final concentration of DMSO in all samples was 10 %. Preincubation reactions were then diluted 1:4 into the assay mixture containing 5 μM UDP-3-O-acyl-N-acetylglucosmine, 1 mg/mL BSA, and 25 mM sodium phosphate, pH 7.4, and assayed for activity. The final concentration of CHIR-090 in the reaction, initiated with AaLpxC preincubated with inhibitor, was 0.005 μg/mL (11.5 nM), and the protein concentration was 0.8 nM. Control reactions were performed in which inhibitor was included in the assay mixture at 0.005 μg/mL (11.5 nM), and the reaction was initiated with the addition of AaLpxC preincubated with DMSO (but with no inhibitor during the preincubation). The activity of the control reaction was compared to the activity of the reaction initiated with AaLpxC preincubated with inhibitor.
Dialysis of inhibitor-protein complexes
Samples containing CHIR-090 and AaLpxC were preincubated as described above and tested for reversibility of inhibition. CHIR-090 was included at a 1.5-fold molar excess over AaLpxC and incubated at 30 °C. Control samples were prepared that contained either no inhibitor or no protein. A portion of each sample was removed after one hour and stored overnight at −80 °C. The remaining sample was subjected to overnight dialysis at 4 °C in 25 mM sodium phosphate buffer, pH 7.4, and 10 % glycerol. Dialysis was performed in micro Tube-O-dialyzer with a molecular weight exclusion of 8000 Da. The previously frozen preincubated samples and the dialyzed samples were diluted into the assay mixture, containing 5 μM UDP-3-O-acyl-N-acetylglucosmine, 1 mg/mL BSA, and 25 mM sodium phosphate, pH 7.4, so that the final enzyme concentration was 0.6 - 0.9 nM, and assayed accordingly.
Electrospray ionization mass spectrometry of the CHIR-090-AaLpxC complex
AaLpxC and CHIR-090 were incubated at a 2.5-fold molar excess of protein (12.6 μM AaLpxC and 5.0 μM CHIR-090) or at a 67-fold molar excess of CHIR-090 (15 μM AaLpxC and 1 mM CHIR-090) for 2 h at 30 °C in 25 mM sodium phosphate, pH 7.4, and 10 % DMSO. Control samples were prepared that contained either AaLpxC or CHIR-090 alone. All samples were subjected to LC followed by ESI/MS. A Shimadzu LC system (comprising a solvent degasser, two LC-10A pumps and a SCL-10A system controller) was coupled to a QSTAR XL quadrupole time-of-flight tandem mass spectrometer (ABI/MDS-Sciex), equipped with an electrospray source. The LC was operated at a flow rate of 200 μL/min with a linear gradient as follows: 100 % solvent A was held isocratically for 2 min, linearly increased to 60 % solvent B over 18 min, and then increased to 100 % solvent B over 5 min. The mobile solvent A consists of water:acetonitrile (98:2 vol/vol) with 0.1 % acetic acid. The mobile solvent B consists of acetonitrile:water (90:10 vol/vol) with 0.1 % acetic acid. A Zorbax reverse-phase column (SB-C8, 2.1 × 50 mm) (Agilent Technology) was used for all LC-MS analyses.
TU-514 protection assays
AaLpxC (1 μM) was preincubated with equimolar CHIR-090 (1 μM) or 100-fold molar excess TU-514 (100 μM) or both for 15 min at 30 °C. The preincubated samples were then diluted 800-fold into the assay mixture, containing 5 μM UDP-3-O-acyl-N-acetylglucosmine, 1 mg/mL BSA, and 25 mM sodium phosphate, pH 7.4, so that the final enzyme concentration was 1.25 nM. Control reactions were prepared that contained 1.25 nM CHIR-090 or 125 nM TU-514, or both, in the assay mixture only, and the reactions were initiated with AaLpxC preincubated with DMSO alone. Their remaining activity was quantified by comparison to an inhibitor-free reaction, initiated with AaLpxC that had been preincubated with DMSO alone.
IC50 assays of AaLpxC
Purified AaLpxC (0.7 nM) was assayed with increasing concentrations of TU-514. Assays at pH 5.5 were performed at 30 °C in 40 mM bis-Tris, pH 5.5, with 1 mg/mL BSA, 5 μM UDP-3-O-acyl-N-acetylglucosamine, and 10 % DMSO. Assays at pH 7.4 were performed at 30 °C in 25 mM sodium phosphate, pH 7.4, with 1 mg/mL BSA, 5 μM UDP-3-O-acyl-N-acetylglucosamine, and 10 % DMSO.
Results
Inhibition of LpxC Activity by a Novel N-Aroyl-L-Threonine Hydroxamic Acid
CHIR-090 (Fig. 1) was initially examined for LpxC inhibitory activity by assaying it at 0.5 μg/mL (1.15 μM) against purified LpxC from either E. coli (EcLpxC) or A. aeolicus (AaLpxC) (Table 1). For comparison, the previously reported competitive inhibitors, L-161, 240 (8) and TU-514 (10) (Fig. 1), were also tested. L-161, 240 is a phenyloxazoline hydroxamic acid that inhibits EcLpxC in a competitive manner with a Ki of 50 nM (Fig. 1) (8, 10), and it is bactericidal against E. coli. This compound shows no inhibitory activity against AaLpxC (Table 1) (10), and it is at least an order of magnitude less effective against LpxC from Pseudomonas aeruginosa than from E. coli (A. L. McClerren, A. Barb and C. R. H. Raetz, unpublished). TU-514 is a substrate-analogue (Scheme 1 and Fig. 1), containing a hydroxamic acid moiety, which inhibits both EcLpxC and AaLpxC with a Ki of ∼ 650 nM, when assayed at pH 5.5 (10). TU-514 does not exhibit significant antibacterial activity, since it probably cannot penetrate the cell envelope (Scheme 1). CHIR-090 (Fig. 1), from the new class of recently described compounds (31), inhibited both EcLpxC and AaLpxC by 98 % when tested at 0.5 μg/mL (1.1 μM) in the standard assay system (Table 1).
Table 1.
LpxC inhibition and/or antibacterial activity of selected compounds.
| Compound | % inhibition EcLpxC | % inhibition AaLpxC | G17S mm killing c |
R477 mm killing c |
PAO1 mm killing c |
Time Dependent d |
|---|---|---|---|---|---|---|
| L-161,240 a | 96 | 3 | 30 | 19 | < 6 | no |
| TU-514 a | 50 | > 99 | < 6 | < 6 | < 6 | no |
| CHIR-090 b | 98 | 98 | 38 | 36 | 34 | yes |
| tobramycin | Not tested | Not tested | 28 | 26 | 24 | N/A |
| ciprofloxacin | Not tested | Not tested | 37 | 35 | 40 | N/A |
Tested at 1 μg/mL in the assay at pH 7.4
Tested at 0.5 μg/mL (1.15 μM) in the assay at pH 7.4
Tested at 20 μg/disc (disc diameter = 6 mm)
Tested by preincubation of AaLpxC (3.3 nM) with 0.02 μg/mL inhibitor
Antibiotic Activity of LpxC Inhibitors Compared to Tobramycin and Ciprofloxacin
Selected compounds were evaluated by a simple disc diffusion assay for their antibacterial activity against strains of E. coli and P. aeruginosa. A small volume of inhibitor (2 μl from a 10 mg/mL stock solution) was spotted onto a sterile filter paper disc, placed on top of a lawn of wild-type E. coli R477, an E. coli R477 mutant (LpxC G17S), or wild-type P. aeruginosa (PAO1). Compounds with antibiotic activity against a given strain exhibited a halo of killing after overnight incubation at 37 °C. Previously characterized LpxC inhibitors in other structural series (8, 11, 22, 23, 34), like L-161,240 (Fig. 3 and Table 1), were not effective against P. aeruginosa under these conditions. In contrast, CHIR-090 showed remarkable antibiotic potency against both E. coli and P. aeruginosa (Fig. 3 and Table 1). CHIR-090 activity was comparable to that of ciprofloxacin and greater than that of tobramycin (Fig. 3 and Table 1), both of which are used clinically to treat Gram-negative infections. Ciprofloxacin is one of the most potent antibiotics of the quinolone class that targets bacterial DNA gyrase (35). Tobramycin is an aminoglycoside that inhibits bacterial protein synthesis (36). As previously reported, L-161,240 showed modest antibiotic activity against wild-type E. coli (Fig. 3) (8). The hypersensitivity of an E. coli strain, producing the partially-inactivated LpxC mutant G17S (8, 32), to L-161,240 was also confirmed (Fig. 3 and Table 1).
Figure 3. Antibiotic activities of CHIR-090 and L-161,240 compared to tobramycin and ciprofloxacin.
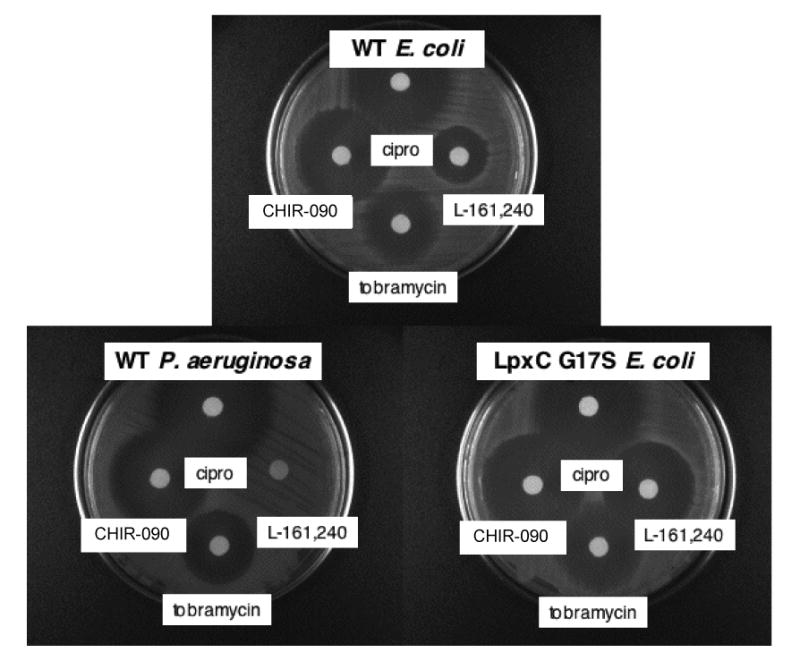
Disc diffusion tests were used to evaluate antibacterial activity. Wild-type E. coli (R477), the LpxC inhibitor hypersensitive E. coli R477 (LpxC G17S), and wild-type P. aeruginosa (PAO1) were tested for killing by L-161,240, CHIR-090, tobramycin, and ciprofloxacin. Each disc contained 20 μg of compound dissolved in DMSO.
Time-dependent Inhibition of AaLpxC Activity
In order to study its mechanism and quantify its potency, CHIR-090 was tested for time-dependent inhibition of AaLpxC activity. AaLpxC was chosen because of the wealth of structural and mechanistic information available for this ortholog (Fig. 2) (28-30, 37, 38). As an initial test for time-dependent inhibition, CHIR-090 was preincubated at 0.02 μg/mL (46 nM) for 1 minute at 30 °C with 3.3 nM AaLpxC before dilution (1:4) into the standard assay mixture, which contained the substrate but no additional inhibitor. A control preincubation was prepared in parallel that also contained 3.3 nM AaLpxC and DMSO, but no inhibitor. The control was then diluted 1:4 into the standard assay system, containing either the substrate alone or the substrate and the inhibitor (at a final concentration of 0.005 μg/mL or 11.5 nM). The rate of deacetylation catalyzed by AaLpxC, preincubated with the inhibitor, was then compared to the rates catalyzed by AaLpxC, preincubated with DMSO, either in the absence or the presence of 11nM inhibitor in the assay mixture. Under these conditions, CHIR-090 partially inhibited AaLpxC (25 % remaining activity) when it was included in the assay at 11.5 nM, versus in the absence of inhibitor. However, CHIR-090 completely inhibited AaLpxC following preincubation and dilution to the same final inhibitor concentration of 11.5 nM (< 3 % remaining activity). This result suggested that CHIR-090 is a time-dependent inhibitor, which was not noted in the recent patent disclosure (31).
We next examined the reversibility of the inhibition exhibited by CHIR-090. AaLpxC was preincubated with CHIR-090 at 1.5-fold molar excess for 1 hour at 30 °C to inactivate the enzyme completely. Controls were prepared in parallel that contained either no inhibitor or no protein. All preincubated samples were dialyzed overnight at 4 °C and assayed for activity. The dialyzed sample, containing only 0.6-0.9 nM AaLpxC, retained 80 - 100% of its activity when compared to the activity of fresh AaLpxC (data not shown). No activity was observed in the dialyzed CHIR-090/AaLpxC mixture, indicating that the inhibition of AaLpxC by CHIR-090 results in the formation of an extremely stable EI complex with a half-life greater than 16 h. The dialyzed sample, containing only CHIR-090, was diluted into a reaction mixture so that its final concentration in the assay (assuming it was not removed by dialysis) would be 2 nM. This reaction mixture was then assayed by the addition of untreated AaLpxC. No inhibition of LpxC activity was observed (data not shown), demonstrating that unbound CHIR-090 can be removed by overnight dialysis.
Mechanism of Time-dependent Inhibition
The data presented so far demonstrate that CHIR-090 is an extraordinarily potent LpxC inhibitor with a possible time-dependent mechanism. An interaction between an enzyme and an inhibitor to yield a single enzyme-inhibitor complex that then exhibits time-dependent inhibition can occur in one of two ways (39). In both cases, the association of the enzyme and substrate is described by k1, while the dissociation of the enzyme-substrate complex is described by k2. The first mechanism of inhibition involves a single step and can be described by the following equation where E represents the enzyme and I is the inhibitor:
| (Mechanism 1) |
Mechanism 2, which is the more common scenario (39), involves two steps. The first is the rapid formation of the initial enzyme-inhibitor complex (EI) followed by the slow conversion of EI to form the final EI* complex. This sequence of events is shown below:
| (Mechanism 2) |
In Mechanism 1, the kinetics can result from the slow association of E and I or, in some cases, the slow dissociation of EI. In contrast, the formation of EI is rapid in Mechanism 2, and it is the conversion to reach EI* that is slow. These two inhibition models presume that the slow-binding inhibition step is reversible. Since we have shown that CHIR-090 is essentially an irreversible inhibitor of Aquifex LpxC, we can modify these models such that k4 (Mechanism 1) or k6 (Mechanism 2) is set to zero. These are the same equations used to describe covalent, slow-binding inhibitors. Using this kinetic model does not necessarily imply that the inhibition is the result of a covalent modification of the enzyme. The possibility of covalent modification of LpxC by CHIR-090 is addressed below.
In order to distinguish between Mechanisms 1 and 2, we measured the progress curves of AaLpxC-catalyzed product formation in the presence of increasing amounts of CHIR-090 (Fig. 4A). AaLpxC (0.7 nM), which was used to start the reaction, was assayed in the presence of 0 to 4.55 nM CHIR-090. For a slow, tight-binding inhibitor, progress curves are biphasic, exhibiting an initial burst of activity, followed by a slower, steady-state rate. This type of inhibition is described by Equation 1,
Figure 4. Progress curves for AaLpxC in the presence of increasing concentrations of CHIR-090.

Panel A) Time-dependent LpxC inhibition by CHIR-090. Wild-type AaLpxC (0.7 nM), which had not been preincubated with inhibitor, was used to start the assay in the presence of 0 (solid squares), 0.57 (solid circles), 1.14 (solid triangles), 1.7 (solid half-squares), 2.28 (open squares), 2.85 (open circles), 3.7 (open triangles), or 4.55 (open half-squares) nM CHIR-090. Panel B) Initial velocity as a function of CHIR-090 concentrations. Initial velocities were calculated from fitting the data obtained from the progress curves, shown in Panel A, to Equation 1 before the onset of time-dependent inhibition. These re-plotted data were fit to Equation 2 to determine the inhibition constant (Ki) for the initial binding of CHIR-090 to AaLpxC. Panel C) First order rate constant kobs as a function of CHIR-090 concentration. The kobs values were calculated from fitting the progress curves obtained in Panel A to Equation 1.
| Eq. 1 |
where P is the concentration of product formed at any time t, v0 is the initial velocity, vS is the final steady-state velocity, and kobs is the apparent first-order rate constant for the conversion from v0 to vS (39). C is a constant, non-zero y-intercept term.
The initial velocity may vary as a function of inhibitor concentration, while the steady-state velocity will decrease at increasing concentrations of inhibitor. In the case of mechanism 1, time-dependent inhibition occurs in a single step such that the initial velocity measured at increasing concentrations of inhibitor would remain the same. In the case of Mechanism 2, the two-step inhibition process is evident by the decrease in the initial velocity of the reaction at increasing inhibitor concentrations. Analysis of the LpxC progress curves in the presence of CHIR-090 follows the pattern in which enzyme and inhibitor form an initial EI complex rapidly, followed by slow conversion to a tighter EI* complex (Mechanism 2). This result is demonstrated upon examination of the initial rates of the progress curves (Fig. 4A). Each curve exhibits a time range in which the initial rate does not deviate from linearity (at least three time points at each inhibitor concentration tested) before the onset of the slower, time-dependent inhibition. CHIR-090 shows a measurable effect on these initial rates of catalysis by decreasing the rate of product formation at increasing inhibitor concentrations. The value of the inhibition constant Ki (k4/k3 from Mechanism 2) associated with the reversible formation of EI can be determined from these initial rates of product formation, when the formation of EI* is so small as to be negligible, using Equation 2.
| Eq. 2 |
The value of Ki determined from the actual data was 1.7 nM (Fig. 4B).
The observed first-order rate constant kobs describes the time-dependent depletion of the initial EI encounter complex to form EI*. Fixing vs at zero, in agreement with complete inhibition of activity in the EI* complex, kobs was determined by fitting to Equation 1 (Fig. 4C). This apparent rate constant follows a hyperbolic function, supporting the two-step mechanism, and can be deconvoluted to yield k5 by Equation 3, the microscopic rate constant describing the conversion of EI to EI*.
| Eq. 3 |
For the inhibition of AaLpxC by CHIR-090, setting k6 to zero, k5 was determined to be 0.0031 s-1, yielding a half-life for the formation of EI* of 56 s.
The Initial Stage of AaLpxC Inhibition by CHIR-090 is Competitive
To confirm the inhibition constant (Ki) for the initial binding of CHIR-090 to AaLpxC and to demonstrate competitive binding with respect to substrate, we measured the initial velocity (v0) of AaLpxC in the presence of increasing concentrations of CHIR-090 (0, 1.7, 2.85, and 3.6 nM) at various concentrations of substrate (1, 2, 3, 10, and 20 μM). Data were collected for times prior to EI* formation, and thus product accumulation was linear with respect to time. The initial rates obtained were plotted at different CHIR-090 concentrations as a function of substrate concentration (Fig. 5). The double-reciprocal plot of CHIR-090 inhibition exhibits a pattern consistent with competitive inhibition in which all lines converged on the y-axis at the same point. The data were fit to Equation 2 for competitive inhibition and yielded a Ki value of 1.00 ± 0.15 nM, in good agreement with 1.7 nM obtained above from the global time-dependent fit. The KM of AaLpxC for UDP-3-O-acyl-N-acetylglucosmine determined from this data set was 1.6 ± 0.2 μM, consistent with published results (9, 21).
Figure 5. Double-reciprocal plots of the global fit of the initial velocities of the AaLpxC initiated reactions at increasing concentrations of CHIR-090.

These data were collected in an independent experiment in which the concentration of substrate was also varied. The Ki value for CHIR-090 determined from this experiment was 1.00 ± 0.15 nM, and the KM for UDP-3-O-acyl-N-acetylglucosamine was 1.6 ± 0.2 μM, using Equation 2. The values calculated are the averages from to two separately fit sets of data.
Time-dependent Inhibition is Not Due to Irreversible Covalent Modification
Because the inhibition of AaLpxC by CHIR-090 appears to be irreversible on our experimental time scale, we considered the possibility that CHIR-090 covalently modifies AaLpxC. We incubated AaLpxC and CHIR-090 at a 2.5-fold molar excess of AaLpxC (or at a 140-fold molar excess of CHIR-090) for up to two h at 30 °C. The complex was then subjected to LC/ESI-MS, as described in Materials and Methods. The ESI mass spectra exhibited two peaks after deconvolution, consistent with the native molecular weight of CHIR-090 and of AaLpxC, but with no evidence of covalent adduct formation (Fig. 6). Furthermore, the molecular weights obtained for CHIR-090 and AaLpxC in the preincubated samples were the same as those determined for the respective control samples of each. These results rule out an acid-labile modification that would have altered the masses of the individual components seen by mass spectrometry and suggest that CHIR-090 does not covalently modify AaLpxC.
Figure 6. LC/ESI-MS analysis of AaLpxC pre-incubated with CHIR-090.
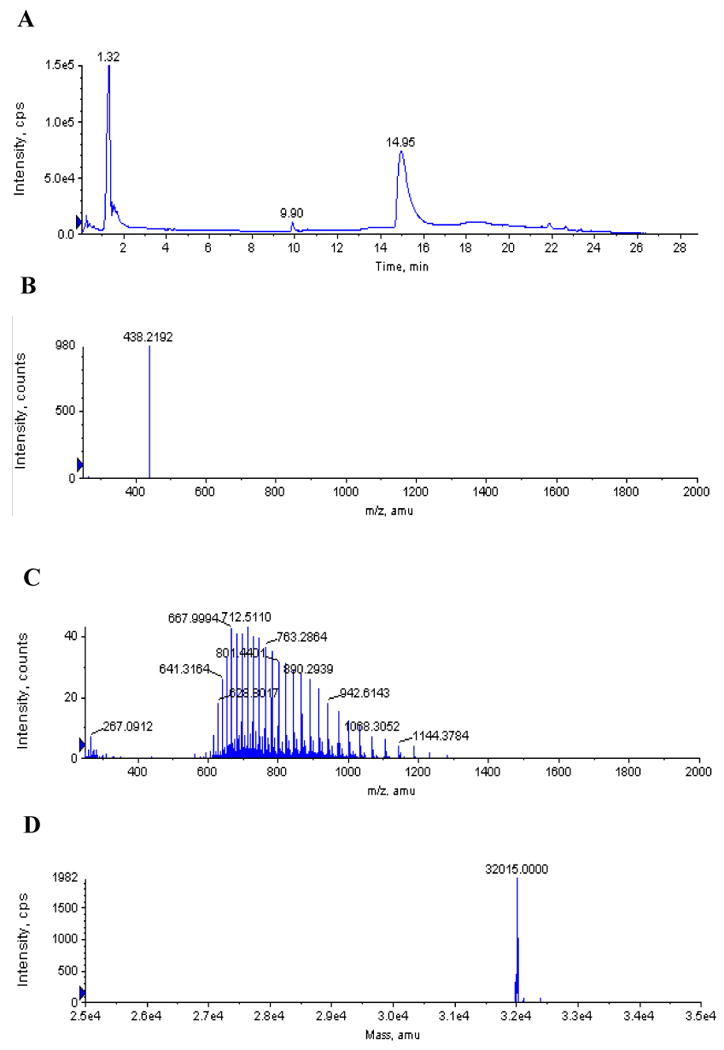
AaLpxC (12.6 μM) and CHIR-090 (5 μM) were incubated for two h at 30 °C and then subjected to LC/ESI-MS analysis. Panel A) Total ion chromatogram of the LC. Panel B) Positive ion mass spectrum collected at 9.9 min. The ion at m/z 438.2 matches the expected [M + H]+ for unmodified CHIR-090. Panel C) Positive ion mass spectrum collected at 14.95 min, showing the multiple charge states of the LpxC protein. Panel D) Deconvoluted spectrum from panel C. The calculated average neutral mass (32,015) is within experimental error of the molecular weight of AaLpxC, lacking a methionine residue (32,013) (41). There was no evidence for a CHIR-090 adduct with AaLpxC.
The model of slow, tight-binding (but non-covalent) inhibition of AaLpxC by CHIR-090 is further supported by assays with several active site mutants. Previous site-directed mutagenesis demonstrated that the E73A, H253A, K227A, H19A, and D234N substitutions each reduced the catalytic activity of AaLpxC by several orders of magnitude (21, 27-29, 37, 38). If the inhibition of AaLpxC by CHIR-090 resulted in a covalent modification, one or more of these active site residues might be essential for the chemistry of this modification. Because of the low residual activity of these mutants, assays at low micromolar concentrations of protein precluded direct Ki determinations in the nanomolar range. Instead, each mutant protein at ∼ 0.5 μM was assayed with a 1.5-fold molar excess of CHIR-090. If any of the mutant proteins were less sensitive to CHIR-090 inhibition versus the wild-type protein, some residual activity might be retained under these experimental conditions. However, all the mutant proteins were inhibited completely (data not shown). This result rules out the possibility that these active site residues are targets for covalent modification by CHIR-090. It furthermore suggests that contact between CHIR-090 and E73, H253, H19, K227, or D234 is not crucial for inhibition.
CHIR-090 Binds to the Same Region of LpxC as Does TU-514
The NMR solution structure of AaLpxC was previously solved in complex with TU-514 (Fig. 2), a substrate-mimetic, hydroxamic acid inhibitor of both EcLpxC and AaLpxC (Fig. 1) (29). The binding region for TU-514 is well defined in the refined AaLpxC NMR structure (Fig. 2), revealing a hydrophobic tunnel that encloses the acyl chain of TU-514, as well as the contacts between the protein and the hexose ring of TU-514 near the catalytic Zn2+ ion (29).
Under our current assay conditions at pH 7.4 (Fig. 7), we observed significantly greater inhibition of AaLpxC by TU-514 than would have been expected based on the previously published assays at pH 5.5 (10). We therefore examined the pH-dependence of TU-514 inhibition of AaLpxC by determining the IC50 of this inhibitor at pH 7.4 and pH 5.5. The resulting data were fit to Equation 4,
Figure 7. TU-514 protection of AaLpxC against irreversible inhibition by CHIR-090.

AaLpxC was preincubated alone or in the presence of TU-514, CHIR-090, or both TU-514 and CHIR-090. Samples were then assayed for activity. Control reactions were performed in which AaLpxC was assayed alone or with TU-514, CHIR-090, or both TU-514 and CHIR-090, but without a preincubation. The remaining activity is expressed as the percent of the uninhibited control.
| (Eq. 4) |
where vi represents the initial velocity at a given concentration of inhibitor, vc represents the initial velocity of a control reaction containing no inhibitor, and I is the concentration of inhibitor. Remarkably, the IC50 of this compound is more than 1000-fold lower at pH 7.4 than at pH 5.5 (Supplemental Fig. 1). This effect is specific for the LpxC protein from A. aeolicus, since the IC50 of TU-514 with E. coli LpxC is approximately the same at pH 7.4 to 5.5 (data not shown). In the initial stage of inhibition CHIR-090 binds AaLpxC competitively with substrate, as previously shown for TU-514 (10). Together, these results suggest that CHIR-090 and TU-514 both bind to AaLpxC within the same substrate-binding pocket. The addition of TU-514 to a preincubation mixture of CHIR-090 and AaLpxC might therefore be expected to confer protection against the time-dependent inhibition by CHIR-090. In order to test this idea, samples containing 1 μM AaLpxC and 100 μM TU-514, 1 μM AaLpxC and 1 μM CHIR-090, or 1 μM AaLpxC and both 100 μM TU514 and 1 μM CHIR-090 were prepared and preincubated prior to dilution and the assay. Each sample was incubated at 30 °C for 1 hour before dilution into a standard assay mixture containing 5 μM substrate. A control reaction, initiated with AaLpxC that had been preincubated in the absence of inhibitors, was used to normalize the relative residual activities. Preincubation of AaLpxC with TU-514 showed ∼ 25 % activity compared to the no inhibitor control (Fig. 7). Preincubation of AaLpxC with DMSO alone and assyed with TU-514 shows the same extent of inhibition, demonstrating that TU-514 does not inhibit AaLpxC in a time-dependent manner, as expected from previously published results (10). In contrast, an equimolar preincubation of AaLpxC with CHIR-090 resulted in a complete loss of activity when compared to the no inhibitor control, consistent with the time-dependent inhibition mechanism described above. For comparison, a preincubation of AaLpxC with DMSO alone and assayed with an equimolar amount of CHIR-090 in the final reaction mixture retained 75 % of the control activity when measured before the onset of the slow, tight-binding inhibition. Strikingly, preincubation of AaLpxC with both TU-514 and CHIR-090 resulted in a preparation that retained 25 % residual activity when compared to the control (Fig. 7). This is the same rate seen for the preincubation of AaLpxC and TU-514, suggesting that the inhibition observed after dilution into the assay mixture is due to TU-514 alone. Thus, a 100-fold molar excess of TU-514 over CHIR-090 appears to protect AaLpxC from time-dependent inhibition by CHIR-090, presumably by competing for the same binding site on the enzyme.
Discussion
In contrast to classical competitive inhibitors, slow, tight-binding inhibitors take several seconds or min to inhibit their targets (39). Time-dependent inhibitors frequently display longer effective half-lives in biological systems than do classical competitive inhibitors, because of slow dissociation from their enzyme targets (39). Accumulation of substrate has no effect on this slow dissociation process and cannot overcome the inhibition (39). In recent years many time-dependent inhibitors have been characterized for enzyme targets with clinical applications, some accompanied by covalent modification but many without (40). This stability makes these time-dependent inhibitors very desirable as therapeutic agents (40).
We have now discovered that CHIR-090 (Fig. 1), a member of a new class of LpxC inhibitors recently reported in the patent literature (31), inhibits AaLpxC (Fig. 4) and other LpxC orthologs (data not shown) in a time-dependent manner. This unexpected finding, together with its low nM potency, may account for the remarkable antibacterial activity observed with CHIR-090 against E. coli and P. aeruginosa (Fig. 3). The mechanism of CHIR-090 inhibition has been characterized in depth. In contrast to the previously reported competitive inhibitors, TU-514 or L-161,240 (Fig. 1), CHIR-090 binds to AaLpxC in a two-step manner. The first step is rapid and reversible. The second step involves slow conversion to a tighter enzyme-inhibitor complex, which has a half-life of 56 s for formation. This time-dependent step is essentially irreversible on the scale of a typical steady-state kinetics experiment, given the immeasurable off-rate during overnight dialysis. The initial binding constant (Ki) in the first step is 1 - 2 nM, which is at least one order of magnitude more potent than the Kis of previously reported LpxC inhibitors (Fig. 1). However, the conversion to the tighter binding complex unmasks the true potency of CHIR-090, as the formation of the final enzyme-inhibitor complex is apparently irreversible.
The AaLpxC-inhibitor complex was examined for evidence of a covalent modification by CHIR-090. We were unable to detect an enzyme-inhibitor adduct by electrospray-ionization mass spectrometry (Fig. 6). The spectra exhibited two peaks, one consistent with the native molecular weight of the inhibitor and the other consistent with the mass of unmodified AaLpxC. This result shows that CHIR-090 does not modify AaLpxC covalently in an irreversible manner. The absence of an irreversible covalent complex between CHIR-090 and LpxC is consistent with the lack of chemically reactive groups in the CHIR-090 molecule. Although the inert acetylenic group of this compound might be activated for covalent modification of an enzyme active site, such activations typically occur in enzymes that generate carbanions as intermediates, perform oxidative reactions, or catalyze oxygen transfer processes. This type of chemistry is not employed by LpxC. However, some unique feature of the structure of CHIR-090 must allow it to bind with extraordinary affinity to the enzyme, leading to essentially irreversible inhibition. CHIR-090 presumably utilizes its hydroxamic acid group to chelate the active site Zn2+ (Fig. 2) and likely hydrogen bonds to other active site residues as well.
We have determined that CHIR-090 binds to AaLpxC competitively with respect to substrate in the initial phase of the interaction (Fig. 5). We have also shown that binding is competitive with respect to the substrate-analog inhibitor TU-514, because adding a large excess of TU-514 to a preincubation mixture of CHIR-090 and AaLpxC can prevent time-dependent inhibition by CHIR-090 (Fig 7). This result suggests that TU-514 and CHIR-090 share the same or overlapping binding pockets on LpxC. From the previous NMR studies of the TU-514/AaLpxC complex (Fig. 2), the binding pocket for TU-514 is well defined (29). The hydroxamic acid moiety of CHIR-090 presumably also binds the active site Zn2+ ion, displacing a solvent water molecule. This interaction may orient the side chain of CHIR-090 correctly for insertion into the same hydrophobic passage in which the acyl chain of TU-514 is located (Fig. 2). However, the conformation of the CHIR-090/AaLpxC complex, including the shape of the binding pocket, may differ from what is seen in the TU-514/AaLpxC complex. Previous structural work has suggested that the residues that form the hydrophobic passage and active site may undergo substantial motion in the absence of ligand or inhibitor (28). It might be this dynamic property that allows LpxC to clamp down on CHIR-090, leading to the potent time-dependent inhibition reported herein. Furthermore, an interaction of CHIR-090 with the putative UDP binding site of LpxC cannot be excluded, because the UDP site has not been identified either by NMR or crystallography (28-30). Additional structural studies of the CHIR-090-AaLpxC complex are clearly needed to help define the binding pocket of CHIR-090 in greater detail and to explain the irreversible nature of the inhibition.
Implications for Antibiotic Development
The overall effectiveness of CHIR-090 can be described in terms of an initial binding affinity (Ki) and a rate of inactivation (k5). Understanding and improving the features of CHIR-090 that contribute to both the initial and tight binding steps should facilitate the further development of LpxC inhibitors as clinically useful antibiotics. Other important inhibitor modifications to consider in future designs include alternative zinc binding ligands, such as sulfhydryl, carboxyl or phosphinic acid groups (5, 10, 34) and structural motifs to enhance bacterial penetration and pharmacokinetics in animals. The remarkable inhibitory activity, antibiotic potency and antibiotic spectrum displayed by CHIR-090 is at least an order of magnitude greater than for any previously reported LpxC inhibitor (Fig. 1). The slow, tight-binding feature of CHIR-090 (Fig. 4) is without precedent and is an extremely desirable feature in an antibiotic that is subject to extrusion by bacterial export pumps. In summary, the recent synthesis of CHIR-090 (31) and our discovery of its unexpected slow, tight-binding inhibition kinetics greatly enhance the likelihood that compounds with clinical utility can be developed around the LpxC reaction of the lipid A biosynthetic pathway (Scheme 1).
Supplementary Material
Acknowledgments
We thank Allan S. Wagman, Small Molecule Drug Discovery, Chiron Corporation, 4560 Horton Street, M/S 4.5, Emeryville, CA 94608-2916 [Office (510) 923-7796, FAX (510) 923-3360, email: allan_wagman@chiron.com] for his careful reading of this manuscript. Chiron and the University of Washington are the sole inventors of CHIR-090 and the other compounds listed in reference 31.
Funding: All studies of the slow, tight-binding inhibition of LpxC by CHIR-090 were supported by NIH Grant GM-51310 to C. R. H. Raetz and conducted at Duke University. The mass spectrometry facility in the Department of Biochemistry of the Duke University Medical Center and Dr. Z. Guan were supported by the LIPID MAPS Large Scale Collaborative Grant number GM069338 from NIH. NMR studies of 19F-labeled inhibitors bound to E. coli or Pseudomonas LpxC, and a wide range of LpxC inhibitor studies by a consortium that included Pathogenesis, Chiron, the University of Washington and Duke University were supported by a grant from the Cystic Fibrosis Foundation. N-aroyl-L-Thr hydroxamic acids, including the lead for CHIR-090 (which lacked substituents on the aroyl unit), were prepared in the Andersen lab during this period. CHIR-090 was prepared at the University of Washington in a synthetic effort that was fully funded by a grant from Chiron.
Abbreviations
- ESI-MS
electrospray ionization mass spectrometry
- LC
liquid chromatography
- TLC
thin layer chromatography
Footnotes
S. Endsley, A. L. McClerren, C. R. H. Raetz and N. H. Andersen, in preparation.
References
- 1.Neu HC. The crisis in antibiotic resistance. Science. 1992;257:1064–1073. doi: 10.1126/science.257.5073.1064. [DOI] [PubMed] [Google Scholar]
- 2.Projan SJ. New (and not so new) antibacterial targets - from where and when will the novel drugs come? Curr Opin Pharmacol. 2002;2:513–522. doi: 10.1016/s1471-4892(02)00197-2. [DOI] [PubMed] [Google Scholar]
- 3.Projan SJ, Youngman PJ. Antimicrobials: new solutions badly needed. Curr Opin Microbiol. 2002;5:463–465. doi: 10.1016/s1369-5274(02)00364-8. [DOI] [PubMed] [Google Scholar]
- 4.Walsh CT. Where will new antibiotics come from? Nat Rev Microbiol. 2003;1:65–70. doi: 10.1038/nrmicro727. [DOI] [PubMed] [Google Scholar]
- 5.Patchett AA, Cordes EH. The design and properties of N-carboxyalkyldipeptide inhibitors of angiotensin-converting enzyme. Adv Enzymol Relat Areas of Mol Biol. 1985;57:1–83. doi: 10.1002/9780470123034.ch1. [DOI] [PubMed] [Google Scholar]
- 6.Muri EM, Nieto MJ, Sindelar RD, Williamson JS. Hydroxamic acids as pharmacological agents. Curr Med Chem. 2002;9:1631–1653. doi: 10.2174/0929867023369402. [DOI] [PubMed] [Google Scholar]
- 7.White RJ, Margolis PS, Trias J, Yuan Z. Targeting metalloenzymes: a strategy that works. Curr Opin Pharmacol. 2003;3:502–507. doi: 10.1016/s1471-4892(03)00115-2. [DOI] [PubMed] [Google Scholar]
- 8.Onishi HR, Pelak BA, Gerckens LS, Silver LL, Kahan FM, Chen MH, Patchett AA, Galloway SM, Hyland SA, Anderson MS, Raetz CRH. Antibacterial agents that inhibit lipid A biosynthesis. Science. 1996;274:980–982. doi: 10.1126/science.274.5289.980. [DOI] [PubMed] [Google Scholar]
- 9.Jackman JE, Raetz CRH, Fierke CA. UDP-3-O-(R-3-hydroxymyristoyl)-N-acetylglucosamine deacetylase of Escherichia coli is a zinc metalloenzyme. Biochemistry. 1999;38:1902–1911. doi: 10.1021/bi982339s. [DOI] [PubMed] [Google Scholar]
- 10.Jackman JE, Fierke CA, Tumey LN, Pirrung M, Uchiyama T, Tahir SH, Hindsgaul O, Raetz CRH. Antibacterial agents that target lipid A biosynthesis in gram-negative bacteria. Inhibition of diverse UDP-3-O-(R-3-hydroxymyristoyl)-N-acetylglucosamine deacetylases by substrate analogs containing zinc binding motifs. J Biol Chem. 2000;275:11002–11009. doi: 10.1074/jbc.275.15.11002. [DOI] [PubMed] [Google Scholar]
- 11.Clements JM, Coignard F, Johnson I, Chandler S, Palan S, Waller A, Wijkmans J, Hunter MG. Antibacterial activities and characterization of novel inhibitors of LpxC. Antimicrob Agents Chemother. 2002;46:1793–1799. doi: 10.1128/AAC.46.6.1793-1799.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Anderson MS, Robertson AD, Macher I, Raetz CRH. Biosynthesis of lipid A in Escherichia coli: identification of UDP-3-O-(R-3-hydroxymyristoyl)-α-D-glucosamine as a precursor of UDP-N2-O3-bis-(R-3-hydroxymyristoyl)-α-D-glucosamine. Biochemistry. 1988;27:1908–1917. doi: 10.1021/bi00406a017. [DOI] [PubMed] [Google Scholar]
- 13.Young K, Silver LL, Bramhill D, Cameron P, Eveland SS, Raetz CRH, Hyland SA, Anderson MS. The envA permeability/cell division gene of Escherichia coli encodes the second enzyme of lipid A biosynthesis. J Biol Chem. 1995;270:30384–30391. doi: 10.1074/jbc.270.51.30384. [DOI] [PubMed] [Google Scholar]
- 14.Anderson MS, Bull HS, Galloway SM, Kelly TM, Mohan S, Radika K, Raetz CRH. UDP-N-acetylglucosamine acyltransferase of Escherichia coli: the first step of endotoxin biosynthesis is thermodynamically unfavorable. J Biol Chem. 1993;268:19858–19865. [PubMed] [Google Scholar]
- 15.Raetz CRH, Whitfield C. Lipopolysaccharide endotoxins. Annu Rev Biochem. 2002;71:635–700. doi: 10.1146/annurev.biochem.71.110601.135414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Stover CK, Pham XQ, Erwin AL, Mizoguchi SD, Warrener P, Hickey MJ, Brinkman FS, Hufnagle WO, Kowalik DJ, Lagrou M, Garber RL, Goltry L, Tolentino E, Westbrock-Wadman S, Yuan Y, Brody LL, Coulter SN, Folger KR, Kas A, Larbig K, Lim R, Smith K, Spencer D, Wong GK, Wu Z, Paulsen IT. Complete genome sequence of Pseudomonas aeruginosa PA01, an opportunistic pathogen. Nature. 2000;406:959–964. doi: 10.1038/35023079. [DOI] [PubMed] [Google Scholar]
- 17.Blattner FR, Plunkett G, Bloch CA, Perna NT, Burland V, Riley M, Collado-Vides J, Glasner JD, Rode CK, Mayhew GF, Gregor J, Davis NW, Kirkpatrick HA, Goeden MA, Rose DJ, Mau B, Shao Y. The complete genome sequence of Escherichia coli K-12. Science. 1997;277:1453–1474. doi: 10.1126/science.277.5331.1453. [DOI] [PubMed] [Google Scholar]
- 18.Holmes MA, Matthews BW. Binding of hydroxamic acid inhibitors to crystalline thermolysin suggests a pentacoordinate zinc intermediate in catalysis. Biochemistry. 1981;20:6912–6920. doi: 10.1021/bi00527a026. [DOI] [PubMed] [Google Scholar]
- 19.Natesh R, Schwager SL, Sturrock ED, Acharya KR. Crystal structure of the human angiotensin-converting enzyme-lisinopril complex. Nature. 2003;421:551–554. doi: 10.1038/nature01370. [DOI] [PubMed] [Google Scholar]
- 20.Becker A, Schlichting I, Kabsch W, Schultz S, Wagner AF. Structure of peptide deformylase and identification of the substrate binding site. J Biol Chem. 1998;273:11413–11416. doi: 10.1074/jbc.273.19.11413. [DOI] [PubMed] [Google Scholar]
- 21.McClerren AL, Zhou P, Guan Z, Raetz CRH, Rudolph J. Kinetic analysis of the zinc-dependent deacetylase in the lipid A biosynthetic pathway. Biochemistry. 2005;44:1106–1113. doi: 10.1021/bi048001h. [DOI] [PubMed] [Google Scholar]
- 22.Kline T, Andersen NH, Harwood EA, Bowman J, Malanda A, Endsley S, Erwin AL, Doyle M, Fong S, Harris AL, Mendelsohn B, Mdluli K, Raetz CRH, Stover CK, Witte PR, Yabannavar A, Zhu S. Potent, novel in vitro inhibitors of the Pseudomonas aeruginosa deacetylase LpxC. J Med Chem. 2002;45:3112–29. doi: 10.1021/jm010579r. [DOI] [PubMed] [Google Scholar]
- 23.Pirrung MC, Tumey LN, McClerren AL, Raetz CRH. High-throughput catch-and-release synthesis of oxazoline hydroxamates. Structure-activity relationships in novel inhibitors of Escherichia coli LpxC: in vitro enzyme inhibition and antibacterial properties. J Am Chem Soc. 2003;125:1575–86. doi: 10.1021/ja0209114. [DOI] [PubMed] [Google Scholar]
- 24.Beall B, Lutkenhaus J. Sequence analysis, transcriptional organization, and insertional mutagenesis of the envA gene of Escherichia coli. J Bacteriol. 1987;169:5408–5415. doi: 10.1128/jb.169.12.5408-5415.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Herring CD, Blattner FR. Conditional lethal amber mutations in essential Escherichia coli genes. J Bacteriol. 2004;186:2673–2681. doi: 10.1128/JB.186.9.2673-2681.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Li X, Uchiyama T, Raetz CRH, Hindsgaul O. Synthesis of a carbohydrate-derived hydroxamic acid inhibitor of the bacterial enzyme (LpxC) involved in lipid A biosynthesis. Org Lett. 2003;5:539–541. doi: 10.1021/ol027458l. [DOI] [PubMed] [Google Scholar]
- 27.Jackman JE, Raetz CRH, Fierke CA. Site directed mutagenesis of the bacterial metalloamidase UDP-(3-O-acyl)-N-acetylglucosamine deacetylase (LpxC). Identification of the zinc binding site. Biochemistry. 2001;40:514–523. doi: 10.1021/bi001872g. [DOI] [PubMed] [Google Scholar]
- 28.Coggins BE, Li X, McClerren AL, Hindsgaul O, Raetz CRH, Zhou P. Structure of the LpxC deacetylase with a bound substrate-analog inhibitor. Nat Struct Biol. 2003;10:645–651. doi: 10.1038/nsb948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Coggins BE, McClerren AL, Jiang L, Li X, Rudolph J, Hindsgaul O, Raetz CRH, Zhou P. Refined solution structure of the LpxC-TU-514 complex and pKa analysis of an active site histidine: insights into the mechanism and inhibitor design. Biochemistry. 2005;44:1114–11126. doi: 10.1021/bi047820z. [DOI] [PubMed] [Google Scholar]
- 30.Whittington DA, Rusche KM, Shin H, Fierke CA, Christianson DW. Crystal structure of LpxC, a zinc-dependent deacetylase essential for endotoxin biosynthesis. Proc Natl Acad Sci U S A. 2003;100:8146–8150. doi: 10.1073/pnas.1432990100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Andersen NH, Bowman J, Erwin A, Harwood E, Kline T, Mdluli K, Pfister KB, Shawar R, Wagman A, Yabannavar A. Antibacterial Agents. Patent WO 2004/062601 A2. Chiron; Emeryville California: World Intellectual Property Organization. 2004:324.
- 32.Jackman JE. PhD Thesis. Department of Biochemistry, Duke Univeristy; Durham, North Carolina: 1999. Metal ion requirement and inhibition of the UDP-3-O-acyl-GlcNAc deacetylases of Escherichia coli and Aquifex aeolicus; p. 203. [Google Scholar]
- 33.Miller JR. Experiments in Molecular Genetics. Cold Spring Harbor Laboratory; Cold Spring Harbor, NY: 1972. [Google Scholar]
- 34.Pirrung MC, Tumey LN, Raetz CRH, Jackman JE, Snehalatha K, McClerren AL, Fierke CA, Gantt SL, Rusche KM. Inhibition of the antibacterial target UDP-(3-O-acyl)-N-acetylglucosamine deacetylase (LpxC): isoxazoline zinc amidase inhibitors bearing diverse metal binding groups. J Med Chem. 2002;45:4359–70. doi: 10.1021/jm020183v. [DOI] [PubMed] [Google Scholar]
- 35.Emmerson AM, Jones AM. The quinolones: decades of development and use. J Antimicrob Chemother. 2003;51 1:13–20. doi: 10.1093/jac/dkg208. [DOI] [PubMed] [Google Scholar]
- 36.Mingeot-Leclercq MP, Glupczynski Y, Tulkens PM. Aminoglycosides: activity and resistance. Antimicrob Agents Chemother. 1999;43:727–737. doi: 10.1128/aac.43.4.727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hernick M, Fierke CA. Zinc hydrolases: the mechanisms of zinc-dependent deacetylases. Arch Biochem Biophys. 2005;433:71–84. doi: 10.1016/j.abb.2004.08.006. [DOI] [PubMed] [Google Scholar]
- 38.Hernick M, Gennadios HA, Whittington DA, Rusche KM, Christianson DW, Fierke CA. UDP-3-O-((R)-3-hydroxymyristoyl)-N-acetylglucosamine deacetylase functions through a general acid-base catalyst pair mechanism. J Biol Chem. 2005;280:16969–16978. doi: 10.1074/jbc.M413560200. [DOI] [PubMed] [Google Scholar]
- 39.Morrison JF, Walsh CT. The behavior and significance of slow-binding enzyme inhibitors. Adv Enzymol Relat Areas Mol Biol. 1988;61:201–301. doi: 10.1002/9780470123072.ch5. [DOI] [PubMed] [Google Scholar]
- 40.Robertson JG. Mechanistic basis of enzyme-targeted drugs. Biochemistry. 2005;44:5561–5571. doi: 10.1021/bi050247e. [DOI] [PubMed] [Google Scholar]
- 41.Deckert G, Warren PV, Gaasterland T, Young WG, Lenox AL, Graham DE, Overbeek R, Snead MA, Keller M, Aujay M, Huber R, Feldman RA, Short GM, Olsen GJ, Swanson RV. The complete genome of the hyperthermophilic bacterium Aquifex aeolicus. Nature. 1998;392:353–358. doi: 10.1038/32831. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.