

Abstract
Regulation of intracellular calcium is an important signaling mechanism for cell proliferation in both normal and cancerous cells. In normal epithelial cells, free calcium concentration is essential for cells to enter and accomplish the S phase and the M phase of the cell cycle. In contrast, cancerous cells can pass these phases of the cell cycle with much lower cytoplasmic free calcium concentrations, indicating an alternative mechanism has developed for fulfilling the intracellular calcium requirement for an increased rate of DNA synthesis and mitosis of fast replicating cancerous cells. The detailed mechanism underlying the altered calcium loading pathway remains unclear; however, there is a growing body of evidence that suggests the T-type Ca2+ channel is abnormally expressed in cancerous cells and that blockade of these channels may reduce cell proliferation in addition to inducing apoptosis. Recent studies also show that the expression of T-type Ca2+ channels in breast cancer cells is proliferation state dependent, i.e. the channels are expressed at higher levels during the fast-replication period, and once the cells are in a non-proliferation state, expression of this channel is minimal. Therefore, selectively blocking calcium entry into cancerous cells may be a valuable approach for preventing tumor growth. Since T-type Ca2+ channels are not expressed in epithelial cells, selective T-type Ca2+ channel blockers may be useful in the treatment of certain types of cancers.
Keywords: T-type calcium channels, Cancer, Cell cycle, Calcium
INTRODUCTION
Calcium is an essential signal transduction element involved in the regulation of many eukaryotic cellular functions including cell cycle progression[1]. Control of intracellular Ca2+ ([Ca2+]i) is crucial for the orderly progression of the cell cycle and plays a vital role in the regulation of cell proliferation and growth[2]; however, excessive calcium or loss of control in calcium signaling can lead to cell death[3]. Therefore, careful control of calcium signaling is required for cell survival. Upon stimulation, the intracellular calcium concentration can increase dramatically, often reaching micromolar amounts. This increase in cytoplasmic calcium can occur via release from intracellular stores or influx through a variety of plasma membrane ion channels. Voltage-gated and ligand-gated Ca2+ channels in the plasma membrane, along with ryanodine receptors (RynR) and inositol triphosphate receptors (InsP3R) at the intracellular calcium stores, provide fluxes of Ca2+ to the cytoplasm. The driving force for calcium entry is the result of an electrochemical gradient between the extracellular concentration (1.3 × 10-3-2 × 10-3 mol/L) of calcium and the intracellular concentration (< 10-8 mol/L).
In general, non-excitable tissues, including the epithelium, do not express voltage gated Ca2+ channels. This is partly because the ranges of membrane potential changes in these cells are too small to activate these channels. However, recent studies show that T-type Ca2+ channels are expressed in cancerous cells, although their functional role has only begun to be investigated. Furthermore, there is a growing body of evidence suggesting that tumor cell proliferation can be halted by the use of ion channel blockers. T-type calcium channels are a class of calcium permeable low voltage activated (LVA) ion channels which open after small depolarizations of the membrane. Molecular biology has revealed the existence of three different T-type calcium channel subunits, the α1G, α1H and α1I. The α1 designation refers to the channels primary ion conducting protein, which consists of four domains each containing six transmembrane segments. There are other auxiliary calcium channel subunits; however, the LVA alpha1 subunits can function as stand alone complexes. The unique low voltage dependent activation/inactivation and slow deactivation of T-type Ca2+ channels indicate that these channels may play a physiological role in carrying depolarizing current at low membrane potentials. Therefore, these channels may play a direct role in regulating [Ca2+]i, especially in non-excitable tissues, including some cancerous cells. At low voltages, T-type Ca2+ channels are known to mediate a phenomenon known as “window current”[4-6]. The term “window” refers to the voltage overlap between the activation and steady state inactivation at low or resting membrane potentials. As a result, there is a sustained inward calcium current carried by a small portion of channels that are not completely inactivated. Window current allows T-type Ca2+ channels to regulate Ca2+ homeostasis under non-stimulated or resting membrane conditions[7]. The most direct evidence of T-type Ca2+ channel mediated Ca2+ window current is from a study conducted in HEK-293 cells expressing the T-type isoform α1G[8], which demonstrated window current peaked at -48 mV. Membrane potentials around this voltage can occur in un-stimulated non-excitable cells.
CALCIUM SIGNALING AND CELL CYCLING
As shown in Figure 1, the cell cycle is divided into four stages: G1, S, G2 and M. DNA replication occurs in the S phase and mitosis occurs in the M phase. Cells must pass through a restriction point between the G1 and S phases before continuing proliferation; otherwise, they exit the cell cycle to G0 and differentiate or terminate. Another checkpoint in the cell cycle is between phases G2 and M. For cells to pass through these various points, one of the most prominent messengers is Ca2+, as demonstrated by the induction of mitotic events by injection of exogenous Ca2+ in a fertilized egg model[9]. Steinhardt et al[10] observed transient increases in cytosolic Ca2+ during late G1, prior to the initiation of the S phase and during G2 before entry into the M phase that were dependent upon external physiological Ca2+ concentration. In the transition from G1 to S phase, cells require external Ca2+ in addition to functional calcium channels in order to directly or indirectly trigger a myriad of critical downstream enzymes such as thymidine kinase, thymidylate synthase, ribonucleotide reductase and DNA polymerase and begin DNA replication. In the transition from G2 to M phase, Ca2+ flashes activate enzymes that are critical for microtubule rearrangement and microfilament contraction. In order to confirm the significant role that Ca2+ plays in the cell cycle, researchers have blocked the progression of the cell cycle via injection of Ca2+ chelators into the same fertilized eggs[11]. The influence of Ca2+ channels on cell growth is clearly demonstrated in pharmacological studies using Ca2+ channel antagonists. In a study by Zeitler et al[12], various Ca2+ channel blockers, including verapamil, nifedipine, diltiazem and isradipine, caused G0/G1 cell-cycle arrest in growth factor induced human umbilical arterial endothelial cells (HUAEC) during proliferation. The Ca2+ signal has also been linked to activation of immediate early genes (e.g. c-fos) that are responsible for inducing resting cells in G0 to re-enter the cell cycle, an attribute most frequently up-regulated in rapidly proliferating cells[10].
Figure 1.

Ca2+ signaling pathways differ between cancerous and non-cancerous cells (adapted from[23]).
At the end of the cycle, cells can undergo suicide through a process known as apoptosis or active cell death, which is a genetic program specifically designed to shape organs during development and adjust cell population levels to appropriate values. The key players of apoptosis are a killer Ca2+ surge and the nuclear membrane Ca2+ activated endonuclease, which terminates the cell by cutting chromatin into fragments (Figure 1). Underlying mechanisms for Ca2+ mediated effects in cell proliferation may involve a wide variety of other intracellular signal transduction pathways such as G-proteins, protein kinase C (PKC), calmodulin, m-calpain, MAP kinase, phospholipase A2 and others[13,14]. Although the details of each pathway is beyond the scope of this discussion, there are several notable mechanisms that act to amplify [Ca2+]i for activation of gene transcription or cell migration. One mechanism is the hydrolysis of inositol lipids by the enzyme phospholipase C, the activation of which is itself dependent on an initial rise in [Ca2+]i, producing diacylglycerol (DAG) and InsP3. Resulting from the activation of G protein-linked or tyrosine-kinase linked receptors[15], InsP3 thus causes a form of Ca2+ dependent Ca2+ release from intracellular stores. Described as “calcium puffs”, which propagate into a local or global Ca2+ signal, this Ca2+ release is important for converting the cytoplasm into an excitable medium that can support repetitive Ca2+ oscillations[16]. The resulting amplification of [Ca2+]i contributes to the signal for mitosis and DNA synthesis.
In addition to InsP3, sensory proteins also play a role in maintaining the calcium signaling system. Calmodulin is a Ca2+ binding protein that acts as a Ca2+ sensor in the cell cycle. High expression of calmodulin has been observed during the S phase and mitosis, while inhibition of its activity by administration of calmodulin monoclonal antibodies is shown to block DNA synthesis[17]. Another sensory mechanism occurs through an extracellular calcium ion concentration sensing receptor (CaR) and calbindin, a high affinity Ca2+-binding regulatory protein belonging to the same family as calmodulin. Parkash et al observed that CaR plays a role in sensing and responding to changes in extracellular Ca2+ ([Ca2+]o). Upon activation by increased [Ca2+]o, CaR interacts with phospholipase C (PLC) via G proteins to produce DAG and InsP3[18]. The subsequent increase in [Ca2+]i is regulated by calbindin, which was previously found to bind to L-type HVA Ca2+ channels in pancreatic islets-cells[19]. It has also been shown that calbindin/CaR co-localization occurs in the estrogen receptor positive breast cancer cell line MCF-7[20]. Activated CaR also increases parathyroid hormone related protein (PTHrP), which appears to exacerbate cell metastasis in MCF-7 cells[21]. A study by Lewalle et al[22] shows that an increase in [Ca2+]i mediates tumor cell transendothelial migration in vitro. By associating the upregulation of these mechanisms in cancerous cells, increases in [Ca2+]i are shown to provide an important and pronounced signal for cell growth.
Calcium signaling in cancerous cells, however, uses an altered pathway during cell cycling[23]. Whitfield has shown that colon carcinomas undergoing carcinogenesis, which have lost their tumor-suppressing genes, have dramatically altered calcium signal mechanisms, and have ignored normal calcium-dependent restrictions by overproducing calcium-binding signal proteins (Figure 1)[18]. Attempts to terminate the mutant cells with Ca2+ signal surges are futile, as the cells are no longer responsive to Ca2+ signals: instead, they produce and respond to their own renegade growth factors. Ca2+ and the signaling enzymes that are directly activated by Ca2+ or by Ca2+-binding proteins play crucial roles in most cell signals and programs and must be understood and implemented in any future differentiation therapies. Since cancerous cells express T-type Ca2+ channels, it is possible that these channels provide an altered Ca2+ influx pathway in responding to the increasing demand of Ca2+ during rapid cell proliferation.
T-TYPE CALCIUM CHANNELS IN CANCEROUS CELL PROLIFERATION
T-type Ca2+ channels and non-cancerous cell cycling
The function of regulating Ca2+ homeostasis may allow T-type Ca2+ channels to play an important role in controlling cell proliferation and differentiation in many tissues. In primary cultured rat aortic smooth muscle cells, a T-type Ca2+ current was found to be present in cells during the G1 and S phases but decreased or absent in all other phases of the cell cycle[24-26]. It was shown that cultured smooth muscle cells exhibited an increased T-type Ca2+ current during stages of proliferation and this current decreased as the cells became confluent or when they came into contact with one another[27]. T-type Ca2+ currents are also present in freshly dissociated or 1-2 d cultured neonatal rat ventricular myocytes when they are still able to proliferate, but are not observed in cells cultured greater than 3 d[28]. Likewise, older tissues under pathological conditions, such as cardiomyopathic hamster heart[29], hypertrophied adult feline left ventricular myocytes[30], and rat neointimal formation after vascular injury[31] have increased T-type Ca2+ current activity. These studies suggest that T-type calcium channels may play a vital role in regulating proliferation under specialized conditions.
T-type Ca2+ channels are broadly expressed in tumor cells
If these channels do participate in proliferation under abnormal conditions, cells must first maintain control of the expression of α1G T-type Ca2+ channel messenger RNA in order to prevent functional expression of the protein. Otherwise, loss of this control may lead to aberrant cell growth and tumor progression. A recent study revealed the presence of T-type calcium channel mRNA expressed in breast tumor tissue that was removed from human biopsies (Figure 2)[32]. In this case, the tumor was later diagnosed as malignant and estrogen receptor positive by pathological examination. Expression of these channels in tumor cells has been reported broadly, as shown in Table 1[1,33-49]. For example, MCF-7 cells, a cell line derived from a human breast adenocarcinoma that has been shown to express α1G and α1H T-type Ca2+ channel mRNA and current transiently (Table 2)[33]. T-type Ca2+ channels have also been suggested as a potential therapeutic target for intracranial tumor and prostate cancer. Mibefradil was found to inhibit human astrocytoma (U87-MG) and neuroblastoma (N1E-115) proliferation and that over-expression of T-type Ca2+ channel protein doubled the proliferation rate while antisense treatment reduced the proliferation rate of these cells[37]. Human prostate cancer epithelial cells (LNCap) have also been shown to express increased T-type Ca2+ channel (α1H) current and mRNA. Similarly, increased T-type Ca2+ channel protein doubled proliferation while antisense treatment reduced the proliferation rate of these cells[1,43]. It was also shown that these channels were found to regulate intracellular calcium in LNCap cells. Another study examined the role of T-type Ca2+ channels in esophageal carcinoma cell proliferation; these data suggested that T-type Ca2+ channels may have a functional role in proliferation that can be reduced by inhibition of T-type Ca2+ channels[44]. Given the role that T-type calcium channels play in cell cycle progression and the relatively recent findings that show the functional expression of these channels in many different cancerous cell types, researchers have now been given the opportunity to investigate the potential of an entirely new target in the fight against cancer. Developing new compounds that target these proteins may hold the key to controlling certain types of cancer.
Figure 2.

T-type Ca2+ channel expression in human malignant breast cancer tissue.
Table 1.
Cancerous cells that expresses T-type Ca2+ channels
| Cell type | Cell line | T-type isoform | Reference |
| Breast carcinoma | MCF-7, MDA-435 | α1G, α1H | [33-36] |
| MDA-231, MDA-361 MB-468, MB-474, BT-20, CAMA1, SKBR-3 | α1G | [34-36] | |
| Neuroblastoma | SK-N-SH, | α1G | [34,37-40] |
| NG 108-15, SK-N-MC | |||
| N1E-115 | α1G, α1H | [37] | |
| Retinoblastoma | Y-79, WERI-Rb1 | α1G, α1H, α1I | [41,42] |
| Glioma | Primary (biopsy) | α1G | [36] |
| U87-MG | α1G, α1H | [37] | |
| Prostate carcinoma | TSU-PRL, DUPRO | α1G | [35,43] |
| LNCaP | α1H | [1,34,35] | |
| PC-3, DU-145 | α1G, α1H | [34,35] | |
| Esophageal carcinoma | TE1, TE10, TE12, KYSE150, KYSE180, KYSE450 | α1H | [44] |
| SKGT4, TE3, TE7, KYSE70 | α1G, α1H | [44] | |
| COLO-680N, SEG1, TE8, TE11, KYSE30, KYSE410, KYSE510 | α1G, α1H, α1I | [44] | |
| Fibrosarcoma | HT1080 | α1G | [45] |
| Colorectal carcinoma | Caco2, DLD-1, Lovo, SW837 | α1G | [35] |
| Pheochromocytoma | MPC 9/3L | α1G | [46] |
| PC-12 | α1H | [47] | |
| Adenocarcinoma | H295R | α1H | [48] |
| Insulinoma | INS-1 | α1G | [49] |
Table 2.
Q-RT-PCR detected T-type Ca2+ channels expression in non-confluent cultures of breast cancer cell lines
| Cell types | T-channels | Non-confluent Δct | Confluent Δct |
| MDA-MB-231 | α1H | 14.28 ± 0.16 | NA, ct > 40 |
| MDA-MB-231 | α1G | 9.45 ± 0.87 | NA, ct > 40 |
| MCF-7 | α1H | 13.43 ± 0.24 | NA, ct > 40 |
| MCF-7 | α1G | 7.32 ± 0.3 | NA, ct > 40 |
NA: Not applicable.
EFFECT OF T-TYPE Ca2+ CHANNEL BLOCKERS ON BREAST CANCER CELL PROLIFERATION
The function of T-type Ca2+ channels with regards to tumor cell proliferation was also reviewed[50,51]. One study found the T-type Ca2+ channel to be particularly effective in controlling oscillations in intracellular Ca2+ as the result of the channels unique activation/inactivation properties. It was concluded that new selective antagonists may become helpful as a therapeutic approach against tumors in which proliferation depends on T-type Ca2+ channel expression[51]. A study performed in knockout animals found that selective inhibition of T-type Ca2+ channels may have impact upon the treatment of cancer[52].
Studies have shown an inhibition in breast cancer proliferation by the channel blockers pimozide, thioridazine[53] and mibefradil[42]. The endogenous cannabinoid anandamide has also been shown to block T-type Ca2+ channels[54], in addition to inhibition of breast cancer cell proliferation[55], an effect that may be due to blockage of T-type Ca2+ channels.
The anti-cancer effect of a T-type Ca2+ channel antagonists on tumor cells in vivo has been investigated[32]. MCF-7 cells were implanted into nude mice, athymic nude BSLB/c, and then either mibefradil (0.5 mg/100 μL) or saline (0.5 mg/100 μL) was injected locally at the tumor sites (s.c) twice a week. After 30 d of the treatment, mice were sacrificed and the tumors were removed for histochemistry examination.
As shown in Figure 3A and B, in the saline injected tissue the proliferation of the malignant tumor cells formed nodules in subcutis. The tumor cells were malignant as indicated by hyperchromatic nuclei with enlarged nuclei, irregular nuclear membrane, prominent nucleoli, and many mitotic features. No signs of degeneration and necrosis were detected. In contrast, the mibefradil injected tissue showed large areas of tumor degeneration and necrosis (Figure 3C and D). The tumor necrosis was accompanied by prominent edema. Furthermore, as shown in Figure 3C, mibefradil more potently destroyed breast cancer cells (indicated by the black arrows) than non-cancerous cells at adjacent areas (indicated by the white arrows), including fibroblasts, endothelial cells and keratinocytes. These results indicate that a local injection of mibefradil induces necrosis of human breast carcinoma cells implanted into subcutaneous adipose tissue in mice.
Figure 3.

Mibefradil destroys tumor cells implanted in a nude mouse. HE stain was applied to show the nuclei and cytoplasm of the cells (A and C, × 100; B and D, × 400).
More recently, the antiproliferative effect of the T-type calcium channel inhibitor NNC 55-0396[56] has been examined in cell lines derived from breast epithelial tissue, MCF-7, MDA-MB-231(ER-α), and an adriamycin resistant cell line ADR. All three of these cell types express α1G and α1H Ca2+ channel mRNA and their proliferation was suppressed by NNC 55-0396, with IC50 of about 1-2 μmol/L[32,33]. The specificity of NNC 55-0396 antagonism on cancerous cell proliferation was investigated in a prostate epithelial cell line (RWPE-1) that does not express T-type Ca2+ channels[32]. As shown in Figure 4, NNC 55-0396 exhibited neither dose-dependent (up to 20 μmol/L Figure 4A) nor time-dependent (up to 60 h, Figure 4B) inhibitory effects on RWPE-1 cell growth, suggesting that the anti-proliferation effect of NNC 55-0396 most likely resulted from blocking T-type Ca2+ channels of breast cancer cells. It also suggested that the general toxicity of NNC 55-0396 is minimal at the concentration that induces suppression of proliferation. T-type Ca2+ channel antisense treatment in these cells reduced the proliferation rate by 45% and antisense had no effect on proliferation on tumor cells not expressing T-type Ca2+ channels.
Figure 4.

Effect of NNC 55-0396 on RWPE-1 cell proliferation (Error bars represent SE; n = 3).
The role of T-type Ca2+ channels in cancerous cell proliferation has also been examined with specific siRNA antagonism[33]. Specifically, MCF-7 cells were treated with siRNA targeting both α1G and α1H (α1G/H). The cells were treated with scrambled (siRNA-S, 100 pmol/L), α1G/H-1 (siRNA-1) or α1G/H-2 (siRNA-2) for 48 h and subjected to MTT assay. The effects of siRNAs on cell proliferation were shown as percent (%) of vehicle control. As shown in Figure 5, scrambled siRNA was not significantly different than the control. However, both siRNA-1 and siRNA-2 treated cells had significantly lower proliferation rates compared to the scrambled and vehicle control siRNAs. These results strongly support the role of T-type Ca2+ channels in breast cancer cell proliferation and indicate that the effect of NNC 55-0396 on the breast cancer cell proliferation is due to the blockade of these channels.
Figure 5.
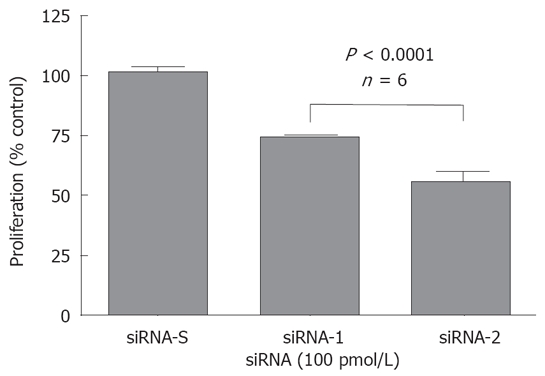
Effect of siRNAs on MCF-7 cell proliferation.
CONCERNS
Since T-type Ca2+ channels are normally expressed in the brain, heart and endocrine tissues of the human body, the potential side-effects of T-type Ca2+ channel blockers to these systems are of concern for therapeutic applications. Although T-type Ca2+ channel blockers have been used clinically for the treatment of neurological disorders (e.g. ethosuximide for absence seizures), the adverse effects of these drugs on the cardiovascular and central nervous systems are still unclear. Specifically, it is important to determine the possible arrhythmic and sedative effects of these drugs.
Human blood cells do not express T-type Ca2+ channels; therefore, it is advantageous to apply T-type Ca2+ channel blockers in the hemopoietic system, since current chemotherapeutic drugs have displayed severe side effects on this system. If we can locally deliver T-type Ca2+ channel blocker into the hemopoietic system, the compound should be very selective in eliminating the breast cancer cells in the blood stream. Thus, T-type Ca2+ channel blockers can be potential anti-metastasis drugs for adjuvant therapy of breast cancer.
PERSPECTIVES
The function of T-type Ca2+ channels may not be restricted to cancerous cell proliferation. These channels may also play roles in cancerous cell colonization, invasion, secretion and angiogenesis. The growing number of proliferating cells need to attract blood vessels (angiogenesis) in order to receive nutrients, O2, etc. to sustain themselves. The transformed cells are able to enter the blood stream and survive there, and colonize (metastasize) other tissues. Invasive growth or cell migration is a highly regulated process in which the migrating cells must secrete matrix proteases that disrupt the extracellular matrix (ECM) and permit easier transit through the surrounding environment. In addition, they must also profoundly reshape their structure, which involves massive cytoskeletal rearrangement. Precise regulation of intracellular calcium concentration is crucial for all of these processes. It is very possible that T-type Ca2+ channels also play significant roles in these processes[45].
An expansion of the list of ion channels implicated in cancer development is expected, and the tools needed to investigate this issue are more readily available. As is the case with other protein families, it will be probably difficult to ascribe tumor development to the malfunction of a single ion channel. Rather, defects in T-type Ca2+ channels probably contribute to the neoplastic phenotype through complex interactions with other ion channels, most of which have not been properly identified. For instance, regulation of K+ channels can affect the membrane potential, which in turn regulates the window currents mediated by T-type Ca2+ channels. However, since in many cases there are already known pharmacological modulators (blockers and activators) of ion channels, identification of a single defective ion channel in a particular cancer could provide a ready-to-go therapeutic approach.
Footnotes
Peer reviewers: Anna S Gukovskaya, Professor, VA Greater Los Angeles Health Care System, University of California, Los Angeles, 11301 Wilshire Blvd, Los Angeles 91301, United States; Zong-Jie Cui, PhD, Professor, Institute of Cell Biology, Beijing Normal University, 19 Xinjiekou Waidajie, Beijing 100875, China
S- Editor Zhong XY L- Editor Roberts SE E- Editor Yin DH
References
- 1.Mariot P, Vanoverberghe K, Lalevee N, Rossier MF, Prevarskaya N. Overexpression of an alpha 1H (Cav3.2) T-type calcium channel during neuroendocrine differentiation of human prostate cancer cells. J Biol Chem. 2002;277:10824–10833. doi: 10.1074/jbc.M108754200. [DOI] [PubMed] [Google Scholar]
- 2.Ciapa B, Pesando D, Wilding M, Whitaker M. Cell-cycle calcium transients driven by cyclic changes in inositol trisphosphate levels. Nature. 1994;368:875–878. doi: 10.1038/368875a0. [DOI] [PubMed] [Google Scholar]
- 3.Choi DW. Ionic dependence of glutamate neurotoxicity. J Neurosci. 1987;7:369–379. doi: 10.1523/JNEUROSCI.07-02-00369.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cohen CJ, McCarthy RT, Barrett PQ, Rasmussen H. Ca channels in adrenal glomerulosa cells: K+ and angiotensin II increase T-type Ca channel current. Proc Natl Acad Sci USA. 1988;85:2412–2416. doi: 10.1073/pnas.85.7.2412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tsien RW, Clozel J-P, Nargeot J. Low-voltage-activated T-type Ca2+ channels. Chester: Adis International Ltd; 1998. pp. 1–394. [Google Scholar]
- 6.Crunelli V, Toth TI, Cope DW, Blethyn K, Hughes SW. The 'window' T-type calcium current in brain dynamics of different behavioural states. J Physiol. 2005;562:121–129. doi: 10.1113/jphysiol.2004.076273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bean BP, McDonough SI. Two for T. Neuron. 1998;20:825–828. doi: 10.1016/s0896-6273(00)80463-8. [DOI] [PubMed] [Google Scholar]
- 8.Chemin J, Monteil A, Briquaire C, Richard S, Perez-Reyes E, Nargeot J, Lory P. Overexpression of T-type calcium channels in HEK-293 cells increases intracellular calcium without affecting cellular proliferation. FEBS Lett. 2000;478:166–172. doi: 10.1016/s0014-5793(00)01832-9. [DOI] [PubMed] [Google Scholar]
- 9.Trump BF, Berezesky IK. Calcium-mediated cell injury and cell death. FASEB J. 1995;9:219–228. doi: 10.1096/fasebj.9.2.7781924. [DOI] [PubMed] [Google Scholar]
- 10.Steinhardt RA, Alderton J. Intracellular free calcium rise triggers nuclear envelope breakdown in the sea urchin embryo. Nature. 1988;332:364–366. doi: 10.1038/332364a0. [DOI] [PubMed] [Google Scholar]
- 11.Zucker RS, Steinhardt RA. Prevention of the cortical reaction in fertilized sea urchin eggs by injection of calcium-chelating ligands. Biochim Biophys Acta. 1978;541:459–466. doi: 10.1016/0304-4165(78)90155-1. [DOI] [PubMed] [Google Scholar]
- 12.Zeitler H, Ko Y, Glodny B, Totzke G, Appenheimer M, Sachinidis A, Vetter H. Cell-cycle arrest in G0/G1 phase of growth factor-induced endothelial cell proliferation by various calcium channel blockers. Cancer Detect Prev. 1997;21:332–339. [PubMed] [Google Scholar]
- 13.Ariyoshi H, Okahara K, Sakon M, Kambayashi J, Kawashima S, Kawasaki T, Monden M. Possible involvement of m-calpain in vascular smooth muscle cell proliferation. Arterioscler Thromb Vasc Biol. 1998;18:493–498. doi: 10.1161/01.atv.18.3.493. [DOI] [PubMed] [Google Scholar]
- 14.Akagi K, Nagao T, Urushidani T. Correlation between Ca(2+) oscillation and cell proliferation via CCK(B)/gastrin receptor. Biochim Biophys Acta. 1999;1452:243–253. doi: 10.1016/s0167-4889(99)00137-8. [DOI] [PubMed] [Google Scholar]
- 15.Berridge MJ. Inositol trisphosphate and calcium signalling. Nature. 1993;361:315–325. doi: 10.1038/361315a0. [DOI] [PubMed] [Google Scholar]
- 16.Lechleiter JD, Clapham DE. Molecular mechanisms of intracellular calcium excitability in X. laevis oocytes. Cell. 1992;69:283–294. doi: 10.1016/0092-8674(92)90409-6. [DOI] [PubMed] [Google Scholar]
- 17.Reddy GP, Reed WC, Sheehan E, Sacks DB. Calmodulin-specific monoclonal antibodies inhibit DNA replication in mammalian cells. Biochemistry. 1992;31:10426–10430. doi: 10.1021/bi00158a002. [DOI] [PubMed] [Google Scholar]
- 18.Kifor O, Diaz R, Butters R, Brown EM. The Ca2+-sensing receptor (CaR) activates phospholipases C, A2, and D in bovine parathyroid and CaR-transfected, human embryonic kidney (HEK293) cells. J Bone Miner Res. 1997;12:715–725. doi: 10.1359/jbmr.1997.12.5.715. [DOI] [PubMed] [Google Scholar]
- 19.Parkash J, Chaudhry MA, Amer AS, Christakos S, Rhoten WB. Intracellular calcium ion response to glucose in beta-cells of calbindin-D28k nullmutant mice and in betaHC13 cells overexpressing calbindin-D28k. Endocrine. 2002;18:221–229. doi: 10.1385/ENDO:18:3:221. [DOI] [PubMed] [Google Scholar]
- 20.Parkash J, Chaudhry MA, Rhoten WB. Calbindin-D28k and calcium sensing receptor cooperate in MCF-7 human breast cancer cells. Int J Oncol. 2004;24:1111–1119. [PubMed] [Google Scholar]
- 21.Sanders JL, Chattopadhyay N, Kifor O, Yamaguchi T, Butters RR, Brown EM. Extracellular calcium-sensing receptor expression and its potential role in regulating parathyroid hormone-related peptide secretion in human breast cancer cell lines. Endocrinology. 2000;141:4357–4364. doi: 10.1210/endo.141.12.7849. [DOI] [PubMed] [Google Scholar]
- 22.Lewalle JM, Cataldo D, Bajou K, Lambert CA, Foidart JM. Endothelial cell intracellular Ca2+ concentration is increased upon breast tumor cell contact and mediates tumor cell transendothelial migration. Clin Exp Metastasis. 1998;16:21–29. doi: 10.1023/a:1006555800862. [DOI] [PubMed] [Google Scholar]
- 23.Whitfield JF. Calcium signals and cancer. Crit Rev Oncog. 1992;3:55–90. [PubMed] [Google Scholar]
- 24.Kuga T, Kobayashi S, Hirakawa Y, Kanaide H, Takeshita A. Cell cycle--dependent expression of L- and T-type Ca2+ currents in rat aortic smooth muscle cells in primary culture. Circ Res. 1996;79:14–19. doi: 10.1161/01.res.79.1.14. [DOI] [PubMed] [Google Scholar]
- 25.Guo W, Kamiya K, Kodama I, Toyama J. Cell cycle-related changes in the voltage-gated Ca2+ currents in cultured newborn rat ventricular myocytes. J Mol Cell Cardiol. 1998;30:1095–1103. doi: 10.1006/jmcc.1998.0675. [DOI] [PubMed] [Google Scholar]
- 26.Li M, Zhang M, Huang L, Zhou J, Zhuang H, Taylor JT, Keyser BM, Whitehurst RM Jr. T-type Ca2+ channels are involved in high glucose-induced rat neonatal cardiomyocyte proliferation. Pediatr Res. 2005;57:550–556. doi: 10.1203/01.PDR.0000155756.89681.3C. [DOI] [PubMed] [Google Scholar]
- 27.Richard S, Neveu D, Carnac G, Bodin P, Travo P, Nargeot J. Differential expression of voltage-gated Ca(2+)-currents in cultivated aortic myocytes. Biochim Biophys Acta. 1992;1160:95–104. doi: 10.1016/0167-4838(92)90042-c. [DOI] [PubMed] [Google Scholar]
- 28.Gomez JP, Potreau D, Branka JE, Raymond G. Developmental changes in Ca2+ currents from newborn rat cardiomyocytes in primary culture. Pflugers Arch. 1994;428:241–249. doi: 10.1007/BF00724503. [DOI] [PubMed] [Google Scholar]
- 29.Bkaily G, Sculptoreanu A, Jacques D, Jasmin G. Increases of T-type Ca2+ current in heart cells of the cardiomyopathic hamster. Mol Cell Biochem. 1997;176:199–204. [PubMed] [Google Scholar]
- 30.Nuss HB, Houser SR. T-type Ca2+ current is expressed in hypertrophied adult feline left ventricular myocytes. Circ Res. 1993;73:777–782. doi: 10.1161/01.res.73.4.777. [DOI] [PubMed] [Google Scholar]
- 31.Schmitt R, Clozel JP, Iberg N, Buhler FR. Mibefradil prevents neointima formation after vascular injury in rats. Possible role of the blockade of the T-type voltage-operated calcium channel. Arterioscler Thromb Vasc Biol. 1995;15:1161–1165. doi: 10.1161/01.atv.15.8.1161. [DOI] [PubMed] [Google Scholar]
- 32.Taylor JT, Rider B, Huang L, Keyser B, Agrawal K, Li M. A selective T-type calcium channel antagonist inhibits breast cancer cell growth (abstract) FASEB. 2004;15:A996. [Google Scholar]
- 33.Taylor JT, Huang L, Pottle JE, Liu K, Yang Y, Zeng X, Keyser BM, Agrawal KC, Hansen JB, Li M. Selective blockade of T-type Ca(2+) channels suppresses human breast cancer cell proliferation. Cancer Lett. 2008;267:116–124. doi: 10.1016/j.canlet.2008.03.032. [DOI] [PubMed] [Google Scholar]
- 34.Gray LS, Perez-Reyes E, Gomora JC, Haverstick DM, Shattock M, McLatchie L, Harper J, Brooks G, Heady T, Macdonald TL. The role of voltage gated T-type Ca2+ channel isoforms in mediating "capacitative" Ca2+ entry in cancer cells. Cell Calcium. 2004;36:489–497. doi: 10.1016/j.ceca.2004.05.001. [DOI] [PubMed] [Google Scholar]
- 35.Toyota M, Ho C, Ohe-Toyota M, Baylin SB, Issa JP. Inactivation of CACNA1G, a T-type calcium channel gene, by aberrant methylation of its 5' CpG island in human tumors. Cancer Res. 1999;59:4535–4541. [PubMed] [Google Scholar]
- 36.Latour I, Louw DF, Beedle AM, Hamid J, Sutherland GR, Zamponi GW. Expression of T-type calcium channel splice variants in human glioma. Glia. 2004;48:112–119. doi: 10.1002/glia.20063. [DOI] [PubMed] [Google Scholar]
- 37.Panner A, Cribbs LL, Zainelli GM, Origitano TC, Singh S, Wurster RD. Variation of T-type calcium channel protein expression affects cell division of cultured tumor cells. Cell Calcium. 2005;37:105–119. doi: 10.1016/j.ceca.2004.07.002. [DOI] [PubMed] [Google Scholar]
- 38.Leuranguer V, Bourinet E, Lory P, Nargeot J. Antisense depletion of beta-subunits fails to affect T-type calcium channels properties in a neuroblastoma cell line. Neuropharmacology. 1998;37:701–708. doi: 10.1016/s0028-3908(98)00060-4. [DOI] [PubMed] [Google Scholar]
- 39.Wyatt CN, Page KM, Berrow NS, Brice NL, Dolphin AC. The effect of overexpression of auxiliary Ca2+ channel subunits on native Ca2+ channel currents in undifferentiated mammalian NG108-15 cells. J Physiol. 1998;510(Pt 2):347–360. doi: 10.1111/j.1469-7793.1998.347bk.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Assandri R, Egger M, Gassmann M, Niggli E, Bauer C, Forster I, Gorlach A. Erythropoietin modulates intracellular calcium in a human neuroblastoma cell line. J Physiol. 1999;516(Pt 2):343–352. doi: 10.1111/j.1469-7793.1999.0343v.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hirooka K, Bertolesi GE, Kelly ME, Denovan-Wright EM, Sun X, Hamid J, Zamponi GW, Juhasz AE, Haynes LW, Barnes S. T-Type calcium channel alpha1G and alpha1H subunits in human retinoblastoma cells and their loss after differentiation. J Neurophysiol. 2002;88:196–205. doi: 10.1152/jn.2002.88.1.196. [DOI] [PubMed] [Google Scholar]
- 42.Bertolesi GE, Shi C, Elbaum L, Jollimore C, Rozenberg G, Barnes S, Kelly ME. The Ca(2+) channel antagonists mibefradil and pimozide inhibit cell growth via different cytotoxic mechanisms. Mol Pharmacol. 2002;62:210–219. doi: 10.1124/mol.62.2.210. [DOI] [PubMed] [Google Scholar]
- 43.Wang YQ, Brooks G, Zhu CB, Yuan WZ, Li YQ, Wu XS. [Functional analysis of the human T-type calcium channel alpha 1H subunit gene in cellular proliferation] . Yi Chuan Xue Bao. 2002;29:659–665. [PubMed] [Google Scholar]
- 44.Lu F, Chen H, Zhou C, Liu S, Guo M, Chen P, Zhuang H, Xie D, Wu S. T-type Ca2+ channel expression in human esophageal carcinomas: a functional role in proliferation. Cell Calcium. 2008;43:49–58. doi: 10.1016/j.ceca.2007.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Huang JB, Kindzelskii AL, Clark AJ, Petty HR. Identification of channels promoting calcium spikes and waves in HT1080 tumor cells: their apparent roles in cell motility and invasion. Cancer Res. 2004;64:2482–2489. doi: 10.1158/0008-5472.can-03-3501. [DOI] [PubMed] [Google Scholar]
- 46.Harkins AB, Cahill AL, Powers JF, Tischler AS, Fox AP. Expression of recombinant calcium channels support secretion in a mouse pheochromocytoma cell line. J Neurophysiol. 2003;90:2325–2333. doi: 10.1152/jn.00425.2003. [DOI] [PubMed] [Google Scholar]
- 47.Del Toro R, Levitsky KL, Lopez-Barneo J, Chiara MD. Induction of T-type calcium channel gene expression by chronic hypoxia. J Biol Chem. 2003;278:22316–22324. doi: 10.1074/jbc.M212576200. [DOI] [PubMed] [Google Scholar]
- 48.Lesouhaitier O, Chiappe A, Rossier MF. Aldosterone increases T-type calcium currents in human adrenocarcinoma (H295R) cells by inducing channel expression. Endocrinology. 2001;142:4320–4330. doi: 10.1210/endo.142.10.8435. [DOI] [PubMed] [Google Scholar]
- 49.Zhuang H, Bhattacharjee A, Hu F, Zhang M, Goswami T, Wang L, Wu S, Berggren PO, Li M. Cloning of a T-type Ca2+ channel isoform in insulin-secreting cells. Diabetes. 2000;49:59–64. doi: 10.2337/diabetes.49.1.59. [DOI] [PubMed] [Google Scholar]
- 50.Lee JY, Park SJ, Park SJ, Lee MJ, Rhim H, Seo SH, Kim KS. Growth inhibition of human cancer cells in vitro by T-type calcium channel blockers. Bioorg Med Chem Lett. 2006;16:5014–5017. doi: 10.1016/j.bmcl.2006.07.046. [DOI] [PubMed] [Google Scholar]
- 51.Panner A, Wurster RD. T-type calcium channels and tumor proliferation. Cell Calcium. 2006;40:253–259. doi: 10.1016/j.ceca.2006.04.029. [DOI] [PubMed] [Google Scholar]
- 52.Lory P, Chemin J. Towards the discovery of novel T-type calcium channel blockers. Expert Opin Ther Targets. 2007;11:717–722. doi: 10.1517/14728222.11.5.717. [DOI] [PubMed] [Google Scholar]
- 53.Strobl JS, Kirkwood KL, Lantz TK, Lewine MA, Peterson VA, Worley JF 3rd. Inhibition of human breast cancer cell proliferation in tissue culture by the neuroleptic agents pimozide and thioridazine. Cancer Res. 1990;50:5399–5405. [PubMed] [Google Scholar]
- 54.Chemin J, Monteil A, Perez-Reyes E, Nargeot J, Lory P. Direct inhibition of T-type calcium channels by the endogenous cannabinoid anandamide. EMBO J. 2001;20:7033–7040. doi: 10.1093/emboj/20.24.7033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.De Petrocellis L, Melck D, Palmisano A, Bisogno T, Laezza C, Bifulco M, Di Marzo V. The endogenous cannabinoid anandamide inhibits human breast cancer cell proliferation. Proc Natl Acad Sci USA. 1998;95:8375–8380. doi: 10.1073/pnas.95.14.8375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Li M, Hansen JB, Huang L, Keyser BM, Taylor JT. Towards selective antagonists of T-type calcium channels: design, characterization and potential applications of NNC 55-0396. Cardiovasc Drug Rev. 2005;23:173–196. doi: 10.1111/j.1527-3466.2005.tb00164.x. [DOI] [PubMed] [Google Scholar]