

Abstract
The potential of mesenchymal stem cells (MSCs) as a viable cell source for cartilage repair hinges on the development of engineered scaffolds that support adequate cartilage tissue formation. Evolving networks (hydrogels with mesh sizes that change over time due to crosslink degradation) may provide the control needed to enhance overall tissue formation when compared to static scaffolds. In this study, MSCs were photoencapsulated in combinations of hydrolytically and enzymatically degradable hyaluronic acid (HA) hydrogels to investigate the tunability of these hydrogels and the influence of network evolution on neocartilage formation. In MSC-laden HA hydrogels, compressive mechanical properties increased when degradation complemented extracellular matrix deposition and decreased when degradation was too rapid. In addition, dynamic hydrogels that started at a higher wt% and decreased to a lower wt% were not equivalent to static hydrogels that started at the higher or lower wt%. Specifically, evolving 2wt% hydrogels (2 wt% degrading to 1 wt%) expressed up-regulation of type II collagen and aggrecan, and exhibited increased glycosaminoglycan content over non-evolving 2 and 1 wt% hydrogels. Likewise, mechanical properties and size maintenance were superior in the dynamic system compared to the static 2 wt% and 1 wt% hydrogels, respectively. Thus, hydrogels with dynamic properties may improve engineered tissues and help translate tissue engineering technology to clinical application.
Keywords: hyaluronic acid, hydrogel, mesenchymal stem cell, degradation, crosslinking
1. Introduction
In efforts to develop clinically translatable approaches for cartilage repair, mesenchymal stem cells (MSCs) have been shown to undergo chondrogenesis and deposit neocartilage in a variety of tissue engineering scaffolds[1]. However, we are still limited in recapitulating the properties of native tissues. For example, Mauck et al[2] showed that the biochemical and mechanical properties of matured MSC-laden agarose scaffolds are lower than those containing donor matched chondrocytes. The diminished ability of MSCs to produce functional cartilage tissue is troubling, as the quality of tissue they produce determines their success as a viable cell source for cartilage repair and regeneration. Beyond the amount and type of matrix produced, the distribution of this matrix is essential for the optimization of tissue properties (e.g., mechanics). Thus, the design of biomaterials that support the distribution of formed tissue is crucial for the optimization of neocartilage formation by MSCs.
Hyaluronic acid (HA) hydrogels are formed by the simple addition of a reactive group to the HA backbone and subsequent crosslinking[3]. These hydrogels are tunable, where hydrogel parameters can be varied by degree of methacrylation, macromer molecular weight, and macromer concentration, providing a wide range of hydrogel properties (e.g., volumetric swelling ratios, mechanical properties)[4], and undergo enzymatic degradation via hyaluronidases. For applications in cartilage repair, we have shown that these HA hydrogels not only support and maintain chondrocyte viability and phenotype when cultured in vitro and in vivo [5, 6], but also that HA hydrogel chemistry supports and promotes the chondrogenic differentiation of MSCs[7, 8]. However, ECM distribution is limited without adequate space for diffusion in these slow enzymatically degrading hydrogels and techniques to better control network evolution with culture are needed.
Ideally, scaffold degradation should coincide with ECM deposition and accumulation. In engineered hydrogel scaffolds, degradation can alter the diffusion of nutrients and waste, cell-scaffold interactions, and the distribution and retention of ECM proteins. Therefore, to tailor temporal degradation of a scaffold, others have introduced hydrolytically degradable components[9], matrix metalloproteinase (MMP)-sensitive peptides[10, 11], and/or exogenous enzymes[12, 13] into scaffold designs. Recently, we engineered a hydrolytically degradable HA hydrogel with the inclusion of repeat units of poly(lactic acid) between the HA backbone and the polymerizing moiety (e.g., methacrylate)[14]. Short-term in vitro studies demonstrated cytocompatibility and increased distribution of chondroitin sulfate in hydrogels with increased hydrolytically degradable components by encapsulated human MSCs, in agreement with others[9]. However, these gels degraded very quickly (e.g., 4wt% hydrogel degraded in < 7 days), exhibited cell clustering within the gel, altered cell morphology, and were not useful for long-term matrix development investigations.
In this study, the degradation of these gels was slowed with the inclusion of poly(caprolactone) units and the long-term effects of temporal network structure on scaffold properties and neocartilage formation by MSCs were investigated. Thus, we present a system that exploits both the advantages of HA in cartilage regeneration, as well as tunable degradation for the optimization of engineered tissue properties.
2. Materials and Methods
2.1 Macromer Syntheses
Methacrylated HA (MeHA) was synthesized as previously reported[3]. Briefly, methacrylic anhydride (Sigma) was added to a solution of 1 wt% HA (Lifecore, MW = 74 kDa) in deionized water, adjusted to a pH of 8 with 5 N NaOH, and reacted on ice for 24 hours. The macromer solution was purified via dialysis (MW cutoff 6-8k) against deionized water for a minimum of 48 hours with repeated changes of water. The final product was obtained by lyophilization and stored at -20°C in powder form prior to use. Methacrylated caprolactone HA (MeCLHA) was synthesized as previously reported[14], with modifications (Figure 1A). Briefly, 2-hydroxyethyl methacrylate (HEMA) (Acros organics) was reacted with ε-caprolactone (Sigma) via a ring opening polymerization in the presence of stannous octoate (Sigma) at 130°C for 1hr. The end group was then functionalized into a carboxylic acid (MeCL-COOH) via reaction with succinic anhydride (Sigma) in the presence of N-methylimidazole at 65°C in dichloroethane for 13 hrs. The sodium salt form of HA was converted to a tetrabutylammonium (TBA) salt by acidic ion exchange with Dowex 50 W × 8-200 resin, followed by resin filtration and neutralization with aqueous TBA hydroxide for solubilization in dimethyl sulfoxide (DMSO). MeCL-COOH was coupled to TBA-HA via an esterification reaction with di-t-butyl dicarbonate (BOC2O) as an activating agent with dimethylaminopyridine (DMAP)[15] for 20 hrs at 45°C. The final product (MeCLHA) was precipitated and washed in acetone, dissolved in DI water, dialyzed (MW cutoff 6-8k) for 24 hours at 4°C, lyophilized, and stored at -20°C in powder form prior to use. The intermediate and final macromer products were confirmed by 1H NMR. Lyophilized macromers were dissolved in phosphate buffered saline (PBS) containing 0.05 wt% 2-methyl-1-[4-(hydroxyethoxy)phenyl]-2-methyl-1-propanone (I2959, Ciba) for polymerization.
Figure 1.
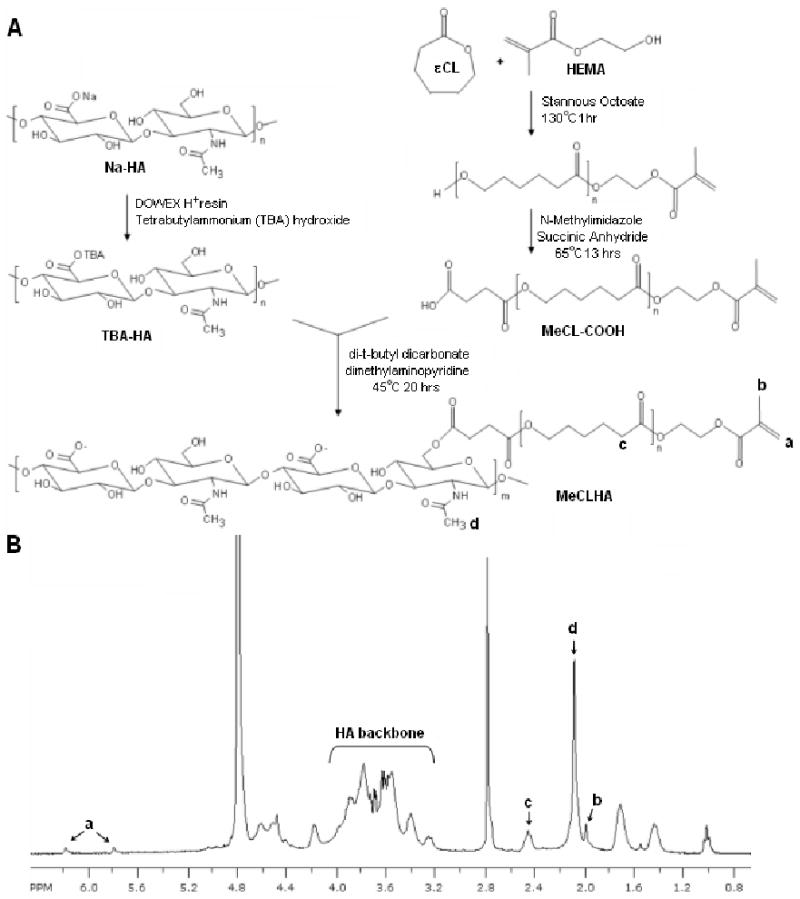
Synthetic scheme for the MeCLHA macromer (A). 1H NMR of the MeCLHA macromer (n= ∼3.8, ∼7.5% modification) (B). % modification was determined by the integration of methacrylate peaks and the peaks associated with the HA backbone.
2.2 Acellular Characterization
Acellular hydrogels (50μl) of varying macromer concentrations (e.g., 5, 2, and 1 wt%) and type (e.g., MeHA, MeCLHA) were polymerized with exposure to ultraviolet light (Eiko, ∼1.9 mW/cm2) for 10 minutes in cylindrical molds. The mechanical properties of homopolymer and copolymer hydrogels consisting of 5:0, 4:1, 3:2, 2:3, 2:0, 1.5:0.5, 1:1, and 1:0 MeHA wt%: MeCLHA wt% were analyzed at various time points for up to 8 weeks in unconfined compression (Dynamic Mechanical Analyzer Q800, TA Instruments) in a PBS bath. Hydrogels were compressed at a rate of 10%/min until 60% of the initial thickness was reached. The modulus was determined as the slope of the stress versus strain profile at low strains (<20%). Degradation in PBS at 37°C was monitored using a uronic acid assay[4, 16] and the hydrogel wet and dry weights were also recorded at each time point.
2.3 MSC Photoencapsulation and Culture
For cell encapsulation, macromers were sterilized using a germicidal lamp in a laminar flow hood for 30 minutes prior to dissolving in a sterile solution of PBS containing 0.05 wt% I2959 for polymerization. Human MSCs (Lonza) were expanded to passage 4 in growth media consisting of α-MEM with 16.7% FBS and 1% penicillin/streptomycin. MSCs were photoencapsulated (20 million cells/mL) in hydrogels (50 μl) of varying macromer concentration (e.g., 5, 2, and 1wt%) and type (e.g., MeHA, MeCLHA). Hydrogels consisting of 5:0, 4:1, 3:2, 2:3, 2:0, 1.5:0.5, 1:1, and 1:0 MeHA wt%: MeCLHA wt% were cultured in DMEM supplemented with 1% penicillin/streptomycin, 1% ITS+, 1mM sodium pyruvate, 40mg/ml L-proline, 100nM dexamethasone, 50μg/ml ascorbic acid 2-phosphate, and 10 ng/ml of TGF-β3 (chondrogenic media) for up to 8 weeks in vitro.
2.4 Cellular Characterization
The viability of MSCs in HA hydrogels was assessed using a live/dead cytotoxicity kit (Molecular Probes) at 1, 7 and 14 days and an Alamar Blue assay (Invitrogen) (n=3) at 7 and 14 days of in vitro culture according to manufacturer's instructions.
For short term gene expression analysis (3 and 14 days of culture), samples were homogenized in Trizol Reagent (Invitrogen) with a tissue grinder, RNA was extracted according to the manufacturer's instructions, and RNA concentration was determined using an ND-1000 spectrophotometer (Nanodrop Technologies). One microgram of RNA from each sample was reverse transcribed into cDNA using reverse transcriptase (Superscript II, Invitrogen) and oligoDT (Invitrogen). Polymerase chain reaction (PCR) was performed on an Applied Biosystems 7300 Real-Time PCR system using a 25μl reaction volume for Taqman (5′-nuclease) reactions. Primers and probes specific for glyceraldehydes 3-phosphate dehydrogenase (GAPDH, housekeeping gene), type I and type II collagen, and aggrecan are listed in Table 1. Relative gene expression was calculated using the ΔΔCT method, where fold difference was calculated using the expression 2-ΔΔCt. Results (n=4-8) from two replicate experiments were combined and samples with poor RNA quality were excluded.
Table 1.
Quantitative PCR primers and probes.
| Gene | Forward Primer | Reverse Primer | Probe |
|---|---|---|---|
| GAPDH | AGGGCTGCTTTTAACTCTGGTAAA | GAATTTGCCATGGGTGGAAT | CCTCAACTACATGGTTTAC |
| Type I Collagen | AGGACAAGAGGCATGTCTGGTT | GGACATCAGGCGCAGGAA | TTCCAGTTCGAGTATGGC |
| Type II Collagen | GGCAATAGCAGGTTCACGTACA | CGATAACAGTCTTGCCCCACTT | CTGCACGAAACATAC |
| Aggrecan | TCGAGGACAGCGAGGCC | TCGAGGGTGTAGCGTGTAGAGA | ATGGAACACGATGCCTTTCACCACGA |
Additional samples (n=5) were measured (diameter and height), weighed (wet weight,) and tested in unconfined compression on a custom mechanical tester[17, 18] after 1, 14, 35, and 56 days of culture. The mechanical tester consists of a computer-controlled stepper motor, a linear variable differential transformer to measure displacement, and a 250g load cell to measure load. Labview software (National Instruments) was used for stepper motor control and data acquisition. Samples were loaded between impermeable glass plates in creep with a 2g tare load until equilibrium (∼300s), followed by stress relaxation with a single ramp displacement of 10% strain at a rate of 10%/minute. The samples were allowed to relax to equilibrium (∼1200s), and the equilibrium confined compression moduli were calculated by dividing the equilibrium load by the area loaded and the % strain for each sample.
Mechanically tested samples were then lyophilized, weighed (dry weight), and digested in a proteinase K solution (200 μg/ml proteinase K (Roche), 100mM ammonium acetate, pH 7.0) overnight at 60°C. Proteinase K was then inactivated at 100°C for 5 min. Total DNA, GAG, and collagen contents (n=5) were determined using the PicoGreen dsDNA Assay[19], the dimethylmethylene blue dye method[20] with chondroitin sulfate as a standard, and the hydroxyproline assay[21] using a collagen to hydroxyproline ratio of 7.25[22, 23], respectively. The proteinase K digestion solution was used as a negative control.
Samples for histological analysis were fixed in 10% formalin for 24 hours, embedded in paraffin, and processed using standard histological procedures. The histological sections (7 μm thick) were stained for chondroitin sulfate and collagen distributions using the Vectastain ABC kit (Vector Labs) and the DAB Substrate kit for peroxidase (Vector Labs). Sections were predigested in 0.5 mg/ml hyaluronidase for 30 min at 37°C and incubated in 0.5 N acetic acid for 4 hours at 4°C to swell the samples prior to overnight incubation with primary antibodies at dilutions of 1:100, 1:200, and 1:3 for chondroitin sulfate (mouse monoclonal anti-chondroitin sulfate, Sigma), and type I (mouse monoclonal anti-collagen type 1, Sigma) and type II collagen antibodies (mouse monoclonal anti-collagen type II, Developmental Studies Hybridoma Bank), respectively. Non-immune controls underwent the same procedure without primary antibody incubation.
2.5 Statistical Analysis
ANOVA with Tukey's post-hoc test was used to determine significant differences (p < 0.05). All value are represented as the mean ± standard deviation.
3. Results
1H NMR confirmed the successful synthesis of hydrolytically degradable HA macromers with ∼7.5-13% methacrylate modification containing ∼3.8 repeat units of ε-caprolactone between the methacrylate and the HA backbone (Figure 1B). MeCLHA with ∼13% modification was used for initial degradation studies to illustrate differences in the degradation of hydrolytically and enzymatically degradable HA hydrogels, while ∼7.5% modification was used for all other experiments. Hydrolytic degradation of MeCLHA was confirmed with the complete degradation of 2 wt% MeCLHA hydrogels within 7 days of incubation in PBS at 37°C without the addition of exogenous enzymes, while 2 wt% MeHA hydrogels, with ∼31.5% modification, remained relatively stable even up to 56 days with only 40% ± 8% HA released prior to hyaluronidase addition to trigger complete degradation (Figure 2A). Copolymerization of MeHA and MeCLHA allowed for a wider range of degradation profiles as the MeCLHA macromer is presumed to be released more rapidly than the MeHA macromer. For instance, 2:3 MeHA wt%: MeCLHA wt% hydrogels exhibited significantly greater HA release (72% ± 16% HA released) compared to 5:0 MeHA wt%: MeCLHA wt% hydrogels with only 21% ± 8% HA released after 56 days of incubation in PBS prior to hyaluronidase addition (Figure 2B).
Figure 2.
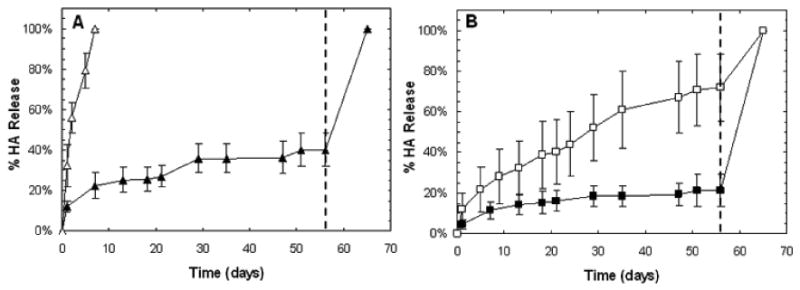
Degradation (n=3) of acellular 2 wt% MeHA (black) and 2 wt% MeCLHA (white) hydrogels (A) measured by uronic acid release. Degradation of acellular 5:0 (black) and 2:3 (white) MeHA wt%: MeCLHA wt% hydrogels (B). Hyaluronidase addition at 56 days is denoted by the dashed line. Values are reported as mean ± standard deviation.
The elastic moduli of acellular hydrogels were dictated by total macromer concentration, macromer composition, and hydrogel degradation. Initial moduli (1 day after polymerization) significantly decreased with decreasing macromer wt% (Table 2) and decreases in moduli were observed coinciding with increased hydrogel degradation (Figure 3). In addition, slight temporal decreases in the elastic moduli of MeHA only (5:0, 2:0, 1:0) hydrogels were also observed. The volumetric swelling ratios (Qv) were calculated from wet and dry weights of the hydrogel at specific time points and were found to be inversely related to total macromer concentration and increased with incubation time, ranging from 32.5 ± 2.5 to 68.5 ± 3.6 at day 1 (Table 2) and increasing to 40.8 ± 3.3 to 70.1 ± 8.2 by day 56 (data not shown).
Table 2.
Compositions, volumetric swelling ratios (n=3) and elastic moduli (n=5) of acellular hydrogels, and metabolic activity (n=3) as measured by Alamar Blue assay for cell-seeded hydrogels. Values are reported as mean ± standard deviation.
| [Macromer] | wt% MeHA: wt% MeCLHA | Qv, t = 1 day | E (kPa), t = 1 day | Metabolic Activity (Fluorescence), t = 14 days |
|---|---|---|---|---|
| 5 wt% | 5:0 | 36 ± 2 | 34 ± 2 | 3800 ± 440 |
| 4:1 | 37 ± 4 | 31 ± 1 | 3200 ± 170 | |
| 3:2 | 33 ± 3 | 25 ± 1 | 3300 ± 470 | |
| 2:3 | 36 ± 2 | 22.4 ± 0.8 | 3300 ± 77 | |
| 2 wt% | 2:0 | 51 ± 14 | 5.6 ± 0.2 | 3700 ± 540 |
| 1.5:0.5 | 47 ± 5 | 4.5 ± 0.2 | 4200 ± 310 | |
| 1:1 | 67 ± 3 | 3.6 ± 0.1 | 3900 ± 540 | |
| 1 wt% | 1:0 | 68 ± 4 | 3.0 ± 0.2 | 2900 ± 120 |
Figure 3.
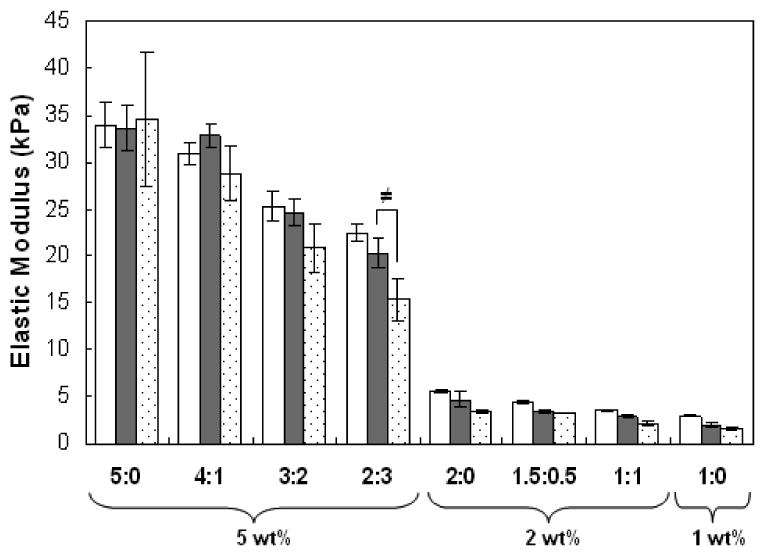
Elastic moduli (n=5) of acellular HA hydrogels after 1 (white), 7 (gray), and 14 days (shaded) of incubation in PBS at 37°C. Statistical analysis of relevant comparisons: ≠ denotes significant difference over time between bracketed groups. Values are reported as mean ± standard deviation.
The photoencapsulated MSCs retained rounded cell morphology in all hydrogels. Viability assessed by live/dead staining indicated greater than 90% viability in hydrogels for cultures up to 14 days in vitro with only a slight increase in the fraction of dead cells for 5:0 hydrogels at day 14 (Figure 4). All groups showed comparable metabolic activity as measured by the Alamar blue assay up to 14 days of in vitro culture with the exception of a slight decrease in metabolic activity in 1:0 hydrogels at day 14 (Table 2). There were no significant increases or decreases in metabolic activity between day 7 and day 14 in all hydrogels, suggesting limited proliferation in these environments (data not shown).
Figure 4.

Representative live (green)/dead (red) staining of HA hydrogels after 1 and 14 days of culture. Scale bar = 200μm.
Short-term gene expression for type I and type II collagen and aggrecan was assessed after 3 and 14 days of in vitro culture. Type II collagen and aggrecan are positive markers for chondrogenic differentiation, while type I collagen indicates transformation to a more fibrochondrocytic phenotype. Each sample was internally normalized to GAPDH, and each group was normalized to expression of MSCs isolated at the time of encapsulation (i.e., after expansion and before differentiation); thus, relative expression greater than 1 represents up-regulation with culture, while relative expression less than 1 represents down-regulation of that gene compared to that of initially encapsulated MSCs. No significant differences between groups were observed for type I collagen at either of the two time points, and overall expression of type I collagen was not significantly different compared to initially encapsulated MSCs (Figure 5A). Up-regulation of type II collagen (note that the plot is on a log-scale) and aggrecan expression was indicative of chondrogenic differentiation in all hydrogels and was found to be statistically higher at day 3 for the 1:1 and 1:0 hydrogels compared to all other groups (Figure 5). At day 14, an increasing trend of aggrecan expression in 2 wt% hydrogels was observed with increased MeCLHA content. Furthermore, the 1:1 hydrogels exhibited significantly higher type II collagen and aggrecan expression over both 2:0 and 1:0 hydrogels.
Figure 5.
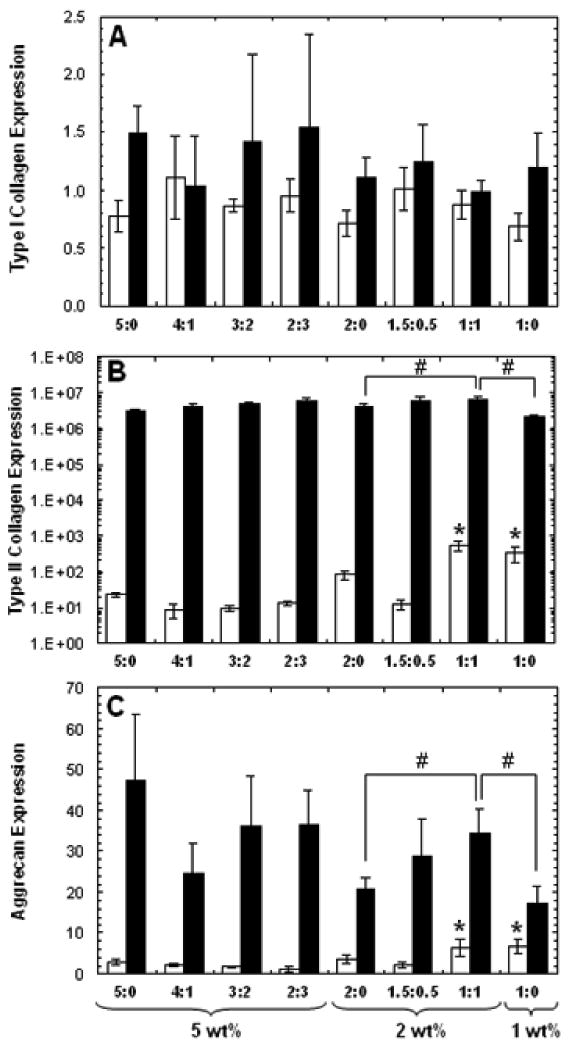
Relative gene expression of (A) type I collagen, (B) type II collagen, and (C) aggrecan for all HA hydrogel formulations after 3 (white) and 14 (black) days of culture. Each sample was internally normalized to GAPDH, and each group was normalized to expression of MSCs isolated at the time of encapsulation. Statistical analysis of relevant comparisons: * denotes significant difference between starred groups and all other groups for the specified time point and # denotes significant difference between bracketed groups. Relative gene expression of type II collagen for 5, 2, and 1 wt% MeHA only hydrogels were also all significantly different from each other at day 14. Values are reported as mean ± standard deviation.
With longer culture, samples demonstrated varying degrees of neocartilage formation; macroscopically, 2 and 1 wt% hydrogels were more opaque in appearance after 8 weeks of in vitro culture compared to higher wt% hydrogels (Figure 6 top). Significant differences in hydrogel diameter were observed among groups at various time points and within individual formulations with culture time. The initial hydrogel diameters were dependent on total macromer concentration, with significant decreases in diameter with decreased macromer wt%. No significant differences were found within each 5, 2 and 1 wt% groups initially, regardless of the formulation; however, at later time points, a general trend of increased diameter was noticed in 5 wt% hydrogels with increased MeCLHA content (Figure 6A). With culture, all 5 and 2 wt% hydrogel diameters increased with culture time, while slight but insignificant increases in height for some compositions were also observed (results not shown). Overall, the most notable difference in hydrogels dimensions was seen with 1:0 hydrogels, as these hydrogels were significantly smaller in diameter (Figure 6A) and height compared to all other groups at every time point. Trends in hydrogel dimensions were also mirrored by hydrogel wet weight (Figure 6B). Again, wet weights of 1:0 hydrogels were significantly lower than all other groups at all time points. Also, a general trend in 5 wt% hydrogels of increased wet weight with increased MeCLHA content was observed, where at day 35 the wet weight of 5:0 hydrogels were significantly lower than that of all other 5 wt% hydrogels at day 35.
Figure 6.
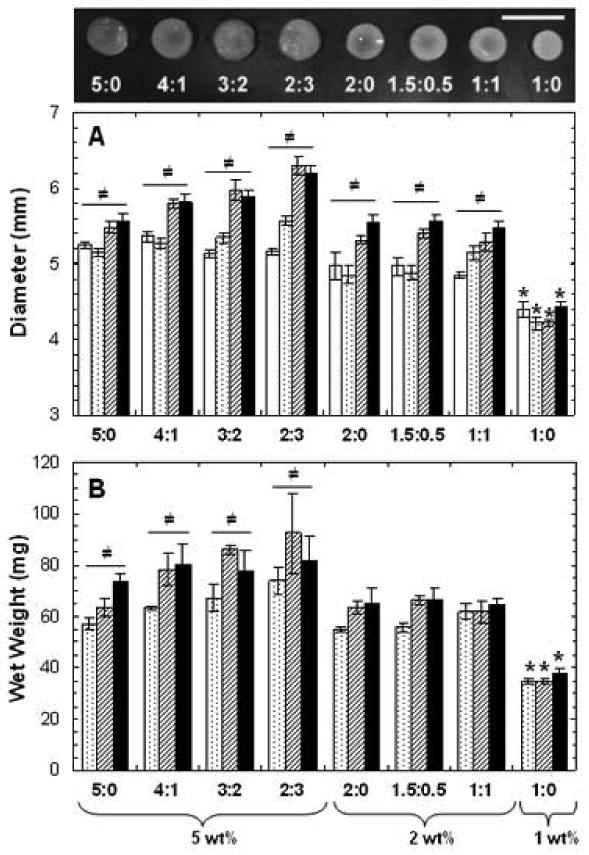
Macroscopic appearance of hydrogels after 8 weeks of in vitro culture (scale bar = 1cm) (top). Diameter (A) and wet weight (B) of hydrogels after 1 (white), 14 (dotted), 35 (striped), and 56 (black) days of in vitro culture (n=5). Values are reported as mean ± standard deviation. Statistical analysis of relevant comparisons: * denotes significant difference between starred groups and all other groups for the specified time point and ≠ denotes significant difference over time within each group.
When mechanically tested in unconfined compression, changes in the compressive equilibrium modulus (Figure 7A) and the peak stress (Figure 7B) were observed among groups at each time point and within specific formulations over time. While initial moduli (day 1) was dictated by macromer concentration (similar to acellular hydrogels), moduli at day 35 and 56 was dictated by ECM production. 2 and 1 wt% samples exhibited an increase in moduli over time with significant increases at days 35 and 56. At day 35, 2 wt% hydrogels containing MeCLHA content (i.e., 1.5:0.5 and 1:1) exhibited moduli (30.4 ± 2.3 kPa and 32.3 ± 7.4 kPa, respectively) that were both significantly higher than that of the MeHA only (i.e., 2:0 had moduli of 14.4 ± 2.3 kPa) samples. By day 56, moduli of 1:1 (49 ± 7 kPa) and 1:0 (57 ± 10 kPa) samples were significantly greater than all other groups. However, there was little change in the mechanics among the 5 wt% formulations and the 4:1 hydrogels had the highest moduli of 27 ± 8 kPa after 56 days. Furthermore, decreases in moduli for the 3:2 and 2:3 hydrogels were observed from day 35 to day 56. Peak stresses obtained during mechanical testing indicated similar trends as the compressive equilibrium moduli. Peak stress reflects water retention within the sample, and is usually an earlier indicator of functional matrix development. Again, peak stress increased with increased MeCLHA content in 2 wt% hydrogels. Peak stress obtained in 1:1 and 1:0 hydrogels were significantly greater than all other groups at days 35 and 56.
Figure 7.
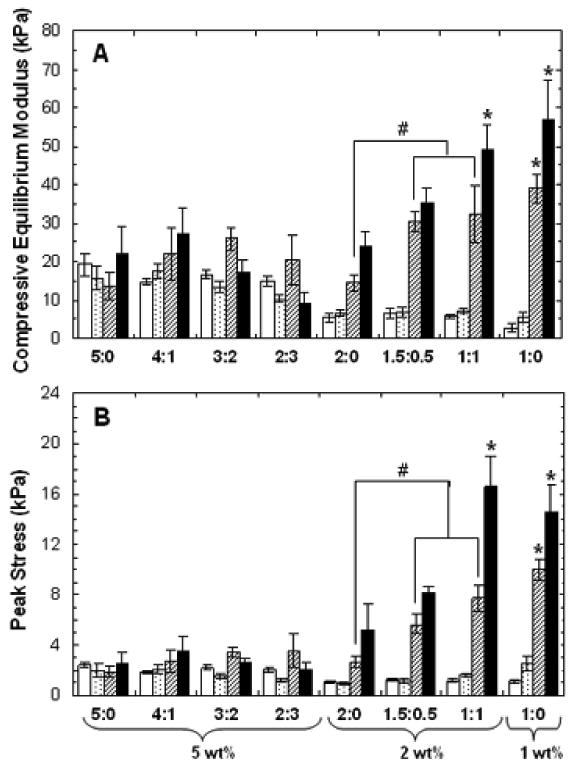
Compressive equilibrium moduli (A) and peak stresses (B) of hydrogels at 1 (white), 14 (dotted), 35 (striped), and 56 (black) days of in vitro culture (n=5). Values are reported as mean ± standard deviation. Statistical analysis or relevant comparisons: * denotes significant difference between starred groups and all other groups for the specified time point and # denotes significant difference between bracketed groups. Significant increases in both moduli and peak stresses over time within each formulation were also observed for all 2 and 1 wt% groups (not denoted). Significant decrease in moduli for 2:3 hydrogels from day 35 to day 56 was also observed (not denoted).
Mechanically tested samples were digested to determine DNA, sGAG, and collagen contents. DNA content remained relatively constant among all groups at each time point and within groups over time (Figure 8A). The only significant changes in DNA content were observed at day 56 with a decrease in DNA content for 3:2 and 2:3 groups. When normalized to DNA content, the GAG content in the samples showed significant changes between groups at each time point and within formulations over time (Figure 8B). Generally, GAG content increased within wt% formulations with increased MeCLHA content, with trends seen as early as 14 days. By day 35, MeHA only (5:0 and 2:0) hydrogels had significantly lower GAG content compared to their copolymer counterparts. By the end of the 8 week culture, the 1:1 hydrogels had the greatest GAG content and was significantly higher in GAG/DNA content (201 ± 4.3 mg/mg) than both 2:0 (169 ± 22 mg/mg) and 1:0 hydrogels (137 ± 4.3 mg/mg). When normalized to wet weight, an increase from 2.0 ± 0.3% to 2.5 ± 0.2% sGAG/wet weight in the 2wt% hydrogels was observed with increased MeCLHA content. No significant differences in collagen content were observed between groups at each time point. However, collagen content increased with culture time in all formulations, with 1.5:0.5 hydrogels attaining the highest collagen content (654 ± 189 μg/sample) after 56 days.
Figure 8.
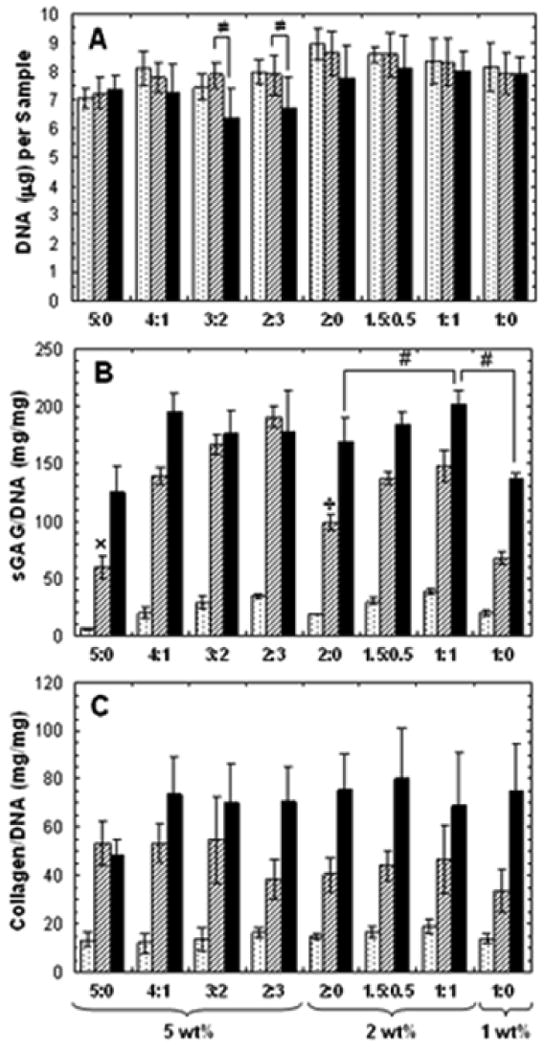
DNA (A), sulfated GAG/DNA (B), and collagen/DNA (C) contents of hydrogels at 14 (dotted), 35 (striped), and 56 (black) days of in vitro culture (n=5). Values are reported as mean ± standard deviation. Statistical analysis of relevant comparisons: * denotes significant difference between starred groups and all other groups for the specified time point, # denotes significant difference between bracketed groups, ≠ denotes significant difference over time between bracketed groups, ÷ denotes significant difference between marked group and all other 2wt% groups, and × denotes significant difference between marked group an all other 5 wt% groups. Significant increases in collagen/DNA content over time was observed for all groups (not denoted).
Immunohistochemical staining of type I and type II collagen and chondroitin sulfate was performed on all groups at 14 and 56 days of culture. At day 14, pericellular staining of chondroitin sulfate was observed for MeHA only (5:0 and 2:0) hydrogels while greater distribution of staining was observed for their copolymer counterparts with increased distribution in hydrogels with increased MeCLHA content (Figure 9). By 56 days of culture, chondroitin sulfate was evenly distributed throughout the hydrogels for all groups. Hydrogels also stained positive for type II collagen in varying degrees for all groups with 5:0 hydrogels showing the least amount of staining compared to copolymer hydrogels showing the most staining (Figure 9). In general, type II collagen staining increased from 14 days to 56 days in all constructs. Clustering of type II collagen in copolymers was also observed. Little to no staining of type I collagen was present in all hydrogels and non-immune controls also stained negative.
Figure 9.
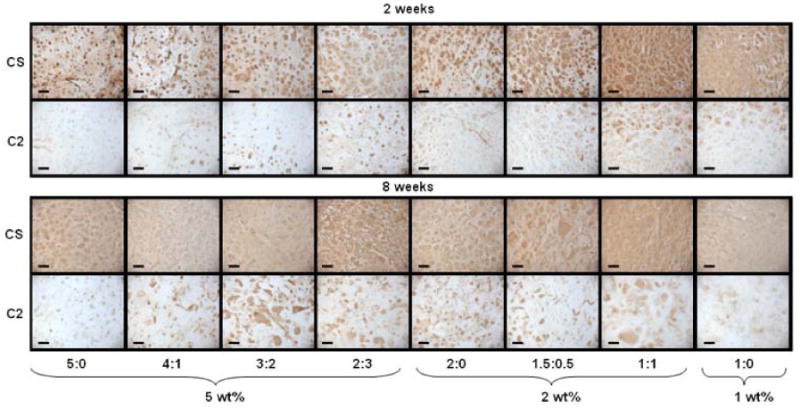
Immunohistochemical staining of chondroitin sulfate (CS) and type II collagen (C2) for 2 and 8 weeks of culture. Scale bar = 100μm.
4. Discussion
The use of MSCs as a cell source for cartilage repair depends on the ability to form adequate neocartilage with the use of engineered materials. Due to the avascular nature of cartilage, native cartilage tissue and engineered constructs rely mainly on diffusion for nutrient supply and metabolic waste removal. With hydrogels, both the initial and temporal crosslinking densities control the diffusion of nutrients, wastes, and newly synthesized ECM proteins[4, 24]. Thus, by developing techniques to better control network evolution with time, we may be able to improve overall tissue formation by encapsulated MSCs.
In this study, we introduced hydrolytically degradable repeat units of caprolactone between the HA backbone and methacrylate groups to control hydrogel degradation. With inclusion of this hydrolytically degradable moiety, photocrosslinked HA hydrogel properties can now be tuned by altering the molecular weight of the HA, total macromer concentration, degree of methacrylation, and now type and the number of degradable repeat units incorporated. Previously, we showed that changes in HA molecular weight affect the viscosity of the macromer solution and that total macromer concentration alters crosslinking density, affecting volumetric swelling, mechanical properties, and enzymatic degradation of the hydrogel[6]. We also synthesized a hydrolytically degradable HA hydrogel with the inclusion of repeat units of poly(lactic acid) between the HA backbone and the methacrylate group[14]. However, these gels degraded very quickly, exhibited cell clustering within the gel, and altered cell morphology. In addition, tunability and processing of this HA macromer was limited and more sensitive, due to the presence of the fast degrading lactic acid unit. Therefore, with the synthesis of MeCLHA, greater control over hydrogel degradation was obtained. These hydrogels could be temporally controlled without the addition of enzyme, and the altering of MeCLHA content in copolymer hydrogels could provide fine-tuning of degradation profiles. The influence of these features on long-term in vitro cultures of MSC-laden hydrogels in chondrogenic media were investigated in this study. The various macromer concentrations (1, 2, and 5 wt%) chosen for this study were based on cell viability, which was compromised at high crosslinking densities (greater than 5wt%), and the formation of a stable scaffold after photopolymerization, where concentrations lower than 1 wt% did not form a hydrogel. In addition, the solubility of MeCLHA dictated an upper limit to the amount of MeCLHA that could be included in the various ratios with MeHA.
The degradation of acellular MeHA hydrogels exhibit an initial release of HA (potentially due to uncrosslinked macromer) which then plateaus over time. Also, the greater percentage of HA release in hydrogels of lower macromer concentrations prior to enzyme addition may be due to a more loosely crosslinked network, which can result in a decreased probability of macromer incorporation into the network and a decrease in the number of bonds that must be broken to release HA into the surrounding PBS solution. Overall, however, these hydrogels are very slowly degrading within the time-scale of these experiments. In contrast, MeCLHA only hydrogels exhibit continual HA release after polymerization until complete degradation, without enzyme addition. Similar to their enzymatically degrading counterparts, total MeCLHA concentration can be altered to increase or decrease the time for complete degradation. In addition, the number of caprolactone repeat units can also be altered (not performed in this study), where decreasing the number of repeat units would result in slower hydrolytic cleavage. Over time, these hydrolytically degradable hydrogels swell, suggesting a bulk degradation mechanism. Furthermore, as HA is released from the hydrogels, the mechanical properties of the hydrogels decrease accordingly. Differences in elastic moduli within wt% groups (5, 2, and 1 wt%) can be attributed to different degrees of HA macromer modification, where MeHA modification was ∼3 times greater than that of MeCLHA for this study, or from degradation differences within the first 24 hours. On the other hand, initial volumetric swelling ratios within each wt% groups were insignificantly different, suggesting that macromer wt% (rather than macromer type) plays a prominent role in swelling, where higher concentrations of the negatively charged HA macromer result in greater retention of water within the hydrogel. However, as HA is released from the hydrogel via hydrolytic degradation, swelling ratios increase with the increased mesh size. In general, the copolymerization of the MeHA and MeCLHA macromers provides a simple method to tune temporal hydrogel properties.
In cellular studies, we show that not only the physical properties of the hydrogel are altered by the network structure, but also cellular responses. Previously, we showed that MSCs, encapsulated at 20 million cells/ml, retained good viability and underwent chondrogenesis in HA hydrogels[7]. However, cell numbers could be altered to further tailor neocartilage production. In this study, the slight decrease in viability in the 5:0 hydrogels (the most densely crosslinked of the hydrogels investigated) may have resulted from nutrient and waste diffusion limitations or increased radical concentrations during crosslinking, while the inclusion of a hydrolytically degradable component in copolymer counterparts allowed for the maintenance of cell viability. In addition, noticeable and significant differences in hydrogel dimensions between wt% groups (5, 2, and 1 wt%) may result from cell-scaffold interactions, where MSCs are able to undergo cellular condensation in the less crosslinked and weaker 1 wt% MeHA hydrogels. Increased hydrogel diameters in 5 and 2 wt% groups over time reflect hydrogel degradation, as temporal decreases in crosslinking density allow for increased volumetric swelling, as well as ECM elaboration and neocartilage formation. Changes in height were less noticeable as height dimensions were smaller than corresponding diameter measurements and may have been limited by the measuring technique. However, it is important to note that the timing and rate of degradation is crucial for functional tissue development. If the degradation rate is faster than ECM deposition, large void spaces created by degradation can result in the loss of cells and ECM proteins into the culture medium. Specifically, the loss of GAGs from the hydrogel can result in compromised mechanical properties, as GAG content is highly correlated to the compressive modulus of native and engineered cartilage tissues[25]. In the case of the 5 wt% hydrogels, the compressive equilibrium moduli decrease in hydrogels with increased MeCLHA content (3:2 and 2:3 hydrogels) and there is no increased GAG accumulation within the hydrogels from day 35 to day 56 that is seen for all other groups. However, when degradation rate compliments the rate of ECM deposition and allows for the distribution of cartilaginous proteins within the hydrogel, compressive moduli increase, as observed in the 2 wt% hydrogels that show an increasing trend in moduli with increased MeCLHA content. Increased distribution of ECM proteins is visualized with immunohistochemical staining. Specifically, at 14 days of culture, staining clearly depicts increased distribution and quantity of chondroitin sulfate in hydrogels with increased MeCLHA content, and at 56 days of culture, increased type II collagen distribution reflects diffusion of larger ECM proteins. Faster and increased distribution of chondroitin sulfate over type II collagen reflect differences in protein size, where the smaller chondroitin sulfate diffuses with greater ease compared to the larger type II collagen fibers[26]. The decrease in acellular elastic moduli and the increase in biochemical content, equilibrium compressive moduli, and ECM distribution in the MSC encapsulated 2wt% copolymer hydrogels reflect a cooperative match in hydrogel degradation rate and ECM deposition.
Furthermore, this study strongly emphasizes the importance of network evolution, where a hydrogel that starts at a higher wt% and decreases to a lower wt% is not equivalent to static hydrogels that start at the higher or lower wt%. Specifically, the 1:1 hydrogels were shown to express up-regulation of type II collagen and aggrecan over the 2:0 and 1:0 hydrogels. Also of importance is hydrogel size and shape throughout culture time. Unlike the dramatic decreases in height and diameter of 1:0 hydrogels, which can pose problems in translation to clinical applications, the 1:1 hydrogels more closely retain their size and shape. With increased GAG/DNA content and size retention, the 1:1 hydrogels show greater promise as an MSC-laden scaffold for cartilage repair compared to 2:0 and 1:0 hydrogels. In addition, although matrix distribution is fairly consistent across groups by 8 weeks, early changes in gel structure are obviously important with respect to final construct properties.
To increase ECM deposition and distribution, others have tailored the degradation of engineered scaffold. Poly(ethylene glycol) dimethacrylate (PEGDM) has been copolymerized with poly(lactic acid)-b-poly(ethylene glycol)-b-poly(lactic acid) encapped with acrylate groups (PEG-LA-DA) to form degradable PEG hydrogels. These chondrocyte-laden constructs showed increased collagen content and distribution in hydrogels with greater degradable group content[9]. In addition, cell-dictated degradation via MMP-based matrix remodeling has been shown to up-regulate expression of type II collagen and aggrecan in MMP-sensitive PEG hydrogels with encapsulated bovine chondrocytes[11]. More recently, degradation triggered by exogenous addition of enzyme has also been explored. In a study by Ng et al, enzyme (agarase) treatment applied to agarose hydrogels resulted in elevated collagen content and dynamic compressive modulus after an initial loss of scaffold properties immediately after enzyme treatment[12]. Furthermore, Rice et al[13] showed that the timing and duration of enzyme (lipase) addition to PEG hydrogels with caprolactone groups greatly affected cartilaginous matrix properties.
Like others, we show that the inclusion of hydrolytically degradable linkages in crosslinked hydrogels results in increased ECM deposition and distribution by encapsulated cells. However, this study focuses specifically on the use of MSCs, rather than isolated chondrocytes. We show clear increasing trends in sGAG/DNA content with degradable content for the 2 wt% groups compared to trends in collagen/DNA content. While sGAG and collagen content still pale in comparison to native cartilage tissue (composed of 2-10% sGAG/wet weight and 5-30% collagen/wet weight[27]), we show improvements in sGAG content/wet weight and increasing equilibrium moduli in the 2wt% hydrogels with increasing MeCLHA content. Although comparisons with other studies are often difficult due to a variety of differing parameters and testing conditions, our results show that the properties of the 2wt% hydrogels improve (increased biochemical content and moduli) during the course of this study, rather than reaching a plateau that has been seen in MSC-laden agarose hydrogels[2]. This could suggest that the dynamic HA hydrogel environment may enhance the potential for neocartilage maturation.
By developing dynamic hydrogels based on HA, a linear polysaccharide found natively in cartilage, we also capitalize on the biological advantages of this molecule. During development, HA regulates a variety of cellular functions (e.g., gene expression, signaling, proliferation, motility, adhesion, and morphogenesis)[28], where HA interactions are MW dependent and mediated through cell surface receptors (e.g., CD44, ICAM-1, and RHAMM)[29, 30]. Thus, HA-based hydrogels have the potential to interact with encapsulated cells via cell surface receptors, and HA macromers released from the hydrogel can serve as biological cues that have the potential to initiate cell signaling pathways or sequester proteoglycan aggregates. The ability to tailor HA macromer release through controlled hydrogel degradation during cartilage repair may play an important role in mimicking the natural time course of cartilage formation and maturation. In native cartilage, HA turns over rapidly with a half-life of 1-3 weeks[31, 32]. Primarily degraded by hyaluronidases (HYAL1, HYAL2, and HYAL3), HA macromers are broken down into HA fragments, which can also induce the expression of MMP-3[33] and nitric oxide synthase[34], synergizing the breakdown and remodeling process. With HA-based hydrogels, these enzymes and other cell-generated reactive oxygen species can assist in the complete breakdown and remodeling of the scaffold over time. Though the degradation products of these hydrogels may yield HA coupled to kinetic chain fragments, we believe that some HA biological cues are imparted on encapsulated MSCs, as we have previously shown that HA chemistry can enhance chondrogenesis[7]. Thus, the HA presentation as a crosslinked network, rather than that presented to cells in native tissue (e.g., within aggrecan molecules) still has biological relevance. The soluble delivery of HA chains or fragments from these hydrogels may also control cellular interactions, but was not specifically explored within this study. However, in this study, we do show that temporal network structure can influence neocartilage formation within HA hydrogels in vitro. Thus, we have developed a photocrosslinkable scaffold with a dynamic network structure, biological relevance, and the ability to support MSC chondrogenesis, as a potential candidate for cartilage repair.
5. Conclusions
This study indicates that the tuning of temporal scaffold properties can be used to control neocartilage production by MSCs in HA hydrogels. The faster degrading MeCLHA component of the hydrogel increases the mesh size and creates void spaces, allowing for the deposition and enhanced distribution of newly synthesized ECM proteins, while the MeHA component provides structural support, maintaining the size and shape of the scaffold, until it is eventually degraded and remodeled by the encapsulated cells. The timing of hydrogel degradation is important since rapid degradation may result in the reduced retention of ECM proteins, whereas hydrogels that degrade too slowly can inhibit tissue formation and distribution. Thus, a careful balance of slow and fast degrading components is needed for optimal growth. Here, the 1:1 hydrogels, with increased mechanical properties and biochemical content over time, while retaining construct size, show great potential as a scaffold to support the production of functional cartilage tissue by MSCs.
Acknowledgments
Support for this research was provided through NIH grants (K22 DE015761 and R01EB008722), an NSF Graduate Research Fellowship (CC), and an NRSA T32 Musculoskeletal Research Training Grant (CC). Authors would like to acknowledge Joshua S. Katz for assistance in running 1H NMR.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Chung C, Burdick JA. Engineering cartilage tissue. Adv Drug Deliver Rev. 2008;60(2):243–62. doi: 10.1016/j.addr.2007.08.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mauck RL, Yuan X, Tuan RS. Chondrogenic differentiation and functional maturation of bovine mesenchymal stem cells in long-term agarose culture. Osteoarthritis and Cartilage. 2006;14(2):179–89. doi: 10.1016/j.joca.2005.09.002. [DOI] [PubMed] [Google Scholar]
- 3.Smeds KA, Pfister-Serres A, Miki D, Dastgheib K, Inoue M, Hatchell DL, et al. Photocrosslinkable polysaccharides for in situ hydrogel formation. J Biomed Mater Res. 2001;54(1):115–21. doi: 10.1002/1097-4636(200101)54:1<115::aid-jbm14>3.0.co;2-q. [DOI] [PubMed] [Google Scholar]
- 4.Burdick JA, Chung C, Jia XQ, Randolph MA, Langer R. Controlled degradation and mechanical behavior of photopolymerized hyaluronic acid networks. Biomacromolecules. 2005;6(1):386–91. doi: 10.1021/bm049508a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chung C, Erickson IE, Mauck RL, Burdick JA. Differential behavior of auricular and articular chondrocytes in hyaluronic acid hydrogels. Tissue Eng Pt A. 2008;14(7):1121–31. doi: 10.1089/ten.tea.2007.0291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chung C, Mesa J, Randolph MA, Yaremchuk M, Burdick JA. Influence of gel properties on neocartilage formation by auricular chondrocytes photoencapsulated in hyaluronic acid networks. J Biomed Mater Res A. 2006;77A(3):518–25. doi: 10.1002/jbm.a.30660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chung C, Burdick JA. Influence of three-dimensional hyaluronic Acid microenvironments on mesenchymal stem cell chondrogenesis. Tissue Eng Pt A. 2009;15(2):243–54. doi: 10.1089/ten.tea.2008.0067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Erickson IE, Huang AH, Chung C, Li RT, Burdick JA, Mauck RL. Differential Maturation and Structure-Function Relationships in MSC- and Chondrocyte-Seeded Hydrogels. Tissue Eng Pt A. 2009 doi: 10.1089/ten.tea.2008.0099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bryant SJ, Anseth KS. Controlling the spatial distribution of ECM components in degradable PEG hydrogels for tissue engineering cartilage. J Biomed Mater Res A. 2003;64A(1):70–9. doi: 10.1002/jbm.a.10319. [DOI] [PubMed] [Google Scholar]
- 10.Lutolf MP, Lauer-Fields JL, Schmoekel HG, Metters AT, Weber FE, Fields GB, et al. Synthetic matrix metalloproteinase-sensitive hydrogels for the conduction of tissue regeneration: Engineering cell-invasion characteristics. P Natl Acad Sci USA. 2003;100(9):5413–8. doi: 10.1073/pnas.0737381100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Park Y, Lutolf MP, Hubbell JA, Hunziker EB, Wong M. Bovine primary chondrocyte culture in synthetic matrix metalloproteinase-sensitive poly(ethylene glycol)-based hydrogels as a scaffold for cartilage repair. Tissue Eng. 2004;10(34):515–22. doi: 10.1089/107632704323061870. [DOI] [PubMed] [Google Scholar]
- 12.Ng KW, Kugler LE, Doty SB, Ateshian GA, Hung CT. Scaffold degradation elevates the collagen content and dynamic compressive modulus in engineered articular cartilage. Osteoarthritis and Cartilage. 2008 doi: 10.1016/j.joca.2008.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rice MA, Anseth KS. Controlling cartilaginous matrix evolution in hydrogels with degradation triggered by exogenous addition of an enzyme. Tissue Eng. 2007;13(4):683–91. doi: 10.1089/ten.2006.0142. [DOI] [PubMed] [Google Scholar]
- 14.Sahoo S, Chung C, Khetan S, Burdick JA. Hydrolytically degradable hyaluronic acid hydrogels with controlled temporal structures. Biomacromolecules. 2008;9(4):1088–92. doi: 10.1021/bm800051m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Goossen LJ, Dohring A. A convenient protocol for the esterification of carboxylic acids with alcohols in the presence of di-t-butyl dicarbonate. Synlett. 2004;(2):263–6. [Google Scholar]
- 16.Bitter T, Muir HM. A modified uronic acid carbazole reaction. Anal Biochem. 1962;4(4):330–4. doi: 10.1016/0003-2697(62)90095-7. [DOI] [PubMed] [Google Scholar]
- 17.Mauck RL, Wang CCB, Oswald ES, Ateshian GA, Hung CT. The role of cell seeding density and nutrient supply for articular cartilage tissue engineering with deformational loading. Osteoarthritis and Cartilage. 2003;11(12):879–90. doi: 10.1016/j.joca.2003.08.006. [DOI] [PubMed] [Google Scholar]
- 18.Soltz MA, Ateshian GA. Experimental verification and theoretical prediction of cartilage interstitial fluid pressurization at an impermeable contact interface in confined compression. J Biomech. 1998;31(10):927–34. doi: 10.1016/s0021-9290(98)00105-5. [DOI] [PubMed] [Google Scholar]
- 19.Singer VL, Jones LJ, Yue ST, Haugland RP. Characterization of PicoGreen reagent and development of a fluorescence-based solution assay for double-stranded DNA quantitation. Anal Biochem. 1997;249(2):228–38. doi: 10.1006/abio.1997.2177. [DOI] [PubMed] [Google Scholar]
- 20.Farndale RW, Sayers CA, Barrett AJ. A direct spectrophotometric microassay for sulfated glycosaminoglycans in cartilage cultures. Connect Tissue Res. 1982;9(4):247–8. doi: 10.3109/03008208209160269. [DOI] [PubMed] [Google Scholar]
- 21.Stegemann H, Stalder K. Determination of hydroxyproline. Clin Chim Acta. 1967;18(2):267–73. doi: 10.1016/0009-8981(67)90167-2. [DOI] [PubMed] [Google Scholar]
- 22.Herbage D, Bouillet J, Bernengo JC. Biochemical and physiochemical characterization of pepsin-solubilized type-II collagen from bovine articular cartilage. Biochem J. 1977;161(2):303–12. doi: 10.1042/bj1610303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Williamson AK, Chen AC, Sah RL. Compressive properties and function-composition relationships of developing bovine articular cartilage. J Orthop Res. 2001;19(6):1113–21. doi: 10.1016/S0736-0266(01)00052-3. [DOI] [PubMed] [Google Scholar]
- 24.Bryant SJ, Nuttelman CR, Anseth KS. The effects of crosslinking density on cartilage formation in photocrosslinkable hydrogels. Biomed Sci Instrum. 1999;35:309–14. [PubMed] [Google Scholar]
- 25.Pfeiffer E, Vickers SM, Frank E, Grodzinsky AJ, Spector M. The effects of glycosaminoglycan content on the compressive modulus of cartilage engineered in type II collagen scaffolds. Osteoarthritis and Cartilage. 2008;16(10):1237–44. doi: 10.1016/j.joca.2008.02.014. [DOI] [PubMed] [Google Scholar]
- 26.Buxton AN, Zhu J, Marchant R, West JL, Yoo JU, Johnstone B. Design and characterization of poly(ethylene glycol) photopolymerizable semi-interpenetrating networks for chondrogenesis of human mesenchymal stem cells. Tissue Eng. 2007;13(10):2549–60. doi: 10.1089/ten.2007.0075. [DOI] [PubMed] [Google Scholar]
- 27.Muir H. The chemistry of the ground substance of a joint cartilage. In: Sokoloff L, editor. The Joints and Synovial Fluid. New York: Academic Press; 1980. pp. 27–94. [Google Scholar]
- 28.Kogan G, Soltes L, Stern R, Gemeiner P. Hyaluronic acid: a natural biopolymer with a broad range of biomedical and industrial applications. Biotechnol Lett. 2007;29(1):17–25. doi: 10.1007/s10529-006-9219-z. [DOI] [PubMed] [Google Scholar]
- 29.Knudson CB, Knudson W. Hyaluronan-Binding Proteins in Development, Tissue Homeostasis, and Disease. Faseb J. 1993;7(13):1233–41. [PubMed] [Google Scholar]
- 30.Menzel EJ, Farr C. Hyaluronidase and its substrate hyaluronan: biochemistry, biological activities and therapeutic uses. Cancer Lett. 1998;131(1):3–11. doi: 10.1016/s0304-3835(98)00195-5. [DOI] [PubMed] [Google Scholar]
- 31.Morales TI, Hascall VC. Correlated Metabolism of Proteoglycans and Hyaluronic-Acid in Bovine Cartilage Organ-Cultures. J Biol Chem. 1988;263(8):3632–8. [PubMed] [Google Scholar]
- 32.Ng CK, Handley CJ, Preston BN, Robinson HC. The Extracellular Processing and Catabolism of Hyaluronan in Cultured Adult Articular-Cartilage Explants. Arch Biochem Biophys. 1992;298(1):70–9. doi: 10.1016/0003-9861(92)90095-e. [DOI] [PubMed] [Google Scholar]
- 33.Ohno S, Ohno-Nakahara M, Knudson CB, Knudson W. Induction of MMP-3 by hyaluronan oligosaccharides in temporomandibular joint chondrocytes. J Dent Res. 2005;84(11):1005–9. doi: 10.1177/154405910508401107. [DOI] [PubMed] [Google Scholar]
- 34.Iacob S, Knudson CB. Hyaluronan fragments activate nitric oxide synthase and the production of nitric oxide by articular chondrocytes. Int J Biochem Cell B. 2006;38(1):123–33. doi: 10.1016/j.biocel.2005.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]