

Introduction
Cancer drug discovery has undergone a paradigm change over the past few years, from predominantly cytotoxic agent-based therapy to therapy aimed at genetic and molecular targets, thanks to a growing understanding of the genes and pathways responsible for cancer initiation and progression and to new drug discovery technologies. The success of drugs like trastuzumab, imatinib, gefitinib, and erlotinib has demonstrated that the targeting of specific oncogenic signal transduction pathways can be clinically useful.1 One such pathway, the Hedgehog-Glioma-associated oncogene homolog zinc finger protein (Hh-Gli)a signaling pathway, has attracted drug discovery scientists for the past decade. Hh-Gli signaling plays an important role in the embryonic patterning and development of many tissues and somatic structures as well as maintaining and repairing mature tissues in adults.2-4 Uncontrolled activation of the Hh-Gli pathway has been implicated in several cancers, including medulloblastoma, rhabdomyosarcoma, melanoma, basal cell carcinoma, and breast, lung, liver, stomach, prostate, and pancreatic cancers. 2, 5-8 Inhibition of the aberrant Hh-Gli pathway (Figure 1) has thus emerged as an attractive target for anticancer therapy.9-11 One Hh pathway inhibitor has shown promising results in phase I clinical trials and is proceeding to phase II12 studies, and two other compounds have entered phase I clinical trials.13, 14 In this article, we review the medicinal chemistry efforts to identify and design inhibitors of Hh-Gli signaling and present a perspective of future developments in this dynamic field. We also present a brief overview of the role of Hh-Gli signaling pathway in normal development and cancer.
Figure 1. Hedgehog pathway activators and inhibitors.

The Hh receptor Ptch is a transmembrane protein that inhibits the activity of Smo in the absence of Hh. Binding of Hh results in activation of Smo, which then modulates Gli transcription factors to initiate transcription of Hh target genes. Proteins involved in modulating signal relay between Smo and Gli are as indicated. Pathway suppressors are indicated in red and activators in green color. The molecules in this pathway that are targets for chemical inhibitors include Smo and Gli.
The hedgehog (Hh) gene was first identified during a search for embryonic lethal mutants of Drosophila melanogaster, which found that mutation of Hh resulted in altered segment patterning of the larva.15 Subsequently the gene was identified in many other invertebrates and vertebrates, including humans. Three mammalian counterparts of the Hh gene, termed Sonic hedgehog (Shh), Dessert hedgehog (Dhh), and Indian hedgehog (Ihh), were identified by combined screening of mouse genomic and cDNA libraries.16 Hh undergoes multiple processing events, including autocatalytic cleavage of the C-terminal domain combined with addition of a cholesterol moiety at the cleavage site, and an N-terminal palmitoylation, to generate the active ligand.17-19
The receptor of secreted Hh protein is the multipass transmembrane protein Patched (Ptch). Of the two vertebrate homologs of Ptch, Ptch1 and Ptch2, the role of Ptch1 is better understood. In the absence of Hh ligand, Ptch inhibits the activity of the downstream effector Smoothened (Smo). The binding of Hh inactivates Ptch, resulting in activation of Smo.20 In Drosophila, a complex of proteins comprising Fused (Fu), Suppressor of Fused (SuFu), and Costal-2 (Cos2) mediates signaling downstream of Smo and is aided by several kinases, such as protein kinase A (PKA), glycogen synthase kinase 3 (GSK3), and casein kinase 1 (CK1). Mammalian homologs of Fu and Cos2 have not yet been identified, suggesting that the signaling mechanisms differ in mammals and Drosophila. 21, 22 Several mammalian-specific kinases that are required for Shh signaling have been identified.23-25 These proteins modulate the function of Gli (Ci in Drosophila), the only transcription factor identified to date that operates directly downstream of Hh.
The first vertebrate Gli gene to be discovered was human Gli1, which was amplified about 50-fold in a malignant glioma.26 Vertebrates have three Gli proteins (Gli1, Gli2 and Gli3), all of which have five highly conserved tandem zinc fingers, a fairly conserved N-terminal domain, several potential PKA sites, and a number of additional small conserved regions in the C-terminal end. Despite these similarities, the functions of the Gli subtypes differ. Both Gli2 and Gli3 contain activation and repressor domains. Consequently, in the absence of upstream Hh signal, full-length Gli3 and, to a lesser extent, Gli2, are constitutively cleaved to generate a truncated repressor form.27-29 Hh signaling inhibits this cleavage, resulting in full-length Gli2 and Gli3, which have activator function. Gli1, in contrast, does not undergo proteolytic cleavage and acts as a constitutive activator.27 The transcription of Gli1 gene is initiated by Hh and is also controlled by Gli3.27 Target genes of the Hh pathway other than Gli1 include Ptch, several Wnt and TGFβ superfamily proteins, cell cycle proteins such as cyclin D, and stem-cell marker genes such as NANOG and SOX2.30, 31 Investigators are now attempting to comprehensively identify the Gli1-target genes.32, 33
Hh signaling in development and tissue maintenance
The Hh signaling pathway is crucial for proper embryonic development.34 It is also essential for restraining growth in the nervous system and other tissues, and in maintenance of stem cells in adults.35-37 The expression and roles of Hh in vertebrate tissues/organs has been extensively described in the recent reviews.34, 38
Two of the functions of Hh in vertebrate embryonic development are both crucial and relatively well understood: neural tube differentiation and anteroposterior limb patterning. The predominant mechanism of Hh signaling in these functions is paracrine signaling, in which the Hh molecules act in a gradient fashion. For example, in vertebrate limb buds, exposure to different concentrations of Shh modulates patterning of the interdigital mesenchyme, which influences the proper growth of digits in a specific pattern.39 In neural tube development, Shh produced by the floor plate causes dorsoventral patterning, the specification of ventral cell populations, and general cellular proliferation in the brain.40 Holoprosencephaly, a disorder involving the development of forebrain and midface in which ventral cell types are lost, is caused in humans by mutations that lead to loss of Shh activity.41
Another important feature of Shh signaling is that the Gli subtypes have both unique and overlapping functions. While ectopic expression of Gli1 in the midbrain and hindbrain of transgenic mice results in expression of some ventral cell types, mice homozygous for a mutation in the region encoding the zinc finger domain of Gli1 develop normally.42, 43 However, Gli1/Gli2 double mutant mice have phenotypes with severe multiple defects, including variable loss of the ventral spinal cord, and smaller lungs; therefore, Gli2 plays a more important role in spinal cord and lung development than does Gli1. In contrast, Gli1/Gli3 double mutant mice did not have these phenotypes.43 Gli2 and Gli3 have both been implicated in skeletal development, with each subtype serving specific functional roles.44, 45 Gli2 mutant mice exhibit severe skeletal abnormalities including cleft palate, tooth defects, absence of vertebral body and intervertebral discs, and shortened limb and sternum.45 Gli3 appears to be the major mediator of Shh effect in the limbs, as Gli1/Gli2 double mutant mice had a normal digit number and pattern while Gli3 mutant mice showed polydactyly.43, 46
Genetic analyses of Gli mutants revealed that requirement for Gli subtypes development is quite divergent even among vertebrates. In zebrafish, both detour (dtr) mutations (encoding loss-of-function alleles of Gli1) and you-too (yot) mutations (encoding C-terminally truncated Gli2) have defects in body axis formation and expression of Hh-target genes in the brain,47 suggesting divergent requirements for Gli1 and Gli2 in mouse and zebrafish.
In adults, the Hh pathway is essential for restraining growth in the nervous system and other tissues, and in maintenance of stem cells. Zhang and Kalderon have shown that Hh acts specifically on stem cells in Drosophila ovaries and that these cells cannot proliferate in the absence of Hh.48 Other studies showed that Hh signaling in the postnatal telencephalon both promotes proliferation and maintains populations of neural progenitors, suggesting that Shh signaling in the mammalian telencephalon may participate in the maintenance of a neural stem cell niche.35 The role of Hh in proliferation of adult neural progenitor cells was confirmed by a study in which Shh was overexpressed and proliferation was inhibited by using a Smo antagonist.49
Hh signaling in cancer
Hh genes have the ability to induce tissue proliferation. This function is important in embryogenesis and tissue maintenance, but inappropriate activation of the pathway can result in tumorigenesis.50 Tumors in about 25% of all cancer deaths are estimated to involve aberrant Hh pathway activation.4 Tumorigenesis or tumor growth can result from abnormal up-regulation of Hh ligand or from deregulation of the expression or function of downstream components by, for example, loss of Ptch,51-53 activating mutations of Smo,54 loss of SuFu,55 amplification or chromosomal translocation of Gli1,26 or Gli2 gene amplification or stabilization of Gli2 protein.56
The first Hh pathway gene found to be amplified in cancers was Gli1, which was expressed at high levels in human glioblastoma and derived cell lines.26 Subsequently, Gli1 was found to be consistently expressed in a variety of glial tumors, and Gli1 overexpression was shown to induce central-nerves system hyperproliferation.57 Gli1 overexpression has also been observed in a panel of brain tumors ranging from low-grade to high-grade in a study that identified Gli1 expression as the only reliable marker of Hh pathway activity.31 Further, cell proliferation in primary cultures of many of these tumors was inhibited by Gli1 small-interfering RNA.31 Gli1 expression was correlated with tumor grade in PDGF-induced gliomagenesis in mice.58 Hh signaling components other than Gli1 also contribute to tumorigenesis in specific subsets of glioblastomas. In PDGF-induced tumors, expression level of Shh was correlated with the tumor grade. However, other studies found only a subset of gliomas to contain high levels of Shh.31
Another cancer with defects in Hh pathway regulation is basal cell carcinoma (BCC). Human Ptch was first identified by virtue of its mutation in patients with Gorlin Syndrome (GS), a genetic disease that gives rise to sporadic BCC.59 The mutations of Ptch identified in BCC include deletions producing truncated proteins and insertion or nonsense mutations accompanied by loss of heterozygosity (LOH) or mutations in the other allele.51, 52 These mutations inhibit the ability of Ptch to suppress Smo, resulting in constitutive Hh signaling. While Ptch1 abnormalities are detected in the majority of BCC patients, it is now clear that a subset of BCC is also driven by a mutation in Smo that decreases its sensitivity to inhibition by Ptch.54 In addition, overexpression of Gli1 protein causes BCC-like tumors in mice, establishing the importance of Gli1 transcription in BCC tumorigenesis.60 The level of Gli1 transcript can be used to discriminate BCC from certain other skin tumors.61 However, blocking of Gli-based transcription has not yet been shown to arrest BCC growth.
Medulloblastoma, the most common malignant pediatric brain tumor, is linked with mutations in Ptch and Smo and mutations in other Hh pathway genes such as SuFu and Gli.62 Inactivation of the Ptch locus by deletion and mutation has been found in about 10% of sporadic medulloblastomas.53 Shh pathway involvement in these tumors was further confirmed by studies in which treatment of murine medulloblastomas with Smo inhibitors inhibited cell proliferation and reduced tumor growth in mice.63-65 Taylor et al.55 identified SuFu as a tumor-suppressor gene whose mutation predisposes individuals to medulloblastoma. They found that a subset of children with medulloblastoma carry germline and somatic mutations in SuFu, accompanied by loss of heterozygosity of the wild-type allele.55 Several of these mutations encoded truncated SuFu proteins that are unable to export Gli protein from the nuclei. In addition, the tumor-suppressor REN has also been linked with medulloblastoma in which the allelic deletion and reduced expression of REN is frequently observed. It is suggested to inhibit medulloblastoma growth by negatively regulating the Hh pathway. 66-68
Hh has also been shown to be an early and late mediator of pancreatic cancer tumorigenesis. Shh was not detected in normal adult human pancreata but was aberrantly expressed in 70% of pancreatic adenocarcinoma specimens.69 Participation of Shh signaling has been indicated at multiple stages of pancreatic carcinogenesis and is accompanied by multiple oncogenic factors, including K-Ras, one of the most frequently mutated genes in pancreatic cancer.70, 71 Activated Hh signaling was detected in cell lines established from primary and metastatic pancreatic adenocarcinomas, and the Smo inhibitor cyclopamine induced apoptosis in a subset of the pancreatic cancer cell lines both in culture and mice.72
Numerous studies indicate that Hh signaling is involved in prostate cancer. Sanchez and others reported the expression of Shh-Gli pathway components in adult human prostate cancer.8 Treatment of primary prostate tumor cultures and metastatic prostate cancer cell lines with Smo inhibitors blocked the pathway and proliferation. Increased expression of Shh in prostate cancer cells upregulates Gli1 expression and dramatically accelerates the growth of prostate tumor xenografts.73 Elevated Shh activity distinguished metastatic from localized prostate cancer, and manipulation of this pathway modulated the invasiveness and metastasis of these tumors.72, 74
Hh signaling has also been implicated in various other cancers, such as lung, colorectal, bladder, endometrial, ovarian, and esophageal carcinomas and rhabdomyosarcoma.75-83 The role of Hh-Gli signaling pathway in cancer and it potential as therapeutic target has been reviewed in more detail in recent articles.10, 11, 30, 84
Small Molecule Inhibitors of the Hh-Gli Pathway
The aberrant activation of Hh-Gli signaling in several cancers has made it an attractive target for anticancer drug discovery. Here, we summarize the medicinal chemistry efforts to discover Hh-Gli pathway inhibitors. The compounds known to date (January 2009) are classified as inhibitors of Shh, Smo, class IV alcohol dehydrogenase (Alcohol dehydrogenase 7, Adh7), and Gli transcription.
The first small molecule inhibitor that blocks the Shh signaling by binding to Shh protein was recently reported by Stanton et al.85 A macrocycle 1 (Figure 2) was discovered by screening a 10,000 small molecule diversity-oriented library in small-molecule microarry for binding to bacterially expressed Shh N-terminus fragment (ShhN). The hit compounds were further evaluated in Shh-LIGHT II cells, a clonal NIH 3T3 cell lines stably incorporated with a Gli-responsive Firefly luciferase reporter. The activity was measured as relative luciferase activity after incubation with the compound in the presence of N-palmitoylated ShhN. The structure-activity relationship (SAR) studies led to robotnikinin (2), a 12-membered macrocycle with a 3.1 μM Kd for the ShhN binding and a dose dependent inhibitor of ShhN-induced pathway activation in Shh-LIGHT II cells. It did not show any substantial cytotoxicity in cell viability assay. In a mouse Ptch1-/- cell line, in which the Ptch1 alleles were replaced with a β-galactosidase (β-gal) reporter, compound 2 did not inhibited the β-gal levels, thus indicating that it inhibits the Shh pathway upstream of Ptch1. It also inhibited alkaline phosphatase induction in a dose-dependent manner in C3H10/T1/2 cells in the presence of ShhN, which was rescued by purmorphamine,86 a Smo agonist (C3H10/T1/2 cells differentiate to osteoblasts upon treatment with N-palmitoylated ShhN, an event measured by alkaline phosphatase as a marker of the transformation87, 88). Compound 2 inhibited Shh-induced expression of Gli1 and Gli2 in primary human keratinocytes and human-derived skin tissue.85
Figure 2.
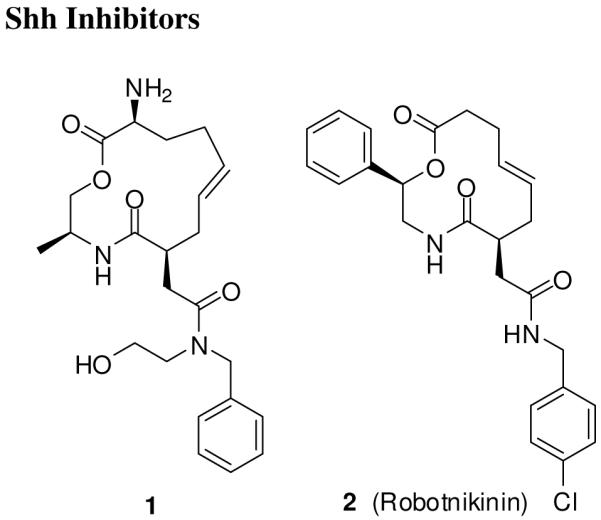
Shh inhibitors.
Smoothened Inhibitors
The Hh pathway is initiated by binding of Hh to the transmembrane protein Ptch, which releases the suppression of Smo and initiates a cascade of events resulting in expression of Hh-responsive genes (Figure 1). Cyclopamine (3), the prototype inhibitor of the Shh pathway, is currently in preclinical and clinical studies as an anticancer agent.89 Cyclopamine inactivates Smo by binding to its heptahelical bundle.74, 90 A number of Smo inhibitors have now been reported and can be classified as cyclopamine analogues or synthetic Smo antagonists.
Cyclopamine Analogues
Cyclopamine (3), a teratogenic steroidal alkaloid derived from the plant Veratrum californicum, has been known since the 1960s. It was first reported to block the Shh signaling pathway in 1998 by Cooper et al.91 The alkaloids cyclopamine and jervine (5) (Figure 3) from this plant were associated with holoprosencephaly and other teratogenic effects in lambs born to pregnant ewes in the 1960s.92, 93 Incardona et al.94 further showed that cyclopamine-induced teratogenesis is independent of cholesterol metabolism and results from inhibition of Shh signaling.
Figure 3.

Cyclopamine derivatives and related steroidal alkaloids.
Incardona et al.95 also studied the SAR of cyclopamine derivatives and related steroidal alkaloids (Figure 3) in a chick embryo neural plate explant assay by measuring Shh inhibition as loss of HNF3β, a floor plate marker. For cyclopamine to be active, its E ring should be perpendicular to rings A-D. Opening of this ring, as in veratramine (8), resulted in much weaker activity. Oxidation of cyclopamine to cyclopamine-4-en-3-one (7) improved potency by at least 2-fold in the explant assay. Jervine (5), a C11 ketone analogue of cyclopamine, was 5- to 10-fold less potent, although it was metabolically more stable in vivo. Reduction of jervine to tetrahydrojervine (6) further decreased the activity by 3-fold. The glycoalkaloid analogue of cyclopamine, cyloposine (4), was equipotent to jervine (5). The steroidal alkaloids solasodine (9) and tomatidine (10), with E rings co-planar to the A-D rings, were 15- to 20-fold less active.95
The modification of cyclopamine at the 3-position (Figure 4) by addition of a carbamate group (11, 12) decreased activity 100-fold. The secondary amine in the ring F of cyclopamine and jervine tolerates modifications (Figure 4), if its basicity is preserved. A small- to medium-size substitution at this position (13-15) improved the inhibitory activity, which is lost by large-size substitution, possibly due to cell impermeability or steric hindrance. Any branched substitution (16) close to the cyclopamine scaffold also decreases the activity. Oxidation of the hydroxyl at the 3-position in the N-substituted derivatives improved activity by 2-fold over that of the corresponding hydroxyl analogues (14 vs. 13). Compound 14 (3-keto-N-aminoethyl aminocaproyl digyrocinnamoyl (KAAD)- cyclopamine) was identified as the most potent inhibitor in this series.96 It showed 10-20-fold better potency but similar or lower toxicity as compared to cyclopamine. Taipale et al.97 reported that the mechanism of action of cyclopamine and its analogues is through reversal of oncogenic activation of the Hh pathway by antagonism of Smo. Photo-affinity studies with a fluorescent-tagged derivative of cyclopamine confirmed the direct binding of cyclopamine to the heptahelical bundle of Smo, inducing a conformation that was structurally similar to that induced by Ptch.90 Compound 14 is currently in preclinical development.89
Figure 4.

Cyclopamine analogues with substitution at 3-position or secondary amine in F ring.
Researchers at Infinity Pharmaceuticals and Johns Hopkins carried out further SAR studies of cyclopamine by making changes in rings A, D, and F (Figure 5) to overcome poor aqueous solubility and chemical instability in acid.98-100 Tremblay et al.99 synthesized the D-homocyclopamine analogue 17 (IPI-611) which was more stable than cyclopamine at low pH, but its hydrochloride salt was less soluble in water and showed weaker activity than cyclopamine. Oppenaeur oxidation of 17 to the corresponding conjugated enone yielded a potent compound, 18 (IPI-269609/IPI-609)98-100 that inhibited differentiation of C3H10/T1/2 cells to osteoblasts (a Shh dependent event87, 88 measured by alkaline phosphatase assay) with an EC50 of 200 nM. It also showed improved aqueous solubility and stability.98-100 Compound 18 can be prepared from cyclopamine with an overall yield of 25-30%.100 Removal of the keto group and saturation of the 4,5-double bond (19) decreased activity.98 The N-acetamide derivative, 20, showed activity similar to that of the parent compound, contrary to earlier reports,96 suggesting importance of basicity of the nitrogen. Bulky groups such as benzyl (21) reduced potency while the glycolate analogue (22) displayed activity similar to that of 18. Compound 23, with a side chain similar to that of 14 (KAAD-cylopamine), showed a 20-fold increase in activity over that of 18, while its truncated analogue 24 was 10-fold more potent. However, 23 and 24 are less soluble than 18.100 Replacement of the 3-keto group with a methanesulfonamide group afforded 25 that potently inhibits C3H10/T1/2 cell differentiation, and cell growth of human multiple myeloma, acute myeloid leukemia, myelodysplastic syndrome, non-Hodgkin’s lymphoma, Hodgkin’s lymphoma, and pre-B cell acute lymphoblastic leukemia cell lines. It also suppressed tumor growth in pancreatic cancer and medulloblastoma mouse models and showed clear tumor regression in a medulloblastoma model at higher doses.98
Figure 5.

D-Homocyclopamine analogues.
Compound 18 showed inhibitory effect in a Gli-responsive reporter assay to a degree comparable to that of cyclopamine. It diminishes the migration and colony formation of pancreatic cancer cells, suppressed growth of a subcutaneous xenograft of pancreatic cancer E3LZ10.7, abrogates metastasis in orthotopic pancreatic cancer xenografts of E3LZ10.7 and Capan-1 cell lines. The results indicated that Smo inhibitors could be effective in inhibiting metastasis in pancreatic cancers.101 Pretreatment with 18 diminishes tumorigenicity in vivo.101 The hydrochloride salt of 18 formulated in water containing 30% of 2-hydropropyl-β-cylodextrin displayed 80% oral bioavailability and a plasma half-life of 3.2 h in CD-1 mice.100
Infinity Pharmaceuticals designed 26 (IPI-926)102 (structure not disclosed) as a second-generation cyclopamine analogue that is orally bioavailable, has a long half-life in plasma (10-24 h in multiple species) and tumors, and is active in vivo. Once-daily oral administration of 26 at 40 mg/kg resulted in complete tumor regression in a murine medulloblastoma model.102 Compound 26 also delays or prevents tumor recurrence after completion of chemotherapy in a small-cell lung cancer xenograft model.103 It entered phase I clinical trials for advanced and/or metastatic solid tumor malignancies in September 2008.13
The cyclopamine lactam analogues (Figure 6) have also been reported to inhibit differentiation of C3H10/T1/2 cells to osteoblasts in a alkaline phosphatase assay.104 The replacement of the ring A of 18 with a γ-lactam ring and a trans A/B ring junction gave compound 27 with potent activity (IC50 < 20 nM) in alkaline phosphatase assay. Change in the A/B ring junction to cis configuration (28) resulted in > 5-fold decrease in the activity. Expansion of ring A to a seven membered ring (29) restored the activity, although it was still 2-5-fold less active than 27. Reversing the amide group of 29 (33) resulted in a significant loss in activity. The ring A-methyl amide 30 was equipotent to 29, while the ring F-methanesulfonamide analogue 31 showed improvement in activity to match the potency of 27. However, incorporating both the methyl group in ring A and the methanesulfonyl group in ring F togeter resulted in significant loss in activity in 32. An unsaturated analogue (34) of 30 showed significant loss in activity. Compound 29 effectively reduced the Gli-responsive luciferase reporter activity in pre-B cell acute lymphocytic leukemia cell lines, and inhibited growth of primary cultures derived from patients with multiple myeloma, acute myeloid leukemia, myelodysplastic syndrome, non-Hodgkin’s lymphoma and Hodgkin’s disease. It was suggested that 29 affects a tumor-stroma interaction in human pancreatic xenograft in mice, as it downregulated the mRNA levels of murine Gli1 but not of human Gli1. It showed ED50< 7.5 mg/kg with once daily oral dose in an allograft model of medulloblastoma from a transgenic mouse with loss of function mutations in Ptch1 and Hypermethylated in Cancer (Hic1).104
Figure 6.

Cyclopamine lactam analogues.
Infinity Pharmaceuticals also reported cyclopamine analogues that have a heterocyclic ring fused to ring A (Figure 7).105 Compound 35 showed potent inhibition (IC50 <100 nM) in an alkaline phosphatase assay using C3H10/T1/2 cells in the presence of Shh-N. Ring D expansion further improved the activity to yield the most potent compound, 36 (IC50 <20 nM). Any further substitution on the pyrazole ring (37-39) or ring F (40-43), however, decreased the activity. Replacement of the pyrazole ring with an oxazole (44) decreased the activity 1-5-fold, and replacement of the ring with a 2-methylthiazole ring (45) decreased activity more than 5-fold as compared to that of 36. Compound 36 significantly suppressed the growth of human pancreatic cancer xenografts in mice at 40 mg/kg/day.105
Figure 7.

Cyclopamine analogues with a fused heterocyclic ring.
Cyclopamine is poorly soluble in water, hindering its development as a drug. Zhang et al.106 recently reported carbohydrate-cyclopamine analogues (Figure 8) aimed at improving solubility and introducing structural diversity. They utilized click chemistry (copper-catalyzed azide-alkyne coupling) to synthesize a library from an alkyne-modified cyclopamine and diverse azidosugars. Compound 46 showed activity against a lung cancer cell line (A549) comparable to that of cyclopamine in a standard viability assay, with an IC50 of 33 μM as compared to 49 μM for cyclopamine; however its aqueous solubility was superior. Another β-linked analogue (47) was less potent than cyclopamine. They also suggested that α-linked L-pyranose might be essential for activity.106
Figure 8.

Carbohydrate cyclopamine analogues. Compound 46 is 89/11 mixture of α/β.
In an effort to decrease the adverse effects of cyclopamine, Kumar et al.107 synthesized prodrugs of cyclopamine (Figure 9) by coupling cyclopamine to the peptide carriers that are proteolytically removed in cancers. They designed the peptide carriers as substrates of the tissue-specific serine protease prostate-specific antigen (PSA), which is expressed at high levels in prostate but not in other organs. Two such carriers, His-Ser-Ser-Lys-Leu-Gln (HSSKLQ) and Ser-Ser-Lys-Tyr-Gln (SSKYQ), were coupled with cyclopamine to yield conjugates 48 and 49, respectively.107
Figure 9.

Cyclopamine prodrugs.
Compounds 48 and 49 are converted to cyclopamine in the presence of PSA, with half-lives of 3.2 h and 22 h, respectively. Both showed minimal activity in the DU145 prostate cancer cell line, which lacks PSA, but addition of PSA to the medium resulted in a 7-fold increase in efficacy over that of cyclopamine. In vivo studies of these compounds are ongoing.107
Non-cyclopamine-Scaffold Compounds
Several pharmaceutical companies have identified new Smo inhibitors with drug-like properties by optimization of high-throughput screen hits. One such small molecule, 101 (GDC-0449, Curis and Genentech, Figure 14), is currently in phase I/II clinical trials for advanced BCC and solid epithelial tumor.108 Development of another candidate, 54 (CUR-61414, Figure 11), was suspended after phase I.89
Figure 14.

Pyridyl and quinoxaline Smo inhibitors
Figure 11. Aminoprolines.

Aminoproline Smo inhibitors.
The structurally diverse compounds 50 (SANT-1), 51 (SANT-2), 52 (SANT-3), and 53 (SANT-4) (Figure 10) were discovered by Chen et al.109 by screening 10,000 small molecules for Hh signaling inhibition (IC50 values for 50-53 were 20, 30, 100, and 200 nM, respectively, in a Shh-LIGHT II assay). These molecules have different mechanisms of action.109
Figure 10.

Compounds discovered by screening with a Shh-reporter.
Another high-throughput screen of a library of 100,000 small molecules using a Hh-reporter cell line (s12) led to discovery of the lead compound 54 (Figure 11), an aminoproline with an IC50 of 100-200 nM, as compared to 500-700 nM for jervine (5).110 Compound 54 showed a physicochemical profile suitable as a drug candidate and had no toxicity in preclinical testing. It suppressed proliferation and induced apoptosis in BCC cells without affecting normal skin cells.110 Curis prepared a library of derivatives of compound 54 to elucidate the SAR of the amide group at the 4-postion of aminoproline, the substitution at the N1 position of the pyrrolidine ring, and the piperazine ring. The cis isomers exhibited more than 100-fold greater activity than the trans isomers. A cyclopropyl or isopropyl substitution of the amide at the 4-position of the proline in 55 and 56, respectively, resulted in similar potency (IC50 < 100 nM). Compounds 57 and 58, with a cyclopentane ring and CH(CH3)CF3 respectively, showed weaker activity than did 55 and 56. Replacement of amide with urea (59) also decreased activity. 3,4,5-Trimethoxybenzyl (61) or (2,3-dihydrobenzo[b][1,4]dioxin-6-yl)methyl (62) substitutions at the 1-position of proline were better tolerated than benzo[d][1,3]dioxol-5-ylmethyl (54) or 3-methoxybenzyl (60). The piperazine ring on proline carbamide is essential for activity. Its replacement with a methyl piperazine (64), piperidine (65), or aliphatic amine (66) decreases activity by several-fold.111 Clinical trials of 54 were halted after a phase I study as a topical agent in patients with BCC.89
Brunton et al. at Evotec and Curis increased the potency of the screening hit 67 (IC50 1.4 μM) to the nanomolar level by lead optimization. They carried out SAR studies in a reporter assay focusing on the urea group, the 4-fluorophenyl, and the quinazolinone ring (Figure 12). The bis-desmethyl analogue of 67 (68) showed a 2-fold improvement in activity, indicating that both methyl groups are not necessary. The methyl derivative 69 was 3-fold less active than 67. The 3-trifluoromethyl-4-chlorophenyl-substituted desmethyl analogue 70 showed a 20-fold improvement in activity over 67, exhibiting an IC50 of 70 nM. Replacement of the 4-fluorophenyl with an isopropyl group in 71 decreased activity 3-fold, indicating the importance of the phenyl ring.112
Figure 12. Quinazolinones and quinazolines.

Exelixis has described quinazolines (72-75) and pyridopyrimidine (76) (Figure 12).113 The tetrahydroisoquinoline analog 72 exhibited an IC50 of 5.8 nM in a reporter assay in the Shh LIGHT II cell line. The cyclopropyl-substituted 74 was most potent, with an IC50 of 2.8 nM. Replacement of the quinazoline with pyridopyrimidine yielded the equipotent 76, with an IC50 of 3.6 nM.113 Exelixis and Bristol-Myers Squibb are co-developing 77 (XL139/BMS-833923)14, 114 (structure not disclosed) as an inhibitor of the Hh pathway. Compound 77 is in phase I clinical trials for advanced or metastatic cancers.14, 114
Peukert et al. from Novartis reported a series of biarylcarboxamides (Figure 13) based on an initial micromolar screening hit 78, which was originally designed as a microsomal triglyceride transfer protein (MTP) antagonist.115, 116 These compounds were subjected to several screens to identify ones that bind directly to Smo. SAR studies revealed that benzylamines (79) were more potent than carboxamides or sulfonamides. The S-enantiomers (81 and 84) were significantly more potent Smo inhibitors than R-enatiomers (80 and 83) which had more potent MTP inhibitory activity. The trifluoromethyl group is essential for activity. The phenyl-substituted 79 was 5-7-fold more potent than pyridyl analogs 81 and 82. The 5-membered hetrocyclic analogues 84-86 showed the most potent activity in TM3Hh12 cells in a transcription assay with approximately 10-fold selectivity over MTP inhibition, and also showed direct Smo binding in membranes from CHO-K1 cells stably expressing HA-tagged mouse or human Smo.115
Figure 13.

Biarylcarboxamide, bisamide, benzimidazole, and sulfonamide derivatives.
Biarylcarboxamides, such as 87,117 and structurally similar bisamide compounds (88-91)118 also are active in the Gli- responsive luciferase reporter assay.
Curis has reported benzimidazole derivatives (92-96) (Figure 13) that are structurally similar to 51.119 SAR study showed that addition of a methyl or chloro group at the 5-position of the benzimidazole ring of 93 and 94, respectively, resulted in activity 50-fold greater than that of 92. Replacement of the methyl group in 93 with a N,N-dimethyl (95) resulted in a further 10-fold increase in activity, to an IC50 <1 nM. Compound 96 (HhAntag/Hh-Antag691)120 inhibited Shh activity with an EC50 of 40 nM as compared to 500 nM for cyclopamine in a Gli-responsive luciferase reporter assay, although the two compounds exhibited similar (IC50 10-30 μM) inhibition of growth of medulloblastoma cultures.65, 119 Twice-daily oral administration of 96 at 100 mg/kg for 2 weeks eliminated medulloblastomas that spontaneously form in Ptch1+/-p53-/- mice,65 suggesting that 96 may penetrate the blood-brain barrier, at least partially. No side effects were observed during a 50-day study. Compound 96 inhibited the expression of several genes including Gli1 and Gli2 in the tumors of the mice,65 and eliminated medulloblastoma allografts with an active Shh pathway but was ineffective against allografts with an inactive Shh pathway; thus showing specificity for medulloblastomas in which the Shh pathway is upregulated.121 Compound 96 (100 mg/kg twice daily for 4 days) also decreased the volume of medulloblastoma allografts from another mice that had Cxcr6 mutation, over-expression of Shh pathway target genes, and low levels of Ptch1.122 However, treatment of young mice (postnatal day 10-14, a period of extremely rapid bone growth) with two doses of 100 mg/kg of 96 within 24 h resulted in permanent defects in bone structure and growth, despite restoration of the somatic Hh reporter activity 48 h after withdrawing 96. The inhibition of Hh pathway by 96 resulted in differentiation of chondrocytes, expansion of hypertrophic zone, and breakdown in columnar organization leading to permanent defects in joint structures. The complete inhibition of the Hh pathway in tumor cells is essential for tumor elimination while even a transient exposure of proliferating chondrocytes to 96 can result in terminal differentiation thus suggesting that these bone defects occurs within the therapeutic window in the young mice. This finding raises a concern about using 96 in pediatric patients, although, it is not known if the bone defect will occur in humans also.123 An earlier study in adult mice did not show any major side effects,65 but the transient inhibition of Hh pathway might also compromise the regenerative capacity of some adult tissues.123
Compounds that have a urea, thiourea, or sulfonamide linker in place of an amide linker were also synthesized and evaluated.119 Sulfonamides 97-100 (Figure 13) also showed potent activity.124
Genentech and Curis have reported pyridyl (101-103)125 and quinoxaline (104-106)126 compounds (Figure 14). Compound 101 is currently in a phase I study for locally advanced or metastatic solid tumors and phase II clinical trials for advanced BCC, metastatic colorectal cancer, and advanced ovarian cancer.12, 108, 127 Compound 101 (IC50, 3 nM in a Gli-responsive luciferase reporter assay) is an once-daily orally active, potent inhibitor of Smo that has an extremely long half-life (10-14 days) with sustained high-micromolar plasma concentrations. Daily dosages of 150, 270, and 540 mg of 101 were tested in phase I studies in patients with BCC or pancreatic, colorectal, or adenocystic carcinomas. It showed at least 2-fold down-regulation of Gli1 expression in skin punch biopsies, irrespective of the dosage level. Two of nine patients with BCC on treatment with 101 had a confirmed response, four had a confirmed partial response, two had stable disease, and one had progressive disease. No dose-limiting toxicity was observed. The side effects included fatigue, asymptomatic hyponatremia, dysgeusia, and alopecia.12, 128
Novartis reported phtalazine analogues (107-114, Figure 15) as Smo antagonists.129 The compounds were evaluated for inhibition of Gli-responsive luciferase activity in TMHh12 cells in the presence of a small molecule Smo agonist. Compounds which showed shift in IC50 with increase in concentration of Smo agonist were judged to be directly interacting with Smo. The compounds were also screened in a Smo binding assay to determine IC50 for displacement of a radio-labeled small molecule Smo agonist from a filter-bound mouse and human Smo.129
Figure 15.
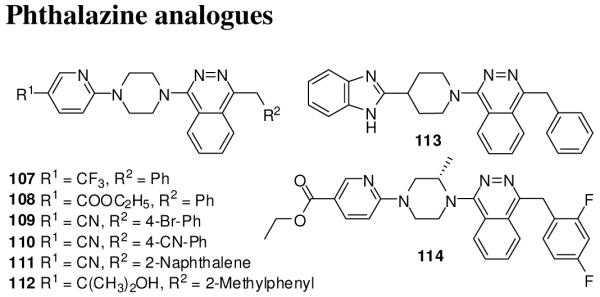
Phthalazine Smo inhibitors.
Merck had discovered triazole derivatives as 11-β-hydroxysteroid dehydrogenase type-1 inhibitors for metabolic disorders.130 Some of these derivatives, 115-120 (Figure 16), also showed Smo antagonist activity in the Shh-Light II reporter assay, and Smo binding assay and suppressed medulloblastoma xenograft growth in mice models.131
Figure 16.
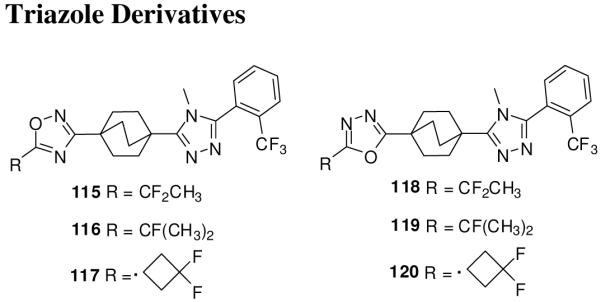
Phthalazine Smo inhibitors.
Smo Intracellular-Loop Analogues
Smo resembles a G protein-coupled receptor (GPCR) in general topology. Remsberg et al.132 designed structural analogues of the Smo intracellular loop. Unlike inhibitors designed for GPCRs using this strategy that were often non-specific (due to significant sequence homology in the intracellular loop), the peptides derived from the Smo intracellular loop were expected to be specific due to the unique structure of Smo, which is conserved among species.132 The peptide analogues of the intracellular loop of Smo were evaluated for growth inhibition in the MCF-7 breast cancer cell line. The second and third loop derivatives were further optimized by C-terminal and N-terminal truncation to identify a 10-amino-acid derivative of the N-terminal half of the second intracellular loop, 121 (SMOi2-12, Palmitoyl-LTYAWHTSFK), with a nanomolar IC50. The retroinverso peptide of 121 with all-d-amino acids and reverse sequence, 122 (SMOi2-20, Ac-KFSTHWAYTLK-e-Pal (all-d-)), was metabolically more stable and 200-fold more potent than 121, with an IC50 of 0.3 nM in an MTT assay in SK-Mel2 melanoma cells.132
Inhibitors of Adh7
Inhibitors of class IV alcohol dehydrogenase 7 (Adh7) were also reported as the Hh signaling antagonists, though the exact mechanism of action of these compounds for the Hh pathway is not understood. The Hh signaling has been shown to be affected by the deletion mutation of retinal dehydrogenase 2 (Raldh2).133 Raldh2 converts retinal, which is produced by alcohol dehydrogenase from retinol, to retinoic acid. Thus Hh signaling can be partially affected by Adh7.134
A high-throughput screen of 20,000 compounds using a transcription assay driven by Gli-responsive elements in the presence of Shh N-terminus led to identification of 2,4-disubstitutued thiazole compounds as Hh pathway inhibitors (e.g. 123 and 124, Figure 17). 2-Aminothiazole 123 (JK184) inhibited the Shh-N-mediated Gli-transcription with an IC50 of 30 nM. Inhibition of the pathway was confirmed also by RT-PCR, in which 123 showed suppression of Gli1 and Ptch1 expression. In a cytotoxicity assay, compound 123 inhibited the growth of cell lines with abnormally activated Hh signaling, including the pancreatic cancer cell lines L3.6pl, Panc 05.04, and BxPC3 and the medulloblastoma cell lines D283 med and Daoy, with GI50 of 3-21 nM, while showing no toxicity to normal human dermal fibroblast cells.134, 135 Compounds with arylamide substitution at the 2-position of thiazole (124) were less active than 123. Compound 123 induced 30-50% inhibition of tumor growth in xenograft models of human L3.6sl and BxPC3 pancreatic cancers when administered orally, but its efficacy was comparable to that of cyclopamine when it was administered subcutaneously, indicating poor oral bioavailability (16%, t1/2 = 1.3 h). It did not compete with BODIPY-cyclopamine for binding with Smo, indicating that it acts by a mechanism other than the direct Smo binding. Compound 123 was shown to inhibit the class IV alcohol dehydrogenase, Adh7.134 Hh pathway antagonists structurally similar to 123 were also discovered by screening another chemical library.136
Figure 17.
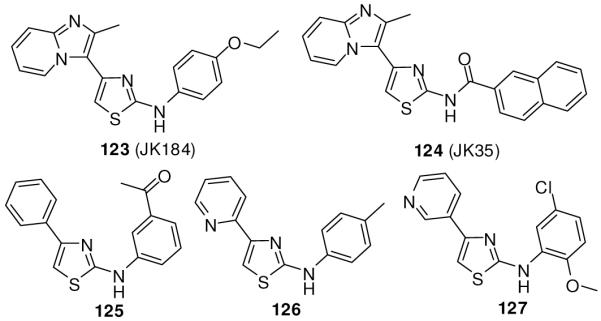
2-Aminothiazoles.
Licentia Oy screened a diversity-oriented library of 6000 small molecules in a Gli-responsive luciferase reporter assay in the Shh-LIGHT2 cell line. They identified 12 compounds with IC50 values ranging from 100 nM to 1 μM, including 125-127 (Figure 17), which share the 2-aminothiazole structure with 123 and 124.124
Inhibitors of Gli-mediated Transcription
Most of the medicinal chemistry efforts involving the Hh-Gli pathway have focused on targeting Smo. However, some cancers have alternative mechanisms for activating the Hh-Gli signaling through effectors that are downstream of Smo, thus rendering Smo inhibitors ineffective. For example, mutation or over-expression of SuFu,21, 55, 137 REN,66, 67 Gli1,6, 30, 138 and Gli2,56, 139 have been shown to activate this pathway. Thus, inhibitors of Gli-transcription, the final event in the pathway, would have broader applicability in cancers, irrespective of the component responsible for the activation of the Hh-Gli signaling.
Lauth et al.140 reported two small-molecule inhibitors of the Gli-transcription, 128 (GANT61) and 129 (GANT58) (Figure 18), discovered in a screen based on inhibition of Gli1-transcription in HEK293 cells transiently transfected with plasmid cDNAs encoding Gli1 and a Gli-responsive luciferase reporter. Since they block the reporter signal induced by exogenously upregulated Gli1, they are expected to be active against cancers in which Gli1 is over-expressed.74, 138 Both compounds also inhibited endogenous Hh signaling at an IC50 of 5 μM in an NIH 3T3 cell line in which the Gli reporter was stably incorporated and induced with a Smo agonist. Unlike cyclopamine, compounds 128 and 129 decreased the expression of Gli1 and Hip1 in Sufu-/- cells, indicating that they act downstream of SuFu. Gli1-positive 22Rv1 prostate cancer xenografts in mice were eradicated by subcutaneous injection of 50 mg/kg of compound 128 every second day for 18 days.140
Figure 18.
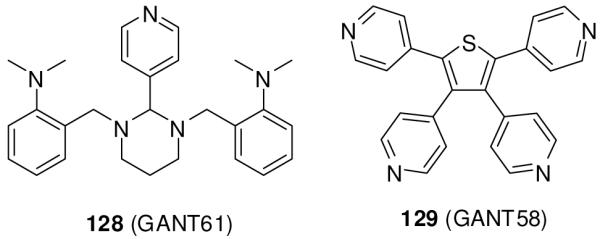
Small molecule Gli-transcription inhibitors from a high-throughput screen.
Natural Products for Inhibition of Gli-Transcription
Hosoya et al.141 designed a cell-based assay to determine Gli1-mediated transcription (Gli-responsive luciferase reporter assay under presence of exogenously over-expressed Gli1) and cell viability (using a fluorimetric microculture cytotoxicity assay) to screen a library of 94 natural products and 192 plant extracts for ability to inhibit the Gli1-transcription without affecting cell viability. Cyclopamine was inactive in this assay at 40 μM, indicating that the assay selectively targeted signaling downstream of Smo. Active compounds were also evaluated for inhibition of Gli2-transcription activity. Three novel classes of inhibitors were identified (Figure 19).141
Figure 19.

Natural product inhibitors of Gli-transcription.
The sesquiterpene zerumbone (130), isolated from Zingiber zerumbet, inhibited Gli1- and Gli2-transcription with IC50 values of 7.1 μM and 0.91 μM, respectively, showing 7-fold selectivity for Gli2 over Gli1. The enone moiety of 130 is important for the inhibition. Other terpenoids, flavonoids, phenylpropanoids, and their glycosides showed weak or no inhibition. Staurosporinone (131) was most active among the bisindole alkaloids 131-134, inhibiting Gli1- and Gli2-transcription with IC50 values of 1.8 and 2.7 μM, respectively. The extract of Physalis minima was the most active of 15 plant extracts. The 13, 14-seco-16, 24-cyclosteroids physalin F (135) and physalin B (136) isolated from this plant extract inhibited Gli1-mediated transcription with IC50 values of 0.66 and 0.62 μM, respectively, without affecting the cell viability. Both compounds showed 2-2.5-fold selectivity for Gli1 over Gli2. Compounds 131, 135, and 136 had 3-5-fold greater cytotoxicity in the PANC1 human pancreatic carcinoma cell line, which has activated Hh-Gli signaling, than in the C3H10/T1/2 cell line.141
The same group also evaluated the active component of an extract of Zizyphus cambodiana (Rhamnaceae). The pentacyclic triterpenes colubrinic acid (137), betulinic acid (138), and alphitolic acid (139) inhibited Gli1-mediated transcription with IC50 values of 38, 32, and 42 μM, respectively. All three compounds showed cytotoxicity against the PANC1 and DU145 cell lines, and compound 137 also inhibited Ptch expression in PANC1 in a dose-dependent manner. Compounds 137 and 138 induced apoptosis and decreased the expression of Bcl2, an anti-apoptotic target gene of Gli1-transcription.142
Pyrazoline compounds 140 and 141 (Figure 20) downregulated expression of Ptch1 and Gli1 in the non-small cell lung carcinoma (NSCLC) cell lines H460 and A549 at 30 μM concentration.143 These compounds also induced apoptosis in NSCLC, melanoma, mesothelioma, sarcoma, human hepatocellular carcinoma, colon, prostate, pancreatic, breast, gastric, nasopharyngeal, and glioma cell lines. Alteration of the length of the carbon chain (142) or replacement of the hydroxyl group on the amide substituent with a carboxylic acid (143) or alkyl (144) group decreased or eliminated the cytotoxicity. Compound 145, with isopropyl groups on the pyrazole ring in place of phenyl, showed loss of the cytotoxicity. Compound 141 suppressed the growth of NSCLC H460, melanoma LOX, and NSCLC A549 in mouse xenograft models.143
Figure 20.

Triarylpyrazolines. Compound 140-146 and 148-149 are racemic trans isomers.
We further analyzed the SAR of 141 analogues by synthesizing 146-151 (unpublished results, Figure 20). Removal of the phenolic hydroxyl of 141 (146) substantially decreased the inhibitory activity for Gli1-transcription in C3H10/T1/2 cells that were transiently co-transfected with plasmid cDNAs encoding Gli1 and a Gli-responsive luciferase reporter. Aromatization of the pyrazole ring (147) resulted in loss of activity. Alteration of the linker element in 141 by reversing the amide group afforded an equipotent compound 148. The methoxy analog 149 and the aromatized analogues 150 and 151 showed weaker inhibition. We also have replaced the pyrazoline moiety with another scaffold and will describe the results in a forthcoming report.
Conclusion and Perspective
Involvement of the Hh-Gli pathway in many cancers has motivated pharmaceutical companies and academic drug discovery groups to develop inhibitors for this pathway.145 These discovery efforts have led to several lead compounds and clinical candidates (Table 1). Cyclopamine proved to be an excellent prototype Hh pathway inhibitor and helped in elucidating the biology of the pathway. A part of the drug discovery efforts have focused on cyclopamine analogues which have resulted in one compound with an improved profile that is currently in phase I clinical trials.13 High-throughput screening, the other main focus of the drug discovery efforts, and subsequent medicinal chemistry studies have also led to discovery of a diverse set of small molecules targeting Smo. Table 2 enlists the screening assays used for evaluation of compounds presented in this review. Compound 54 was the first Smo inhibitor to enter the clinical trials as a topical agent in patients with BCC, but trials were subsequently halted after the phase I study.89 Results of the phase I trial of another small molecule Smo inhibitor 101 have provided a proof-of-concept for Hh-Gli pathway inhibitors as anticancer agents (six of nine patients with BCC showed a confirmed or a confirmed partial response and two had a stable disease).128, 146
Table 1.
Hh-Gli pathway inhibitors in clinical and preclinical studies.*
| Compound | Target | Stage of Development |
|---|---|---|
| Cyclopamine (3)144 | Smo | Preclinical/Clinical |
| KAAD-Cyclopamine (14)97 | Smo | Preclinical |
| 18 101 | Smo | Preclinical |
| 26 13 | Smo | Phase I |
| 54 89 | Smo | Halted after Phase I |
| 7714, 114 | Smo | Phase I |
| 9665, 123 | Smo | Preclinical |
| 101 108 | Smo | Phase I/II |
| 128 140 | Gli | Preclinical |
as of December 2008.
Table 2.
Assay methods used in each discovery program
| Assay method | Compounds |
|---|---|
| Chick embryo neural plate explant | 3-10 95 |
| Shh-responsive luciferase reporter assay in a colonal cell line derived from NIH-3T3 | 11-16 96 |
| Alkaline phostphatase | 1-2,85 17-24,100 27-34,104 35-45105 |
| Shh-LIGHT-II | 1-2,85 50-53,109 72-76,113 115-120,131 125-127124 |
| Gli-responsive luciferase reporter (no exogenous Gli) | 54-66,110, 111 67-71,112 78-86,115 87,117, 88-91,118 92-96,119 101-103,125 104-106,126 107-114,129 123-124,134 128-129,140 |
| Smo binding | 78-86115 |
| Gli-responsive luciferase reporter with exogenous Gli1 or Gli2 | 130-139,141, 142 140-145,143 146-151 |
Most drug discovery efforts till now have been focused on targeting Smo. However, recently compounds targeting Gli-mediated transcription,140-143 a downstream event in the Hh-Gli pathway, have also been reported and can be good prototypes for medicinal chemistry studies. Furthermore, inhibitors targeting the downstream events could provide a broader spectrum of activity against cancers where Hh-Gli pathway components downstream of Smo including SuFu,21, 55, 137 REN,66, 67 Gli1,6, 30, 138 and Gli2,56,139 (Figure 1) have been implicated in deregulation. Smo inhibitors might not be effective in such cases, for example, Gli proteins in isolated SuFu(-/-) mouse embryonic fibroblasts, having cell-autonomous downstream activation of the Hh signaling, showed a constitutive Gli activity that could not be inhibited by cyclopamine as activation occurs downstream of Smo.147, 148 Drugs specifically designed to modulate these downstream components would provide personalized medicine for the patients with specific mutations in this pathway.
The Hh-Gli pathway has been reported to play a role in metastasis.74, 81, 149 Gli1 has been shown to induce expression of Snail resulting in reduced expression of proteins, such as E-cadherin that maintains epithelial organization, thus resulting in a metastatic phenotype which can be inhibited by cyclopamine.74 Gli1 and Snail were also significantly overexpressed in 50% and 75% of the metastatic foci, respectively, in samples obtained from patients with disseminated pancreatic cancer.150 The local or systemic interference in Hh-Gli signaling was reported to inhibit melanoma growth and prevent metastasis in mice.149 Therefore, inhibitors of the Hh-Gli pathway may be effective in preventing metastasis.
The Hh-Gli pathway is essential in development and tissue maintenance; therefore great care should be taken to assess the safety of drugs that act through this pathway. Phase I clinical study of 101 indicates a satisfactory safety profile in adults,12 but permanent defects in bone structure were observed in young mice (10-14 days old) exposed to another Smo inhibitor (96)123 though no serious side effects were observed in adult mice.65 This would also underline, as mentioned in previously, the importance of designing inhibitors of downstream events of this pathway especially for pediatric therapeutics. Gli1-transcription, one such downstream event, plays an important role in several cancers6, 30, 139 and has been suggested as a potential target for development of novel therapies.121, 138 Gli2 expression has also been implicated in the tumorigenicity of human glioma stem cells.30 Gli1 knockout mice have been reported to show no obvious abnormalities, whereas Gli2 and Gli3 knockout mice show abnormal skeletal growth and embryonic or perinatal lethality.5, 43, 45, 138 Gli2 is an important factor for expression of parathyroid hormone-related peptide (PTHrP) in the developing growth plate, while Gli1 and Gli3 do not stimulate the PTHrP promoter.44 An important mediator of bone differentiation, bone-morphogenetic protein (BMP)-2 is regulated by Gli2 but not by Gli1.151 Runx2, an essential transcription factor for bone formation has a down-stream function of BMP2 signaling, and is upregulated by Gli2 in C3H10/T1/2 cells152 that express very little endogenous Gli1. Therefore, selective inhibition of Gli1-transcription might eliminate the permanent bone defects observed with a Smo inhibitor.145
The significance of the Hh-Gli pathway as an anticancer drug target has been supported by numerous experiments using homogenous cancer cell cultures with aberrant Hh-Gli signaling. Recent evidence has shown importance of paracrine mechanism for upregulating the Hh signaling in a tumor animal model.120 This observation raises the need to consider the stroma-tumor interactions in Hh-Gli signaling in the experimental models.153 Other observations also suggest that maintenance of the Hh-Gli pathway in cancers needs such paracrine regulation. For example, in a human pancreatic xenograft mouse model, compound 29 only downregulated the murine Gli1 mRNA levels but not the human Gli1.104 Nolan-Stevaux et al.154 have also suggested that paracrine signaling in adjacent mesenchymal cells via secreted Shh ligand is responsible for pancreatic ductal adenocarcinoma progression. Such stroma-tumor interactions have been well demonstrated in many faces of cancer progression, as pioneered by Judah Folkman’s work in tumor angiogenesis. Secondary screens that mimic the paracrine Hh environment could be used to further evaluate hit compounds. Co-culture of cancer cells and stroma cells have been previously used to study micro-environments involving paracrice Hh controlled angiogenesis.155 The functional role of the Hh-Gli pathway in tumor progression needs to be further studied for creating the next generation of lead compounds with better safety and clinical efficacy.
Acknowledgements
This work was supported by the American Lebanese Syrian Associated Charities (ALSAC), St. Jude Children’s Research Hospital (SJCRH), and NIH Cancer Center Support Grant P30CA021765-30. We thank Ms. Sharon H. Naron for English editing.
Biography
Neeraj Mahindroo obtained his PhD in Pharmaceutical Sciences from Panjab University, Chandigarh, India in 2001 under supervision of Dr. V. K. Kapoor and Dr. K. L. Dhar after receiving his M. Pharma. in Pharmaceutical Chemistry from the same university in 1996. He joined as Lecturer in Pharmaceutical Sciences at Guru Nanak Dev University, Amritsar, India in October 2000. In 2002, he moved to Taiwan to carry out his postdoctoral research in Dr. Hsing-Pang Hsieh’s Lab at National Health Research Institutes on design and synthesis of novel PPAR agonists targeting type 2 diabetes, and anticancer agents targeting microtubules. He joined Dr. Naoaki Fujii’s lab in August 2006 where his current interests include design, synthesis and evaluation of small drug-like molecules to target the oncogenic signaling pathways responsible for pediatric cancers.
Chandanamali Punchihewa received her PhD in Pharmaceutical Sciences at the University of Arizona, Tucson Arizona in 2006, on the investigations of DNA and DNA interacting proteins as anticancer drug targets, and was supported by numerous research Fellowships. She is now doing postdoctoral studies in the laboratory of Dr. Naoaki Fujii at St. Jude Children’s Research Hospital. Her research interests include the investigation of specific protein-protein interactions and their relevance in Wnt and Hh pathway signaling, as well as identification of potential inhibitors of these pathways.
Naoaki Fujii has joined Fujisawa Pharmaceuticals Company and rose through the ranks to become Research Manager in 2001. During this period he received his PhD in synthetic organic chemistry from Osaka University in 1998. He moved to the University of California, San Francisco, USA in 2002 to join Prof. R. Kip Guy’s Lab as Assistant Research. In 2006, he joined as Assistant Member and Director of Medicinal Chemistry Core in the Department of Chemical Biology and Therapeutics at St Jude Children’s Research Hospital. His current research focuses on identifying new chemical entities for pediatric cancers.
Footnotes
Abbreviations: Hh, Hedgehog; Gli, Glioma-associated oncogene homolog zinc finger protein; Shh, Sonic hedgehog; Dhh, Desert hedgehog; Ihh, Indian hedgehog; Smo, Smoothened; Ptch, Patched; Fu, Fused; SuFu, Suppressor of Fused; Cos2, Costal-2; PKA, protein kinase A; GSK3, glycogen synthase kinase 3; CK1, casein kinase 1; BCC, basal cell carcinoma; GS, Gorlin syndrome; Adh7, alcohol dehydrogenase 7; Raldh2, retinal dehydrogenase 2; MTP, microsomal triglyceride transfer protein; PTHrP, parathyroid hormone-related peptide; BMP2, bone-morphogenetic protein-2; Dyrk, dual specificity tyrosine phosphorylation regulated kinase; IFT, intraflagellar transport protein; Ras, rat sarcoma; Akt, protein kinase B; MEK, mitogen-activated protein/extracellular signal-regulated kinase; ERK, extracellular regulated kinases; PKC, protein kinase C.
References
- 1.Workman P, Collins I. Modern Cancer Drug Discovery: Integrating Targets, Technologies and Treatments. In: Neidle S, editor. Cancer Drug Design And Discovery. First ed. Elsevier; New York: 2008. pp. 3–38. [Google Scholar]
- 2.Pasca di Magliano M, Hebrok M. Hedgehog signalling in cancer formation and maintenance. Nat. Rev. Cancer. 2003;3:903–11. doi: 10.1038/nrc1229. [DOI] [PubMed] [Google Scholar]
- 3.Beachy PA, Karhadkar SS, Berman DM. Tissue repair and stem cell renewal in carcinogenesis. Nature. 2004;432:324–31. doi: 10.1038/nature03100. [DOI] [PubMed] [Google Scholar]
- 4.Lum L, Beachy PA. The Hedgehog response network: sensors, switches, and routers. Science. 2004;304:1755–9. doi: 10.1126/science.1098020. [DOI] [PubMed] [Google Scholar]
- 5.Ruiz i Altaba A, Sanchez P, Dahmane N. Gli and hedgehog in cancer: tumours, embryos and stem cells. Nat. Rev. Cancer. 2002;2:361–72. doi: 10.1038/nrc796. [DOI] [PubMed] [Google Scholar]
- 6.Bian YH, Huang SH, Yang L, Ma XL, Xie JW, Zhang HW. Sonic hedgehog-Gli1 pathway in colorectal adenocarcinomas. World J. Gastroenterol. 2007;13:1659–65. doi: 10.3748/wjg.v13.i11.1659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gerber AN, Wilson CW, Li YJ, Chuang PT. The hedgehog regulated oncogenes Gli1 and Gli2 block myoblast differentiation by inhibiting MyoD-mediated transcriptional activation. Oncogene. 2007;26:1122–36. doi: 10.1038/sj.onc.1209891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sanchez P, Hernandez AM, Stecca B, Kahler AJ, DeGueme AM, Barrett A, Beyna M, Datta MW, Datta S, Ruiz i Altaba A. Inhibition of prostate cancer proliferation by interference with SONIC HEDGEHOG-GLI1 signaling. Proc. Natl. Acad. Sci. U. S. A. 2004;101:12561–6. doi: 10.1073/pnas.0404956101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Xie J. Hedgehog signaling pathway: Development of antagonists for cancer therapy. Current Oncology Reports. 2008;10:107–113. doi: 10.1007/s11912-008-0018-7. [DOI] [PubMed] [Google Scholar]
- 10.Rubin LL, de Sauvage FJ. Targeting the Hedgehog pathway in cancer. Nat. Rev. Drug Disc. 2006;5:1026–1033. doi: 10.1038/nrd2086. [DOI] [PubMed] [Google Scholar]
- 11.Borzillo GV, Lippa B. The Hedgehog signaling pathway as a target for anticancer drug discovery. Curr. Top. Med. Chem. 2005;5:147–157. doi: 10.2174/1568026053507732. [DOI] [PubMed] [Google Scholar]
- 12.LoRusso PM, Rudin CM, Borad MJ, Vernillet L, Darbonne WC, Mackey H, DiMartino JF, de Sauvage F, Low JA, Von Hoff DD. A First-in-human, First-in-class, Phase I Study of Systemic Hedgehog Pathway Antagonist, GDC-0449, in Patients with Advanced Solid Tumors. J. Clin. Oncol. (Meeting Abstracts) 2008;26:3516. [Google Scholar]
- 13.A Phase 1 Study of IPI-926 in Patients With Advanced and/or Metastatic Solid Tumor Malignancies. 2008 October 2; http://clinicaltrials.gov/ct2/show/NCT00761696.
- 14.XL139. 2009 January 8; http://www.exelixis.com/pipeline.shtml.
- 15.Nusslein-Volhard C, Wieschaus E. Mutations affecting segment number and polarity in Drosophila. Nature. 1980;287:795–801. doi: 10.1038/287795a0. [DOI] [PubMed] [Google Scholar]
- 16.Echelard Y, Epstein DJ, St-Jacques B, Shen L, Mohler J, McMahon JA, McMahon AP. Sonic hedgehog, a member of a family of putative signaling molecules, is implicated in the regulation of CNS polarity. Cell. 1993;75:1417–1430. doi: 10.1016/0092-8674(93)90627-3. [DOI] [PubMed] [Google Scholar]
- 17.Lee JJ, Ekker SC, von Kessler DP, Porter JA, Sun BI, Beachy PA. Autoproteolysis in hedgehog protein biogenesis. Science. 1994;266:1528–1537. doi: 10.1126/science.7985023. [DOI] [PubMed] [Google Scholar]
- 18.Porter JA, Young KE, Beachy PA. Cholesterol Modification of Hedgehog Signaling Proteins in Animal Development. Science. 1996;274:255–259. doi: 10.1126/science.274.5285.255. [DOI] [PubMed] [Google Scholar]
- 19.Pepinsky RB, Zeng C, Wen D, Rayhorn P, Baker DP, Williams KP, Bixler SA, Ambrose CM, Garber EA, Miatkowski K, Taylor FR, Wang EA, Galdes A. Identification of a Palmitic Acid-modified Form of Human Sonic hedgehog. J. Biol. Chem. 1998;273:14037–14045. doi: 10.1074/jbc.273.22.14037. [DOI] [PubMed] [Google Scholar]
- 20.Stone DM, Hynes M, Armanini M, Swanson TA, Gu Q, Johnson RL, Scott MP, Pennica D, Goddard A, Phillips H, Noll M, Hooper JE, de Sauvage F, Rosenthal A. The tumour-suppressor gene patched encodes a candidate receptor for Sonic hedgehog. Nature. 1996;384:129–134. doi: 10.1038/384129a0. [DOI] [PubMed] [Google Scholar]
- 21.Svärd J, Henricson KH, Persson-Lek M, Rozell B, Lauth M, Bergström Å, Ericson J, Toftgård R, Teglund S. Genetic elimination of Suppressor of fused reveals an essential repressor function in the mammalian Hedgehog signaling pathway. Dev. Cell. 2006;10:187–97. doi: 10.1016/j.devcel.2005.12.013. [DOI] [PubMed] [Google Scholar]
- 22.Varjosalo M, Li SP, Taipale J. Divergence of hedgehog signal transduction mechanism between Drosophila and mammals. Dev. Cell. 2006;10:177–86. doi: 10.1016/j.devcel.2005.12.014. [DOI] [PubMed] [Google Scholar]
- 23.Varjosalo M, Bjorklund M, Cheng F, Syvanen H, Kivioja T, Kilpinen S, Sun Z, Kallioniemi O, Stunnenberg HG, He WW, Ojala P, Taipale J. Application of active and kinase-deficient kinome collection for identification of kinases regulating hedgehog signaling. Cell. 2008;133:537–48. doi: 10.1016/j.cell.2008.02.047. [DOI] [PubMed] [Google Scholar]
- 24.Mao J, Maye P, Kogerman P, Tejedor FJ, Toftgard R, Xie W, Wu G, Wu D. Regulation of Gli1 transcriptional activity in the nucleus by Dyrk1. J. Biol. Chem. 2002;277:35156–61. doi: 10.1074/jbc.M206743200. [DOI] [PubMed] [Google Scholar]
- 25.Riobo NA, Haines GM, Emerson CP., Jr. Protein kinase C-delta and mitogen-activated protein/extracellular signal-regulated kinase-1 control GLI activation in hedgehog signaling. Cancer Res. 2006;66:839–45. doi: 10.1158/0008-5472.CAN-05-2539. [DOI] [PubMed] [Google Scholar]
- 26.Kinzler KW, Bigner SH, Bigner DD, Trent JM, Law ML, O’Brien SJ, Wong AJ, Vogelstein B. Identification of an amplified, highly expressed gene in a human glioma. Science. 1987;236:70–3. doi: 10.1126/science.3563490. [DOI] [PubMed] [Google Scholar]
- 27.Dai P, Akimaru H, Tanaka Y, Maekawa T, Nakafuku M, Ishii S. Sonic Hedgehog-induced activation of the Gli1 promoter is mediated by GLI3. J. Biol. Chem. 1999;274:8143–52. doi: 10.1074/jbc.274.12.8143. [DOI] [PubMed] [Google Scholar]
- 28.Ruiz i Altaba A. Gli proteins encode context-dependent positive and negative functions: implications for development and disease. Development. 1999;126:3205–16. doi: 10.1242/dev.126.14.3205. [DOI] [PubMed] [Google Scholar]
- 29.Shin SH, Kogerman P, Lindstrom E, Toftgard R, Biesecker LG. GLI3 mutations in human disorders mimic Drosophila cubitus interruptus protein functions and localization. Proc. Natl. Acad. Sci. U. S. A. 1999;96:2880–4. doi: 10.1073/pnas.96.6.2880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ruiz i Altaba A, Mas C, Stecca B. The Gli code: an information nexus regulating cell fate, stemness and cancer. Trends Cell Biol. 2007;17:438–47. doi: 10.1016/j.tcb.2007.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Clement V, Sanchez P, de Tribolet N, Radovanovic I, Ruiz i Altaba A. HEDGEHOG-GLI1 signaling regulates human glioma growth, cancer stem cell self-renewal, and tumorigenicity. Curr. Biol. 2007;17:165–72. doi: 10.1016/j.cub.2006.11.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yoon JW, Kita Y, Frank DJ, Majewski RR, Konicek BA, Nobrega MA, Jacob H, Walterhouse D, Iannaccone P. Gene expression profiling leads to identification of GLI1-binding elements in target genes and a role for multiple downstream pathways in GLI1-induced cell transformation. J. Biol. Chem. 2002;277:5548–55. doi: 10.1074/jbc.M105708200. [DOI] [PubMed] [Google Scholar]
- 33.Yoon JW, Gilbertson R, Iannaccone S, Iannaccone P, Walterhouse D. Defining a role for Sonic hedgehog pathway activation in desmoplastic medulloblastoma by identifying GLI1 target genes. Int. J. Cancer. 2008;124:109–119. doi: 10.1002/ijc.23929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ingham PW, McMahon AP. Hedgehog signaling in animal development: paradigms and principles. Genes Dev. 2001;15:3059–87. doi: 10.1101/gad.938601. [DOI] [PubMed] [Google Scholar]
- 35.Machold R, Hayashi S, Rutlin M, Muzumdar MD, Nery S, Corbin JG, Gritli-Linde A, Dellovade T, Porter JA, Rubin LL, Dudek H, McMahon AP, Fishell G. Sonic Hedgehog Is Required for Progenitor Cell Maintenance in Telencephalic Stem Cell Niches. Neuron. 2003;39:937–950. doi: 10.1016/s0896-6273(03)00561-0. [DOI] [PubMed] [Google Scholar]
- 36.Lavine KJ, Kovacs A, Ornitz DM. Hedgehog signaling is critical for maintenance of the adult coronary vasculature in mice. J. Clin. Invest. 2008;118:2404–14. doi: 10.1172/JCI34561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Balordi F, Fishell G. Mosaic removal of hedgehog signaling in the adult SVZ reveals that the residual wild-type stem cells have a limited capacity for self-renewal. J. Neurosci. 2007;27:14248–59. doi: 10.1523/JNEUROSCI.4531-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Varjosalo M, Taipale J. Hedgehog: functions and mechanisms. Genes Dev. 2008;22:2454–72. doi: 10.1101/gad.1693608. [DOI] [PubMed] [Google Scholar]
- 39.Tabin CJ, McMahon AP. DEVELOPMENTAL BIOLOGY: Grasping Limb Patterning. Science. 2008;321:350–352. doi: 10.1126/science.1162474. [DOI] [PubMed] [Google Scholar]
- 40.Roelink H, Augsburger A, Heemskerk J, Korzh V, Norlin S, Ruiz i Altaba A, Tanabe Y, Placzek M, Edlund T, Jessell TM, Dodd J. Floor plate and motor neuron induction by vhh-1, a vertebrate homolog of hedgehog expressed by the notochord. Cell. 1994;76:761–775. doi: 10.1016/0092-8674(94)90514-2. [DOI] [PubMed] [Google Scholar]
- 41.Belloni E, Muenke M, Roessler E, Traverso G, Siegel-Bartelt J, Frumkin A, Mitchell HF, Donis-Keller H, Helms C, Hing AV, Heng HH, Koop B, Martindale D, Rommens JM, Tsui LC, Scherer SW. Identification of Sonic hedgehog as a candidate gene responsible for holoprosencephaly. Nat. Genet. 1996;14:353–6. doi: 10.1038/ng1196-353. [DOI] [PubMed] [Google Scholar]
- 42.Hynes M, Stone DM, Dowd M, Pitts-Meek S, Goddard A, Gurney A, Rosenthal A. Control of cell pattern in the neural tube by the zinc finger transcription factor and oncogene Gli-1. Neuron. 1997;19:15–26. doi: 10.1016/s0896-6273(00)80344-x. [DOI] [PubMed] [Google Scholar]
- 43.Park HL, Bai C, Platt KA, Matise MP, Beeghly A, Hui CC, Nakashima M, Joyner AL. Mouse Gli1 mutants are viable but have defects in SHH signaling in combination with a Gli2 mutation. Development. 2000;127:1593–605. doi: 10.1242/dev.127.8.1593. [DOI] [PubMed] [Google Scholar]
- 44.Sterling JA, Oyajobi BO, Grubbs B, Padalecki SS, Munoz SA, Gupta A, Story B, Zhao M, Mundy GR. The hedgehog signaling molecule Gli2 induces parathyroid hormone-related peptide expression and osteolysis in metastatic human breast cancer cells. Cancer Res. 2006;66:7548–53. doi: 10.1158/0008-5472.CAN-06-0452. [DOI] [PubMed] [Google Scholar]
- 45.Mo R, Freer AM, Zinyk DL, Crackower MA, Michaud J, Heng HH, Chik KW, Shi XM, Tsui LC, Cheng SH, Joyner AL, Hui C. Specific and redundant functions of Gli2 and Gli3 zinc finger genes in skeletal patterning and development. Development. 1997;124:113–23. doi: 10.1242/dev.124.1.113. [DOI] [PubMed] [Google Scholar]
- 46.Hui CC, Joyner AL. A mouse model of greig cephalopolysyndactyly syndrome: the extra-toesJ mutation contains an intragenic deletion of the Gli3 gene. Nat. Genet. 1993;3:241–6. doi: 10.1038/ng0393-241. [DOI] [PubMed] [Google Scholar]
- 47.Karlstrom RO, Tyurina OV, Kawakami A, Nishioka N, Talbot WS, Sasaki H, Schier AF. Genetic analysis of zebrafish gli1 and gli2 reveals divergent requirements for gli genes in vertebrate development. Development. 2003;130:1549–64. doi: 10.1242/dev.00364. [DOI] [PubMed] [Google Scholar]
- 48.Zhang Y, Kalderon D. Hedgehog acts as a somatic stem cell factor in the Drosophila ovary. Nature. 2001;410:599–604. doi: 10.1038/35069099. [DOI] [PubMed] [Google Scholar]
- 49.Lai K, Kaspar BK, Gage FH, Schaffer DV. Sonic hedgehog regulates adult neural progenitor proliferation in vitro and in vivo. Nat. Neurosci. 2003;6:21–7. doi: 10.1038/nn983. [DOI] [PubMed] [Google Scholar]
- 50.Hunter T. Oncoprotein networks. Cell. 1997;88:333–46. doi: 10.1016/s0092-8674(00)81872-3. [DOI] [PubMed] [Google Scholar]
- 51.Unden AB, Holmberg E, Lundh-Rozell B, Stahle-Backdahl M, Zaphiropoulos PG, Toftgard R, Vorechovsky I. Mutations in the human homologue of Drosophila patched (PTCH) in basal cell carcinomas and the Gorlin syndrome: different in vivo mechanisms of PTCH inactivation. Cancer Res. 1996;56:4562–5. [PubMed] [Google Scholar]
- 52.Gailani MR, Stahle-Backdahl M, Leffell DJ, Glynn M, Zaphiropoulos PG, Pressman C, Unden AB, Dean M, Brash DE, Bale AE, Toftgard R. The role of the human homologue of Drosophila patched in sporadic basal cell carcinomas. Nat. Genet. 1996;14:78–81. doi: 10.1038/ng0996-78. [DOI] [PubMed] [Google Scholar]
- 53.Raffel C. Medulloblastoma: molecular genetics and animal models. Neoplasia. 2004;6:310–22. doi: 10.1593/neo.03454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Xie J, Murone M, Luoh SM, Ryan A, Gu Q, Zhang C, Bonifas JM, Lam CW, Hynes M, Goddard A, Rosenthal A, Epstein EH, Jr., de Sauvage FJ. Activating Smoothened mutations in sporadic basal-cell carcinoma. Nature. 1998;391:90–2. doi: 10.1038/34201. [DOI] [PubMed] [Google Scholar]
- 55.Taylor MD, Liu L, Raffel C, Hui CC, Mainprize TG, Zhang X, Agatep R, Chiappa S, Gao L, Lowrance A, Hao A, Goldstein AM, Stavrou T, Scherer SW, Dura WT, Wainwright B, Squire JA, Rutka JT, Hogg D. Mutations in SUFU predispose to medulloblastoma. Nat. Genet. 2002;31:306–10. doi: 10.1038/ng916. [DOI] [PubMed] [Google Scholar]
- 56.Bhatia N, Thiyagarajan S, Elcheva I, Saleem M, Dlugosz A, Mukhtar H, Spiegelman VS. Gli2 is targeted for ubiquitination and degradation by beta-TrCP ubiquitin ligase. J. Biol. Chem. 2006;281:19320–6. doi: 10.1074/jbc.M513203200. [DOI] [PubMed] [Google Scholar]
- 57.Dahmane N, Sanchez P, Gitton Y, Palma V, Sun T, Beyna M, Weiner H, Ruiz i Altaba A. The Sonic Hedgehog-Gli pathway regulates dorsal brain growth and tumorigenesis. Development. 2001;128:5201–12. doi: 10.1242/dev.128.24.5201. [DOI] [PubMed] [Google Scholar]
- 58.Becher OJ, Hambardzumyan D, Fomchenko EI, Momota H, Mainwaring L, Bleau AM, Katz AM, Edgar M, Kenney AM, Cordon-Cardo C, Blasberg RG, Holland EC. Gli activity correlates with tumor grade in platelet-derived growth factor-induced gliomas. Cancer Res. 2008;68:2241–9. doi: 10.1158/0008-5472.CAN-07-6350. [DOI] [PubMed] [Google Scholar]
- 59.Johnson RL, Rothman AL, Xie J, Goodrich LV, Bare JW, Bonifas JM, Quinn AG, Myers RM, Cox DR, Epstein EH, Jr., Scott MP. Human homolog of patched, a candidate gene for the basal cell nevus syndrome. Science. 1996;272:1668–71. doi: 10.1126/science.272.5268.1668. [DOI] [PubMed] [Google Scholar]
- 60.Nilsson M, Unden AB, Krause D, Malmqwist U, Raza K, Zaphiropoulos PG, Toftgard R. Induction of basal cell carcinomas and trichoepitheliomas in mice overexpressing GLI-1. Proc. Natl. Acad. Sci. U. S. A. 2000;97:3438–43. doi: 10.1073/pnas.050467397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hatta N, Hirano T, Kimura T, Hashimoto K, Mehregan DR, Ansai S, Takehara K, Takata M. Molecular diagnosis of basal cell carcinoma and other basaloid cell neoplasms of the skin by the quantification of Gli1 transcript levels. J. Cutan. Pathol. 2005;32:131–6. doi: 10.1111/j.0303-6987.2005.00264.x. [DOI] [PubMed] [Google Scholar]
- 62.Pomeroy SL, Tamayo P, Gaasenbeek M, Sturla LM, Angelo M, McLaughlin ME, Kim JY, Goumnerova LC, Black PM, Lau C, Allen JC, Zagzag D, Olson JM, Curran T, Wetmore C, Biegel JA, Poggio T, Mukherjee S, Rifkin R, Califano A, Stolovitzky G, Louis DN, Mesirov JP, Lander ES, Golub TR. Prediction of central nervous system embryonal tumour outcome based on gene expression. Nature. 2002;415:436–42. doi: 10.1038/415436a. [DOI] [PubMed] [Google Scholar]
- 63.Berman DM, Karhadkar SS, Hallahan AR, Pritchard JI, Eberhart CG, Watkins DN, Chen JK, Cooper MK, Taipale J, Olson JM, Beachy PA. Medulloblastoma growth inhibition by hedgehog pathway blockade. Science. 2002;297:1559–61. doi: 10.1126/science.1073733. [DOI] [PubMed] [Google Scholar]
- 64.Sanchez P, Ruiz i Altaba A. In vivo inhibition of endogenous brain tumors through systemic interference of Hedgehog signaling in mice. Mech. Dev. 2005;122:223–30. doi: 10.1016/j.mod.2004.10.002. [DOI] [PubMed] [Google Scholar]
- 65.Romer JT, Kimura H, Magdaleno S, Sasai K, Fuller C, Baines H, Connelly M, Stewart CF, Gould S, Rubin LL, Curran T. Suppression of the Shh pathway using a small molecule inhibitor eliminates medulloblastoma in Ptc1(+/-)p53(-/-) mice. Cancer Cell. 2004;6:229–40. doi: 10.1016/j.ccr.2004.08.019. [DOI] [PubMed] [Google Scholar]
- 66.Di Marcotullio L, Ferretti E, De Smaele E, Argenti B, Mincione C, Zazzeroni F, Gallo R, Masuelli L, Napolitano M, Maroder M, Modesti A, Giangaspero F, Screpanti I, Alesse E, Gulino A. REN(KCTD11) is a suppressor of Hedgehog signaling and is deleted in human medulloblastoma. Proc. Natl. Acad. Sci. U. S. A. 2004;101:10833–8. doi: 10.1073/pnas.0400690101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Argenti B, Gallo R, Di Marcotullio L, Ferretti E, Napolitano M, Canterini S, De Smaele E, Greco A, Fiorenza MT, Maroder M, Screpanti I, Alesse E, Gulino A. Hedgehog antagonist REN(KCTD11) regulates proliferation and apoptosis of developing granule cell progenitors. J. Neurosci. 2005;25:8338–46. doi: 10.1523/JNEUROSCI.2438-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Zawlik I, Zakrzewska M, Witusik M, Golanska E, Kulczycka-Wojdala D, Szybka M, Piaskowski S, Wozniak K, Zakrzewski K, Papierz W, Liberski PP, Rieske P. KCTD11 expression in medulloblastoma is lower than in adult cerebellum and higher than in neural stem cells. Cancer Genet. Cytogenet. 2006;170:24–8. doi: 10.1016/j.cancergencyto.2006.04.014. [DOI] [PubMed] [Google Scholar]
- 69.Thayer SP, di Magliano MP, Heiser PW, Nielsen CM, Roberts DJ, Lauwers GY, Qi YP, Gysin S, Fernandez-del Castillo C, Yajnik V, Antoniu B, McMahon M, Warshaw AL, Hebrok M. Hedgehog is an early and late mediator of pancreatic cancer tumorigenesis. Nature. 2003;425:851–6. doi: 10.1038/nature02009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Morton JP, Mongeau ME, Klimstra DS, Morris JP, Lee YC, Kawaguchi Y, Wright CV, Hebrok M, Lewis BC. Sonic hedgehog acts at multiple stages during pancreatic tumorigenesis. Proc. Natl. Acad. Sci. U. S. A. 2007;104:5103–8. doi: 10.1073/pnas.0701158104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Ji Z, Mei FC, Xie J, Cheng X. Oncogenic KRAS activates hedgehog signaling pathway in pancreatic cancer cells. J. Biol. Chem. 2007;282:14048–55. doi: 10.1074/jbc.M611089200. [DOI] [PubMed] [Google Scholar]
- 72.Sheng T, Li C, Zhang X, Chi S, He N, Chen K, McCormick F, Gatalica Z, Xie J. Activation of the hedgehog pathway in advanced prostate cancer. Mol. Cancer. 2004;3:29. doi: 10.1186/1476-4598-3-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Fan L, Pepicelli CV, Dibble CC, Catbagan W, Zarycki JL, Laciak R, Gipp J, Shaw A, Lamm ML, Munoz A, Lipinski R, Thrasher JB, Bushman W. Hedgehog signaling promotes prostate xenograft tumor growth. Endocrinology. 2004;145:3961–70. doi: 10.1210/en.2004-0079. [DOI] [PubMed] [Google Scholar]
- 74.Karhadkar SS, Bova GS, Abdallah N, Dhara S, Gardner D, Maitra A, Isaacs JT, Berman DM, Beachy PA. Hedgehog signalling in prostate regeneration, neoplasia and metastasis. Nature. 2004;431:707–12. doi: 10.1038/nature02962. [DOI] [PubMed] [Google Scholar]
- 75.Chi S, Huang S, Li C, Zhang X, He N, Bhutani MS, Jones D, Castro CY, Logrono R, Haque A, Zwischenberger J, Tyring SK, Zhang H, Xie J. Activation of the hedgehog pathway in a subset of lung cancers. Cancer Lett. 2006;244:53–60. doi: 10.1016/j.canlet.2005.11.036. [DOI] [PubMed] [Google Scholar]
- 76.Watkins DN, Berman DM, Burkholder SG, Wang B, Beachy PA, Baylin SB. Hedgehog signalling within airway epithelial progenitors and in small-cell lung cancer. Nature. 2003;422:313–7. doi: 10.1038/nature01493. [DOI] [PubMed] [Google Scholar]
- 77.Qualtrough D, Buda A, Gaffield W, Williams AC, Paraskeva C. Hedgehog signalling in colorectal tumour cells: induction of apoptosis with cyclopamine treatment. Int. J. Cancer. 2004;110:831–7. doi: 10.1002/ijc.20227. [DOI] [PubMed] [Google Scholar]
- 78.McGarvey TW, Maruta Y, Tomaszewski JE, Linnenbach AJ, Malkowicz SB. PTCH gene mutations in invasive transitional cell carcinoma of the bladder. Oncogene. 1998;17:1167–72. doi: 10.1038/sj.onc.1202045. [DOI] [PubMed] [Google Scholar]
- 79.Feng YZ, Shiozawa T, Miyamoto T, Kashima H, Kurai M, Suzuki A, Ying-Song J, Konishi I. Overexpression of hedgehog signaling molecules and its involvement in the proliferation of endometrial carcinoma cells. Clin. Cancer Res. 2007;13:1389–98. doi: 10.1158/1078-0432.CCR-06-1407. [DOI] [PubMed] [Google Scholar]
- 80.Bhattacharya R, Kwon J, Ali B, Wang E, Patra S, Shridhar V, Mukherjee P. Role of hedgehog signaling in ovarian cancer. Clin. Cancer Res. 2008;14:7659–66. doi: 10.1158/1078-0432.CCR-08-1414. [DOI] [PubMed] [Google Scholar]
- 81.Mori Y, Okumura T, Tsunoda S, Sakai Y, Shimada Y. Gli-1 expression is associated with lymph node metastasis and tumor progression in esophageal squamous cell carcinoma. Oncology. 2006;70:378–89. doi: 10.1159/000098111. [DOI] [PubMed] [Google Scholar]
- 82.Tostar U, Malm CJ, Meis-Kindblom JM, Kindblom LG, Toftgard R, Unden AB. Deregulation of the hedgehog signalling pathway: a possible role for the PTCH and SUFU genes in human rhabdomyoma and rhabdomyosarcoma development. J. Pathol. 2006;208:17–25. doi: 10.1002/path.1882. [DOI] [PubMed] [Google Scholar]
- 83.Hahn H, Wojnowski L, Zimmer AM, Hall J, Miller G, Zimmer A. Rhabdomyosarcomas and radiation hypersensitivity in a mouse model of Gorlin syndrome. Nat. Med. 1998;4:619–22. doi: 10.1038/nm0598-619. [DOI] [PubMed] [Google Scholar]
- 84.Romer J, Curran T. Targeting medulloblastoma: small-molecule inhibitors of the Sonic Hedgehog pathway as potential cancer therapeutics. Cancer Res. 2005;65:4975–8. doi: 10.1158/0008-5472.CAN-05-0481. [DOI] [PubMed] [Google Scholar]
- 85.Stanton BZ, Peng LF, Maloof N, Nakai K, Wang X, Duffner JL, Taveras KM, Hyman JM, Lee SW, Koehler AN, Chen JK, Fox JL, Mandinova A, Schreiber SL. A small molecule that binds Hedgehog and blocks its signaling in human cells. Nat. Chem. Biol. 2009;5:154–156. doi: 10.1038/nchembio.142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Sinha S, Chen JK. Purmorphamine activates the Hedgehog pathway by targeting Smoothened. Nat. Chem. Biol. 2006;2:29–30. doi: 10.1038/nchembio753. [DOI] [PubMed] [Google Scholar]
- 87.van der Horst G, Farih-Sips H, Lowik CW, Karperien M. Hedgehog stimulates only osteoblastic differentiation of undifferentiated KS483 cells. Bone. 2003;33:899–910. doi: 10.1016/j.bone.2003.07.004. [DOI] [PubMed] [Google Scholar]
- 88.Spinella-Jaegle S, Rawadi G, Kawai S, Gallea S, Faucheu C, Mollat P, Courtois B, Bergaud B, Ramez V, Blanchet AM, Adelmant G, Baron R, Roman-Roman S. Sonic hedgehog increases the commitment of pluripotent mesenchymal cells into the osteoblastic lineage and abolishes adipocytic differentiation. J. Cell Sci. 2001;114:2085–94. doi: 10.1242/jcs.114.11.2085. [DOI] [PubMed] [Google Scholar]
- 89.Kolterud Å, Toftgård R. Strategies for Hedgehog inhibition and its potential role in cancer treatment. Drug Discovery Today: Therapeutic Strategies. 2007;4:229–235. [Google Scholar]
- 90.Chen JK, Taipale J, Cooper MK, Beachy PA. Inhibition of Hedgehog signaling by direct binding of cyclopamine to Smoothened. Genes Dev. 2002;16:2743–8. doi: 10.1101/gad.1025302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Cooper MK, Porter JA, Young KE, Beachy PA. Teratogen-mediated inhibition of target tissue response to Shh signaling. Science. 1998;280:1603–7. doi: 10.1126/science.280.5369.1603. [DOI] [PubMed] [Google Scholar]
- 92.Binns W, James LF, Shupe JL, Everett G. A Congenital Cyclopian-Type Malformation in Lambs Induced by Maternal Ingestion of a Range Plant, Veratrum Californicum. Am. J. Vet. Res. 1963;24:1164–75. [PubMed] [Google Scholar]
- 93.Keeler RF, Binns W. Teratogenic compounds of Veratrum californicum (Durand). V. Comparison of cyclopian effects of steroidal alkaloids from the plant and structurally related compounds from other sources. Teratology. 1968;1:5–10. doi: 10.1002/tera.1420010103. [DOI] [PubMed] [Google Scholar]
- 94.Incardona JP, Gaffield W, Kapur RP, Roelink H. The teratogenic Veratrum alkaloid cyclopamine inhibits sonic hedgehog signal transduction. Development. 1998;125:3553–62. doi: 10.1242/dev.125.18.3553. [DOI] [PubMed] [Google Scholar]
- 95.Incardona JP, Gaffield W, Lange Y, Cooney A, Pentchev PG, Liu S, Watson JA, Kapur RP, Roelink H. Cyclopamine inhibition of Sonic hedgehog signal transduction is not mediated through effects on cholesterol transport. Dev. Biol. 2000;224:440–52. doi: 10.1006/dbio.2000.9775. [DOI] [PubMed] [Google Scholar]
- 96.Beachy PA. WO01027135 A3 Regulators of The Hedgehog Pathway, Compostitions and Uses Related Thereto. 2001 April 19;
- 97.Taipale J, Chen JK, Cooper MK, Wang B, Mann RK, Milenkovic L, Scott MP, Beachy PA. Effects of oncogenic mutations in Smoothened and Patched can be reversed by cyclopamine. Nature. 2000;406:1005–9. doi: 10.1038/35023008. [DOI] [PubMed] [Google Scholar]
- 98.Brian A, Janardanannair S, Lescarbeau A, Tremblay M, Grayzel D, Matsui W. WO2008083252 A2 Methods of Use for Cyclopamine Analogs. 2008 July 10;
- 99.Tremblay MR, McGovern K, Nevalainen M, Nair S, Porter JR, Behnke M, Yu LC, Grenier L, Campbell M, Cushing J, El-Assi Z, Castro AC, Curtis M, Foley MA, Wright J, Tong JK, Palombella V, Adams J. Synthesis of Novel, Chemically Stable D-homo-Cyclopamine Analogs via a Cyclopropanation/Ring-Expansion Sequence; Gordon Research Conference; 2007. [Google Scholar]
- 100.Tremblay MR, Nevalainen M, Nair SJ, Porter JR, Castro AC, Behnke ML, Yu L-C, Hagel M, White K, Faia K, Grenier L, Campbell MJ, Cushing J, Woodward CN, Hoyt J, Foley MA, Read MA, Sydor JR, Tong JK, Palombella VJ, McGovern K, Adams J. Semisynthetic Cyclopamine Analogues as Potent and Orally Bioavailable Hedgehog Pathway Antagonists. J. Med. Chem. 2008;51:6646–6649. doi: 10.1021/jm8008508. [DOI] [PubMed] [Google Scholar]
- 101.Feldmann G, Fendrich V, McGovern K, Bedja D, Bisht S, Alvarez H, Koorstra JB, Habbe N, Karikari C, Mullendore M, Gabrielson KL, Sharma R, Matsui W, Maitra A. An orally bioavailable small-molecule inhibitor of Hedgehog signaling inhibits tumor initiation and metastasis in pancreatic cancer. Mol. Cancer Ther. 2008;7:2725–35. doi: 10.1158/1535-7163.MCT-08-0573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Pink M, Proctor J, Briggs K, MacDougall J, Whitebread N, Tremblay MR, Grogan MJ, Palombella V, Castro A, Adams J, Read M, Corcoran-Schwartz IM, Harcke T, Eberhart CG, Watkins DN, Sydor J. Activity of IPI-926, a Potent HH Pathway Inhibitor, in a Novel Model of Medulloblatoma Derived from Ptch/HIC +/- mice. American Association of Cancer Research; San Diego, California, USA: 2008. p Abstract #1588. [Google Scholar]
- 103.Travaglione V, Peacock CD, MacDougall J, McGovern K, Cushing J, Yu LC, Trudeau M, Palombella V, Adams J, Hierman J, Rhodes J, Devereux WL, Watkins DN. A novel Hh pathway inhibitor, IPI-926, delays recurrence post-chemotherapy in a primary human SCLC xenograft model. American Association for Cancer Research; San Diego, California, USA: 2008. p Abstract # 4611. [Google Scholar]
- 104.Gorgan MJ, Tremblay M. WO2008109829 Cyclopamine Lacatam Analogs and Mehtods of Use Thereof. 2008 September 12;
- 105.Grogan MJ, Tremblay M. WO2008109184A1 The Hetrocyclic Cyclopamine Analogs And Methods of Use Thereof. 2008 September 12;
- 106.Zhang J, Garrossian M, Gardner D, Garrossian A, Chang YT, Kim YK, Chang CW. Synthesis and anticancer activity studies of cyclopamine derivatives. Bioorg. Med. Chem. Lett. 2008;18:1359–63. doi: 10.1016/j.bmcl.2008.01.017. [DOI] [PubMed] [Google Scholar]
- 107.Kumar SK, Roy I, Anchoori RK, Fazli S, Maitra A, Beachy PA, Khan SR. Targeted inhibition of hedgehog signaling by cyclopamine prodrugs for advanced prostate cancer. Bioorg. Med. Chem. 2008;16:2764–8. doi: 10.1016/j.bmc.2008.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.2008 October 15; http://www.clinicaltrials.gov/ct2/results?term=GDC-0449&show_flds=Y.
- 109.Chen JK, Taipale J, Young KE, Maiti T, Beachy PA. Small molecule modulation of Smoothened activity. Proc. Natl. Acad. Sci. U. S. A. 2002;99:14071–6. doi: 10.1073/pnas.182542899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Williams JA, Guicherit OM, Zaharian BI, Xu Y, Chai L, Wichterle H, Kon C, Gatchalian C, Porter JA, Rubin LL, Wang FY. Identification of a small molecule inhibitor of the hedgehog signaling pathway: effects on basal cell carcinoma-like lesions. Proc. Natl. Acad. Sci. U. S. A. 2003;100:4616–21. doi: 10.1073/pnas.0732813100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Baxter AD, Boyd EA, Guicherit OM, Price S, Lee R. WO0126644 A2 Mediators of Hedgehog Signaling Pathways, Composition and Uses Related Thereto. 2001 April 19;
- 112.Brunton SA, Stibbard JHA, Rubin LL, Kruse LI, Guicherit OM, Boyd EA, Price S. Potent inhibitors of the Hedgehog signaling pathway. J. Med. Chem. 2008;51:1108–1110. doi: 10.1021/jm070694n. [DOI] [PubMed] [Google Scholar]
- 113.Bahceci S, Bajjalieh W, Chen J, Epshteyn S, Forsyth TP, Huynh TP, Kim BG, Leahy JW, Lee MS, Lewis GL, Mac MB, Mann G, Marlowe CK, Ridgway BH, Sangalang JC, Shi X, Takeuchi CS, Wang Y. WO2008112913 Inhibitors of The Hedgehog Pathway. 2008 September 18;
- 114.A Phase 1 Study of BMS-833923 (XL139) in Subjects With Advanced or Metastatic Cancer. 2009 January 8; http://clinicaltrials.gov/ct2/show/NCT00670189?spons=%22Exelixis%22&spons_ex=Y&rank=38.
- 115.Peukert S, Jain RK, Geisser A, Sun Y, Zhang R, Bourret A, Carlson A, Dasilva J, Ramamurthy A, Kelleher JF. Identification and structure-activity relationships of ortho-biphenyl carboxamides as potent Smoothened antagonists inhibiting the Hedgehog signaling pathway. Bioorg. Med. Chem. Lett. 2009;19:328–31. doi: 10.1016/j.bmcl.2008.11.096. [DOI] [PubMed] [Google Scholar]
- 116.Jain RK, Kelleher J, Peukert S, Sun Y. WO2007120827 A2 Use of Biarylcarboxamides in the Treatment of Hedgehog Pathway-Related Disorders. 2007 October 25;
- 117.Gao W, Jiang J, Wan Y, Cheng D, Han D, Wu X, Pan S. WO2007131201 A2 Compounds and Compositions as Hegehog Pathway Modulators. 2007 November 15;
- 118.Goldsmith RA, Sutherlin DA, Robarge KD, Olivero AG. WO2007059157 Bisamide Inhibitors of Hedgehog Signaling. 2007 May 24;
- 119.Rubin L, Guicherit OM, Price S, Boyd EA. WO03011219 A2 Mediators of Hegehog Signaling Pathways, Composition and Uses Related Thereto. 2003 July 29;
- 120.Yauch RL, Gould SE, Scales SJ, Tang T, Tian H, Ahn CP, Marshall D, Fu L, Januario T, Kallop D, Nannini-Pepe M, Kotkow K, Marsters JC, Rubin LL, de Sauvage FJ. A paracrine requirement for hedgehog signalling in cancer. Nature. 2008;455:406–10. doi: 10.1038/nature07275. [DOI] [PubMed] [Google Scholar]
- 121.Sasai K, Romer JT, Lee Y, Finkelstein D, Fuller C, McKinnon PJ, Curran T. Shh pathway activity is down-regulated in cultured medulloblastoma cells: implications for preclinical studies. Cancer Res. 2006;66:4215–22. doi: 10.1158/0008-5472.CAN-05-4505. [DOI] [PubMed] [Google Scholar]
- 122.Sasai K, Romer JT, Kimura H, Eberhart DE, Rice DS, Curran T. Medulloblastomas derived from Cxcr6 mutant mice respond to treatment with a smoothened inhibitor. Cancer Res. 2007;67:3871–7. doi: 10.1158/0008-5472.CAN-07-0493. [DOI] [PubMed] [Google Scholar]
- 123.Kimura H, Ng JM, Curran T. Transient inhibition of the Hedgehog pathway in young mice causes permanent defects in bone structure. Cancer Cell. 2008;13:249–60. doi: 10.1016/j.ccr.2008.01.027. [DOI] [PubMed] [Google Scholar]
- 124.Oinas A, Taipale J, Lahdenpera J. WO2007054623 A2 Mammalian Hedgehog Signaling Inhibitors. 2007 May 18;
- 125.Gunzner J, Sutherlin D, Stanley M, Bao L, Castanedo G, Lalonde R, Wang S, Reynolds M, Savage S, Malesky K, Dina M. WO2006028958 Pyridyl Inhibitors of Hedgehog Signalling. 2006 March 16;
- 126.Koehler MFT, Goldsmith R, Sutherlin DP. WO2006078283 Quinoxaline Inhibitors of The Hedgehog Signalling Pathway. 2006 July 27;
- 127.Hedgehog Inhibitor. 2008 October 15; http://www.gene.com/gene/pipeline/status/oncology/hedgehog/
- 128.Von Hoff D, Rudin C, LoRusso P, Borad M, Korn R, Heath E, Yauch R, Darbonne W, Kadel E, Zerivitz K, Nelson L, Mackey H, Marsters J, de Sauvage F, Low J. Efficacy data of GDC-0449, a systemic Hedgehog pathway antagonist, in a first-in-human, first-in-class Phase I study with locally advanced, multifocal or metastatic basal cell carcinoma patients; AACR Meeting Abstracts; 2008; 2008. LB-138. [Google Scholar]
- 129.Dai M, He F, Jain RK, Karki R, Kelleher J, Lei J, Llamas L, Mcewan MA, Miller-Moslin K, Perez LA, Peukert S, Yusuff N. WO2008110611 Organic Compounds and Their Uses. 2008 September 18;
- 130.Waddell ST, Balkovec JM, Kevin NJ, Gu X. WO2007047625 A2 Triazole Derivatives as Inhibitors of 11-Beta-Hydroxysteroid Dehydrogenase-1. 2007 April 26;
- 131.Balkovec JM, Thieringer R, Waddell ST. WO2008130552 A1 Triazole Derivatives which are Smo Antagonists. 2008 October 30;
- 132.Remsberg JR, Lou H, Tarasov SG, Dean M, Tarasova NI. Structural analogues of smoothened intracellular loops as potent inhibitors of Hedgehog pathway and cancer cell growth. J. Med. Chem. 2007;50:4534–4538. doi: 10.1021/jm0705657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Ribes V, Wang Z, Dolle P, Niederreither K. Retinaldehyde dehydrogenase 2 (RALDH2)-mediated retinoic acid synthesis regulates early mouse embryonic forebrain development by controlling FGF and sonic hedgehog signaling. Development. 2006;133:351–61. doi: 10.1242/dev.02204. [DOI] [PubMed] [Google Scholar]
- 134.Lee J, Wu X, Di Magliano MP, Peters EC, Wang Y, Hong J, Hebrok M, Ding S, Cho CY, Schultz PG. A small-molecule antagonist of the Hedgehog signaling pathway. ChemBioChem. 2007;8:1916–1919. doi: 10.1002/cbic.200700403. [DOI] [PubMed] [Google Scholar]
- 135.Wu X, Ding S, Schultz PG. WO2006050351 Compounds and Composition as Hedgehog Pathway Modulators. 2006 May 11;
- 136.Beachy PA, Chen JK, Taipale AJ. WO2005033288 Hedgehog Pathway Antagonists. 2005 April 14;
- 137.Lee Y, Kawagoe R, Sasai K, Li Y, Russell HR, Curran T, McKinnon PJ. Loss of suppressor-of-fused function promotes tumorigenesis. Oncogene. 2007;26:6442–7. doi: 10.1038/sj.onc.1210467. [DOI] [PubMed] [Google Scholar]
- 138.Kimura H, Stephen D, Joyner A, Curran T. Gli1 is important for medulloblastoma formation in Ptc1+/-mice. Oncogene. 2005;24:4026–36. doi: 10.1038/sj.onc.1208567. [DOI] [PubMed] [Google Scholar]
- 139.Goyette P, Allan D, Peschard P, Chen CF, Wang W, Lohnes D. Regulation of gli activity by all-trans retinoic acid in mouse keratinocytes. Cancer Res. 2000;60:5386–9. [PubMed] [Google Scholar]
- 140.Lauth M, Bergstrom A, Shimokawa T, Toftgard R. Inhibition of GLI-mediated transcription and tumor cell growth by small-molecule antagonists. Proc. Natl. Acad. Sci. U. S. A. 2007;104:8455–60. doi: 10.1073/pnas.0609699104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Hosoya T, Arai MA, Koyano T, Kowithayakorn T, Ishibashi M. Naturally occurring small-molecule inhibitors of hedgehog/GLI-mediated transcription. ChemBioChem. 2008;9:1082–1092. doi: 10.1002/cbic.200700511. [DOI] [PubMed] [Google Scholar]
- 142.Arai MA, Tateno C, Hosoya T, Koyano T, Kowithayakorn T, Ishibashi M. Hedgehog/GLI-mediated transcriptional inhibitors from Zizyphus cambodiana. Bioorg. Med. Chem. 2008;16:9420–4. doi: 10.1016/j.bmc.2008.09.053. [DOI] [PubMed] [Google Scholar]
- 143.He B, Fujii N, You L, Xu Z, Jablons DM. WO2007067814 Targeting Gli Proteins In Human Cancer By Small Molecules. 2007 June 14;
- 144.Tabs S, Avci O. Induction of the differentiation and apoptosis of tumor cells in vivo with efficiency and selectivity. Eur. J. Dermatol. 2004;14:96–102. [PubMed] [Google Scholar]
- 145.Garber K. Hedgehog drugs begin to show results. J. Natl. Cancer Inst. 2008;100:692–7. doi: 10.1093/jnci/djn169. [DOI] [PubMed] [Google Scholar]
- 146.Epstein EH. Basal cell carcinomas: attack of the hedgehog. Nat. Rev. Cancer. 2008;8:743–54. doi: 10.1038/nrc2503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Lauth M, Bergstrom A, Toftgard R. Phorbol esters inhibit the Hedgehog signalling pathway downstream of Suppressor of Fused, but upstream of Gli. Oncogene. 2007;26:5163–8. doi: 10.1038/sj.onc.1210321. [DOI] [PubMed] [Google Scholar]
- 148.Cheng SY, Yue S. Role and regulation of human tumor suppressor SUFU in Hedgehog signaling. Adv. Cancer Res. 2008;101:29–43. doi: 10.1016/S0065-230X(08)00402-8. [DOI] [PubMed] [Google Scholar]
- 149.Stecca B, Mas C, Clement V, Zbinden M, Correa R, Piguet V, Beermann F, Ruiz IAA. Melanomas require HEDGEHOG-GLI signaling regulated by interactions between GLI1 and the RAS-MEK/AKT pathways. Proc. Natl. Acad. Sci. U. S. A. 2007;104:5895–900. doi: 10.1073/pnas.0700776104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Feldmann G, Dhara S, Fendrich V, Bedja D, Beaty R, Mullendore M, Karikari C, Alvarez H, Iacobuzio-Donahue C, Jimeno A, Gabrielson KL, Matsui W, Maitra A. Blockade of hedgehog signaling inhibits pancreatic cancer invasion and metastases: a new paradigm for combination therapy in solid cancers. Cancer Res. 2007;67:2187–96. doi: 10.1158/0008-5472.CAN-06-3281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Zhao Y, Wang Y, Wang Z, Liu H, Shen Y, Li W, Heller S, Li H. Sonic hedgehog promotes mouse inner ear progenitor cell proliferation and hair cell generation in vitro. Neuroreport. 2006;17:121–4. doi: 10.1097/01.wnr.0000198439.44636.49. [DOI] [PubMed] [Google Scholar]
- 152.Shimoyama A, Wada M, Ikeda F, Hata K, Matsubara T, Nifuji A, Noda M, Amano K, Yamaguchi A, Nishimura R, Yoneda T. Ihh/Gli2 signaling promotes osteoblast differentiation by regulating Runx2 expression and function. Mol. Biol. Cell. 2007;18:2411–8. doi: 10.1091/mbc.E06-08-0743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Curran T, Ng JM. Cancer: Hedgehog’s other great trick. Nature. 2008;455:293–4. doi: 10.1038/455293a. [DOI] [PubMed] [Google Scholar]
- 154.Nolan-Stevaux O, Lau J, Truitt ML, Chu GC, Hebrok M, Fernandez-Zapico ME, Hanahan D. GLI1 is regulated through Smoothened-independent mechanisms in neoplastic pancreatic ducts and mediates PDAC cell survival and transformation. Genes Dev. 2009;23:24–36. doi: 10.1101/gad.1753809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Geng L, Cuneo KC, Cooper MK, Wang H, Sekhar K, Fu A, Hallahan DE. Hedgehog signaling in the murine melanoma microenvironment. Angiogenesis. 2007;10:259–67. doi: 10.1007/s10456-007-9078-9. [DOI] [PubMed] [Google Scholar]