

Abstract
An individual's tendency to show exaggerated or otherwise dysregulated cardiovascular reactions to acute stressors has long been associated with increased risk for clinical and preclinical endpoints of coronary heart disease (CHD). However, the ‘brain-body’ pathways that link stressor-evoked cardiovascular reactions to CHD risk remain uncertain. This review summarizes emerging neuroimaging research indicating that individual differences in stressor-evoked blood pressure reactivity (a particular form of cardiovascular reactivity) are associated with activation patterns in corticolimbic brain areas that are jointly involved in processing stressors and regulating the cardiovascular system. As supported empirically by activation likelihood estimates derived from a meta-analysis, these corticolimbic areas include divisions of the cingulate cortex, insula, and amygdala—as well as networked cortical and subcortical areas involved in mobilizing hemodynamic and metabolic support for stress-related behavioral responding. Contextually, the research reviewed here illustrates how behavioral medicine and health neuroscience methods can be integrated to help characterize the ‘brain-body’ pathways that mechanistically link stressful experiences with CHD risk.
Keywords: amygdala, blood pressure reactivity, coronary heart disease, cingulate cortex, insula, health neuroscience, stress
Introduction
Psychological stress has long been implicated in the development of coronary heart disease (CHD), the leading public health burden in industrialized nations (Brotman et al., 2007; Holmes et al., 2006; Krantz et al., 1988; Manuck et al., 1988; Osler, 1897). As a slowly developing chronic illness, CHD results chiefly from a progressive narrowing of the blood vessels that supply oxygenated blood to the heart. Beginning early in life and progressing over a period of decades, this vessel narrowing can ultimately lead to several clinical endpoints, including an inadequate ejection of blood from the heart (heart failure), non-fatal and fatal heart attacks (myocardial infarctions), irregular cardiac rhythms (arrhythmias), and chest pain (angina) caused by an insufficient supply of blood to cardiac tissue (ischemia). A major contributor to CHD is atherosclerosis, a degenerative and inflammatory syndrome promoting the vascular accumulation of cholesterol and cellular waste products that remodel peripheral blood vessels and impair coronary vessel functioning and blood flow dynamics (Libby, 2002; Libby and Theroux, 2005; Rosamond et al., 2008). It is also important to note here that the clinical expression of many late-stage CHD endpoints and events, particularly acute coronary syndromes, depends not only on the severity of cumulative atherosclerotic progression and vessel narrowing over the lifespan, but also on the vulnerability of metabolically-active atherosclerotic plaques (atheromata) to acute rupture and consequent thrombosis (blood clotting that obstructs vessel blood flow) (Buja and Willerson, 1981; Davies and Thomas, 1985; Falk, 1983; Fuster, 1994; Lefkowitz and Willerson, 2001; Libby, 1995, 2001). Moreover, it is now widely recognized that the likelihood that such vulnerable plaques will rupture and trigger the expression of clinical CHD events may be increased in the context of abnormal vascular and vasomotor dynamics evoked by acute environmental, emotional, and behavioral stressors that are of personal relevance to the individual (Bhattacharyya and Steptoe, 2007; Steptoe and Brydon, 2009; Strike and Steptoe, 2005).
It is well established that the developmental progression of atherosclerosis and the related expression of late-stage CHD endpoints noted above have manifold and interacting genetic, biological, behavioral, social and environmental risk factors. Together, however, many primary and secondary risk factors (e.g., a family history of CHD, smoking, hypertension, dyslipidemia, an imprudent diet, and a lack of regular physical activity) do not fully account for all newly diagnosed (incident) CHD events among men and women, although such risk factors may be prevalent among patients with existing CHD (Anderson et al., 1991; Beaglehole and Magnus, 2002; Greenland et al., 2002; Greenland et al., 2003; Khot et al., 2003; Magnus and Beaglehole, 2001). In part, this has focused attention on other suspected factors that could plausibly influence CHD risk trajectories. Many of these factors include genetic polymorphisms that contribute to the regulation of lipoprotein levels, blood coagulation, blood pressure, immune function, and vessel biology, as well as biomediators of inflammation, hemostasis, thrombosis, fibrinolysis, lipid metabolism, and molecular oxidation processes affecting endothelial function (Lefkowitz and Willerson, 2001; Wood, 2001).
As stated above, psychological stress is another potential CHD risk factor that has received appreciable attention in clinical and epidemiological studies in humans and translational studies in nonhuman animals (for recent reviews, see Brotman et al., 2007; Holmes et al., 2006). A particular stress-related factor long implicated in CHD risk is an individual's tendency to show large-magnitude or so-called ‘exaggerated’ cardiovascular reactions to acute stressors (Krantz and Manuck, 1984; Manuck, 1994; Obrist, 1981; Schwartz et al., 2003; Treiber et al., 2003). More precisely, several (although not unequivocal) lines of evidence suggest that exaggerated stressor-evoked cardiovascular reactions predict (1) an accelerated progression of atherosclerosis in humans and nonhuman primates; (2) the premature development of high blood pressure (hypertension) and other precursors to CHD; and (3) the likelihood of having a future coronary event (e.g., myocardial infarction) (Schwartz et al., 2003; Treiber et al., 2003). As reviewed below, however, the neurobiological or ‘brain-body’ pathways that couple the central nervous system processing of acute stressors with the peripheral expression of cardiovascular reactions implicated in CHD risk remain largely uncertain (Lane et al., 2009a; Lane et al., 2009b; Lovallo, 2005; Lovallo and Gerin, 2003; Soufer et al., 2002). Arguably, delineating these neurobiological pathways may aid not only in developing brain-based strategies for augmenting CHD risk stratification and prediction, but also in furthering a mechanistic understanding of stress-related processes contributing to CHD vulnerability.
In view of these possibilities, this review (1) describes for non-specialists some fundamental conceptual and measurement issues regarding the construct of stressor-evoked cardiovascular reactivity as a biobehavioral dimension of individual differences; (2) highlights clinical, epidemiological, and translational evidence indicating that individual differences in stressor-evoked cardiovascular reactivity are associated with CHD risk; and (3) summarizes emerging neuroimaging evidence suggesting that individual differences in stressor-evoked blood pressure reactivity (a specific form of cardiovascular reactivity) may be mediated in part by alterations in the functionality of corticolimbic brain circuits that are jointly involved in processing acute stressors and regulating the cardiovascular system.
Cardiovascular reactivity: concepts and metrics
Operationally, ‘cardiovascular reactivity’ is defined as a change in hemodynamic activity from a resting (baseline) state to a subsequent behavioral state involving a psychological or physical challenge (Obrist, 1981). In human laboratory studies, such challenges are nominally referred to as stressors, and they often require individuals to complete acutely demanding tasks that (1) entail negative or positive consequences of motivational or personal relevance, and that (2) prompt behavioral coping responses that enable successful task performance and engagement (Kamarck and Lovallo, 2003). Among the most common and frequently standardized psychological stressors used to evoke cardiovascular reactivity are those that require solving difficult mental arithmetic problems; preparing and delivering speeches on interpersonally distressing topics; and engaging in effortful or frustrating cognitive tasks that tax executive control processes (e.g., anagram, working memory, inverted or mirror-image tracing, and Stroop color-word interference tasks). Also common are physical stressors that involve immersing a limb in painfully cold water or squeezing a handgrip dynamometer to maintain a constant amount of muscular tension over a prolonged period of time (isometric exercise). For most individuals, such stressors will evoke measurable cardiovascular changes from a baseline state; however, it has long been recognized that individuals differ markedly in the magnitude, patterning, and duration of these cardiovascular changes. Moreover, some individuals appear to have a trait-like (dispositional) tendency to reliably express relatively large-magnitude (exaggerated) and sometimes prolonged stressor-evoked increases in cardiovascular activity, which are implicated in CHD risk.
It is important to recognize here, however, that cardiovascular reactivity patterns may vary not only as a function of presumptive dispositional tendencies, but also as a function of the experimental dimensions of evocative stressors (Kamarck and Lovallo, 2003). For instance, stressors can require either active or passive coping behaviors, and they can be dimensionally categorized as more or less psychological or physical in nature (Obrist, 1981). Critically, from a neurobiological perspective, such stressor dimensions could plausibly recruit dissociable brain circuitries and pathways supporting divergent or even comparable patterns of observable cardiovascular reactions. Hence, passively coping with the physical pain imposed by immersing one's hand in cold water may evoke a rise in blood pressure because of an increase in vascular resistance due to a temperature-induced stimulation of alpha-adrenergic receptors located on peripheral blood vessels. By contrast, actively coping with the psychological demands imposed by a frustrating cognitive challenge, such as the Stroop color-word interference task, may evoke an equivalent rise in blood pressure because of a mixed increase in cardiac output and vascular resistance resulting from a centrally-orchestrated stimulation of beta- and alpha-adrenergic receptors located on the myocardium and blood vessels, respectively. Thus, selecting stressor tasks and interpreting cardiovascular changes evoked by particular stressors should be grounded by the caveat that different stressors can evoke different (and sometimes comparable) patterns of cardiovascular reactivity (e.g., equivalent rises in blood pressure) via diverse neurobiological and peripheral response mechanisms. In extension, such mechanisms may differ in their predictive relevance for CHD risk (Kamarck and Lovallo, 2003; Obrist, 1981).
Another noteworthy issue in this context is that quantifying cardiovascular reactivity accurately is complicated by physiological factors and statistical issues related to the reliable measurement of change. More precisely, the magnitude and even direction of change from a baseline (or resting) state in a given cardiovascular parameter often depends on the initial level of that parameter. This baseline dependency is conventionally referred to as the principle of initial values, according to which higher absolute levels of a given physiological parameter will tend to predict subsequently smaller and often directionally negative evoked changes in that parameter (Berntson et al., 1994; Stern et al., 2001; Wainer, 1991; Wilder, 1967). Conversely, lower absolute levels of a given physiological parameter will tend to predict larger and often directionally positive evoked changes in that parameter. Hence, some statistical control or quantification of baseline dependency is generally recommended for studies of cardiovascular reactivity. A final salient methodological issue to be noted is the often reduced statistical reliability of change scores, which are routinely used to quantify cardiovascular reactivity. At present, there is cumulative evidence from cardiovascular reactivity research that change scores computed by subtraction procedures (e.g., subtracting baseline from task levels of a cardiovascular parameter) and regression procedures (e.g., regressing task on baseline levels and retaining the residual values) show adequate statistical (test-retest) reliability when such scores are averaged across multiple stressors sharing similar task dimensions (Kamarck et al., 1992; Kamarck and Lovallo, 2003; Llabre et al., 1991; Manuck et al., 1995a). Following psychometric principles, such averaging minimizes nuisance or error variance attributable to the idiosyncratic features of particular stressors, along with measurement and testing occasions; such averaging also maximizes the likelihood of capturing the dispositional properties of a given cardiovascular reactivity measure, which are thought to be of most importance for predicting CHD risk across individuals (Kamarck and Lovallo, 2003). Again, however, averaging procedures implemented to improve the reliability of reactivity measures should be guided by the consideration that each stressor task used for averaging should share as many experiential and standardized experimental aspects as are feasible.
Common parameters of cardiovascular reactivity: importance of blood pressure reactivity in CHD risk
The most common parameters of cardiovascular reactivity assessed in laboratory psychophysiological studies are stressor-evoked changes in blood pressure (systolic, diastolic, and mean arterial pressure), heart rate, stroke volume (the amount of blood ejected from the ventricles with each contraction of the heart), cardiac output (the average amount of blood ejected from the heart per minute), total peripheral resistance (the sum of systemic resistance to blood flow), and time intervals within the cardiac cycle reflecting myocardial performance (e.g., pre-ejection period [PEP] and left ventricular ejection time [LVET])1. In the context of this review, it is important to note that measures of blood pressure and heart rate are presently the most amenable to monitoring in neuroimaging paradigms. This is because of the wide availability of recording equipment compatible with imaging environments, particularly magnetic resonance scanning environments. Measurement of other cardiovascular parameters noted above generally require the use of impedance cardiography or other methodologies that presently pose safety and related challenges for physiological monitoring in the imaging environment (Gray et al., 2009a; Lane et al., 2009a; Lane et al., 2009b). Further, as detailed next, individual differences in blood pressure reactivity in particular appear to be most consistently associated with prospective risk for atherosclerosis, precursors to CHD, and clinical CHD events in clinical and epidemiological studies (Treiber et al., 2003). Accordingly, the remainder of this review will focus largely on stressor-evoked blood pressure reactivity in the context of CHD risk.
Stressor-evoked blood pressure reactivity and CHD risk: epidemiological and translational evidence
A change in blood pressure is a core facet of the prototypical cardiovascular reaction to an acute stressor (for an accessible review, see Herd, 1991). Broadly stated, an acute stressor-evoked blood pressure reaction results from net changes in autonomic (sympathetic and parasympathetic) nervous system activity that alter cardiac output and peripheral vascular resistance among other hemodynamic parameters. Such changes generally serve to shunt blood from the viscera toward large muscle groups, presumably to provide metabolic support for adaptive behavioral action (e.g., the fight-or-flight response) (Cannon, 1928, 1932; McEwen, 1998, 2007; Obrist, 1981; Sapolsky et al., 2000). As noted above, some individuals have a reliable tendency to express relatively large rises in blood pressure that are viewed to exceed the metabolic demands of a given stressor (Obrist, 1981). Indeed, this exaggerated cardiovascular response tendency appears to be a trait-like dispositional characteristic of some individuals (Allen et al., 1987; Cohen and Hamrick, 2003; Fauvel et al., 1996; Gerin et al., 1993; Kamarck et al., 1994; Kamarck and Lovallo, 2003; Llabre et al., 1991; Manuck et al., 1995a; Sherwood et al., 1990), which may be partially heritable (De Geus et al., 2007; Turner and Hewitt, 1992) and predictive of atherosclerotic and CHD risk early in development (Matthews et al., 2006; Roemmich et al., 2009). Moreover, individuals expressing exaggerated stressor-evoked cardiovascular reactions, particularly exaggerated blood pressure reactions, appear to be at elevated risk for precursors to CHD and related endpoints in later life. For example, otherwise healthy individuals who express exaggerated stressor-evoked blood pressure reactions in comparison with their less reactive counterparts are at prospective risk for hypertension (Bedi et al., 2000; Knox et al., 2002; Matthews et al., 2003; Matthews et al., 1993; Menkes et al., 1989; Ming et al., 2004; Stewart et al., 2006), stroke (Everson et al., 2001), myocardial infarction (Alderman et al., 1990; Manuck et al., 1992), and premature atherosclerosis in major arteries (Treiber et al., 2003; Jennings et al., 2004; Matthews et al., 2006). In a recent review of these and other epidemiological observations, it was concluded that “…cardiovascular reactivity can predict the development of some preclinical…states and perhaps even new clinical events in some patients with essential hypertension or coronary heart disease. However, much more information is needed concerning moderating and potentially confounding variables before the robustness of the positive relationships can become clinically useful” (Treiber et al., 2003; p. 46). As detailed below, one such ‘moderating’ variable may entail inter-individual variation in the functionality of corticolimbic brain circuits that are jointly involved in stressor processing and cardiovascular regulation.
Further, although it is presently unknown (and the subject of much debate) whether exaggerated, prolonged, or otherwise dysregulated stressor-evoked blood pressure or additional cardiovascular reactions contribute causally to CHD risk, it is plausible that such reactions may increase CHD risk by promoting structural, particularly atherogenic, changes in vascular tissue (Krantz and Manuck, 1984; Manuck, 1994; Manuck et al., 1986; Manuck et al., 1995b; Schwartz et al., 2003; Treiber et al., 2003). For example, the repeated expression of exaggerated stressor-evoked blood pressure reactions may injure the endothelial (inner) layer of peripheral blood vessels via turbulent (non-laminar) blood flow patterns that promote shear stress, particularly at vessel bifurcations. The atherogenic effects of such injuries could include an increased permeability of the endothelium to circulating lipoproteins; a release of mitogenic substances by newly regenerated endothelial cells; a proliferation of intimal smooth muscle cells; and a disruption of the lipid metabolism of endothelial cells (Beere et al., 1984; Gordon et al., 1981; Shafi et al., 1989). Hence, endothelial injury in the aorta or coronary arteries can (1) be induced by stressors that elicit large rises in blood pressure and can (2) be prevented by beta-adrenergic (sympathetic nervous system) blockade (Strawn et al., 1989; Strawn et al., 1991). Moreover, acute stressors can trigger clinical events in those with preclinical or established CHD by pressor-related hemodynamic or vasoconstrictive factors. For instance, acute rises in blood pressure may rupture vulnerable plaques, leading to thrombus formation or embolization (Kop, 1999, 2003; Krantz et al., 1999; Strike and Steptoe, 2004). In aggregate, it is thus reasonable to speculate that a dispositional tendency to repeatedly express exaggerated or prolonged stressor-evoked cardiovascular—particularly blood pressure—reactions may have damaging effects on the vasculature, possibly increasing atherosclerotic and related CHD risk.
A potential role for corticolimbic systems in stressor-evoked blood pressure reactivity and CHD risk
Despite the clinical and epidemiological evidence reviewed above, little is known about the human neural systems that link the central processing of acute stressors with individual differences in cardiovascular reactivity and associated CHD risk. Hence, only a small number of neuroimaging studies have investigated the functional neural correlates of blood pressure and other forms of stressor-evoked cardiovascular reactivity. Moreover, there are even fewer conceptual frameworks focusing specifically on the putative neural origins of individual differences in cardiovascular reactivity, particularly as formulated within the context of CHD risk. Importantly, such models are necessary for guiding and developing hypothesis-driven studies that extend existing human and translational animal studies on the neural regulation of cardiovascular stress reactivity.
One such conceptual model of cardiovascular reactivity proposed by Lovallo and colleagues (Lovallo, 2005; Lovallo and Gerin, 2003) integrates the cognitive concepts of stressor appraisal theory (Holroyd and Lazarus, 1982; Lazarus and Folkman, 1984) with neurobiological concepts derived from early animal research on the central neural control over the cardiovascular stress response (Bard, 1928; Cannon, 1928, 1932). According to cognitive stressor appraisal theory as it is generally applied in behavioral medicine, ‘stress’ is a transactional process arising from real or perceived demands that can be evaluated (appraised) as threatening or benign, depending on the adaptive coping resources available to an individual (Cohen et al., 2007; Monroe, 2008). Furthermore, the biological, behavioral, and other coping responses and reactions that ensue from such appraisal processes are held to influence risk for and resilience against ill health, including CHD (cf., McEwen, 2007; McEwen and Gianaros, in press). As viewed from Lovallo's conceptual model, the stressor appraisal processes noted above and the resulting neural commands for integrated peripheral physiological (e.g., cardiovascular) and behavioral stress reactions are instantiated in rostral or corticolimbic brain systems that are located above the level of the hypothalamus in the neuroaxis. Hence, according to this model, individual differences in stressor-evoked cardiovascular reactivity could originate specifically from altered stressor-related activity along three reciprocally interacting ‘levels-of-response’, which are heuristically organized from (1) corticolimbic systems to (2) midbrain and brainstem relay pathways and neuromodulatory systems to (3) peripheral target organs (i.e., the heart and vasculature). Here, corticolimbic systems support the evaluative (cognitive) appraisal of self-relevant psychological and environmental demands (stressors). After appraising such demands, corticolimbic systems are hypothesized to reciprocally signal with midbrain and brainstem relay pathways and neuromodulatory systems to generate adaptive coping behaviors that are integrated with metabolically supportive changes in target organ reactivity (e.g., peripheral changes in cardiac output and vasoconstriction underlying net increases in blood pressure). An individual's tendency to express increased stressor-evoked neural activation in corticolimbic systems could therefore mediate the peripheral expression of exaggerated cardiovascular reactivity. The specific corticolimbic systems implicated by Lovallo's model in the regulation (generation, representation, and termination) of stressor-evoked cardiovascular reactions include networked divisions of the cingulate and medial prefrontal cortices, amygdala, and septal nuclei. Specific relay pathways and neuromodulatory systems by which the above corticolimbic systems may interactively regulate the acute expression of cardiovascular reactions include the hypothalamus, ventral tegmentum, pontine raphe, and locus ceruleus. It is important to note again here, though, that Lovallo's conceptual model was derived from an inferential synthesis of cognitive stressor appraisal theory with laboratory work on cardiovascular reactivity: The model was not based explicitly on evidence from human in vivo neuroimaging and patient lesion studies of stressor-evoked cardiovascular and autonomic nervous system reactivity. By comparison, much of this neuroimaging and lesion evidence was incorporated into a complementary neurobiological model of emotional and cognitive integration developed by Critchley and colleagues (Critchley, 2005; Critchley et al., 2000; Critchley et al., 2003).
According to this latter model, functional subdivisions of several corticolimbic brain systems emphasized by Lovallo and colleagues, chiefly the cingulate and medial prefrontal cortices, insula, and amygdala, are posited to play instrumental and interactive roles in calibrating autonomic and cardiovascular reactions with contextually adaptive behavior to meet the metabolic demands of emotional and cognitive challenges. This model has been substantiated by several neuroimaging studies showing that behaviorally-evoked changes in cardiovascular (e.g., blood pressure, heart rate) and cardiac-autonomic (e.g., heart rate variability) activity are correlated directly with neural activity within areas of the cingulate and medial prefrontal cortices, often in interaction with activity in the insula, amygdala, and relay regions of the thalamus, hypothalamus, midbrain and brainstem (e.g., Critchley, 2005; Critchley et al., 2000; Critchley et al., 2003; Gianaros et al., 2005a; Gianaros et al., 2007; Gianaros et al., 2008; Gianaros et al., 2004; Gray et al., 2009b; Lane et al., 2001; Matthews et al., 2004; Mujica-Parodi et al., 2009; O'Connor et al., 2007; Wager et al., 2009a; Wager et al., 2009b). Providing additional support for this model, parallel studies of patients with lesions to the cingulate cortex further implicate this particular corticolimbic system as being critical for coordinating autonomic and cardiovascular adjustments with emotional and cognitive behaviors (Critchley, 2005; Critchley et al., 2003), which are relevant to stress-related processes.
In synthesis of the models above and prior work in nonhuman animal models, there are thus conceptual and empirical grounds to posit that corticolimbic systems—particularly the cingulate, insula, and amygdala—are involved in regulating stressor-evoked blood pressure and other cardiovascular reactions. In extension, it is reasonable to speculate that an individual's dispositional tendency to express increased stressor-evoked activity in these corticolimbic systems could relate to the peripheral expression of exaggerated cardiovascular reactions, possibly conferring heightened CHD risk. Although the complex and interacting variables contributing to the expression of such centrally mediated dispositional response tendencies are likely numerous, they are broadly assumed here to arise from heritable factors, cumulative and unique developmental life experiences, and personal learning histories impacting stress sensitivity and reactivity over the lifespan (cf., Boyce and Ellis, 2005; Ellis et al., 2005; Lane et al., 2009a; Lovallo, 2005; McEwen, 2007).
Elaborating on these notions, we next focus on how areas of the cingulate cortex, insula, and amygdala may be functionally involved in mediating individual differences in stressor-evoked cardiovascular reactivity, particularly by highlighting their putative support of stress-related cognitive and behavioral response processes. We also speculate on some of the circuitries and pathways by which these systems may generate, represent, and terminate stressor-evoked cardiovascular reactions. We conclude with a quantitative summary of the available neuroimaging studies suggesting that stressor-evoked blood pressure reactivity is associated with concurrent functional neural activity in cortical and subcortical areas presumably involved in evaluative stressor-processing, adaptive behavioral responding, and concurrent cardiovascular control.
Functional roles of the cingulate, insula, and amygdala in stressor-evoked blood pressure reactivity
Cingulate cortex
The cingulate cortex is a medial cortical brain system that supports cognitive, emotional, nociceptive, skeletal-motor, and visceromotor processes. Regional cingulate differences in cellular architecture and projections to and from other brain areas define three putatively distinct functional subdivisions, nominally labeled as (1) a rostral (perigenual) affective division encompassing cytoarchitectual areas 24a-c, 25 & 32; (2) a dorsal (supragenual) cognitive-motor division encompassing areas 24b′-c′ & 32′; and (3) a caudal (retrosplenial) evaluative-monitoring division encompassing areas 23, 29, 30, & 31 (Bush et al., 2000; Devinsky et al., 1995; Paus, 2001; Vogt, 2005; Vogt et al., 1992; Vogt et al., 1995). In addition to supporting cognitive- and emotion-related functions, evidence summarized below implicates cingulate subdivisions in mediating stressor-evoked blood pressure reactivity.
The perigenual anterior cingulate cortex (pACC) is viewed to support several stress-related functions, including the appraisal of salient environmental events, the subjective experience of aversive behavioral states, and the regulation of behavioral and autonomic responses to aversive stimuli (Bush et al., 2000; Critchley, 2005; Paus, 2001; Phillips et al., 2003; Vogt, 2005; Wager et al., 2009a; Wager et al., 2009b). For example, imaging evidence demonstrates that the pACC is engaged by negative mood induction procedures, anticipatory anxiety, and contexts involving the threat of negative social evaluation (George et al., 1995; Mayberg et al., 1999; Straube et al., 2009; Wager et al., 2009a; Wager et al., 2009b), by the presence of distracting emotional information during demanding cognitive task performance (Mohanty et al., 2007), and by committing self-relevant and negatively evaluated errors during frustrating cognitive tasks (Kiehl et al., 2000). Complementing this work, both animal and human findings document an important role for the pACC in supporting stressor-evoked autonomic and cardiovascular reactivity. This role of the pACC is instantiated through its reciprocal circuitry with the orbital and medial prefrontal cortex, insula, amygdala, and cell groups in the thalamus, hypothalamus, periaqueductal grey (PAG), pons, medulla, and the pre-sympathetic intermediolateral (IML) cell column of the spinal cord (Barbas, 2000; Barbas et al., 2003; Buchanan and Powell, 1993; Chiba et al., 2001; Critchley, 2005; Freedman et al., 2000; Öngür et al., 1998; Öngür and Price, 2000; Vogt, 2005). As such, the pACC—along with networked areas of the dorsal and posterior cingulate cortex discussed next—may provide for an interface between self-relevant stressor appraisal processes and concurrent cardiovascular control.
Areas in the dorsal anterior cingulate (dACC) are conventionally viewed to support processes related to attention, effortful executive control, and conflict and error monitoring. These processes are instantiated by dense reciprocal circuitry with the lateral prefrontal cortex, motor and supplementary motor cortex, and posterior parietal cortex (Vogt and Pandya, 1987). A conventional view is that dACC areas monitor for conflicts between competing streams of incompatible information, which foster the potential for behavioral error (Botvinick et al., 2001; Hester et al., 2004; Holroyd and Coles, 2002; Ridderinkhof et al., 2004a; Ridderinkhof et al., 2004b). After conflict detection, dACC areas engage prefrontal, motor, and parietal cortices to resolve conflicts and minimize behavioral error by modulating attention, working memory, and motor control processes (Koski and Paus, 2000; Paus, 2001; Paus et al., 1998). Growing evidence also implicates dACC areas in stress-related behavioral processes associated with physiological reactivity. For example, dACC areas are engaged by states of pain-related anxiety (Ochsner and Gross, 2005; Vogt et al., 2003), intentional regulation of autonomic activity (Critchley et al., 2001, 2002), awareness of subjective emotional experiences (Lane et al., 1998), and even social rejection associated with activation of the hypothalamic-pituitary-adrenal stress response axis (Eisenberger et al., 2007). Hence, consistent with the neuroimaging evidence reviewed below and the conceptual frameworks reviewed above (Critchley, 2005; Lovallo, 2005), the dACC may be important for generating autonomic and cardiovascular responses via projections to networked cortical and subcortical areas to support volitional, cognitive, and emotional behaviors, all processes that are key for mediating stressor-evoked blood pressure reactivity.
The posterior cingulate cortex (pCC) is viewed to support self-relevant evaluative processes related to cognition and emotion, including (a) maintaining a general representation of the environment, (b) appraising the emotional salience of environmental events, and (c) monitoring the environment for threatening or otherwise stressful stimuli (Gusnard et al., 2001; Maddock, 1999; Vogt and Laureys, 2005; Vogt et al., 2006). These processes are supported principally by reciprocal circuitry between the pCC, pACC, and parahippocampal cortices (Vogt and Laureys, 2005; Vogt et al., 2006). In support of its putative role in evaluative appraisal processing, a recent meta-analysis (Maddock, 1999) implicates the pCC in association with the automatic appraisal of unpleasant, particularly self-relevant, stimuli. Considerable human neuroimaging evidence also indicates that the pCC is a major component of a distributed network of functionally and anatomically connected brain systems (including dorsal and ventral medial prefrontal cortex, medial and lateral parietal cortex, and areas of the medial and lateral temporal cortex) that all show coherent and relatively high levels of metabolic activity during resting states (Buckner et al., 2008; Fox and Raichle, 2007; Fox et al., 2007; Greicius et al., 2003; Greicius et al., in press; Gusnard et al., 2001). Activity in components of this so-called ‘default mode network’ is thought to foster inwardly directed attention to self-referential multimodal and interoceptive (e.g., autonomic) information (Nagai et al., 2004). In addition, there is substantial evidence that when cognitive effort and attentional resources are redirected to the external environment to support goal-directed behaviors, activity in the pCC and other components of the default mode network is markedly curtailed, possibly as a means of attenuating incompatible neural activity supporting a self-referential focus in order to meet the demands of environmental challenges and execute outwardly directed action (Buckner et al., 2008; Gusnard et al., 2001).
Although the pCC does issue light projections to areas such as the PAG (An et al., 1998), it generally lacks the more direct connections with pre-autonomic and cardiovascular regulatory cell groups that are characteristic of other corticolimbic areas emphasized here; namely, the pACC, dACC, insula, and amygdala. Nevertheless, several neuroimaging studies demonstrate that stressor-evoked autonomic and cardiovascular reactions are observed in conjunction with changes in pCC activity (e.g., Gianaros et al., 2007; Gianaros et al., 2005b; Gianaros et al., 2008; Wong et al., 2007). For example, individuals classified as stable and high blood pressure reactors have been shown to express enhanced pCC activation to a Stroop color-word interference stressor, as compared with their less reactive counterparts who consistently showed expected patterns of deactivation in the pCC during effortful task performance (Gianaros et al., 2005b). One interpretation of these particular findings is that high blood pressure reactors may be unable to effectively curtail pCC activity when faced with behavioral challenges, facilitating a generalized form of environmental processing and monitoring for threatening or otherwise stressful stimuli. While a linear relationship between inter-individual variation in stressor-evoked blood pressure reactivity and pCC activation (or lesser deactivation) has also been observed in two subsequent studies (Gianaros et al., 2007; Gianaros et al., 2008), the functional significance of the pCC in autonomic and cardiovascular control remains uncertain. An interesting hypothesis compatible with the interpretation of pCC functionality outlined above was recently posited by Wong and colleagues (2007) in an fMRI study of exercise related autonomic cardiovascular control. Specifically, Wong et al. demonstrated that an effortful isometric handgrip exercise evoked both (1) transient increases in heart rate and blood pressure and (2) decreases in the activity of the pCC and ventromedial prefrontal cortex among healthy young men and women. However, fine-grained analyses of the blood-oxygenated level dependent (BOLD) time series data revealed that activity changes in the ventromedial prefrontal cortex, but not the pCC, were directly associated with time-related and exercise-induced changes in heart rate. Thus, Wong et al. posited that pCC activity may be suspended during effortful behavioral tasks, such as isometric exercise, and that this activity change is not instrumental for associated task-related changes in autonomic or cardiovascular function. Hence, in view of existing evidence and known neuroanatomical connections of the pCC, activity in this region is likely to correspond to the evaluative appraisal of self-referential information, and possibly environmental contexts and stressors, which may indirectly (or spuriously) relate to autonomic and cardiovascular functioning because of concurrent changes in the activity of ventromedial and other visceromotor cortices, which do project to subcortical areas and circuitries proximally involved in autonomic and cardiovascular control (cf., O'Connor et al., 2007).
Insula
Broadly stated, the insula expresses efferent and afferent connections that are comparable to those of the anterior cingulate and other regions of the orbital and medial prefrontal cortex, including connections with the amygdala, hypothalamus, thalamus, PAG, pons, nucleus tractus solitarius (NTS), and medullary and brainstem areas that control pre-autonomic nuclei innervating peripheral target organs (e.g., the heart and vasculature) (Augustine, 1996; Cechetto, 1994; Öngür and Price, 2000; Verberne and Owens, 1998). Further, multi-synaptic afferent relays from all peripheral target organs project to the insula along a caudal-to-rostral extent. These afferent projections are routed via the NTS, parabrachial pontine nuclei, ventral posterior and mediodorsal thalamic nuclei, and the lateral hypothalamic area, thus providing the insula with an ascending or ‘viscerotopic’ map of the body (Craig, 2003, 2005). Such a map has been posited to support the integration of interoceptive information with the appraisal of emotion-related stimuli and contextually-adaptive behavioral and autonomic responses (Craig, 2005; Critchley, 2005; Paulus and Stein, 2006). Similar to areas of the cingulate, the insula is engaged by behavioral challenges that elicit aversive errors (Klein et al., 2007) and by negative emotional stimuli and associated behavioral states (Feldman-Barrett and Wager, 2006; Phan et al., 2002; Taylor et al., 2003).
Longstanding lesion, stimulation, neuroanatomical tracing, and in vivo brain imaging evidence derived from humans and animals strongly implicates the insula in autonomic and cardiovascular regulation, particularly via the efferent and modulatory afferent sympathetic, parasympathetic, and baroreflex pathways described below (Allen and Cechetto, 1992, 1993; Allen et al., 1991; Cechetto, 1994; Cechetto and Chen, 1990; Oppenheimer, 1993; Ruggiero et al., 1987; Verberne and Owens, 1998; Yasui et al., 1991). Moreover, clinical evidence from patient populations has revealed that ischemic strokes selectively involving the insula elevate risk for cardiac arrhythmias (Cheung and Hachinski, 2000; Colivicchi et al., 2004, 2005), directly implicating the insula in the expression of clinically relevant late-stage CHD endpoints. Across prior human and animal studies, there is also some evidence to suggest that the insular regulation of autonomic and cardiovascular function may be lateralized, with the left insula being more involved in regulating parasympathetic cardiovascular control and in mediating depressor responses and the right insula in sympathetic cardiovascular control and in mediating pressor responses (Craig, 2005; Kimmerly et al., 2005; Oppenheimer et al., 1992; Oppenheimer et al., 1996). However, not all human neuroimaging evidence on stressor-evoked blood pressure reactivity is consistent with this generalization. For example, bilateral (Gianaros et al., 2005a; Gianaros et al., 2008) and left unilateral (Gianaros et al., 2007) insular activation has been associated with blood pressure reactivity evoked by a Stroop color-word interference stressor. Also, there is recent evidence that heightened levels of resting neural activity in the right insula prospectively predict subsequently greater stressor-evoked blood pressure reactions across individuals, possibly reflecting an individual's “preparedness” to exhibit heightened sympathetically-mediated pressor reactivity (Gianaros et al., 2009). In view of this evidence, the existing neuroimaging literature hinders precise predictions regarding a potential lateralized insular regulation of stressor-evoked blood pressure reactivity per se. Nonetheless, it is clear from both human and animal work that the insula is involved in regulating stressor-evoked blood pressure changes via efferent and afferent signaling with networked cortical and subcortical areas important for autonomic and cardiovascular control (cf., Harper et al., 1998; Harper et al., 2000; Henderson et al., 2002; King et al., 1999; Lamb et al., 2007).
Amygdala
As a complex cell assembly, a critical function of the amygdala in stressor-related processing involves the assignment of behavioral salience or relevance to environmental events (Davis and Whalen, 2001; LeDoux, 2003; Sah et al., 2003b; Zald, 2003). The amygdala supports such processing by integrating multimodal sensory inputs from distributed cortical, thalamic, and brainstem afferent relays. More precisely, sensory input is relayed through thalamic and cortical-thalamic pathways to the basolateral area via the lateral nucleus, basolateral nucleus, and accessory basal nucleus (LeDoux, 2003; Sah et al., 2003a, b). From the basolateral nucleus, behaviorally-relevant sensory signals are relayed to the central nucleus. As a primary output nucleus, the central nucleus relays commands for adaptive changes in behavior and supporting physiological adjustments via the stria terminalis to lateral and paraventricular hypothalamic nuclei and to periaqueductal, medullary, and pre-autonomic nuclei. Importantly, the central nucleus is also networked with cortical areas that are involved in stressor-related processing and autonomic-cardiovascular control, specifically areas of the pACC, dACC, and insula (Amaral and Price, 1984; McDonald, 1998; Morecraft et al., 2007; Price, 2003). Hence, the amygdala is broadly thought to be instrumental for interrelating cortical processes supporting the coordination of stressor-evoked changes in behavior and cardiovascular reactivity (Berntson et al., 1998; Dampney, 1994; Saper, 2002; Smith and DeVito, 1984; Smith et al., 1984; Westerhaus and Loewy, 2001). Also noteworthy in the context of CHD risk, central nucleus lesions blunt exaggerated stressor-evoked blood pressure reactions in rats genetically prone to hypertension (Galeno et al., 1984; Sanders et al., 1994) and prevent the development of hypertension induced by chronic stress (Fukumori et al., 2004) and fear conditioning (Baklavadzhyan et al., 2000).
A specific pathway by which the amygdala can regulate blood pressure reactivity is via its influence over the baroreflex (Berntson et al., 1998; Dampney, 1994; Saha, 2005). The baroreflex is a negative-feedback control mechanism that constrains arterial pressure around a regulatory set point by modulating efferent autonomic outflow (for review, see Eckberg, 1992). Specifically, the baroreflex controls beat-by-beat changes in blood pressure by adjusting parasympathetic and sympathetic control over heart rate, cardiac output, and vascular resistance. As a negative-feedback loop, the baroreflex relies on afferent projections from stretch-sensitive cardiopulmonary mechanoreceptors (baroreceptors) and chemoreceptors that signal changes in blood pressure to the NTS (see Figure 1). Afferent activation of the NTS in turn activates vagal nuclei in the medulla and, via signaling with the caudal ventrolateral medulla, inhibits pre-sympathetic nuclei in the rostroventrolateral medulla and IML column. In effect, these dynamic changes in autonomic control adjust heart rate, cardiac output, and vascular resistance to maintain blood pressure variations within a homeostatic range to match ongoing metabolic demands (Dampney, 1994; Guyenet, 2006).
Figure 1.

Conceptual diagram summarizing selected brain systems wherein the processing of stressor-related information may relate to the expression of stressor-evoked blood pressure reactivity. Although the model is anatomically incomplete, key areas implicated by animal and human evidence are included. PVN, paraventricular nucleus; LHA, lateral hypothalamic area; NTS, nucleus tractus solitarius; DVN, dorsal vagal nucleus; NA, nucleus ambiguous; CVLM, caudal ventrolateral medulla; RVL, rostral ventrolateral medullary; IML, intermediolateral cell column; HR, heart rate; BRS, baroreflex sensitivity; CO, cardiac output; TPR, total peripheral resistance. Blocked endpoints denote inhibitory influences; arrowed endpoints denote excitatory influences. Broken gray lines correspond to ascending (afferent) visceral input, particularly from peripheral baroreceptors. This model was derived explicitly from the work of Berntson et al. (1998), Dampney (1994), Saha (2005), Saper (2002) and Westerhaus and Loewy (2001).
As illustrated in the schematic model depicted in Figure 1, the amygdala can gate or suppress the sensitivity of the baroreflex via projections that inhibit the NTS and that activate the rostroventrolateral medulla (Berntson et al., 1998; Dampney, 1994; Saha, 2005; Saper, 2002). These projections are routed partly through the hypothalamus, PAG, and pons—core components of the circuitry emphasized in the models of Lovallo and Critchley (Critchley, 2005; Lovallo, 2005). Important in the present context, these amygdala projections are paralleled by similar projections from areas within the cingulate and medial prefrontal cortex and insula, which can also gate the baroreflex on exposure to acute stressors, allowing blood pressure to exceed its regulatory set point via descending (efferent) central mechanisms (Berntson et al., 1998; Dampney, 1994; Saper, 2002). In view of this control circuitry, it has been hypothesized that the amygdala and networked corticolimbic areas could partly underlie individual differences in cardiovascular reactivity and related CHD risk by linking stressor-related processing with mechanisms such as baroreflex gating (Berntson et al., 1998). Indeed, the sensitivity of the baroreflex is suppressed (gated) by psychological stressors in humans (Reyes del Paso et al.; Steptoe and Sawada, 1989), and suppressed baroreflex sensitivity has been associated with the severity of preclinical atherosclerosis in otherwise healthy adults (e.g., Gianaros et al., 2002) and with prospective risk for clinical CHD events in epidemiological and patient samples (De Ferrari et al., 2007; La Rovere et al., 1998; La Rovere et al., 2008; Schwartz et al., 1992).
Putative role of ascending mechanisms in stressor-evoked blood pressure reactivity
In addition to the stressor-evoked efferent (or descending) corticolimbic control mechanisms and associated clinical observations reviewed above, it is important to consider the modulatory role of visceral afferent (or ascending) influences of baroreflex and related interoceptive information on the neural circuitry depicted in Figure 1. In particular, there is translational evidence consistent with early speculations by James (1884) that visceral afferent, including ascending autonomic and baroreceptor, activity can influence a range of centrally-mediated cognitive, emotional, and behavioral processes via delimited feedback mechanisms (Adam, 1998; Berntson et al., 2003; Cameron, 2002; Craig, 2003; Critchley, 2005; Dworkin, 1993). In rats, for instance, baroreceptor activation can decrease cortical arousal (Adam, 1998; Dworkin, 1993) and inhibit the processing of nociceptive stimuli (Dworkin et al., 1979). In humans, baroreceptor activation may similarly influence nociceptive processing (Edwards et al., 2002), particularly via corticolimbic and brainstem pathways (Gray et al., 2009b). Functionally, these cardiovascular and visceral feedback mechanisms have been implicated not only in adaptive stressor responding, but also risk for hypertension, a primary precursor of CHD (McCubbin, 1993; Rau and Brody, 1994; Zamir and Maixner, 1986). More broadly, ascending cardiovascular and visceral afferent information arising from both the sympathetic and parasympathetic autonomic branches may impact other central processes supporting stressor-related cognitive functions and behavioral responses, including amygdala-mediated attention, memory and arousal processes (Cahill and McGaugh, 1998; Kapp et al., 1992; McGaugh et al., 1996) and cortically-mediated decision-making processes important for guiding adaptive behaviors (Bechara et al., 1999).
The primary node in the brainstem that is instrumental for relaying ascending visceral afferent information, particularly baroreceptor information, to higher-level corticolimbic systems involved in blood pressure control is the NTS (Berntson et al., 2003; Dampney, 1994; Guyenet, 2006), as noted above (Figure 1). Specifically, afferent traffic originating from blood pressure changes that are detected by peripheral arterial baroreceptors in the carotid sinus and aortic arch is transmitted via the glossopharyngeal and vagal nerves, which terminate within the NTS. From the NTS, multi-synaptic projections are issued not only to medullary and pre-autonomic brainstem, midbrain, and hypothalamic cell groups, but also higher-level corticolimbic systems, particularly the amygdala, insula, and areas of the cingulate, medial and orbital prefrontal cortices (Allen and Cechetto, 1992, 1993; Allen et al., 1991; Berntson et al., 2003; Buchanan and Powell, 1993; Dampney, 1994; Dampney et al., 2002; Dampney et al., 2003; Verbene and Owens, 1998). Viewed as components of an integrated control circuitry relying on nested feedforward and feedback pathways, these higher-level corticolimbic systems may thus integrate and represent afferent visceral information regarding dynamic (e.g., stressor-evoked) blood pressure changes in the service of adaptive and dynamic cardiovascular regulation (Dampney, 1994; Dampney et al., 2002). For example, afferent traffic triggered by stressor-evoked rises in blood pressure may be relayed via the NTS for representation by higher-level corticolimbic areas. In turn, this stressor-evoked visceral afferent information may modulate ongoing corticolimbic activity in a positive or negative feedback manner, which could further modulate descending corticolimbic signaling with midbrain and brainstem circuits. This modulated signaling resulting from ascending visceral feedback could serve to adjust ongoing autonomic, neuroendocrine, and cardiovascular functioning—which, in aggregate, could impact the magnitude and even duration of stressor-evoked blood pressure changes. Thus, higher-level corticolimbic areas should not be viewed only in light of their putative descending influences over stressor-evoked blood pressure reactivity, but also in view of ascending processes and the possibility that visceral afferent traffic may serve to affect the magnitude and duration (e.g., recovery to baseline) of stressor-evoked blood pressure reactions through positive and negative feedback mechanisms. In this respect, one speculative possibility is that individuals with a dispositional tendency to show exaggerated or prolonged stressor-evoked blood pressure or other cardiovascular and autonomic reactions linked to CHD risk may also exhibit dysregulated patterns of feedback and feedforward connectivity between higher-level corticolimbic systems and lower-level midbrain and brainstem circuits, which may reflect impairments in efferent and visceral afferent regulatory mechanisms (Gianaros et al. 2008; see below). Notably, such dysregulated connectivity may correspond to CHD relevant impairments in what has been referred to recently as neurovisceral integration (Thayer and Lane, 2007, 2009).
In view of the frameworks and concepts reviewed above, we next review available human neuroimaging studies that have specifically correlated stressor-evoked blood pressure reactions with neural activation in the corticolimbic brain systems and circuitries emphasized herein. We note that the activation patterns reported in these correlational studies are likely to reflect integrated aspects of ascending and descending mechanisms underlying stressor-evoked blood pressure reactivity. Following this review, we close by underscoring open questions, methodological developments, and empirical studies that are needed to further develop a mechanistic understanding of the neural pathways that bind psychological stress, cardiovascular reactivity processes, and CHD risk.
Neuroimaging studies of stressor-evoked blood pressure reactivity and CHD risk
As reviewed above and illustrated in Figure 1, divisions of the cingulate, insula, and amygdala may be viewed as core—albeit not exclusive—components of network of corticolimbic systems involved in mediating stressor-evoked changes in cardiovascular activity, particularly changes in blood pressure. To our knowledge, however, there have been only four human neuroimaging studies published to date that offer direct, but provisional, support for this view (Critchley et al., 2000; Gianaros et al., 2005a; Gianaros et al., 2007; Gianaros et al., 2008; see Table 1). In these studies, stressor-evoked changes in blood pressure were directly correlated with concurrent changes in functional neural activity. Two studies reviewed first (Critchley et al., 2000; Gianaros et al., 2005a) tested for correlations between concurrent changes in stressor-evoked blood pressure and functional neural activity on a within-person basis. In other words, multiple (repeated) observations of blood pressure and functional neural activity were obtained over the course of a scanning session, correlated with one another on an individual basis, and then aggregated across individuals. The remaining two studies (Gianaros et al., 2007; Gianaros et al., 2008) tested for correlations between stressor-evoked blood pressure reactivity and functional neural activity on a between-person basis. In these studies, one composite (averaged) measure of blood pressure reactivity was correlated with one composite measure of neural activation across individuals. As yet, no neuroimaging studies to our knowledge have employed mediation or path analytic analyses to statistically and simultaneously model both within- and between-person variation in neural activity linked to stressor-evoked blood pressure reactivity, as has been done for stressor-evoked heart rate reactivity (Wager et al., 2009a; Wager et al., 2009b). Also, we note that many published studies have investigated changes in functional neural activity resulting from exposure to blood pressure challenges involving exercise, respiratory maneuvers, cold pressor stimuli, and sophisticated experimental manipulations, such as baroreceptor unloading (e.g., Harper et al., 1998; Harper et al., 2000; Henderson et al., 2002; Kimmerly et al., 2005; King et al., 1999; Lamb et al., 2007; Williamson et al., 1997; Wong et al., 2007). However, these studies did not specifically employ experimental manipulations that engaged the psychological stress processes noted earlier in this review and generally suspected in stress-related CHD risk. Critically, though, these other studies converge with the meta-analytic findings reviewed below to indicate that activity in areas of the cingulate and medial prefrontal cortices, insula, amygdala, and other networked cortical, midbrain, brainstem, and cerebellar areas is reliably linked to blood pressure and other cardiovascular responses to diverse behavioral and physiological challenges. Hence, it would appear at present that many of the core components of the central network for autonomic and cardiovascular control are commonly involved in mediating reactivity processes evoked by wide ranging environmental, behavioral, and physiological challenges via the efferent and afferent mechanisms noted above and detailed in prior translational work (Berntson et al., 1998, 2003; Cechetto, 1994; Dampney, 1994; Jänig, 2006; Saper, 2002; Verbene and Owens, 1998; Westerhaus and Loewy, 2001).
Table 1.
Brain imaging studies reporting foci where greater functional neural activation covaried with greater concurrent stressor-evoked blood pressure reactivity
| Authors | Reference | Imaging modality | Sample size (n) | Stressor tasks | Blood pressure measure | Imaging processing package | Coordinate space of foci | Number of foci reported | |
|---|---|---|---|---|---|---|---|---|---|
| 1 | Critchley et al. | J Physiol, 2000 | PET | 6 | Isometric exercise & Mental arithmetic | MAP | SPM | Talairach | 11 |
| 2 | Gianaros et al. | Psychophysiology, 2005 | fMRI | 20 | Stroop | MAP | SPM | MNI | 23 |
| 3 | Gianaros et al. | Hypertension, 2007 | fMRI | 46 | Stroop | SBP | SPM | MNI | 6 |
| 4 | Gianaros et al. | J Neurosci, 2008 | fMRI | 32 | Stroop | SBP | SPM | MNI | 8 |
Note. Foci from each study are plotted in Figure 2. These foci were pooled to generate activation likelihood estimates shown as clusters in Figure 3 and reported in Table 2 (see Supplementary Methods online). PET, Positron emission tomography; fMRI, functional magnetic resonance imaging; MAP, mean arterial pressure; SBP, systolic blood pressure; SPM, statistical parametric mapping, MNI, Montreal Neurological Institute.
Meta-analytic summary of neuroimaging studies of stressor-evoked blood pressure reactivity
For a descriptive and quantitative summary, we executed a meta-analysis of the four neuroimaging studies reviewed below (see Tables 1 and 2; Figures 2 and 3; and Supplementary Methods available online). For this meta-analysis, we used the method of activation likelihood estimation (Laird et al., 2005; Turkeltaub et al., 2002) to assess the cross-study concordance of brain areas in which greater stressor-evoked neural activation correlated with greater blood pressure reactivity. We emphasize that this descriptive summary is based on only four studies that differed in terms of the imaging modalities employed, the blood pressure measures obtained, the populations and sample sizes tested, and stressor tasks used to evoke reactivity. Further, this summary focuses only on patterns of neural activation, not deactivation, linked to stressor-evoked blood pressure reactivity. This is because deactivation patterns were not explored in three out of the four studies. We note that patterns of deactivation will be important to assess in future studies in this area, as these deactivation patterns may reflect what have been termed ‘disinhibition’ mechanisms involved in peripheral stress reactivity (Pruessner et al., 2007; Wager et al., 2009a; Wager et al., 2009b). Hence, the summary below should be viewed cautiously as an early abstraction of available neuroimaging results bearing on the neural regulation of stressor-evoked blood pressure reactivity. In aggregate, however, this summary does provide initial support for the notion that the corticolimbic areas emphasized above—namely, the cingulate, insula, and amygdala—may represent core, but clearly not exclusive, components of a broader neural circuitry mediating stressor-evoked blood pressure reactivity. More practically, this meta-analysis (specifically the coordinates provided in Table 2) could be used to aid empirically in targeting specific regions-of-interest in future studies of stressor-evoked blood pressure reactivity and related CHD risk.
Table 2.
Meta-analytic summary of brain regions where greater functional neural activation has covaried with greater stressor-evoked blood pressure reactivity
| Cluster number | Side | Region | BA | MNI (x, y, z) coordinates in mm | Volume (mm3) | Activation Likelihood Estimate (×10-3) | ||
|---|---|---|---|---|---|---|---|---|
| 1 | L | Anterior cingulate cortex | 32 | -8 | 26 | 24 | 120 | 3.86 |
| 2 | R | - | 32 | 11 | 29 | 20 | 504 | 4.17 |
| 3 | R | - | 24 | 13 | 10 | 39 | 112 | 3.77 |
| 4 | R | - | 32 | 18 | 26 | 35 | 96 | 3.77 |
| 5 | R | - | 32 | 13 | 44 | 0 | 88 | 3.85 |
| 6 | L/R | Posterior cingulate cortex | 23/31 | 0 | -36 | 29 | 424 | 4.04 |
| 7 | R | - | 30 | 30 | -73 | 15 | 96 | 3.70 |
| 8 | R | Insula | 13 | 36 | -32 | 16 | 856 | 5.11 |
| 9 | L | - | 44 | -48 | 3 | 15 | 704 | 4.96 |
| 10 | R | - | 21 | 40 | -11 | -20 | 96 | 3.82 |
| 11 | L | - | 47 | -24 | 15 | -19 | 96 | 3.74 |
| 12 | R | - | 6 | 54 | 4 | 10 | 96 | 3.84 |
| 13 | L | - | 38 | -48 | 13 | -12 | 88 | 3.84 |
| 14 | L | Amygdala | -27 | 0 | -15 | 104 | 3.72 | |
| 15 | R | - | 27 | 3 | -12 | 96 | 3.73 | |
| 16 | L | Medial prefrontal cortex | 10 | -9 | 63 | 6 | 104 | 3.69 |
| 17 | R | - | 10 | 21 | 58 | 4 | 448 | 4.07 |
| 18 | L | Lateral prefrontal cortex | 9 | -33 | 30 | 36 | 96 | 3.77 |
| 19 | L | - | 9 | -30 | 49 | 28 | 104 | 3.84 |
| 20 | R | Dorsal prefrontal cortex | 8 | 5 | 28 | 48 | 88 | 3.85 |
| 21 | L | Parietal cortex | 40 | -57 | -46 | 42 | 112 | 3.84 |
| 22 | R | Occipital cortex | 39 | 49 | -74 | 16 | 128 | 3.84 |
| 23 | L | Post-central gyrus | 3 | -31 | -28 | 52 | 128 | 3.79 |
| 24 | R | Cuneus | 17 | 6 | -80 | 18 | 120 | 3.84 |
| 25 | R | Lentiform area | 24 | 5 | 4 | 104 | 3.88 | |
| 26 | R | Caudate | 12 | 10 | 14 | 104 | 3.86 | |
| 27 | R | Thalamus | 16 | -24 | 16 | 128 | 3.84 | |
| 28 | L | Cerebellum | -18 | -66 | -9 | 544 | 4.10 | |
| 29 | R | - | 18 | -41 | -30 | 120 | 3.83 | |
Note. Activation likelihood estimates were thresholded at a corrected false discovery rate of q = 0.05 (or p-value = 0.0038) and a combined cluster size of 80mm3 (see Supplementary Methods online). BA, Brodmann Area; MNI, Montreal Neurological Institute.
Figure 2.
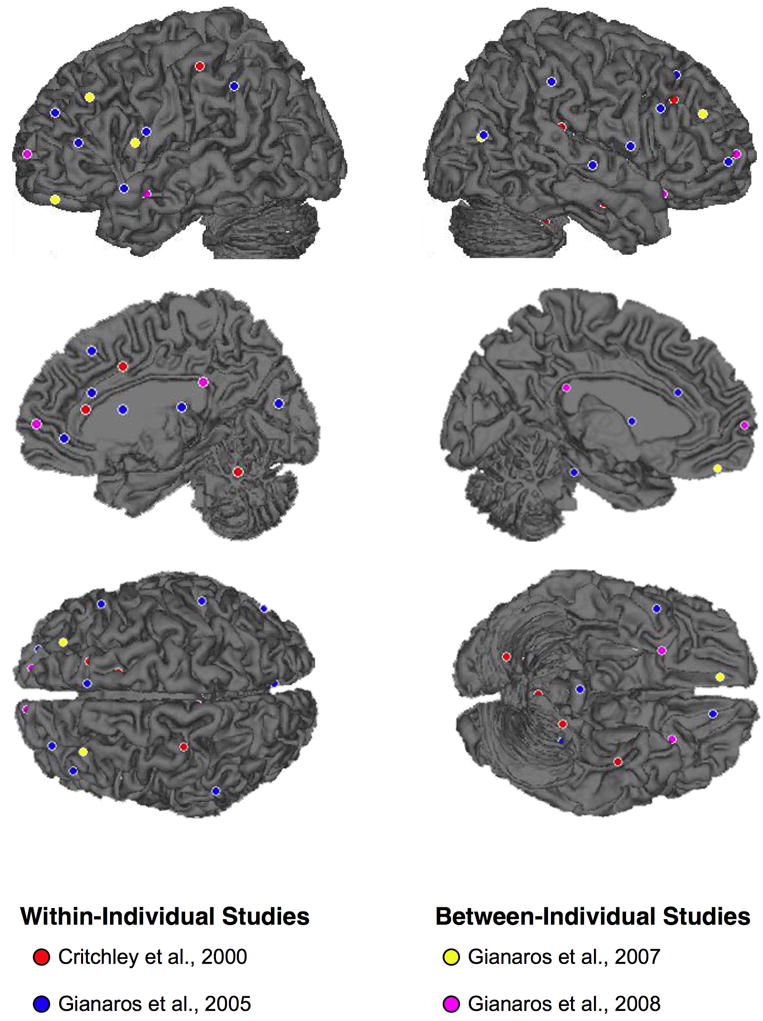
Lateral and medial surface projections of foci where a significant positive correlation between stressor-evoked neural activation and blood pressure reactivity has been reported in neuroimaging studies reviewed in the text (see Tables 1-2). These foci (47 in total, not all visible in this figure) were submitted to a meta-analysis using the activation likelihood estimation method.
Figure 3.

Meta-analytic activation likelihood estimation map thresholded at a corrected false discovery rate of q = 0.05 (or p-value of 0.0038). This map illustrates clusters wherein greater neural activation correlated with greater stressor-evoked blood pressure reactivity in the neuroimaging studies listed in Tables 1-2. Cluster numbering system corresponds to Table 2 labels.
Within-individual neuroimaging studies of stressor-evoked blood pressure reactivity
In a positron emission tomography study of six men (mean age, 35 years), Critchley and colleagues (2000) tested whether changes in mean arterial pressure correlated with concurrent changes in functional neural activation evoked by two stressors, a frustrating mental arithmetic task and an isometric handgrip task. An explicit aim of this study was to isolate neural activity patterns that covaried directly with stressor-evoked blood pressure changes during the performance of both stressor tasks. Results showed that increased mean arterial pressure evoked by the stressors correlated on a within-individual basis with increased cerebral blood flow to the perigenual and mid-anterior areas of the cingulate cortex, the orbitofrontal cortex, postcentral gyrus, insula, and cerebellum (see Table 2 in Critchley et al., 2000)—providing the first human neuroimaging evidence in support of the view that these brain regions may initiate or represent increases in blood pressure to behavioral stressors.
These findings were replicated and extended in a subsequent functional magnetic resonance imaging (fMRI) study of twenty older adults (9 men, 11 women; mean age, 64 years) (Gianaros et al., 2005a). In this study, participants completed a performance-titrated Stroop color-word interference task, which had been adapted specifically from a Stroop task used in previous epidemiological studies of stressor-evoked blood pressure reactivity and CHD risk (Debski et al., 1991; Jennings et al., 2004; Kamarck et al., 1997; Kamarck et al., 1992). Results showed that increased mean arterial pressure evoked by the Stroop stressor correlated on a within-individual basis with greater activation in the perigenual and mid-anterior cingulate cortex (areas 24 and 32), insula, medial and lateral prefrontal cortex, supplementary motor area, and regions of the temporal, inferior parietal, and occipital cortex. Subcortical regions in which greater activation correlated with increased mean arterial pressure included the basal ganglia, lentiform area bordering the extended amygdala and caudate, thalamus, cerebellum, and PAG (see Table 2 of Gianaros et al., 2005).
Collectively, the results of these two studies provided an initial characterization of the brain systems putatively involved in regulating blood pressure reactions to behavioral stressors in humans. Interestingly, both studies provided evidence for an association between stressor-evoked blood pressure reactivity and activation of areas in the cingulate cortex and insula. Further, as motivated by these two particular within-individual studies, the two studies reviewed next directly examined the covariation between individual differences in stressor-evoked blood pressure reactivity and concurrent functional neural activity. From the perspective emphasized in this review, such studies are needed to directly examine the putative neural processes linking stressor-evoked blood pressure reactivity with relative CHD risk across individuals.
Between-individual neuroimaging studies of stressor-evoked blood pressure reactivity
In an fMRI study of individual differences in stressor-evoked blood pressure reactivity, 46 postmenopausal women (mean age, 68 years) performed a similar version of the Stroop color-word interference task described above (Gianaros et al., 2007). Across individuals, a larger task-induced rise in systolic and diastolic blood pressure covaried with heightened activation of the pACC, extending into area 10 of the medial prefrontal cortex. Further, a larger stressor-evoked increase in systolic and diastolic pressure covaried with heightened activation of the insula, pCC (extending from area 31 into the precuneus), the lateral prefrontal cortex, and cerebellum (see Table 2 in Gianaros et al., 2007). Moreover, blood pressure reactions evoked by the Stroop stressor in a separate laboratory testing session correlated with reactions evoked in the fMRI testing session, providing support for the notion that the individual differences in stressor-evoked blood pressure reactivity evoked by this particular stressor task were stable (reliably elicited) across testing settings. In this study, however, no associations were observed between individual differences in stressor-evoked blood pressure reactivity and activation in other corticolimbic regions thought to be involved in cardiovascular regulation, particularly the amygdala, midbrain and brainstem areas. These null findings, however, were attributed to the scanning sequence and field of view coverage used in this particular study. Hence, in a follow-up neuroimaging study described next, this study was extended to show that individual differences in stressor-evoked blood pressure reactivity covaried across individuals not only with amygdala activation, but also patterns of functional connectivity between the amygdala, pACC and pons.
Specifically, in a study of 32 young adults (12 men, mean age 20 years), a region-of-interest approach was implemented to test directly whether individual differences in stressor-evoked blood pressure reactivity covaried with amygdala activation, as well as functional connectivity with corticolimbic and subcortical areas implicated in stressor processing and cardiovascular regulation (Gianaros et al., 2008). Here, mean arterial pressure and concurrent fMRI signal changes were monitored during the performance of the Stroop color-word stressor task used in the two studies described above (Gianaros et al., 2005a; Gianaros et al., 2007). Individuals who exhibited greater stressor-evoked blood pressure reactivity showed greater stressor-evoked pACC, pCC, insula, and amygdala activation and a stronger positive functional connectivity between the amygdala and pACC and between the amygdala and pons. Collectively, these findings supported the notion that individual differences in stressor-evoked blood pressure reactivity are correlated not only with patterns of co-activation in corticolimbic systems, but also the functional interactions between these systems. As noted above, these functional interactions may correspond to efferent feedforward and afferent feedback mechanisms involved in central autonomic and cardiovascular control.
In particular, one set of functional connectivity findings from this study indicated that the pons may represent a relay area that specifically links individual differences in stressor-evoked amygdala activity with the peripheral expression of blood pressure reactions. This notion is supported by neuroanatomical evidence that the amygdala expresses reciprocal connections with pontine cell groups critical for cardiovascular control (Dampney, 1994; Hopkins and Holstege, 1978; Miller et al., 1991). Further, the pons is known to relay afferent cardiovascular information to higher levels of the neuroaxis, including the amygdala (Dampney, 1994). In view of this circuitry, one possibility is that individual differences in blood pressure reactivity could result from differential signaling between the amygdala and pre-autonomic areas such as the pons. Hence, stronger efferent amygdala-pre-autonomic signaling could reflect stronger descending commands for rises in blood pressure during acute stressful experiences. In parallel, stronger afferent pre-autonomic-amygdala signaling could reflect stronger ascending negative-feedback to the amygdala that functionally curtails excessive blood pressure rises. The latter possibility may account for the finding reported by Gianaros et al (2008) that individuals showing more negative amygdala-pons connectivity showed less (smaller magnitude) blood pressure reactivity. A critical limitation hindering support for these inferences, however, was that the connectivity measures obtained in that study did not distinguish efferent from afferent signaling patterns linked to blood pressure changes, which were measured on a minute-by-minute basis. Hence, and important direction for future research will be to employ effective connectivity procedures (Friston, 1994) and continuous (beat-by-beat) blood pressure monitoring methods (Gray et al., 2009b) to parse transient efferent from afferent neural signaling patterns in the amygdala and pre-autonomic areas putatively involved in cardiovascular control.
Also reported by Gianaros et al. (2008) was that greater mean arterial pressure reactivity varied with greater pACC activation and with more positive amygdala-pACC functional connectivity across individuals. As reviewed above, the amygdala and areas of the anterior cingulate cortex, such as the pACC, are densely networked and considered as components of a corticolimbic circuitry that orchestrates adaptive and integrated neurobehavioral and visceromotor stress responses (cf., Bush et al., 2000; Devinsky et al., 1995; Paus, 2001; Vogt, 2005). Furthermore, evidence from fear-conditioning and emotion-regulation literatures suggests that subdivisions of the anterior cingulate cortex, particularly in the perigenual area, may regulate amygdala activity (Etkin et al., 2006; Ochsner and Gross, 2005; Quirk and Beer, 2006). Hence, if regulatory amygdala-pACC signaling modulates visceromotor reactivity to behaviorally-salient stimuli, then the differential coupling between these areas may partly influence individual differences in stressor-evoked blood pressure reactivity. Consistent with this notion, individuals who expressed more negative amygdala-pACC connectivity expressed less stressor-evoked blood pressure reactivity. However, as for the findings linking amygdala-pons connectivity with stressor-evoked blood pressure reactivity, these interpretations should be taken as provisional until future studies replicate both sets of observations and explicate the specific nature of amygdala-pACC and amygdala-pons signaling described above.
Here, it is noteworthy that pursuing such questions regarding corticolimbic connectivity in addition to activation patterns per se may prove useful in understanding the pathophysiological processes underling CHD risk. For example, it was recently demonstrated that individuals who exhibit heightened amygdala activation to threatening emotional stimuli (i.e., facial expressions of fear and anger) also show increased intima-media vessel wall thickness in the carotid arteries, which is a surrogate measure of preclinical atherosclerosis that predicts clinical endpoints of CHD (Gianaros et al., in press). Moreover, in that study, it was demonstrated that increased intima-media thickness was associated with a pattern of functional connectivity between the pACC and amygdala which paralleled the connectivity patterns observed in association with exaggerated blood pressure reactivity described above. Hence, those individuals showing increased intima-media thickness exhibited a stronger and more positive connectivity between the pACC and amygdala. In general, these lines of evidence thus suggest that alterations in corticolimbic functionality, including alterations in co-activation and connectivity, may impact atherosclerotic disease processes and consequent CHD risk via stress-related processes such as blood pressure reactivity.
Summary and future directions
Only recently have neuroimaging methods and theoretical frameworks been directed at understanding the neurobiological or ‘brain-body’ pathways linking the central processing of psychological stressors to the regulation of blood pressure and other cardiovascular reactions implicated in CHD risk (Critchley, 2005; Lane et al., 2009a; Lane et al., 2009b; Lovallo, 2005; Soufer et al., 1998). The early neuroimaging evidence and models summarized here suggest that several corticolimbic brain areas, including areas of the cingulate cortex, insula, and amygdala, are involved in regulating at least one stress-related risk factor for CHD: stressor-evoked blood pressure reactivity. Nevertheless, it is important to re-emphasize that CHD is a slowly developing and chronic illness with many interacting genetic, developmental, biobehavioral, social and environmental risk factors that are still being defined. Moreover, stress-related factors relevant to CHD risk can be conceptualized and measured at multiple levels of analysis, over multiple time scales, and throughout multiple stages of life in different demographic, ethnic, and socioeconomically stratified populations of individuals who vary in their risk for CHD. In this regard, one salient concluding point is that future neuroimaging studies in this area should employ prospective study designs that (1) account for the interactive neurobiological effects of multiple CHD risk factors in large and representative populations and that (2) include assessments of social and developmental factors related to the progression and expression of CHD endpoints across the lifespan. Also important for future neuroimaging studies in this area will be to address the following fundamental, but still open questions:
How are the implicit and explicit appraisal and coping processes that presumably underlie individual differences in stressor-evoked cardiovascular reactivity instantiated in corticolimbic brain systems involved in central cardiovascular regulation?
How do alterations in the functionality and even morphology of corticolimbic systems and circuitries interact with specific genetic, developmental, biobehavioral, social and environmental factors to influence CHD risk?
How might markers of corticolimbic functionality (e.g., stressor-evoked activation and connectivity patterns) and morphology (e.g., volume and structural connectivity) aid in understanding CHD etiology and enhancing CHD risk stratification?
How are efferent vs. afferent neural activity patterns in corticolimbic brain areas and subcortical circuitries differentially associated with blood pressure and other cardiovascular and autonomic changes evoked by behavioral stressors, particularly within the broader context of CHD risk?
Are similar brain systems involved in the expression of (a) other cardiovascular and peripheral physiological reactivity parameters and (b) clinical CHD events, including ischemia and reversible left ventricular dysfunction precipitated by emotional stress (Wittstein et al., 2005)?
Could CHD risk stratification and prediction be aided by the application of (a) cardiovascular and autonomic monitoring methods that allow for fine-grained temporal physiological monitoring (Gray et al., 2009b) and (b) advanced statistical methods that allow for the mediational modeling of both within-person and between-person variance in stressor-evoked neural activation patterns linked to reactivity and recovery processes (Wager et al., 2009a; Wager et al., 2009b)?
At minimum, addressing these interdependent questions with the cross-disciplinary integration of behavioral medicine and health neuroscience methods holds promise for characterizing the neurobiological mechanisms by which stressful experiences affect CHD risk.
Supplementary Material
Acknowledgments
Support for the preparation of this manuscript was provided by National Institutes of Health (NIH) Grants K01 MH070616 and R01 HL 089850 and by The Pittsburgh Mind-Body Center (NIH grants HL076852/076858; http://www.pghmbc.org). We thank Natasha Tokowicz, J. Richard Jennings, and Ikechukwu Onywuenyi for their comments on a draft of this manuscript.
Footnotes
Another common cardiovascular parameter measured in clinical and epidemiological studies of CHD risk is left ventricular ejection fraction (LVEF), or the fraction of the end-diastolic volume that is ejected from the left ventricle with each heartbeat. Most laboratory studies of stressor-evoked cardiovascular reactivity, however, do not typically assess LVEF because it is not generally justified for non-patient samples and because it is estimated by methodologies requiring trained medical technicians (e.g., echocardiography, cardiac MRI, computed axial tomography imaging, ventriculography, single photon emission computed tomography, and other methodologies for the assessment of myocardial performance).
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Adam G. Visceral perception understanding internal cognition. Springer-Verlag; New York: 1998. [Google Scholar]
- Alderman MH, Ooi WL, Madhavan S, Cohen H. Blood pressure reactivity predicts myocardial infarction among treated hypertensive patients. J Clin Epidemiol. 1990;43:859–866. doi: 10.1016/0895-4356(90)90069-2. [DOI] [PubMed] [Google Scholar]
- Allen GV, Cechetto DF. Functional and anatomical organization of cardiovascular pressor and depressor sites in the lateral hypothalamic area: I. Descending projections. J Comp Neurol. 1992;315:313–332. doi: 10.1002/cne.903150307. [DOI] [PubMed] [Google Scholar]
- Allen GV, Cechetto DF. Functional and anatomical organization of cardiovascular pressor and depressor sites in the lateral hypothalamic area. II. Ascending projections. J Comp Neurol. 1993;330:421–438. doi: 10.1002/cne.903300310. [DOI] [PubMed] [Google Scholar]
- Allen GV, Saper CB, Hurley KM, Cechetto DF. Organization of visceral and limbic connections in the insular cortex of the rat. J Comp Neurol. 1991;311:1–16. doi: 10.1002/cne.903110102. [DOI] [PubMed] [Google Scholar]
- Allen MT, Sherwood A, Obrist PA, Crowell MD, Grange LA. Stability of cardiovascular reactivity to laboratory stressors: a 2 1/2 yr follow-up. J Psychosom Res. 1987;31:639–645. doi: 10.1016/0022-3999(87)90043-2. [DOI] [PubMed] [Google Scholar]
- Amaral DG, Price JL. Amygdalo-cortical projections in the monkey (Macaca fascicularis) J Comp Neurol. 1984;230:465–496. doi: 10.1002/cne.902300402. [DOI] [PubMed] [Google Scholar]
- An X, Bandler R, Ongur D, Price J. Prefrontal cortical projections to longitudinal columns in the midbrain periaqueductal gray in macaque monkeys. J Comp Neurol. 1998;410:455–479. [PubMed] [Google Scholar]
- Anderson KM, Wilson PW, Odell PM, Kannel WB. An updated coronary risk profile. A statement for health professionals. Circulation. 1991;83:356–362. doi: 10.1161/01.cir.83.1.356. [DOI] [PubMed] [Google Scholar]
- Augustine JR. Circuitry and functional aspects of the insular lobe in primates including humans. Brain Res Rev. 1996;22:229–244. doi: 10.1016/s0165-0173(96)00011-2. [DOI] [PubMed] [Google Scholar]
- Barbas H. Connections underlying the synthesis of cognition, memory, and emotion in primate prefrontal cortices. Brain Res Bull. 2000;52:319–330. doi: 10.1016/s0361-9230(99)00245-2. [DOI] [PubMed] [Google Scholar]
- Barbas H, Saha S, Rempel-Clower N, Ghashghaei T. Serial pathways from primate prefrontal cortex to autonomic areas may influence emotional expression. BMC Neurosci. 2003;4:25. doi: 10.1186/1471-2202-4-25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bard P. A diencephalic mechanism for the expression of rage with special reference to the sympathetic nervous system. Am J Physiol. 1928;84:490–515. [Google Scholar]
- Beaglehole R, Magnus P. The search for new risk factors for coronary heart disease: occupational therapy for epidemiologists? Int J Epidemiol. 2002;31:1117–1122. 1134–1115. doi: 10.1093/ije/31.6.1117. author reply. [DOI] [PubMed] [Google Scholar]
- Bechara A, Damasio H, Damasio AR, Lee GP. Different contributions of the human amygdala and ventromedial prefrontal cortex to decision-making. J Neurosci. 1999;19:5473–5481. doi: 10.1523/JNEUROSCI.19-13-05473.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bedi M, Varshney VP, Babbar R. Role of cardiovascular reactivity to mental stress in predicting future hypertension. Clin Exp Hypertens. 2000;22:1–22. doi: 10.1081/ceh-100100058. [DOI] [PubMed] [Google Scholar]
- Beere P, Glagov S, Zaains C. Retarding effect of lowered heart rate on coronary atherosclerosis. Science. 1984;226:180–182. doi: 10.1126/science.6484569. [DOI] [PubMed] [Google Scholar]
- Berntson GG, Sarter M, Cacioppo JT. Anxiety and cardiovascular reactivity: The basal forebrain cholinergic link. Behav Brain Res. 1998;94:225–248. doi: 10.1016/s0166-4328(98)00041-2. [DOI] [PubMed] [Google Scholar]
- Berntson GG, Sarter M, Cacioppo JT. Ascending visceral regulation of cortical affective information processing. Eur J Neurosci. 2003;18:2103–2109. doi: 10.1046/j.1460-9568.2003.02967.x. [DOI] [PubMed] [Google Scholar]
- Berntson GG, Uchino BN, Cacioppo JT. Origins of baseline variance and the Law of Initial Values. Psychophysiology. 1994;31:204–210. doi: 10.1111/j.1469-8986.1994.tb01042.x. [DOI] [PubMed] [Google Scholar]
- Bhattacharyya MR, Steptoe A. Emotional triggers of acute coronary syndromes: strength of evidence, biological processes, and clinical implications. Prog Cardiovasc Dis. 2007;49:353–365. doi: 10.1016/j.pcad.2006.11.002. [DOI] [PubMed] [Google Scholar]
- Botvinick MM, Braver TS, Barch DM, Carter CS, Cohen JD. Conflict monitoring and cognitive control. Psychol Rev. 2001;108:624–652. doi: 10.1037/0033-295x.108.3.624. [DOI] [PubMed] [Google Scholar]
- Boyce WT, Ellis BJ. Biological sensitivity to context: I. An evolutionary-developmental theory of the origins and functions of stress reactivity. Dev Psychopathol. 2005;17:271–301. doi: 10.1017/s0954579405050145. [DOI] [PubMed] [Google Scholar]
- Brotman DJ, Golden SH, Wittstein IS. The cardiovascular toll of stress. Lancet. 2007;370:1089–1100. doi: 10.1016/S0140-6736(07)61305-1. [DOI] [PubMed] [Google Scholar]
- Buchanan SL, Powell DA. Cingulothalamic and prefrontal control of autonomic function. In: Vogt BA, Gabriel M, editors. Neurobiology of the cingulate cortex and limbic thalamus: a comprehensive handbook. Birkhauser; Boston: 1993. pp. 381–414. [Google Scholar]
- Buckner RL, Andrews-Hanna JR, Schacter DL. The brain's default network: anatomy, function, and relevance to disease. Ann N Y Acad Sci. 2008;1124:1–38. doi: 10.1196/annals.1440.011. [DOI] [PubMed] [Google Scholar]
- Buja LM, Willerson JT. Clinicopathologic correlates of acute ischemic heart disease syndromes. Am J Cardiol. 1981;47:343–356. doi: 10.1016/0002-9149(81)90407-0. [DOI] [PubMed] [Google Scholar]
- Bush G, Luu P, Posner MI. Cognitive and emotional influences in anterior cingulate cortex. Trends Cog Sci. 2000;4:215–222. doi: 10.1016/s1364-6613(00)01483-2. [DOI] [PubMed] [Google Scholar]
- Cahill L, McGaugh JL. Mechanisms of emotional arousal and lasting declarative memory. Trends Neurosci. 1998;21:294–299. doi: 10.1016/s0166-2236(97)01214-9. [DOI] [PubMed] [Google Scholar]
- Cameron OG. Visceral sensory neuroscience: interoception. Oxford University Press; Oxford, UK: 2002. [Google Scholar]
- Cannon WB. The mechanism of emotional disturbance of bodily functions. N Engl J Med. 1928;198:877–884. [Google Scholar]
- Cannon WB. The wisdome of the body. WW Norton & Company; New York: 1932. [Google Scholar]
- Cechetto DF. Identification of a cortical site for stress-induced cardiovascular dysfunction. Integr Physiol Behav Sci. 1994;29:362–373. doi: 10.1007/BF02691356. [DOI] [PubMed] [Google Scholar]
- Cechetto DF, Chen SJ. Subcortical sites mediating sympathetic responses from insular cortex in rats. Am J Physiol. 1990;258:R245–255. doi: 10.1152/ajpregu.1990.258.1.R245. [DOI] [PubMed] [Google Scholar]
- Cheung RT, Hachinski V. The insula and cerebrogenic sudden death. Arch Neurol. 2000;57:1685–1688. doi: 10.1001/archneur.57.12.1685. [DOI] [PubMed] [Google Scholar]
- Chiba T, Kayahara T, Nakano K. Efferent projections of infralimbic and prelimbic areas of the medial prefrontal cortex in the Japanese monkey, Macaca fuscata. Brain Res. 2001;888:83–101. doi: 10.1016/s0006-8993(00)03013-4. [DOI] [PubMed] [Google Scholar]
- Cohen S, Hamrick N. Stable individual differences in physiological response to stressors: implications for stress-elicited changes in immune related health. Brain Behav Immun. 2003;17:407–414. doi: 10.1016/s0889-1591(03)00110-7. [DOI] [PubMed] [Google Scholar]
- Cohen S, Janicki-Deverts D, Miller GE. Psychological stress and disease. J A M A. 2007;298:1685–1687. doi: 10.1001/jama.298.14.1685. [DOI] [PubMed] [Google Scholar]
- Colivicchi F, Bassi A, Santini M, Caltagirone C. Cardiac autonomic derangement and arrhythmias in right-sided stroke with insular involvement. Stroke. 2004;35:2094–2098. doi: 10.1161/01.STR.0000138452.81003.4c. [DOI] [PubMed] [Google Scholar]
- Colivicchi F, Bassi A, Santini M, Caltagirone C. Prognostic implications of right-sided insular damage, cardiac autonomic derangement, and arrhythmias after acute ischemic stroke. Stroke. 2005;36:1710–1715. doi: 10.1161/01.STR.0000173400.19346.bd. [DOI] [PubMed] [Google Scholar]
- Craig AD. Interoception: the sense of the physiological condition of the body. Curr Opin Neurobiol. 2003;13:500–505. doi: 10.1016/s0959-4388(03)00090-4. [DOI] [PubMed] [Google Scholar]
- Craig AD. Forebrain emotional asymmetry: a neuroanatomical basis? Trends Cogn Sci. 2005;9:566–571. doi: 10.1016/j.tics.2005.10.005. [DOI] [PubMed] [Google Scholar]
- Critchley HD. Neural mechanisms of autonomic, affective, and cognitive integration. J Comp Neurol. 2005;493:154–166. doi: 10.1002/cne.20749. [DOI] [PubMed] [Google Scholar]
- Critchley HD, Corfield DR, Chandler MP, Mathias CJ, Dolan RJ. Cerebral correlates of autonomic cardiovascular arousal: A functional neuroimaging investigation in humans. J Physiol. 2000;523:259–270. doi: 10.1111/j.1469-7793.2000.t01-1-00259.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Critchley HD, Mathias CJ, Josephs O, O'Doherty J, Zanini S, Dewar BK, Cipolotti L, Shallice T, Dolan RJ. Human cingulate cortex and autonomic control: converging neuroimaging and clinical evidence. Brain. 2003;126:2139–2152. doi: 10.1093/brain/awg216. [DOI] [PubMed] [Google Scholar]
- Critchley HD, Melmed RN, Featherstone E, Mathias CJ, Dolan RJ. Brain activity during biofeedback relaxation: a functional neuroimaging investigation. Brain. 2001;124:1003–1012. doi: 10.1093/brain/124.5.1003. [DOI] [PubMed] [Google Scholar]
- Critchley HD, Melmed RN, Featherstone E, Mathias CJ, Dolan RJ. Volitional control of autonomic arousal: a functional magnetic resonance study. Neuroimage. 2002;16:909–919. doi: 10.1006/nimg.2002.1147. [DOI] [PubMed] [Google Scholar]
- Dampney RA. Functional organization of central pathways regulating the cardiovascular system. Physiol Rev. 1994;74:323–364. doi: 10.1152/physrev.1994.74.2.323. [DOI] [PubMed] [Google Scholar]
- Dampney RA, Coleman MJ, Fontes MA, Hirooka Y, Horiuchi J, Li YW, Polson JW, Potts PD, Tagawa T. Central mechanisms underlying short- and long-term regulation of the cardiovascular system. Clin Exp Pharmacol Physiol. 2002;29:261–268. doi: 10.1046/j.1440-1681.2002.03640.x. [DOI] [PubMed] [Google Scholar]
- Dampney RA, Polson JW, Potts PD, Hirooka Y, Horiuchi J. Functional organization of brain pathways subserving the baroreceptor reflex: studies in conscious animals using immediate early gene expression. Cell Mol Neurobiol. 2003;23:597–616. doi: 10.1023/A:1025080314925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies MJ, Thomas AC. Plaque fissuring: the cause of acute myocardial infarction, sudden ischaemic death, and crescendo angina. Br Heart J. 1985;53:363–373. doi: 10.1136/hrt.53.4.363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis M, Whalen PJ. The amygdala: vigilance and emotion. Mol Psychiatry. 2001;6:13–34. doi: 10.1038/sj.mp.4000812. [DOI] [PubMed] [Google Scholar]
- De Ferrari GM, Sanzo A, Bertoletti A, Specchia G, Vanoli E, Schwartz PJ. Baroreflex sensitivity predicts long-term cardiovascular mortality after myocardial infarction even in patients with preserved left ventricular function. J Am Coll Cardiol. 2007;50:2285–2290. doi: 10.1016/j.jacc.2007.08.043. [DOI] [PubMed] [Google Scholar]
- De Geus EJ, Kupper N, Boomsma DI, Snieder H. Bivariate genetic modeling of cardiovascular stress reactivity: does stress uncover genetic variance? Psychosom Med. 2007;69:356–364. doi: 10.1097/PSY.0b013e318049cc2d. [DOI] [PubMed] [Google Scholar]
- Debski T, Kamarck T, Jennings J, Young L, Eddy M, Zhang Y. A computerized test battery for the assessment of cardiovascular reactivity. Int J Biomed Comput. 1991;27:277–289. doi: 10.1016/0020-7101(91)90068-p. [DOI] [PubMed] [Google Scholar]
- Devinsky O, Morrell MJ, Vogt BA. Contributions of anterior cingulate cortex to behaviour. Brain. 1995;118:279–306. doi: 10.1093/brain/118.1.279. [DOI] [PubMed] [Google Scholar]
- Dworkin BR. Learning and physiological regulation. The University of Chicago Press; Chicago, IL: 1993. [Google Scholar]
- Dworkin BR, Filewich RJ, Miller NE, Craigmyle N, Pickering TG. Baroreceptor activation reduces reactivity to noxious stimulation: implications for hypertension. Science. 1979;205:1299–1301. doi: 10.1126/science.472749. [DOI] [PubMed] [Google Scholar]
- Eckberg DL, Sleight P. Human baroreflexes in health and disease. Oxford University Press; Oxford, UK: 1992. [Google Scholar]
- Edwards L, McIntyre D, Carroll D, Ring C, Martin U. The human nociceptive flexion reflex threshold is higher during systole than diastole. Psychophysiology. 2002;39:678–681. doi: 10.1017.S0048577202011770. [DOI] [PubMed] [Google Scholar]
- Eisenberger NI, Taylor SE, Gable SL, Hilmert CJ, Lieberman MD. Neural pathways link social support to attenuated neuroendocrine stress responses. Neuroimage. 2007;35:1601–1612. doi: 10.1016/j.neuroimage.2007.01.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellis BJ, Essex MJ, Boyce WT. Biological sensitivity to context: II. empirical explorations of an evolutionary-developmental theory. Dev Psychopathol. 2005;17:303–328. doi: 10.1017/s0954579405050157. [DOI] [PubMed] [Google Scholar]
- Etkin A, Egner T, Peraza DM, Kandel ER, Hirsch J. Resolving emotional conflict: a role for the rostral anterior cingulate cortex in modulating activity in the amygdala. Neuron. 2006;51:871–882. doi: 10.1016/j.neuron.2006.07.029. [DOI] [PubMed] [Google Scholar]
- Everson SA, Lynch JW, Kaplan GA, Lakka TA, Sivenius J, Salonen JT. Stress-induced blood pressure reactivity and incident stroke in middle-aged men. Stroke. 2001;32:1263–1270. doi: 10.1161/01.str.32.6.1263. [DOI] [PubMed] [Google Scholar]
- Falk E. Plaque rupture with severe pre-existing stenosis precipitating coronary thrombosis. Characteristics of coronary atherosclerotic plaques underlying fatal occlusive thrombi. Br Heart J. 1983;50:127–134. doi: 10.1136/hrt.50.2.127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fauvel J, Bernard N, Laville M, Daoud S, Pozet N, Zech P. Reproducibility of the cardiovascular reactivity to a computerized version of the Stroop stress test in normotensive and hypertensive subjects. Clin Auton Res. 1996;6:219–224. doi: 10.1007/BF02291137. [DOI] [PubMed] [Google Scholar]
- Feldman-Barrett L, Wager T. The structure of emotion: evidence from neuroimaging studies. Curr Dir Psychol Sci. 2006;15:79–83. [Google Scholar]
- Fox MD, Raichle ME. Spontaneous fluctuations in brain activity observed with functional magnetic resonance imaging. Nat Rev Neurosci. 2007;8:700–711. doi: 10.1038/nrn2201. [DOI] [PubMed] [Google Scholar]
- Fox MD, Snyder AZ, Vincent JL, Raichle ME. Intrinsic fluctuations within cortical systems account for intertrial variability in human behavior. Neuron. 2007;56:171–184. doi: 10.1016/j.neuron.2007.08.023. [DOI] [PubMed] [Google Scholar]
- Freedman LJ, Insel TR, Smith Y. Subcortical projections of area 25 (subgenual cortex) of the macaque monkey. J Comp Neurol. 2000;421:172–188. [PubMed] [Google Scholar]
- Friston K. Functional and effective connectivity in neuroimaging: a synthesis. Hum Brain Mapp. 1994;2:56–78. [Google Scholar]
- Fuster V. Lewis A. Conner Memorial Lecture. Mechanisms leading to myocardial infarction: insights from studies of vascular biology. Circulation. 1994;90:2126–2146. doi: 10.1161/01.cir.90.4.2126. [DOI] [PubMed] [Google Scholar]
- George MS, Ketter TA, Parekh PI, Horwitz B, Herscovitch P, Post RM. Brain activity during transient sadness and happiness in healthy women. Am J Psychiatry. 1995;152:341–351. doi: 10.1176/ajp.152.3.341. [DOI] [PubMed] [Google Scholar]
- Gerin W, Pieper C, Pickering TG. Measurement reliability of cardiovascular reactivity change scores: a comparison of intermittent and continuous methods of assessment. J Psychosom Res. 1993;37:493–501. doi: 10.1016/0022-3999(93)90005-z. [DOI] [PubMed] [Google Scholar]
- Gianaros PJ, Derbyshire SW, May JC, Siegle GJ, Gamalo MA, Jennings JR. Anterior cingulate activity correlates with blood pressure during stress. Psychophysiology. 2005a;42:627–635. doi: 10.1111/j.1469-8986.2005.00366.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gianaros PJ, Hariri AR, Sheu LK, Sutton-Tyrrell K, Muldoon MF, Manuck SB. Preclinical atherosclerosis covaries with individual differences in reactivity and functional connectivity of the amygdala. Biol Psychiatry. doi: 10.1016/j.biopsych.2008.10.007. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gianaros PJ, Jennings JR, Olafsson GB, Steptoe A, Sutton-Tyrrell K, Muldoon MF, Manuck SB. Greater intima-media thickness in the carotid bulb is associated with reduced baroreflex sensitivity. Am J Hypertens. 2002;15:486–491. doi: 10.1016/s0895-7061(02)02923-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gianaros PJ, Jennings JR, Sheu LK, Derbyshire SW, Matthews KA. Heightened functional neural activation to psychological stress covaries with exaggerated blood pressure reactivity. Hypertension. 2007;49:134–140. doi: 10.1161/01.HYP.0000250984.14992.64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gianaros PJ, May JC, Siegle GJ, Jennings JR. Is there a functional neural correlate of individual differences in cardiovascular reactivity? Psychosom Med. 2005b;67:31–39. doi: 10.1097/01.psy.0000151487.05506.dc. [DOI] [PubMed] [Google Scholar]
- Gianaros PJ, Sheu LK, Matthews KA, Jennings JR, Manuck SB, Hariri AR. Individual differences in stressor-evoked blood pressure reactivity vary with activation, volume, and functional connectivity of the amygdala. J Neurosci. 2008;28:990–999. doi: 10.1523/JNEUROSCI.3606-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gianaros PJ, Sheu LK, Remo AM, Christie IC, Crtichley HD, Wang J. Heightened resting neural activity predicts exaggerated stressor-evoked blood pressure reactivity. Hypertension. 2009 doi: 10.1161/HYPERTENSIONAHA.108.126227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gianaros PJ, Van Der Veen FM, Jennings JR. Regional cerebral blood flow correlates with heart period and high-frequency heart period variability during working-memory tasks: Implications for the cortical and subcortical regulation of cardiac autonomic activity. Psychophysiology. 2004;41:521–530. doi: 10.1111/1469-8986.2004.00179.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon D, Guyton J, Darnovsky M. Intimal alterations in rat aorta induced by stressful stimuli. Lab Invest. 1981;45:14–27. [PubMed] [Google Scholar]
- Gray MA, Minati L, Harrison NA, Gianaros PJ, Napadow V, Critchley H. Physiological recordings: basic concepts and implementation in the fMRI scanner. Neuroimage. 2009a doi: 10.1016/j.neuroimage.2009.05.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray MA, Rylander K, Harrison NA, Wallin BG, Critchley HD. Following one's heart: cardiac rhythms gate central initiation of sympathetic reflexes. J Neurosci. 2009b;29:1817–1825. doi: 10.1523/JNEUROSCI.3363-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenland P, Gidding SS, Tracy RP. Commentary: lifelong prevention of atherosclerosis: the critical importance of major risk factor exposures. Int J Epidemiol. 2002;31:1129–1134. doi: 10.1093/ije/31.6.1129. [DOI] [PubMed] [Google Scholar]
- Greenland P, Knoll MD, Stamler J, Neaton JD, Dyer AR, Garside DB, Wilson PW. Major risk factors as antecedents of fatal and nonfatal coronary heart disease events. J A M A. 2003;290:891–897. doi: 10.1001/jama.290.7.891. [DOI] [PubMed] [Google Scholar]
- Greicius MD, Krasnow B, Reiss AL, Menon V. Functional connectivity in the resting brain: a network analysis of the default mode hypothesis. Proc Natl Acad Sci USA. 2003;100:253–258. doi: 10.1073/pnas.0135058100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greicius MD, Supekar K, Menon V, Dougherty RF. Resting-state functional connectivity reflects structural connectivity in the default mode network. Cereb Cortex. doi: 10.1093/cercor/bhn059. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gusnard DA, Raichle ME, Raichle ME. Searching for a baseline: functional imaging and the resting human brain. Nat Rev Neurosci. 2001;2:685–694. doi: 10.1038/35094500. [DOI] [PubMed] [Google Scholar]
- Guyenet PG. The sympathetic control of blood pressure. Nat Rev Neurosci. 2006;7:335–346. doi: 10.1038/nrn1902. [DOI] [PubMed] [Google Scholar]
- Harper R, Gozal D, Bandler R, Spriggs D, Lee J, Alger J. Regional brain activation in humans during respiratory and blood pressure challenges. Clin Exp Pharm Physiol. 1998;25:483–486. doi: 10.1111/j.1440-1681.1998.tb02240.x. [DOI] [PubMed] [Google Scholar]
- Harper RM, Bandler R, Spriggs D, Alger JR. Lateralized and widespread brain activation during transient blood pressure elevation revealed by magnetic resonance imaging. J Comp Neurol. 2000;417:195–204. doi: 10.1002/(sici)1096-9861(20000207)417:2<195::aid-cne5>3.0.co;2-v. [DOI] [PubMed] [Google Scholar]
- Henderson LA, Macey PM, Macey KE, Frysinger RC, Woo MA, Harper RK, Alger JR, Yan-Go FL, Harper RM. Brain responses associated with the Valsalva maneuver revealed by functional magnetic resonance imaging. J Neurophysiol. 2002;88:3477–3486. doi: 10.1152/jn.00107.2002. [DOI] [PubMed] [Google Scholar]
- Herd JA. Cardiovascular response to stress. Physiol Rev. 1991;71:305–330. doi: 10.1152/physrev.1991.71.1.305. [DOI] [PubMed] [Google Scholar]
- Hester R, Fassbender C, Garavan H. Individual differences in error processing: a review and reanalysis of three event-related fMRI studies using the GO/NOGO task. Cereb Cortex. 2004;14:986–994. doi: 10.1093/cercor/bhh059. [DOI] [PubMed] [Google Scholar]
- Holmes SD, Krantz DS, Rogers H, Gottdiener J, Contrada RJ. Mental stress and coronary artery disease: a multidisciplinary guide. Prog Cardiovasc Dis. 2006;49:106–122. doi: 10.1016/j.pcad.2006.08.013. [DOI] [PubMed] [Google Scholar]
- Holroyd CB, Coles MG. The neural basis of human error processing: reinforcement learning, dopamine, and the error-related negativity. Psychol Rev. 2002;109:679–709. doi: 10.1037/0033-295X.109.4.679. [DOI] [PubMed] [Google Scholar]
- Holroyd KA, Lazarus RS. Stress, coping, and somatic adaptation. In: Goldberger L, Breznitz S, editors. Handbook of stress: theoretical and clinical aspects. The Free Press; New York, NY: 1982. [Google Scholar]
- Hopkins DA, Holstege G. Amygdaloid projections to the mesencephalon, pons and medulla oblongata in the cat. Exp Brain Res. 1978;32:529–547. doi: 10.1007/BF00239551. [DOI] [PubMed] [Google Scholar]
- James W. What is an emotion? Mind. 1884;9:188–205. [Google Scholar]
- Jänig W. The integrative action of the autonomic nervous system: neurobiology of homeostasis. Cambridge University Press; Cambridge, UK: 2006. [Google Scholar]
- Jennings JR, Kamarck TW, Everson-Rose SA, Kaplan GA, Manuck SB, Salonen JT. Exaggerated blood pressure responses during mental stress are prospectively related to enhanced carotid atherosclerosis in middle-aged Finnish men. Circulation. 2004;110:2198–2203. doi: 10.1161/01.CIR.0000143840.77061.E9. [DOI] [PubMed] [Google Scholar]
- Kamarck T, Everson S, Kaplan G, Manuck S, Jennings J, Salonen R, Salonen J. Exaggerated blood pressure responses during mental stress are associated with enhanced carotid atherosclerosis in middle aged Finnish men: findings from the Kuopio Ischemic Heart Disease Study. Circulation. 1997;96:3842–3848. doi: 10.1161/01.cir.96.11.3842. [DOI] [PubMed] [Google Scholar]
- Kamarck TW, Jennings JR, Debski TT, Glickman-Weiss E, Johnson PS, Eddy MJ, Manuck SB. Reliable measures of behaviorally-evoked cardiovascular reactivity from a PC-based test battery: results from student and community samples. Psychophysiology. 1992;29:17–28. doi: 10.1111/j.1469-8986.1992.tb02006.x. [DOI] [PubMed] [Google Scholar]
- Kamarck TW, Jennings JR, Pogue-Geile M, Manuck SB. A multidimensional measurement model for cardiovascular reactivity: stability and cross-validation in two adult samples. Health Psychol. 1994;13:471–478. doi: 10.1037//0278-6133.13.6.471. [DOI] [PubMed] [Google Scholar]
- Kamarck TW, Lovallo WR. Cardiovascular reactivity to psychological challenge: conceptual and measurement considerations. Psychosom Med. 2003;65:9–21. doi: 10.1097/01.psy.0000030390.34416.3e. [DOI] [PubMed] [Google Scholar]
- Kapp B, Whalen P, Pascoe J, Supple W. Amygdaloid contributions to conditioned arousal and sensory information processing. In: Aggleton J, editor. The amygdala: neurobiological aspects of emotion, memory and mental dysfunction. Wiley-Liss; New York, NY: 1992. pp. 229–254. [Google Scholar]
- Khot UN, Khot MB, Bajzer CT, Sapp SK, Ohman EM, Brener SJ, Ellis SG, Lincoff AM, Topol EJ. Prevalence of conventional risk factors in patients with coronary heart disease. J A M A. 2003;290:898–904. doi: 10.1001/jama.290.7.898. [DOI] [PubMed] [Google Scholar]
- Kiehl KA, Liddle PF, Hopfinger JB. Error processing and the rostral anterior cingulate: an event-related fMRI study. Psychophysiology. 2000;37:216–223. [PubMed] [Google Scholar]
- Kimmerly DS, O'Leary DD, Menon RS, Gati JS, Shoemaker JK. Cortical regions associated with autonomic cardiovascular regulation during lower body negative pressure in humans. J Physiol. 2005;569:331–345. doi: 10.1113/jphysiol.2005.091637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King AB, Menon RS, Hachinski V, Cechetto DF. Human forebrain activation by visceral stimuli. J Comp Neurol. 1999;413:572–582. [PubMed] [Google Scholar]
- Klein TA, Endrass T, Kathmann N, Neumann J, von Cramon DY, Ullsperger M. Neural correlates of error awareness. Neuroimage. 2007;34:1774–1781. doi: 10.1016/j.neuroimage.2006.11.014. [DOI] [PubMed] [Google Scholar]
- Knox SS, Hausdorff J, Markovitz JH. Reactivity as a predictor of subsequent blood pressure: racial differences in the Coronary Artery Risk Development in Young Adults (CARDIA) Study. Hypertension. 2002;40:914–919. doi: 10.1161/01.hyp.0000041417.94797.57. [DOI] [PubMed] [Google Scholar]
- Kop WJ. Chronic and acute psychological risk factors for clinical manifestations of coronary artery disease. Psychosom Med. 1999;61:476–487. doi: 10.1097/00006842-199907000-00012. [DOI] [PubMed] [Google Scholar]
- Kop WJ. The integration of cardiovascular behavioral medicine and psychoneuroimmunology: new developments based on converging research fields. Brain Behav Immun. 2003;17:233–237. doi: 10.1016/s0889-1591(03)00051-5. [DOI] [PubMed] [Google Scholar]
- Koski L, Paus T. Functional connectivity of the anterior cingulate cortex within the human frontal lobe: a brain-mapping meta-analysis. Exp Brain Res. 2000;133:55–65. doi: 10.1007/s002210000400. [DOI] [PubMed] [Google Scholar]
- Krantz DS, Contrada RJ, Hill DR, Friedler E. Environmental stress and biobehavioral antecedents of coronary heart disease. J Consult Clin Psychol. 1988;56:333–341. doi: 10.1037//0022-006x.56.3.333. [DOI] [PubMed] [Google Scholar]
- Krantz DS, Manuck SB. Acute psychophysiologic reactivity and risk of cardiovascular disease: a review and methodologic critique. Psychol Bull. 1984;96:435–464. [PubMed] [Google Scholar]
- Krantz DS, Santiago HT, Kop WJ, Bairey-Merz CN, Rozanski A, Gottdiener JS. Prognostic value of mental stress testing in coronary artery disease. Am J Cardiol. 1999;84:1292–1297. doi: 10.1016/s0002-9149(99)00560-3. [DOI] [PubMed] [Google Scholar]
- La Rovere MT, Bigger JT, Jr, Marcus FI, Mortara A, Schwartz PJ. Baroreflex sensitivity and heart-rate variability in prediction of total cardiac mortality after myocardial infarction. ATRAMI (Autonomic Tone and Reflexes After Myocardial Infarction) Investigators. Lancet. 1998;351:478–484. doi: 10.1016/s0140-6736(97)11144-8. [DOI] [PubMed] [Google Scholar]
- La Rovere MT, Pinna GD, Raczak G. Baroreflex sensitivity: measurement and clinical implications. Ann Noninvasive Electrocardiol. 2008;13:191–207. doi: 10.1111/j.1542-474X.2008.00219.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laird AR, Fox PM, Price CJ, Glahn DC, Uecker AM, Lancaster JL, Turkeltaub PE, Kochunov P, Fox PT. ALE meta-analysis: controlling the false discovery rate and performing statistical contrasts. Hum Brain Mapp. 2005;25:155–164. doi: 10.1002/hbm.20136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamb K, Gallagher K, McColl R, Mathews D, Querry R, Williamson JW. Exercise-induced decrease in insular cortex rCBF during postexercise hypotension. Med Sci Sports Exerc. 2007;39:672–679. doi: 10.1249/mss.0b013e31802f04e0. [DOI] [PubMed] [Google Scholar]
- Lane RD, Reiman EM, Ahern GL, Thayer JF. Activity in the medial prefrontal cortex correlates with vagal component of heart rate variability. Brain and Cognition. 2001;47:97–100. [Google Scholar]
- Lane RD, Reiman EM, Axelrod B, Yun LS, Holmes A, Schwartz GE. Neural correlates of levels of emotional awareness. Evidence of an interaction between emotion and attention in the anterior cingulate cortex. J Cogn Neurosci. 1998;10:525–535. doi: 10.1162/089892998562924. [DOI] [PubMed] [Google Scholar]
- Lane RD, Waldstein SR, Chesney MA, Jennings JR, Lovallo WR, Kozel PJ, Rose RM, Drossman DA, Schneiderman N, Thayer JF, Cameron OG. The rebirth of neuroscience in psychosomatic medicine, part I: historical context, methods and relevant basic science. Psychosomatic Medicine. 2009a;71:117–134. doi: 10.1097/PSY.0b013e31819783be. [DOI] [PubMed] [Google Scholar]
- Lane RD, Waldstein SR, Critchley HD, Derbyshire SW, Drossman DA, Wager TD, Schneiderman N, Chesney MA, Jennings JR, Lovallo WR, Rose RM, Thayer JF, Cameron OG. The rebirth of neuroscience in psychosomatic medicine, part II: clinical applications and implications for research. Psychosomatic Medicine. 2009b;71:135–151. doi: 10.1097/PSY.0b013e318198a11f. [DOI] [PubMed] [Google Scholar]
- Lazarus RS, Folkman S. Stress, appraisal and coping. Guilford; New York: 1984. [Google Scholar]
- LeDoux J. The emotional brain, fear, and the amygdala. Cell Mol Neurobiol. 2003;23:727–738. doi: 10.1023/A:1025048802629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lefkowitz RJ, Willerson JT. Prospects for cardiovascular research. J A M A. 2001;285:581–587. doi: 10.1001/jama.285.5.581. [DOI] [PubMed] [Google Scholar]
- Libby P. Molecular bases of the acute coronary syndromes. Circulation. 1995;91:2844–2850. doi: 10.1161/01.cir.91.11.2844. [DOI] [PubMed] [Google Scholar]
- Libby P. Current concepts of the pathogenesis of the acute coronary syndromes. Circulation. 2001;104:365–372. doi: 10.1161/01.cir.104.3.365. [DOI] [PubMed] [Google Scholar]
- Libby P. Inflammation in atherosclerosis. Nature. 2002;420:868–874. doi: 10.1038/nature01323. [DOI] [PubMed] [Google Scholar]
- Libby P, Theroux P. Pathophysiology of coronary artery disease. Circulation. 2005;111:3481–3488. doi: 10.1161/CIRCULATIONAHA.105.537878. [DOI] [PubMed] [Google Scholar]
- Llabre MM, Spitzer SB, Saab PG, Ironson GH, Schneiderman N. The reliability and specificity of delta versus residualized change as measures of cardiovascular reactivity to behavioral challenges. Psychophysiology. 1991;28:701–711. doi: 10.1111/j.1469-8986.1991.tb01017.x. [DOI] [PubMed] [Google Scholar]
- Lovallo WR. Cardiovascular reactivity: mechanisms and pathways to cardiovascular disease. Int J Psychophysiol. 2005;58:119–132. doi: 10.1016/j.ijpsycho.2004.11.007. [DOI] [PubMed] [Google Scholar]
- Lovallo WR, Gerin W. Psychophysiological reactivity: mechanisms and pathways to cardiovascular disease. Psychosom Med. 2003;65:36–45. doi: 10.1097/01.psy.0000033128.44101.c1. [DOI] [PubMed] [Google Scholar]
- Maddock RJ. The retrosplenial cortex and emotion: new insights from functional neuroimaging of the human brain. Trends Neurosci. 1999;22:310–316. doi: 10.1016/s0166-2236(98)01374-5. [DOI] [PubMed] [Google Scholar]
- Magnus P, Beaglehole R. The real contribution of the major risk factors to the coronary epidemics: time to end the “only-50%” myth. Arch Intern Med. 2001;161:2657–2660. doi: 10.1001/archinte.161.22.2657. [DOI] [PubMed] [Google Scholar]
- Manuck SB. Cardiovascular reactivity in cardiovascular disease: “Once more unto the breach”. Int J Behav Med. 1994;1:4–31. doi: 10.1207/s15327558ijbm0101_2. [DOI] [PubMed] [Google Scholar]
- Manuck SB, Kamarck TW, Kasprowicz AS, Waldstein SR. Stability and patterning of behaviorally evoked cardiovascular reactivity. In: Blascovich J, Katkin ES, editors. Cardiovascular reactivity to psychological stress and disease. APA; Washington, DC: 1995a. [Google Scholar]
- Manuck SB, Kaplan JR, Adams MR, Clarkson TB. Effects of stress and the sympathetic nervous system on coronary artery atherosclerosis in the cynomolgus macaque. Am Heart J. 1988;116:328–333. doi: 10.1016/0002-8703(88)90110-x. [DOI] [PubMed] [Google Scholar]
- Manuck SB, Kaplan JR, Matthews KA. Behavioral antecedents of coronary heart disease and atherosclerosis. Arterioscler Thromb Vasc Biol. 1986;6:2–14. doi: 10.1161/01.atv.6.1.2. [DOI] [PubMed] [Google Scholar]
- Manuck SB, Marsland AL, Kaplan JR, Williams JK. The pathogenicity of behavior and its neuroendocrine mediation: an example from coronary artery disease. Psychosom Med. 1995b;57:275–283. doi: 10.1097/00006842-199505000-00009. [DOI] [PubMed] [Google Scholar]
- Manuck SB, Olsson G, Hjemdahl P, Rehnqvist N. Does cardiovascular reactivity to mental stress have prognostic value in postinfarction patients? A pilot study. Psychosom Med. 1992;54:102–108. doi: 10.1097/00006842-199201000-00003. [DOI] [PubMed] [Google Scholar]
- Matthews KA, Salomon K, Brady SS, Allen MT. Cardiovascular reactivity to stress predicts future blood pressure in adolescence. Psychosom Med. 2003;65:410–415. doi: 10.1097/01.psy.0000057612.94797.5f. [DOI] [PubMed] [Google Scholar]
- Matthews KA, Woodall KL, Allen MT. Cardiovascular reactivity to stress predicts future blood pressure status. Hypertension. 1993;22:479–485. doi: 10.1161/01.hyp.22.4.479. [DOI] [PubMed] [Google Scholar]
- Matthews KA, Zhu S, Tucker DC, Whooley MA. Blood pressure reactivity to psychological stress and coronary calcification in the Coronary Artery Risk Development in Young Adults Study. Hypertension. 2006;47:391–395. doi: 10.1161/01.HYP.0000200713.44895.38. [DOI] [PubMed] [Google Scholar]
- Matthews SC, Paulus MP, Simmons AN, Nelesen RA, Dimsdale JE. Functional subdivisions within anterior cingulate cortex and their relationship to autonomic nervous system function. Neuroimage. 2004;22:1151–1156. doi: 10.1016/j.neuroimage.2004.03.005. [DOI] [PubMed] [Google Scholar]
- Mayberg HS, Liotti M, Brannan SK, McGinnis S, Mahurin RK, Jerabek PA, Silva JA, Tekell JL, Martin CC, Lancaster JL, Fox PT. Reciprocal limbic-cortical function and negative mood: converging PET findings in depression and normal sadness. Am J Psychiatry. 1999;156:675–682. doi: 10.1176/ajp.156.5.675. [DOI] [PubMed] [Google Scholar]
- McCubbin JA. Stress and endogenous opioids: behavioral and circulatory interactions. Biol Psychol. 1993;35:91–122. doi: 10.1016/0301-0511(93)90008-v. [DOI] [PubMed] [Google Scholar]
- McDonald AJ. Cortical pathways to the mammalian amygdala. Prog Neurobiol. 1998;55:257–332. doi: 10.1016/s0301-0082(98)00003-3. [DOI] [PubMed] [Google Scholar]
- McEwen BS. Protective and damaging effects of stress mediators. N Engl J Med. 1998;338:171–179. doi: 10.1056/NEJM199801153380307. [DOI] [PubMed] [Google Scholar]
- McEwen BS. Physiology and neurobiology of stress and adaptation: central role of the brain. Physiol Rev. 2007;87:873–904. doi: 10.1152/physrev.00041.2006. [DOI] [PubMed] [Google Scholar]
- McEwen BS, Gianaros PJ. Central role of the brain in stress and adaptation: Links to socioeconomic status, health, and disease. Ann N Y Acad Sci. doi: 10.1111/j.1749-6632.2009.05331.x. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGaugh JL, Cahill L, Roozendaal B. Involvement of the amygdala in memory storage: interaction with other brain systems. Proc Natl Acad Sci U S A. 1996;93:13508–13514. doi: 10.1073/pnas.93.24.13508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menkes MS, Matthews KA, Krantz DS, Lundberg U, Mead LA, Qaqish B, Liang KY, Thomas CB, Pearson TA. Cardiovascular reactivity to the cold pressor test as a predictor of hypertension. Hypertension. 1989;14:524–530. doi: 10.1161/01.hyp.14.5.524. [DOI] [PubMed] [Google Scholar]
- Miller FJ, Jr, Marcus ML, Brody MJ, Gutterman DD. Activation in the region of parabrachial nucleus elicits neurogenically mediated coronary vasoconstriction. Am J Physiol. 1991;261:H1585–1596. doi: 10.1152/ajpheart.1991.261.5.H1585. [DOI] [PubMed] [Google Scholar]
- Ming EE, Adler GK, Kessler RC, Fogg LF, Matthews KA, Herd JA, Rose RM. Cardiovascular reactivity to work stress predicts subsequent onset of hypertension: the Air Traffic Controller Health Change Study. Psychosom Med. 2004;66:459–465. doi: 10.1097/01.psy.0000132872.71870.6d. [DOI] [PubMed] [Google Scholar]
- Mohanty A, Engels AS, Herrington JD, Heller W, Ringo Ho MH, Banich MT, Webb AG, Warren SL, Miller GA. Differential engagement of anterior cingulate cortex subdivisions for cognitive and emotional function. Psychophysiology. 2007;44:343–351. doi: 10.1111/j.1469-8986.2007.00515.x. [DOI] [PubMed] [Google Scholar]
- Monroe SM. Modern approaches to conceptualizing and measuring human life stress. Annu Rev Clin Psychol. 2008;4:33–52. doi: 10.1146/annurev.clinpsy.4.022007.141207. [DOI] [PubMed] [Google Scholar]
- Morecraft RJ, McNeal DW, Stilwell-Morecraft KS, Gedney M, Ge J, Schroeder CM, van Hoesen GW. Amygdala interconnections with the cingulate motor cortex in the rhesus monkey. J Comp Neurol. 2007;500:134–165. doi: 10.1002/cne.21165. [DOI] [PubMed] [Google Scholar]
- Mujica-Parodi LR, Korgaonkar M, Ravindranath B, Greenberg T, Tomasi D, Wagshul M, Ardekani B, Guilfoyle D, Khan S, Zhong Y, Chon K, Malaspina D. Limbic dysregulation is associated with lowered heart rate variability and increased trait anxiety in healthy adults. Hum Brain Mapp. 2009;30:47–58. doi: 10.1002/hbm.20483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagai Y, Critchley HD, Featherstone E, Trimble MR, Dolan RJ. Activity in ventromedial prefrontal cortex covaries with sympathetic skin conductance level: a physiological account of a “default mode” of brain function. Neuroimage. 2004;22:243–251. doi: 10.1016/j.neuroimage.2004.01.019. [DOI] [PubMed] [Google Scholar]
- O'Connor MF, Gundel H, McRae K, Lane RD. Baseline vagal tone predicts BOLD response during elicitation of grief. Neuropsychopharmacology. 2007;32:2184–2189. doi: 10.1038/sj.npp.1301342. [DOI] [PubMed] [Google Scholar]
- Obrist PA. Cardiovascular Psychophysiology: a Perspective. Plenum Press; New York, NY: 1981. [Google Scholar]
- Ochsner KN, Gross JJ. The cognitive control of emotion. Trends Cogn Sci. 2005;9:242–249. doi: 10.1016/j.tics.2005.03.010. [DOI] [PubMed] [Google Scholar]
- Öngür D, An X, Price JL. Prefrontal cortical projections to the hypothalamus in macaque monkeys. J Comp Neurol. 1998;401:480–505. [PubMed] [Google Scholar]
- Öngür D, Price J. The organization of networks within the orbital and medial prefrontal cortex of rats, monkeys, and humans. Cereb Cortex. 2000;10:206–219. doi: 10.1093/cercor/10.3.206. [DOI] [PubMed] [Google Scholar]
- Oppenheimer S. The anatomy and physiology of cortical mechanisms of cardiac control. Stroke. 1993;24:I3–5. [PubMed] [Google Scholar]
- Oppenheimer SM, Gelb A, Girvin JP, Hachinski VC. Cardiovascular effects of human insular cortex stimulation. Neurology. 1992;42:1727–1732. doi: 10.1212/wnl.42.9.1727. [DOI] [PubMed] [Google Scholar]
- Oppenheimer SM, Kedem G, Martin WM. Left-insular cortex lesions perturb cardiac autonomic tone in humans. Clin Auton Res. 1996;6:131–140. doi: 10.1007/BF02281899. [DOI] [PubMed] [Google Scholar]
- Osler W. Lectures on angina pectoris and allied states. Appleton; New York: 1897. [Google Scholar]
- Paulus MP, Stein MB. An insular view of anxiety. Biol Psychiatry. 2006;60:383–387. doi: 10.1016/j.biopsych.2006.03.042. [DOI] [PubMed] [Google Scholar]
- Paus T. Primate anterior cingulate cortex: where motor control, drive, and cognition interface. Nat Neurosci Rev. 2001;2:417–424. doi: 10.1038/35077500. [DOI] [PubMed] [Google Scholar]
- Paus T, Koski L, Caramanos Z, Westbury C. Regional differences in the effects of task difficulty and motor output on blood flow response in the human anterior cingulate cortex: a review of 107 PET activation studies. Neuroreport. 1998;9:R37–47. doi: 10.1097/00001756-199806220-00001. [DOI] [PubMed] [Google Scholar]
- Phan KL, Wager T, Taylor SF, Liberzon I. Functional neuroanatomy of emotion: a meta-analysis of emotion activation studies in PET and fMRI. Neuroimage. 2002;16:331–348. doi: 10.1006/nimg.2002.1087. [DOI] [PubMed] [Google Scholar]
- Phillips ML, Drevets WC, Rauch SL, Lane R. Neurobiology of emotion perception I: the neural basis of normal emotion perception. Biol Psychiatry. 2003;54:504–514. doi: 10.1016/s0006-3223(03)00168-9. [DOI] [PubMed] [Google Scholar]
- Price JL. Comparative aspects of amygdala connectivity. Ann N Y Acad Sci. 2003;985:50–58. doi: 10.1111/j.1749-6632.2003.tb07070.x. [DOI] [PubMed] [Google Scholar]
- Pruessner JC, Dedovic K, Khalili-Mahani N, Engert V, Pruessner M, Buss C, Renwick R, Dagher A, Meaney MJ, Lupien S. Deactivation of the limbic system during acute psychosocial stress: evidence from positron emission tomography and functional magnetic resonance imaging studies. Biol Psychiatry. 2007;63:234–240. doi: 10.1016/j.biopsych.2007.04.041. [DOI] [PubMed] [Google Scholar]
- Quirk GJ, Beer JS. Prefrontal involvement in the regulation of emotion: convergence of rat and human studies. Curr Opin Neurobiol. 2006;16:723–727. doi: 10.1016/j.conb.2006.07.004. [DOI] [PubMed] [Google Scholar]
- Rau H, Brody S. Psychoneurocardiology: psychosomatic and somatopsychic approaches to hypertension research. Integr Physiol Behav Sci. 1994;29:348–354. doi: 10.1007/BF02691354. [DOI] [PubMed] [Google Scholar]
- Reyes del Paso GA, González I, Hernández JA. Baroreceptor sensitivity and effectiveness varies differentially as a function of cognitive-attentional demands. Biol Psychol. 67:385–395. doi: 10.1016/j.biopsycho.2004.02.001. [DOI] [PubMed] [Google Scholar]
- Ridderinkhof KR, Ullsperger M, Crone EA, Nieuwenhuis S. The role of the medial frontal cortex in cognitive control. Science. 2004a;306:443–447. doi: 10.1126/science.1100301. [DOI] [PubMed] [Google Scholar]
- Ridderinkhof KR, van den Wildenberg WP, Segalowitz SJ, Carter CS. Neurocognitive mechanisms of cognitive control: the role of prefrontal cortex in action selection, response inhibition, performance monitoring, and reward-based learning. Brain Cogn. 2004b;56:129–140. doi: 10.1016/j.bandc.2004.09.016. [DOI] [PubMed] [Google Scholar]
- Roemmich JN, Lobarinas CL, Joseph PN, Lambiase MJ, Archer FD, Iii, Dorn J. Cardiovascular reactivity to psychological stress and carotid intima-media thickness in children. Psychophysiology. 2009;46:293–299. doi: 10.1111/j.1469-8986.2008.00776.x. [DOI] [PubMed] [Google Scholar]
- Rosamond W, Flegal K, Furie K, Go A, Greenlund K, Haase N, Hailpern SM, Ho M, Howard V, Kissela B, Kittner S, Lloyd-Jones D, McDermott M, Meigs J, Moy C, Nichol G, O'Donnell C, Roger V, Sorlie P, Steinberger J, Thom T, Wilson M, Hong Y. Heart disease and stroke statistics--2008 update: a report from the American Heart Association Statistics Committee and Stroke Statistics Subcommittee. Circulation. 2008;117:e25–146. doi: 10.1161/CIRCULATIONAHA.107.187998. [DOI] [PubMed] [Google Scholar]
- Ruggiero DA, Mraovitch S, Granata AR, Anwar M, Reis DJ. A role of insular cortex in cardiovascular function. J Comp Neurol. 1987;257:189–207. doi: 10.1002/cne.902570206. [DOI] [PubMed] [Google Scholar]
- Sah P, Faber ES, Lopez De Armentia M, Power J. The amygdaloid complex: anatomy and physiology. Physiol Rev. 2003a;83:803–834. doi: 10.1152/physrev.00002.2003. [DOI] [PubMed] [Google Scholar]
- Sah P, Faber ES, Lopez De Armentia M, Power J. The amygdaloid complex: anatomy and physiology. Physiol Rev. 2003b;83:803–834. doi: 10.1152/physrev.00002.2003. [DOI] [PubMed] [Google Scholar]
- Saha S. Role of the central nucleus of the amygdala in the control of blood pressure: descending pathways to medullary cardiovascular nuclei. Clin Exp Pharmacol Physiol. 2005;32:450–456. doi: 10.1111/j.1440-1681.2005.04210.x. [DOI] [PubMed] [Google Scholar]
- Saper CB. The central autonomic nervous system: conscious visceral perception and autonomic pattern generation. Annu Rev Neurosci. 2002;25:433–469. doi: 10.1146/annurev.neuro.25.032502.111311. [DOI] [PubMed] [Google Scholar]
- Sapolsky RM, Romero LM, Munck AU. How do glucocorticoids influence stress responses? Integrating permissive, suppressive, stimulatory, and preparative actions. Endocr Rev. 2000;21:55–89. doi: 10.1210/edrv.21.1.0389. [DOI] [PubMed] [Google Scholar]
- Schwartz AR, Gerin W, Davidson KW, Pickering TG, Brosschot JF, Thayer JF, Christenfeld N, Linden W. Toward a causal model of cardiovascular responses to stress and the development of cardiovascular disease. Psychosom Med. 2003;65:22–35. doi: 10.1097/01.psy.0000046075.79922.61. [DOI] [PubMed] [Google Scholar]
- Schwartz PJ, La Rovere MT, Vanoli E. Autonomic nervous system and sudden cardiac death. Experimental basis and clinical observations for post-myocardial infarction risk stratification. Circulation. 1992;85:I77–91. [PubMed] [Google Scholar]
- Shafi S, Cusack N, Born G. Increased uptake of methylated low-density lipoprotien induced by noradrenaline in carotid arteries of anesthetized rabbits. Proc Roy Soc London B. 1989;235:289–298. doi: 10.1098/rspb.1989.0001. [DOI] [PubMed] [Google Scholar]
- Sherwood A, Turner JR, Light KC, Blumenthal JA. Temporal stability of the hemodynamics of cardiovascular reactivity. Int J Psychophysiol. 1990;10:95–98. doi: 10.1016/0167-8760(90)90050-n. [DOI] [PubMed] [Google Scholar]
- Smith OA, DeVito JL. Central neural integration for the control of autonomic responses associated with emotion. Annu Rev Neurosci. 1984;7:43–65. doi: 10.1146/annurev.ne.07.030184.000355. [DOI] [PubMed] [Google Scholar]
- Smith OA, DeVito JL, Astley CA. Organization of central nervous system pathways influencing blood pressure responses during emotional behavior. Clin Exp Hypertens A. 1984;6:185–204. doi: 10.3109/10641968409062560. [DOI] [PubMed] [Google Scholar]
- Soufer R, Arrighi JA, Burg MM. Brain, behavior, mental stress, and the neurocardiac interaction. J Nucl Cardiol. 2002;9:650. doi: 10.1067/mnc.2002.129884. [DOI] [PubMed] [Google Scholar]
- Soufer R, Bremner JD, Arrighi JA, Cohen I, Zaret BL, Burg MM, Goldman-Rakic P. Cerebral cortical hyperactivation in response to mental stress in patients with coronary artery disease. Proc Natl Acad Sci USA. 1998;95:6454–6459. doi: 10.1073/pnas.95.11.6454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steptoe A, Brydon L. Emotional triggering of cardiac events. Neurosci Biobehav Rev. 2009;33:63–70. doi: 10.1016/j.neubiorev.2008.04.010. [DOI] [PubMed] [Google Scholar]
- Steptoe A, Sawada Y. Assessment of baroreceptor reflex function during mental stress and relaxation. Psychophysiology. 1989;26:140–147. doi: 10.1111/j.1469-8986.1989.tb03145.x. [DOI] [PubMed] [Google Scholar]
- Stern RM, Ray WJ, Quigley KS. Psychophysiological recording. 2nd. Oxford University Press; New York: 2001. [Google Scholar]
- Stewart JC, Janicki DL, Kamarck TW. Cardiovascular reactivity to and recovery from psychological challenge as predictors of 3-year change in blood pressure. Health Psychol. 2006;25:111–118. doi: 10.1037/0278-6133.25.1.111. [DOI] [PubMed] [Google Scholar]
- Straube T, Schmidt S, Weiss T, Mentzel HJ, Miltner WH. Dynamic activation of the anterior cingulate cortex during anticipatory anxiety. Neuroimage. 2009;44:975–981. doi: 10.1016/j.neuroimage.2008.10.022. [DOI] [PubMed] [Google Scholar]
- Strawn W, Clarkson T, Kaplan J, Bondjers G, Shively C, Ablad B. Inhibition by beta-adrenergic blockade of psychosocially induced endothelial injury in the coronary arteries of monkeys. Circulation. 1989;30 II:435. [Google Scholar]
- Strawn WB, Bondjers G, Kaplan JR, Manuck SB, Schwenke DC, Hansson GK, Shively CA, Clarkson TB. Endothelial dysfunction in response to psychosocial stress in monkeys. Circ Res. 1991;68:1270–1279. doi: 10.1161/01.res.68.5.1270. [DOI] [PubMed] [Google Scholar]
- Strike PC, Steptoe A. Psychosocial factors in the development of coronary artery disease. Prog Cardiovasc Dis. 2004;46:337–347. doi: 10.1016/j.pcad.2003.09.001. [DOI] [PubMed] [Google Scholar]
- Strike PC, Steptoe A. Behavioral and emotional triggers of acute coronary syndromes: a systematic review and critique. Psychosom Med. 2005;67:179–186. doi: 10.1097/01.psy.0000155663.93160.d2. [DOI] [PubMed] [Google Scholar]
- Taylor SF, Phan KL, Decker LR, Liberzon I. Subjective rating of emotionally salient stimuli modulates neural activity. Neuroimage. 2003;18:650–659. doi: 10.1016/s1053-8119(02)00051-4. [DOI] [PubMed] [Google Scholar]
- Thayer JF, Lane RD. The role of vagal function in the risk for cardiovascular disease and mortality. Biol Psychol. 2007;74:224–242. doi: 10.1016/j.biopsycho.2005.11.013. [DOI] [PubMed] [Google Scholar]
- Thayer JF, Lane RD. Claude Bernard and the heart-brain connection: further elaboration of a model of neurovisceral integration. Neurosci Biobehav Rev. 2009;33:81–88. doi: 10.1016/j.neubiorev.2008.08.004. [DOI] [PubMed] [Google Scholar]
- Treiber FA, Kamarck T, Schneiderman N, Sheffield D, Kapuku G, Taylor T. Cardiovascular reactivity and development of preclinical and clinical disease states. Psychosom Med. 2003;65:46–62. doi: 10.1097/00006842-200301000-00007. [DOI] [PubMed] [Google Scholar]
- Turkeltaub PE, Eden GF, Jones KM, Zeffiro TA. Meta-analysis of the functional neuroanatomy of single-word reading: method and validation. Neuroimage. 2002;16:765–780. doi: 10.1006/nimg.2002.1131. [DOI] [PubMed] [Google Scholar]
- Turner JR, Hewitt JK. Twin studies of cardiovascular response to psychological challenge: a review and suggested future directions. Ann Beh Med. 1992;14:12–20. [Google Scholar]
- Verbene AJM, Owens NC. Cortical modulation of the cardiovasular system. Prog Neurobiol. 1998;54:149–168. doi: 10.1016/s0301-0082(97)00056-7. [DOI] [PubMed] [Google Scholar]
- Verberne AJ, Owens NC. Cortical modulation of the cardiovascular system. Prog Neurobiol. 1998;54:149–168. doi: 10.1016/s0301-0082(97)00056-7. [DOI] [PubMed] [Google Scholar]
- Vogt BA. Pain and emotion interactions in subregions of the cingulate gyrus. Nat Rev Neurosci. 2005;6:533–544. doi: 10.1038/nrn1704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogt BA, Berger GR, Derbyshire SWG. Structural and functional dichotomy of human midcingulate cortex. Eur J Neurosci. 2003;18:3134–3144. doi: 10.1111/j.1460-9568.2003.03034.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogt BA, Finch DM, Olson CR. Functional heterogeneity in cingulate cortex: The anterior executive and posterior evaluative regions. Cerebral Cortex. 1992;2:435–443. doi: 10.1093/cercor/2.6.435-a. [DOI] [PubMed] [Google Scholar]
- Vogt BA, Laureys S. Posterior cingulate, precuneal and retrosplenial cortices: cytology and components of the neural network correlates of consciousness. Prog Brain Res. 2005;150:205–217. doi: 10.1016/S0079-6123(05)50015-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogt BA, Nimchinsky EA, Vogt LJ, Hof PR. Human cingulate cortex: surface features, flat maps, and cytoarchitecture. J Comp Neurol. 1995;359:490–506. doi: 10.1002/cne.903590310. [DOI] [PubMed] [Google Scholar]
- Vogt BA, Pandya DN. Cingulate cortex of the rhesus monkey: II. Cortical afferents. J Comp Neurol. 1987;262:271–289. doi: 10.1002/cne.902620208. [DOI] [PubMed] [Google Scholar]
- Vogt BA, Vogt L, Laureys S. Cytology and functionally correlated circuits of human posterior cingulate areas. Neuroimage. 2006;29:452–466. doi: 10.1016/j.neuroimage.2005.07.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wager T, Van Ast V, Hughes B, Davidson M, Lindquist M, KN O. Brain mediators of cardiovascular responses to social threat, Part II: prefrontal- subcortical pathways and relationship with anxiety. Neuroimage. 2009a doi: 10.1016/j.neuroimage.2009.05.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wager TD, Waugh CE, Lindquist MA, Noll DC, Fredrickson BL, Taylor SF. Brain mediators of cardiovascular responses to social threat, Part I: reciprocal dorsal and ventral sub-regions of the medial prefrontal cortex and heart-rate reactivity. Neuroimage. 2009b doi: 10.1016/j.neuroimage.2009.05.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wainer H. Adjusting for differential base rates: Lord's paradox again. Psychol Bull. 1991;109:147–151. doi: 10.1037/0033-2909.109.1.147. [DOI] [PubMed] [Google Scholar]
- Westerhaus MJ, Loewy AD. Central representation of the sympathetic nervous system in the cerebral cortex. Brain Res. 2001;903:117–127. doi: 10.1016/s0006-8993(01)02453-2. [DOI] [PubMed] [Google Scholar]
- Wilder J. Stimulus and response: the law of initial value. Wright; Bristol: 1967. [Google Scholar]
- Williamson JW, Nobrega AC, McColl R, Mathews D, Winchester P, Friberg L, Mitchell JH. Activation of the insular cortex during dynamic exercise in humans. J Physiol. 1997;503(Pt 2):277–283. doi: 10.1111/j.1469-7793.1997.277bh.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wittstein IS, Thiemann DR, Lima JA, Baughman KL, Schulman SP, Gerstenblith G, Wu KC, Rade JJ, Bivalacqua TJ, Champion HC. Neurohumoral features of myocardial stunning due to sudden emotional stress. N Engl J Med. 2005;352:539–548. doi: 10.1056/NEJMoa043046. [DOI] [PubMed] [Google Scholar]
- Wong SW, Masse N, Kimmerly DS, Menon RS, Shoemaker JK. Ventral medial prefrontal cortex and cardiovagal control in conscious humans. Neuroimage. 2007;35:698–708. doi: 10.1016/j.neuroimage.2006.12.027. [DOI] [PubMed] [Google Scholar]
- Wood D. Established and emerging cardiovascular risk factors. Am Heart J. 2001;141:S49–57. doi: 10.1067/mhj.2001.109951. [DOI] [PubMed] [Google Scholar]
- Yasui Y, Breder CD, Saper CB, Cechetto DF. Autonomic responses and efferent pathways from the insular cortex in the rat. J Comp Neurol. 1991;303:355–374. doi: 10.1002/cne.903030303. [DOI] [PubMed] [Google Scholar]
- Zald DH. The human amygdala and the emotional evaluation of sensory stimuli. Brain Res Brain Res Rev. 2003;41:88–123. doi: 10.1016/s0165-0173(02)00248-5. [DOI] [PubMed] [Google Scholar]
- Zamir N, Maixner W. The relationship between cardiovascular and pain regulatory systems. Ann N Y Acad Sci. 1986;467:371–384. doi: 10.1111/j.1749-6632.1986.tb14641.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.