

Abstract
GM-CSF plays a critical role in innate immunity by stimulating the differentiation of tissue macrophages via the transcription factor PU.1. Previous studies showed that GM-CSF deficient (GM-CSF−/−) mice had susceptibility to and impaired clearance of group B streptococcal bacteria by macrophages. For these studies, we hypothesized that GM-CSF−/− mice have increased susceptibility to peritonitis caused by immune dysfunction of peritoneal macrophages. We examined the role of peritoneal macrophages in pathogen clearance, cytokine responses, and survival in a murine cecal ligation and puncture (CLP) model of peritonitis/sepsis. Surprisingly, CLP minimally affected survival in GM-CSF−/− mice while markedly reducing survival in wild type mice. This was not explained by differences in the composition of microbial flora, rates of bacterial peritonitis or sepsis, all of which were similar in GM-CSF−/− and wild type mice. However, survival correlated with peritoneal and serum TNF-α and IL-6 levels which were significantly lower in GM-CSF−/− than control mice. Following peritoneal LPS instillation, GM-CSF−/− mice also had improved survival and reduced TNF-α and IL-6 responses. In vitro studies demonstrated reduced secretion of TNF-α and IL-6 by peritoneal macrophages isolated from sham GM-CSF−/− mice as compared to macrophages from sham control mice. Peritoneal instillation of GM-CSF−/−/PU.1Positive macrophages, but not GM-CSF−/−/PU.1Negative macrophages into GM-CSF−/− mice conferred susceptibility to death following CLP or peritoneal LPS exposure. These results demonstrate that GM-CSF/PU.1-dependent peritoneal macrophage responses are a critical determinant of survival following experimentally induced peritonitis/sepsis or exposure to LPS and have implications for therapies to treat such infections.
Keywords: Sepsis, cecal ligation and puncture, Granulocyte Macrophage-Colony Stimulating Factor, macrophage
INTRODUCTION
Epidemiologic data on sepsis estimate an annual incidence of 751,000 cases, a mortality rate of 28.8% and related health care costs of nearly 17 billion dollars. These data highlight the tremendous societal burden of sepsis which remains the most common cause of death in the adult ICU population [1]. In response to this challenge, both clinical and basic science investigators have continued to try to elucidate the pathophysiologic mechanisms mediating sepsis with the goal of defining improved therapeutic approaches. Despite advances in the physiologic and pharmacologic management of the critically ill septic patient, few therapeutic innovations have had a dramatic impact on the devastating sequelae of sepsis with the exception of the reported efficacy of activated protein C [2].
The pathophysiology of sepsis is linked to immunobiology of host-pathogen interactions. Organisms possess pathogen-associated molecular patterns (PAMPs) that uniquely differ from the host and are efficiently recognized by receptors on the cells of the innate immune system [3–5]. A key example of this in the pathobiology of sepsis is the recognition of the Gram negative bacterial cell wall product, lipopolysaccharide (endotoxin), by the Toll-like receptor (TLR)-4 complex [6]. As a result of engagement of cell surface receptors, a myriad of key signal transduction events are initiated ultimately resulting in the production of a variety of immune active proteins including cytokines (e.g. tumor necrosis factor (TNF)-α), chemokines (e.g. CXCL8/IL-8) and adhesion molecules (e.g. ICAM-1) all of which have been implicated as important mediators in the systemic inflammatory response syndrome in sepsis [7, 8]. Disappointingly, several clinical trials aimed at modulating the activity of proinflammatory mediators have failed to significantly improve outcome [9]. This has led to further considerations of alternative hypotheses that may explain the high mortality associated with sepsis and the underlying immunobiology responsible for this.
It has been proposed that an over-exuberant counter regulatory response, termed the “Compensatory Anti-inflammatory Response Syndrome” (CARS), may follow the hyper-inflammatory phase of sepsis and leave the host at risk from further infectious complications during the late phase of sepsis [10–12]. For example, increased levels of IL-10 have correlated to decreased expression of HLA-DR [13, 14] and a state of immune paralysis is associated with an increased risk of death from impaired pathogen clearance [15]. Thus, it has been suggested that amplification of the host inflammatory response may assist in eradicating an invading pathogen and may afford a clinical benefit in late sepsis. As a result, administration of the stimulatory growth factor, granulocyte/macrophage-colony stimulating factor (GM-CSF) has attracted attention due to its role in the proliferation and differentiation of granulocytic and monocytic progenitor cells [16–18].
As a growth factor that stimulates proliferation and differentiation of myeloid cells, GM-CSF plays a key in the functional maturity of monocytes and neutrophils [19]. In vitro studies have shown that it modulates mature cells by enhancing antigen presentation, altering leukocyte adhesion, increasing complement and antibody mediated phagocytosis, as well as potentiating cytokine release all of which would aid host pathogen clearance. In corroborative studies, the absence of GM-CSF in the genetically altered null mouse (GM-CSF−/−) was associated with decreased cytokine production, notably TNF-α, in response to highly purified Pseudomonas LPS stimulation [20]. This effect was mediated by GM-CSF influence on the differentiation of monocytes and macrophages that resulted in altered expression of a subset of proteins crucial to this innate immune response, importantly CD14 [20]. This decreased expression could be rescued by the over-expression of the differentiation factor, PU.1. In contrast, we’ve shown that in humans and mice, abrogation of GM-CSFsignaling (by anti-GM-CSF antibodies in patients with pulmonary alveolar proteinosis or the GM-CSF null mice) reduces, but does not completely abolish, multiple neutrophil functions as morphology, cell-surface differentiation markers, and PU.1 expression are not altered [21]. These data suggest that while macrophage-dependent cytokine responses will be altered, neutrophil differentiationmay remain intact. The current studies were designed to test the hypothesis that GM-CSF was critical to host protection from infection by examining the mortality and immune response to cecal ligation and puncture in the GM-CSF null mutant mouse. The deletion of GM-CSF results in multiple immune function abnormalities in macrophages secondary to ineffective differentiation including impairment of phagocytosis, oxygen radical production and cytokine signaling [22, 23]. Thus, we hypothesized that deficiencies in the innate immune function of the GM-CSF null mouse would be associated with defective pathogen clearance and increased mortality in CLP-induced sepsis.
METHODS
Animals
Adult female mice (10–12 weeks/18–22 grams) were used in these experiments. GM-CSF null mice were generated as previously described [24, 25] and were maintained in the C57Bl/6 background. C57Bl/6 wild type mice were obtained from Jackson Laboratories (Bar Harbor, ME) and acclimated in the same husbandry room as GM-CSF knock-out mice for a period of 2 weeks to allow for adaptation to the environment and exclude any animals with pre-existing disease. During this period and throughout the experiments animals were allowed free access to food and water. Mouse colonies were housed in an IACUC-approved, pathogen-free barrier facility and studied under protocols approved by the Institutional Animal Care and Use Committee.
Cecal ligation and Puncture
Anesthesia was maintained with isoflourane delivered via self-scavenging anesthesia machine using a modified murine nose cone apparatus. (Flow 2.25L/min with FIO2 = 0.7). The abdomen was prepped with betadine and alcohol swabs. A 1 cm incision was made in the skin and peritoneum. Blunt dissection was utilized to identify the cecum in situ. The cecum was then delivered to a sterile operative field on abdominal surface. 3-0 silk suture was used to ligate the cecum at its proximal aspect without occlusion of the intestinal lumen. A 21-guage needle was use to puncture the cecum through and through twice and patency of punctures were assured by gentle expression of fecal material. The peritoneum was then closed using a horizontal mattress suture technique and the skin incision closed using Dermabond skin adhesive. Mice were resuscitated with 1cc of warmed, normal saline injected subcutaneously followed by placement in a warming bed until all mice with fully recovered from anesthesia and ambulatory. Of note, although antibiotics have been clearly shown to modify the course of CLP, they were not provided in the course of these experiments in order to specifically determine the effect of GM-CSF knock-out on host containment of pathogens.
Microbiological analysis
Twenty-four hours after CLP, mice were euthanized with isoflourane. The ventral abdominal wall was incised and the peritoneal space was irrigated with 70% (v/v) ethanol in water. The inferior vena cava was cannulated and blood was withdrawn into heparinized tubes and transferred immediately into sterile culture vials. Both qualitative and quantitative microbial cultures were performed by the clinical laboratory at the Cincinnati Children’s Hospital Medical Center clinical laboratory using standard clinical laboratory microbiological protocols.
Lipopolysaccharide (LPS) administration
In a separate set of experiments, all mice were weighed prior to administration of previously described isoflourane anesthesia. Upon adequate sedation, the abdomen was prepped with betadine and alcohol swabs. Lipopolysaccharide (LPS) (E. coli serotype 055:B5; Sigma, St. Louis, MO) was injected intraperitoneally at a dose of 50 μg/kg/body weight corresponding to a mouse LD50 dose as previously observed [26]. Animals were resuscitated under warming lamp and monitored until fully recovered from anesthesia and ambulatory.
Enzyme-linked immunosorbent assay (ELISA)
Serum, peritoneal fluid and culture supernatant levels of IL-6 and TNF-α were measured using commercially available ELISA kits (Biosource International, Camarillo, CA) specific for the cytokines and in accordance with the manufacturer’s recommendations. Absorbance values were read by spectrophotometry and converted to concentrations (pg/ml) by comparison with the standard curves. Limits of detection by ELSIA were 20 pg/ml for both TNF-α and IL-6. Values are expressed as the mean ± standard error of the mean for all groups.
Isolation and culture of peritoneal macrophages
At the designated time points after CLP peritoneal macrophages were obtained from euthanized mice by lavaging the peritoneum with 8–9 mls of warmed DMEM culture media under sterile conditions. Collected media and cells were centrifuged (1000 rpm, 5 mins) and then resuspended in 5 mls fresh DMEM media. Total white blood cell counts were obtained using a Beckman Coulter counter. A differential cell count was obtained from cytospin (Shandon) preparations of two 100 μl cell samples transferred to slides that were fixed and stained with ABC products (Vectastain) and examined by light microscopy by a blinded investigator (B.Z.). This provided the percent of macrophages in the retrieved cells so that the volume containing 1 × 106 macrophages was determined. The remaining peritoneal lavage samples were subjected to centrifugation (1200 rpm × 10 minutes) and the supernatant stored at −70°C for analysis of cytokine levels by ELISA. The cells were washed twice and resuspended in warmed DMEM media supplemented with 8% fetal calf serum (FCS) (Gibco BRL, Grand Island, NY). The volume containing 1 × 106 macrophages (range of volumes 1.4–2.7 mls) was added to each well of a 6-well flat bottom culture plate to allow for adherence of macrophages. After 2 hours of incubation (37°C/5% CO2), the media and non-adherent cells were removed and adherent cells (>95% CD14+ macrophages by FACS) were gently washed and treated with 2 mls fresh DMEM/FCS media (37°C/5% CO2) overnight. This process allowed for a consistent number of macrophages to be examined in the in vitro studies. At 24 hours, cell culture supernatant was collected and stored for analysis of spontaneous cytokine expression by ELISA. Cells were then stimulated with 100 ng/ml media LPS (E. coli serotype 055:B5; Sigma). At 24 hours after LPS stimulation, the cell culture supernatant was collected and stored for analysis of stimulated cytokine production by ELISA. Thus, both spontaneous (non-stimulated) cytokine production and stimulated cytokine production were determined in these experiments.
GM-CSF reconstitution with PU.1
PU.1 is a critical transcription factor expressed selectively in myeloid cells and as a “master” transcription factor, it regulates numerous genes that play an important role in monocyte differentiation. Fully-differentiated functionality of macrophages from the GM-CSF knock out mouse can be reconstituted by transfection with a PU.1 containing retroviral vector [27]. Stably transfected PU.1 expressing or control vector transduced cells were injected into the abdominal compartment of GM-CSF−/− mice at a concentration of 1 × 106 cells/ml 24 hours prior to CLP (21 gauge, 2 punctures) or LPS administration (50 mg/kg body weight). Mortality up to 14 days was tracked and reported as Kaplan-Meier plots.
Statistical Analysis
All continuous numerical data are expressed as the mean± standard error of the mean (SEM) and the number of mice per group (n) indicated. Statistical comparisons of group data were performed using multiple groups ANOVA, followed by Tukey’spost hoc analysis. Survival data is presented as Kaplan-Meier plots and was comparison of survival in different groups was performed using Fisher’s exact test with a value of p < 0.05 considered as significant.
RESULTS
GM-CSF deficiency protects against mortality following CLP
In order to evaluate the role of differentiated peritoneal macrophages during intra-abdominal sepsis induced by CLP, mortality was compared between C57Bl/6 wild type (WT) and GM-CSF null mutant (−/−) mice. In this study, a total of 29 WT and 26 GM-CSF−/− female mice that were matched for age, weight and environmental conditions underwent CLP as described in Methods and followed for mortality. Sham mice (n=6) in each group were similarly tracked. Because we were specifically interested in the endogenous immune response and pathogen clearance, antibiotics were not given; however, all mice received 1 cc (about 50 cc/kg body weight) of normal saline intraperitoneally as fluid resuscitation. All CLP mice appeared lethargic in the early post-operative hours (1–2 hs). General progression of illness was characterized by hunched backs, ruffled fur, diarrhea and ocular discharge within 12 hours; however, these symptoms were qualitatively decreased in GM-CSF−/− mice. Contrary to our hypothesis, the overall mortality was significantly higher in WT mice. Mortality was 93% (27 out of 29 animals) in the WT mice versus 15% (4 out of 26 animals) in the GM-CSF −/− (p<.001) (Figure 1). Sham surgery without CLP did not result in any deaths over a similar period of evaluation in either wild-type or GM-CSF−/− mice (n=6/group; data not shown).
Figure 1. Mortality following cecal ligation and puncture (CLP) is reduced in GM-CSF deficient mice.

Wild-type (n=29) or GM-CSF−/− (n=26) mice were subjected to CLP and then mortality was evaluated every 12 hours for 7 days.
In order to evaluate the possibility that there might be intrinsic differences in the intestinal flora which led to a variable CLP-induced inflammatory response between the two groups, both qualitative and quantitative blood cultures were obtained 24 hours following CLP. WT and GM-CSF−/− mice underwent CLP (n=5 per group) or sham surgery. Qualitative cultures confirmed that CLP resulted in a polymicrobial bacteremia. The presence of group B streptococcus, enteroccocus, and E. coli did not differ between the WT and GM-CSF−/− groups (Table 1). Of note, sham mice which underwent only anesthesia, abdominal incision, and cecal manipulation did not demonstrate bacteremia as determined by blood culture. Furthermore, in a separate set of mice undergoing CLP, quantitative cultures for group B streptococcus and enterococcus demonstrated expected variability, but no significant differences in numbers of colony forming units in blood culture positive WT and GM-CSF−/− mice (Table 2). Intriguingly, these data suggested that the observed difference in mortality appeared to be unrelated to bacterial load or pathogen clearance directly.
Table 1.
Qualitative microbial culture results of blood taken 24 hours after CLP.
| Mouse | Group B strep | E. coli | Enterococcus | Mouse | Group B strep | E. coli | Enterococcus |
|---|---|---|---|---|---|---|---|
| WT sham | − | − | − | GM−/− sham | − | − | − |
| WT CLP-1 | + | − | − | GM−/− CLP-1 | + | + | + |
| WT CLP-2 | + | − | + | GM−/− CLP-2 | + | + | + |
| WT CLP-3 | + | + | + | GM−/− CLP-3 | + | − | − |
| WT CLP-4 | + | + | − | GM−/− CLP-4 | + | − | + |
| WT CLP-5 | + | + | + | GM−/− CLP-5 | + | + | + |
| Total | 5/5 | 3/5 | 3/5 | 5/5 | 3/5 | 4/5 |
Table 2.
Qualitative microbial culture results of blood taken 24 hours after CLP*
| Mouse | Anaerobic strep | Enterococcus | Mouse | Anaerobic strep | Enterococcus |
|---|---|---|---|---|---|
| WT sham | 7 | 0 | GM−/− sham | 0 | 0 |
| WT CLP-1 | 27 | 0 | GM−/− CLP-1 | 10 | 200 |
| WT CLP-2 | 150 | 300 | GM−/− CLP-2 | 22 | 0 |
| WT CLP-3 | 33 | 0 | GM−/− CLP-3 | 18 | 10 |
| WT CLP-4 | 0 | 15 | GM−/− CLP-4 | 0 | 20 |
| WT CLP-5 | 10 | 20 | GM−/− CLP-5 | 120 | 200 |
| Total | 220 4/5 |
335 3/5 |
170 4/5 |
430 4/5 |
Data are expressed as colony forming units (CFU).
GM-CSF deficiency protects against mortality following peritoneal administration of LPS
Contrary to our original hypothesis mortality appeared to be independent of the type and degree of bacteremia. As a result, we surmised that mortality might instead be related to differences in the immune responsiveness of the peritoneal macrophages stimulated by bacteria/bacterial products released during evolving CLP. Thus, to test this hypothesis and exclude pathogen clearance as a factor, the model was simplified to an intraperitoneal challenge with an LD50 dose of endotoxin (LPS). In this experiment, 19 WT and 20 GM-CSF−/− age- and weight-matched female mice were injected intraperitoneally with LPS and observed for 2 weeks to determine mortality. No mice in either group injected with PBS as vehicle control died (data not shown). All LPS-treated WT mice initially appeared lethargic and subsequent manifestations of illness included hunched backs, ruffled fur, diarrhea and ocular discharge prior to death. GM-CSF−/− mice, though initially lethargic, returned to a healthy state within 36 hours. Consistent with the findings with CLP, a significant difference in mortality was observed between the WT (9 out of 19, 47% mortality) and GM-CSF−/− mice in which no deaths were observed (mortality was 0 of 20) (Figure 2). These results suggested that mortality differences between WT and GM-CSF−/− mice induced by either CLP or LPS were similar and independent of the presence of a pathogen. As a result, we hypothesized this was related to a difference in the inflammatory response to the pathogenic challenge rather than the presence of the invasive organisms.
Figure 2. Mortality following intraperitoneal LPS administration is reduced in GM-CSF deficient mice.

Wild-type (n=19) or GM-CSF−/− mice (n=20) received LPS by intraperitoneal injection and then mortality was evaluated every 12 hours over 7 days.
The proinflammatory TNF-α and IL-6 responses to CLP and peritoneal LPS administration are impaired in GM-CSF−/− mice
TNF-α and IL-6 are known to play a significant role in the pathogenesis of septic shock [28, 29]. Therefore, levels of these cytokines in both the serum and peritoneal lavage fluid were measured at 6, 12, 18, and 24 hours post-CLP to evaluate for differences in the kinetics and/or degree of cytokine response between mouse groups (n=4 sham, n=8 CLP per group). At all time points examined GM-CSF−/− mice produced less TNF-α and IL-6 as measured by ELISA when compared to WT mice; however, for TNF-α, statistically significant differences were noted starting at 18 hours and were maximally different in both serum and peritoneal fluid by 24 hours (Figure 3). For IL-6, significant differences in production were noted during the early phase after CLP (6 hours, Figure 4A), a time point which had been previously shown to reflect peak of IL-6 expression [30, 31] and to correlate with CLP-induced mortality [32]. In addition, while IL-6 levels were not significantly different at 12 hours, differences were noted later in the time course at 18 and 24 hours (Figure 4B) in both the serum and peritoneal fluid. These data suggested that maturational defects in peritoneal macrophages of GM-CSF null mice result in an attenuated proinflammatory cytokine response following CLP-induced peritoneal sepsis and may explain the differences in mortality despite similar bacterial burdens.
Figure 3. The proinflammatory TNFα response to CLP is reduced in GM-CSF deficient mice.
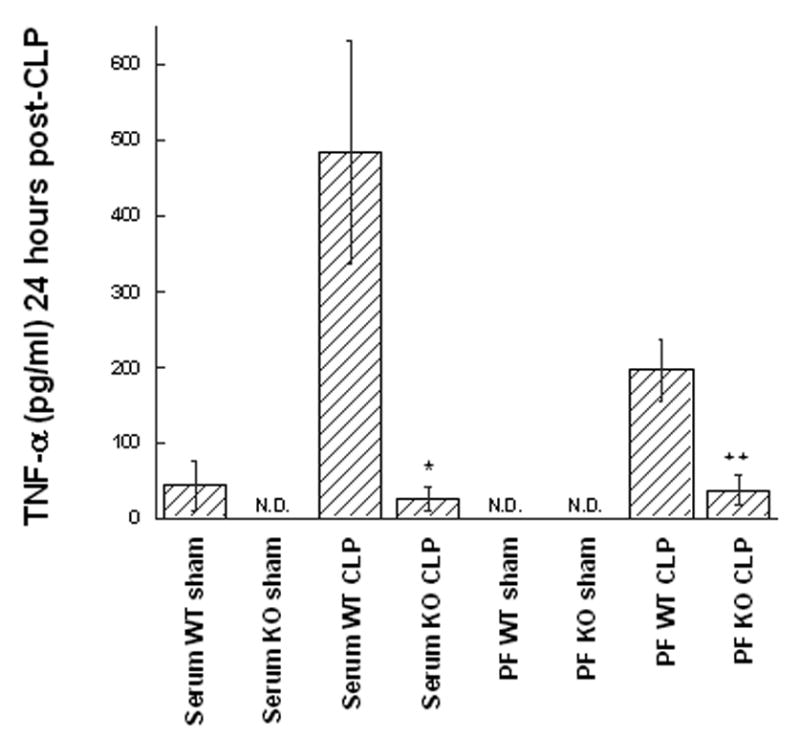
Wild type (WT) or GM-CSF−/− (KO) mice were subjected to the CLP procedure (n=8/group) or sham surgery (n=4/group) and the TNF-α levels were quantified in peritoneal fluid (PF) or serum after 24 hr. For statistical comparisons between corresponding values for GM-CSF−/− and WT mice, p values of <0.01 (*) and <0.001 (**) are indicated.
Figure 4. The IL-6 response to CLP is reduced in GM-CSF deficient mice.
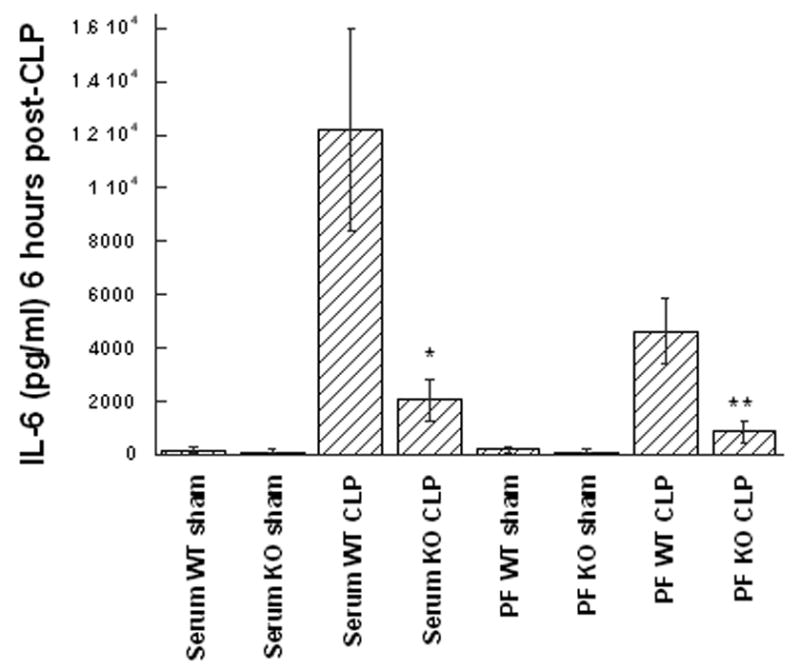
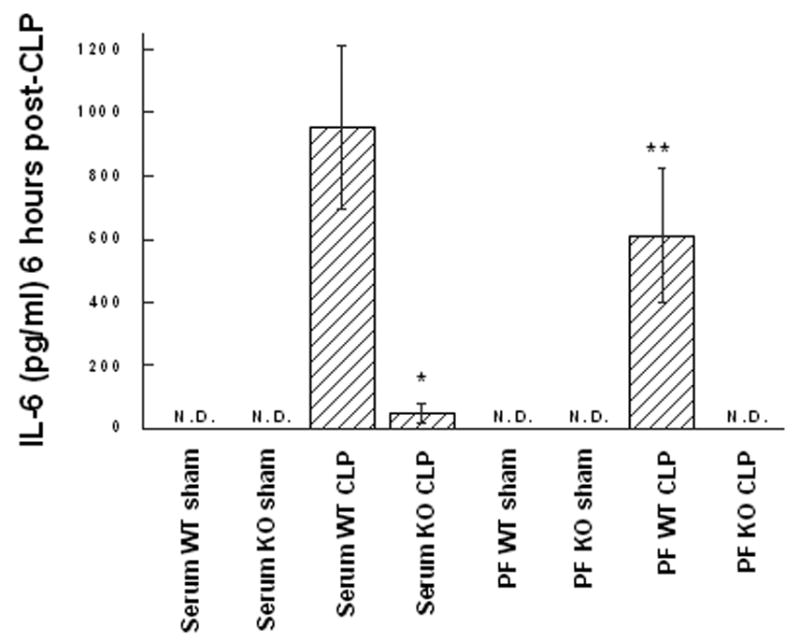
Wild type (WT) or GM-CSF−/− (KO) mice were subjected to the CLP procedure (n=8/group) or sham surgery (4/group) and the TNF-α levels were quantified in peritoneal fluid (PF) or serum after 6 hr (A), or 24 hr (B). For statistical comparisons between corresponding values for GM-CSF and wild type mice, p values of <0.01 (*) and <0.001 (**) are indicated.
Ex vivo response to LPS in peritoneal macrophages isolate from WT and GM-CSF−/− mice following CLP
To further elucidate the role of the peritoneal macrophage in the generation of inflammatory cytokines following CLP, cells isolated by peritoneal lavage at 6, 12, 18, and 24 hours after sham surgery or CLP in either WT or GM-CSF−/− (n=8 per group, per time point) were analyzed using an in vitro cell culture model. Cell counts and differentials on isolated peritoneal macrophages were performed so as to plate 1 × 106 cells per well as described in Methods.
Consistent with prior observations in alveolar macrophages [20], ex vivo analysis of peritoneal macrophages isolated from GM-CSF−/− mice at 24 hours following sham surgery demonstrated an attenuated response to endotoxin stimulation as compared to WT mice. Similar to the reported alveolar macrophage response to Pseudomonas LPS [20], LPS stimulation (100 ng/ml media) of peritoneal macrophages from sham GM-CSF−/− mice produced significantly less TNF-α as compared to peritoneal macrophages from sham WT mice which showed a robust response (Figure 5). Thus, the characteristically robust ex vivo responsiveness of these naive peritoneal macrophages from WT mice was significantly attenuated in the GM-CSF−/− mice. In contrast, macrophages retrieved from WT mice 24 hours following CLP, when intraperitoneal cytokine levels were substantially elevated, (see Figures 3 and 4) showed a significantly attenuated response to LPS stimulation as compared to sham operated WT mice (Figure 5). This attenuated ex vivo response to LPS following a prior exposure to an inflammatory stimulus (here equated to the cytokine milieu within the peritoneal cavity) is characteristic of the phenotypic response of endotoxin-tolerant cells. Conversely, macrophages from CLP GM-CSF−/− mice, in which the peritoneal cytokine levels where significantly less compared to WT, had little change in the response to ex vivo LPS stimulation (Figure 5) consistent with an absence of a “tolerized” or “reprogrammed” response in the GM-CSF−/− mice peritoneal macrophage.
Figure 5. Ex vivo LPS response of peritoneal macrophages is altered in GM-CSF deficient mice.
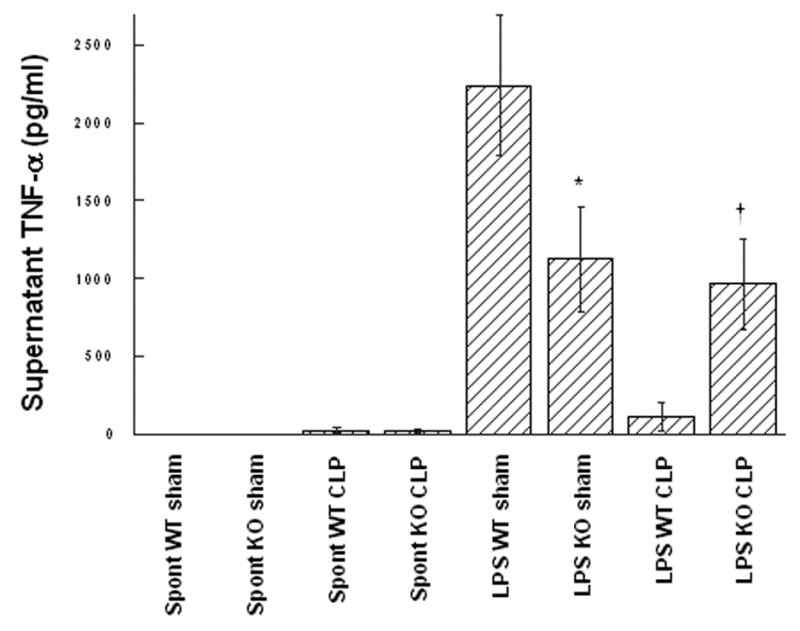
Wild type (WT) or GM-CSF−/− mice (KO) were subjected to the CLP procedure (n=9 in WT group/n=16 in GM-CSF−/− group) or sham surgery (n=3 in WT group/n=7 in GM-CSF−/− group) and 24 hours later, peritoneal macrophages were evaluated ex vivo for spontaneous (spont), or LPS-stimulated (LPS) release of TNF-α as described in Materials and Methods. For statistical comparisons between corresponding values for macrophages from GM-CSF and wild type mice, p values of 0.35 (*) and <0.01 (†) are indicated.
Mortality in response to CLP or peritoneal LPS administration requires the presence of PU.1-expressing, differentiated peritoneal macrophages
Together, the data above suggested that deficiency in maturation of peritoneal macrophages present in GM-CSF−/− mice resulted in an attenuated cytokine response to pathogenic challenge and favored improved mortality. In order to provide more conclusive data regarding the role of the peritoneal macrophage, we aimed to reverse this cellular abnormality in the peritoneal cavity of the GM-CSF−/− mice. We hypothesized that restoring a pool of differentiated macrophages would reverse the mortality discrepancy between the WT and GM-CSF−/− mice. To achieve this, GM-CSF−/−-derived macrophages that were stably transfected with the differentiating factor, PU.1, to reverse the maturational defect were injected into the abdominal cavity at a concentration of 1 × 106 cells/ml 24 hs prior to CLP. Experimental groups consisted of: 1) GM-CSF−/− mice (n=15) transplanted with PU.1-reconstituted GM-CSF null macrophage (MAM-PU.1) cells which had been shown to reverse the functional maturational defects in GM-CSF null cells [27, 33]; 2) GM-CSF−/− mice (n=15) transplanted with MAM cells (MAM-empty) that were stably transfected with the empty vector (differentiation defects not corrected); and 3) WT mice (n=18) also transplanted with MAM-empty cells. After CLP in GM-CSF−/− mice transplanted with MAM-empty macrophages mortality was low (6%; 1/18) (Figure 6A) and not different from unmanipulated GM-CSF−/− mice (see Figure 1). Also similar to previous results, WT mice transplanted with MAM-empty cells demonstrated 7 day mortality of 53% (8/15) consistent with results obtained with unmanipulated WT mice (see Figure 1). However, mortality in GM-CSF−/− mice transplanted with PU.1-reconstituted MAM cells was significantly higher (mortality of 27%; 4/15; p<0.05) than that observed in the MAM-empty transplanted GM-CSF−/− mice, but still not to the full degree observed in MAM-empty transplanted WT mice (Figure 6A).
Figure 6. Mortality following CLP or peritoneal LPS administration is dependent on expression of PU.1 in peritoneal macrophages.

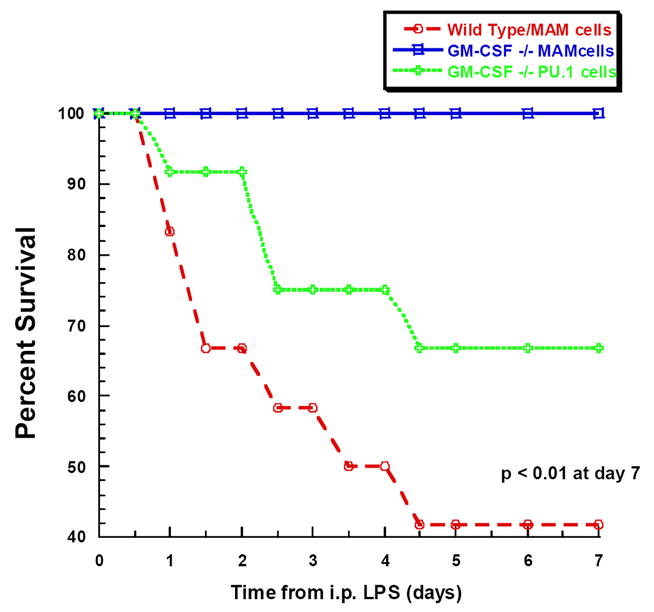
A. GM-CSF−/− mice received GM-CSF−/−/PU.1Negative macrophages (mAM cells) or GM-CSF−/−,/PU.1Positive macrophages (mAMPU.1+ cells) and WT mice received mAM cells, all by passive intraperitoneal transfusion (n= 15, 15, 18 per group, respectively). Twenty-four hours later, all mice underwent CLP and mortality was evaluated every 12 hr for 7 days. B. GM-CSF−/− and WT mice were injected with either mAM or mAMPU.1+ cells as above (A) (n=12 mice/group). Twenty-four hours later all mice received LPS by intraperitoneal injection and mortality was evaluated every 12 hr for 7 days.
To again evaluate whether this was related to pathogen load or clearance, these same experimental groups consisting of MAM-empty transplanted WT mice, MAM-empty transplanted GM-CSF−/− mice, and GM-CSF−/− mice transplanted with PU.1-reconstituted MAM cells (n=12 per group) were challenged with intraperitoneal LPS. Similar to previous results shown in Figure 2, mortality after LPS in PU.1-transplanted GM-CSF−/− mice was 58% (7/12) as compared to 0% (0/12) in MAM-empty transplanted GM-CSF−/− mice (Figure 6B). Mortality in the GM-CSF−/− mice transplanted with PU.1-reconstituted MAM cells was 33% (4/12) which was significantly greater than the GM-CSF−/− mice transplanted with undifferentiated MAM cells, but again not to the full degree observed in WT mice.
These data suggest that the presence of differentiated macrophages in the peritoneal compartment appear to mediate cytokine production and contribute to a significant, though not full percentage of the mortality associated with either CLP or intraperitoneal LPS. However, it appears that additional factors such as a second compartment of immune active cells affected in the GM-CSF null mice, but not replaced by intraperitoneal transplantation strategy, mediate an additional and significant percentage of the observed mortality.
DISCUSSION
In the surgical patient, intra-abdominal infection is one of the major causes of septic shock and multiple organ failure; however, the mechanism of disease progression and etiology of mortality remains unclear in both clinical and animal models. One of the most commonly employed animal models used to model this biology of sepsis is the cecal ligation and puncture (CLP) model [34]. CLP induces peritonitis by mixed intestinal bacterial flora and is thought to be more consistent with the pathophysiology of intra-abdominal sepsis than administration of various isolates. This model can be manipulated in a number of ways in order to provide a predictable degree of inflammatory responses and mortality by varying the size gauge needle used, number of punctures, expression of fecal material, provision of fluid resuscitation, and provision of antibiotics [35]. In the present studies, incomplete maturation of macrophages in the GM-CSF knock-out mouse allowed us to examine the role of the peritoneal macrophage in the inflammatory response induced by CLP.
Previous work has identified GM-CSF to be an important inducer of macrophage differentiation [36]. Cell isolated from these mice had been shown to display defects in endocytic internalization of adenovirus and non-specific opsonization suggesting the absence of GM-CSF may have important consequences on pathogen clearance. In light of these observations, we hypothesized that deficiencies in innate immune function related to an undifferentiated macrophage phenotype in the GM-CSF null mouse would lead to inadequate pathogen clearance and increased mortality in CLP-induced sepsis. To our surprise, a significant decrease in mortality was noted in GM-CSF−/− mice as compared to WT mice from the same genetic background. In elucidating causal factors contributing to this finding, it was noted that both the type (qualitative cultures) and burden (quantitative cultures) of bacteria isolated from the blood 24 hours following CLP did not differ between the two groups of mice. Our experiments were designed to emphasize alterations in pathogen clearance that might differ between the groups so that antibiotics were not given in these experiments. While we acknowledge this would likely change the degree of mortality we observed, because the bacterial burden was similar and mortality continued to differ dramatically in a “sterile” model of sepsis, it is unlikely the addition of antibiotics to these studies would have provided any additional mechanistic insight.
In utilizing the two models (CLP and i.p. LPS), it became clear that activation of peritoneal inflammation, regardless of pathogen clearance capacity, differed greatly between GM-CSF−/− and WT mice. The results suggested that pathogen clearance in the model employed had less of an impact on mortality than the ability to mount a robust, proinflammatory cytokine response. As previous investigations had suggested that increased IL-6 production was highly predictive of mortality in CLP [30], our mortality results were not surprising as significant differences in the IL-6 responses were observed regardless of the stimulus (CLP or LPS). These studies emphasized the need to understand the regulation of cytokine response in peritoneal macrophages isolated from the GM-CSF−/− mice.
Prior studies using the GM-CSF−/− mice have begun to elucidate the potential mechanisms that would explain the attenuation of cytokine production focusing on at least one of the canonical pathways involved in the response to LPS, TLR4 signaling [20]. In these studies, alveolar macrophages isolated from the GM-CSF−/− mice showed significantly decreased TNF-α production following Pseudomonas-derived LPS stimulation that was related to altered expression of components of the TLR4 signaling pathway, notably CD14, though TLR4 expression was unaffected [20]. Interestingly, in these studies decreased expression of IRAK-M was also seen in alveolar macrophages from the GM-CSF−/− mice. These data are relevant to our observations of what we have described as an “endotoxin tolerant” phenotype of the peritoneal macrophages isolated from WT mice subjected to CLP. Using classical terminology, peritoneal macrophages from sham operated WT mice could be described as “naive” cells and maintained a robust, ex vivo response to LPS stimulation (Figure 5). In considering the cytokine milieu of the peritoneal cavity as the “prior stimulus”, we argue that the attenuated response to LPS stimulation of macrophages from WT CLP mice (Figure 5) is consistent with an endotoxin tolerant phenotype. Conversely, macrophages isolated from the GM-CSF−/− mice subjected to CLP showed no evidence of tolerization to the ex vivo stimulation with LPS. This is notable as IRAK-M has been strongly implicated in the establishment of endotoxin tolerance [37–39]; thus, a decreased expression of IRAK-M in peritoneal macrophages from GM-CSF−/− mice could explain the failure to acquire this phenotype following CLP in these cells. However, given the complex nature of cellular recruitment to sites of inflammation (i.e. the peritoneal cavity) and expected release of differentially mature circulating cells from the bone marrow following the CLP trigger, it is difficult to exclude the possibility that different macrophage phenotypes are being compared.
In the absence of GM-CSF, reconstitution of differentiation and full functionality of macrophages derived from the mice can be achieved in two ways. First, prior studies have shown that alveolar macrophages isolated from these mice display similar functional defects, which have been successfully corrected by restoring GM-CSF to the lungs by inclusion of a germline transgene that expresses GM-CSF only in the lungs, by adenovirus-mediated, pulmonary GM-CSF gene delivery and expression, and by direct aerosol administration of GM-CSF to the lungs. In all cases, the delivery of protein stimulated maturation of the macrophages in vivo. In the present studies, we utilized a second approach of transfecting null cells with a PU.1 containing retroviral vector [27]. PU.1 is a critical transcription factor expressed selectively in myeloid cells and as a “master” transcription factor is able to regulate those genes that are key to monocyte differentiation. The ability to utilize a PU.1 stably transfected GM-CSF−/− cell line afforded the opportunity to see what role the peritoneal macrophage played in this response by transplanting the PU.1 expressing (or control vector) transduced cells into the abdominal cavity to determine whether full mortality (that is the percentage observed in GM-CSF+/+ mice) could be restored. In both models of peritoneal-induced inflammation, a significant increase in mortality was observed in the GM-CSF−/− mice transplanted with PU.1-differentiated cells as compared to the null mice transplanted with PU.1 null cells; however, in neither the CLP nor the i.p. LPS model was mortality restored to the level observed in WT mice. That full reconstitution of the macrophage responsiveness in the GM-CSF−/− to that of the WT mice could not be achieved cannot be excluded in the current studies. So that whether these findings reflect a failure to fully restore macrophage functionality in the peritoneal cavity via this transplantation route, or that a second pool of macrophage-derived cells contribute to the lethality associated with both models remains unknown. In a limited number of GM-CSF−/− mice reconstituted with PU.1-differentiated cells the “dose” of transplanted cells was increased to 5 × 106 cells, however these mice did not demonstrate a mortality rate significantly different from those given 1 × 106 cells that were reported in Figure 6. In our experience with routine lavage of an unmanipulated peritoneal cavity in mice, this range of cell number (1–5 × 106 cells) is a typical amount recovered. Thus, we speculate that the inability to restore mortality to the degree seen in WT mice more likely reflects a second pool of immunoactive cells derived from the monocyte/macrophage lineage that are effected by the absence of GM-CSF. Kuppfer cells in the liver, circulating monocytes and alveolar macrophages could all be affected in the GM-CSF−/− mice and would have been unaffected by intraperitoneal transplantation in the short term. On-going studies are aimed at identifying a second set of cells potentially contributing to this incomplete reconstitution of mortality.
It was surprising to observe that there was a lack of correlation between the degree and/or type of bacteremia and mortality. The cecal ligation and puncture model has previously been shown to result in a polymicrobial bacteremia as was observed in the current studies. Admittedly, antibiotics have clearly been demonstrated to affect the mortality rate, particularly at when higher lethality is achieved with larger bore punctures [34]. Though antibiotics were not administered in these studies, it appeared that in many cases, GM-CSF−/− mice were capable of ultimately containing and clearing the ischemic cecum that served as the early nidus of infection. From these studies it appears that a diminished cytokine response to bacterial infection plays a more dominant physiologic role in subsequent mortality in this mouse CLP model while some neutrophil function targeting bacterial clearance is maintained. Relevant to our observation with the pathogen challenge inherent to the CLP model, abrogation of GM-CSFsignaling has been shown to reduce, but not completely abolish neutrophil function [21]. These data are consistent with our findings of diminished macrophage-dependent cytokine responses in the GM-CSF−/− mice, but predict that neutrophil differentiation and some functional containment of the pathogens remain intact.
These studies may hold important clinical implications as GM-CSF has been increasingly considered for broad use in accentuating the host immune response in the setting of clinical infections. The largest experience to date involves its use in neonatal intensive care where large trials have examined the usefulness of GM-CSF in preventing neonatal sepsis. Several developmental immunologic deficiencies have been identified in neonates that put them at increased risk for the infections and subsequent sepsis as compared to the rest of the pediatric population [40]. As a means to augment the host immune response, investigators have considered the addition of GM-CSF to the management of preterm neonates at high risk of developing sepsis or with suspected sepsis [41–44]. Results of these studies have been mixed with regards to various outcomes and dependent upon the specific population studies. For example, when used in the setting of neutropenic neonates with established sepsis, administration of GM-CSF was associated with improved outcome as assessed by mortality [45]. However, when considered either for prophylactic use in neutropenic neonates or for use in non-neutropenic neonates with sepsis, stimulation of the immune system has been observed [46], but no significant effect on survival was observed [47]. Similar studies have not been conducted in an older pediatric population. Recently, the effect of administering GM-CSF to non-neutropenic, critically-ill adults with sepsis was evaluated [48]. In this study, GM-CSF therapy was associated with the upregulation of functional markers of innate immune function on circulating neutrophils and monocytes, but without a concomitant exacerbation of sepsis-related organ injury as might have been predicted by our animal model data. Numerous additional studies have demonstrated the ability of GM-CSF to reverse functional immune deactivation of circulating monocytes in a variety of settings including trauma [49], sepsis [18] and burn [50]. However, despite the apparent beneficial effect of GM-CSF in augmenting functional immune responses, an overall benefit with regards to survival advantage has not been consistently observed. An important reason for these differences may have much to do with the homogeneity of the population being examined. In neonates, it is likely that functional immune deficiency is the more important risk factor for infection. In contrast, in adults, the course of sepsis can altered by immune dissonance from either pro- or anti-inflammatory predominance thereby negating the beneficial effect of GM-CSF in those whose pathophysiology is resulting from too much inflammation.
In light of these clinical controversies, the data from the present studies must be in interpreted in the context of certain limitations. First, the mouse models of sepsis, while providing fundamentally important insight as to the pathobiology of sepsis have never correlated well with human clinical outcome studies. In that vein, we have reported our data with an emphasis on pathophysiologic understanding of the role of peritoneal macrophages, rather than any predictor of the clinical response to GM-CSF therapy in humans. Second, extrapolation to the clinical setting needs to be tempered in that standard therapies utilized in human bacterial peritonitis (e.g. broad spectrum antibiotics, fluid resuscitation and surgical drainage) were not employed in the model studied. As stated above, our intent was to focus on the contribution of the peritoneal macrophage in sepsis, rather than precisely model the clinical setting. Finally, GM-CSF genetic deletion may be affecting other arms of the immune system unrecognized to date. That full mortality could not be reconstituted when limited to the peritoneal cavity suggests this is the case. Therefore future studies are aimed at trying to identify the additional cell population and/or mediator that may be responsible for the additional percentage of mortality observe in the wild type mice. Such studies are likely to extend our understanding of the biologic mechanisms responsible for peritonitis-induced morbidity and mortality.
Acknowledgments
The authors wish to thank Dr. Daniel G. Remick with his assistance in establishing the CLP model. This work was supported by NIH Grant RO1 GM66839 to T.P.S.
References
- 1.Angus DC, Wax RS. Epidemiology of sepsis: an update. Crit Care Med. 2001;29:S109–116. doi: 10.1097/00003246-200107001-00035. [DOI] [PubMed] [Google Scholar]
- 2.Bernard GR, Vincent JL, Laterre PF, LaRosa SP, Dhainaut JF, Lopez-Rodriguez A, Steingrub JS, Garber GE, Helterbrand JD, Ely EW, Fisher CJ., Jr Efficacy and safety of recombinant human activated protein C for severe sepsis. N Engl J Med. 2001;344:699–709. doi: 10.1056/NEJM200103083441001. [DOI] [PubMed] [Google Scholar]
- 3.Read RC, Wyllie DH. Toll receptors and sepsis. Curr Opin Crit Care. 2001;7:371–375. doi: 10.1097/00075198-200110000-00010. [DOI] [PubMed] [Google Scholar]
- 4.Brightbill HD, Modlin RL. Toll-like receptors: molecular mechanisms of the mammalian immune response. Immunology. 2000;101:1–10. doi: 10.1046/j.1365-2567.2000.00093.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Aderem A, Ulevitch RJ. Toll-like receptors in the induction of the innate immune response. Nature. 2000;406:782–787. doi: 10.1038/35021228. [DOI] [PubMed] [Google Scholar]
- 6.Beutler B, Du X, Poltorak A. Identification of Toll-like receptor 4 (Tlr4) as the sole conduit for LPS signal transduction: genetic and evolutionary studies. J Endotoxin Res. 2001;7:277–280. [PubMed] [Google Scholar]
- 7.Bone RC. Sepsis syndrome. New insights into its pathogenesis and treatment. Infect Dis Clin North Am. 1991;5:793–805. [PubMed] [Google Scholar]
- 8.Parent C, Eichacker PQ. Neutrophil and endothelial cell interactions in sepsis. The role of adhesion molecules. Infect Dis Clin North Am. 1999;13:427–447. x. doi: 10.1016/s0891-5520(05)70084-2. [DOI] [PubMed] [Google Scholar]
- 9.Bone RC. Why sepsis trials fail. JAMA. 1996;276:565–566. [PubMed] [Google Scholar]
- 10.Dinarello CA. Role of pro- and anti-inflammatory cytokines during inflammation: experimental and clinical findings. J Biol Regul Homeost Agents. 1997;11:91–103. [PubMed] [Google Scholar]
- 11.Dinarello CA. Proinflammatory and anti-inflammatory cytokines as mediators in the pathogenesis of septic shock. Chest. 1997;112:321S–329S. doi: 10.1378/chest.112.6_supplement.321s. [DOI] [PubMed] [Google Scholar]
- 12.Bone RC. Sir Isaac Newton, sepsis, SIRS, and CARS. Crit Care Med. 1996;24:1125–1128. doi: 10.1097/00003246-199607000-00010. [DOI] [PubMed] [Google Scholar]
- 13.Woiciechowsky C, Asadullah K, Nestler D, Schoning B, Glockner F, Docke WD, Volk HD. Diminished monocytic HLA-DR expression and ex vivo cytokine secretion capacity in patients with glioblastoma: effect of tumor extirpation. J Neuroimmunol. 1998;84:164–171. doi: 10.1016/s0165-5728(97)00236-1. [DOI] [PubMed] [Google Scholar]
- 14.Asadullah K, Woiciechowsky C, Docke WD, Egerer K, Kox WJ, Vogel S, Sterry W, Volk HD. Very low monocytic HLA-DR expression indicates high risk of infection--immunomonitoring for patients after neurosurgery and patients during high dose steroid therapy. Eur J Emerg Med. 1995;2:184–190. doi: 10.1097/00063110-199512000-00003. [DOI] [PubMed] [Google Scholar]
- 15.Hynninen M, Pettila V, Takkunen O, Orko R, Jansson SE, Kuusela P, Renkonen R, Valtonen M. Predictive value of monocyte histocompatibility leukocyte antigen-DR expression and plasma interleukin-4 and -10 levels in critically ill patients with sepsis. Shock. 2003;20:1–4. doi: 10.1097/01.shk.0000068322.08268.b4. [DOI] [PubMed] [Google Scholar]
- 16.Volk HD, Thieme M, Heym S, Docke WD, Ruppe U, Tausch W, Manger D, Zuckermann S, Golosubow A, Nieter B, et al. Alterations in function and phenotype of monocytes from patients with septic disease--predictive value and new therapeutic strategies. Behring Inst Mitt. 1991:208–215. [PubMed] [Google Scholar]
- 17.Flohe S, Lendemans S, Selbach C, Waydhas C, Ackermann M, Schade FU, Kreuzfelder E. Effect of granulocyte-macrophage colony-stimulating factor on the immune response of circulating monocytes after severe trauma. Crit Care Med. 2003;31:2462–2469. doi: 10.1097/01.CCM.0000089640.17523.57. [DOI] [PubMed] [Google Scholar]
- 18.Nierhaus A, Montag B, Timmler N, Frings DP, Gutensohn K, Jung R, Schneider CG, Pothmann W, Brassel AK, Schulte Am Esch J. Reversal of immunoparalysis by recombinant human granulocyte-macrophage colony-stimulating factor in patients with severe sepsis. Intensive Care Med. 2003;29:646–651. doi: 10.1007/s00134-003-1666-6. [DOI] [PubMed] [Google Scholar]
- 19.Hamilton JA, Anderson GP. GM-CSF Biology. Growth Factors. 2004;22:225–231. doi: 10.1080/08977190412331279881. [DOI] [PubMed] [Google Scholar]
- 20.Berclaz PY, Carey B, Fillipi MD, Wernke-Dollries K, Geraci N, Cush S, Richardson T, Kitzmiller J, O’Connor M, Hermoyian C, Korfhagen T, Whitsett JA, Trapnell BC. GM-CSF regulates a PU.1-dependent transcriptional program determining the pulmonary response to LPS. Am J Respir Cell Mol Biol. 2007;36:114–121. doi: 10.1165/rcmb.2006-0174OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Uchida K, Beck DC, Yamamoto T, Berclaz PY, Abe S, Staudt MK, Carey BC, Filippi MD, Wert SE, Denson LA, Puchalski JT, Hauck DM, Trapnell BC. GM-CSF autoantibodies and neutrophil dysfunction in pulmonary alveolar proteinosis. N Engl J Med. 2007;356:567–579. doi: 10.1056/NEJMoa062505. [DOI] [PubMed] [Google Scholar]
- 22.Shibata Y, Berclaz PY, Chroneos ZC, Yoshida M, Whitsett JA, Trapnell BC. GM-CSF regulates alveolar macrophage differentiation and innate immunity in the lung through PU.1. Immunity. 2001;15:557–567. doi: 10.1016/s1074-7613(01)00218-7. [DOI] [PubMed] [Google Scholar]
- 23.Trapnell BC, Whitsett JA. Gm-CSF regulates pulmonary surfactant homeostasis and alveolar macrophage-mediated innate host defense. Annu Rev Physiol. 2002;64:775–802. doi: 10.1146/annurev.physiol.64.090601.113847. [DOI] [PubMed] [Google Scholar]
- 24.Dranoff G, Mulligan RC. Activities of granulocyte-macrophage colony-stimulating factor revealed by gene transfer and gene knockout studies. Stem Cells. 1994;12 Suppl 1:173–182. discussion 182–174. [PubMed] [Google Scholar]
- 25.Huffman JA, Hull WM, Dranoff G, Mulligan RC, Whitsett JA. Pulmonary epithelial cell expression of GM-CSF corrects the alveolar proteinosis in GM-CSF-deficient mice. J Clin Invest. 1996;97:649–655. doi: 10.1172/JCI118461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zingarelli B, Hasko G, Salzman AL, Szabo C. Effects of a novel guanylyl cyclase inhibitor on the vascular actions of nitric oxide and peroxynitrite in immunostimulated smooth muscle cells and in endotoxic shock. Crit Care Med. 1999;27:1701–1707. doi: 10.1097/00003246-199909000-00001. [DOI] [PubMed] [Google Scholar]
- 27.Berclaz PY, Shibata Y, Whitsett JA, Trapnell BC. GM-CSF, via PU.1, regulates alveolar macrophage Fcgamma R-mediated phagocytosis and the IL-18/IFN-gamma -mediated molecular connection between innate and adaptive immunity in the lung. Blood. 2002;100:4193–4200. doi: 10.1182/blood-2002-04-1102. [DOI] [PubMed] [Google Scholar]
- 28.Cerami A, Beutler B. The role of cachectin/TNF in endotoxic shock and cachexia. Immunol Today. 1988;9:28–31. doi: 10.1016/0167-5699(88)91353-9. [DOI] [PubMed] [Google Scholar]
- 29.Calandra T, Gerain J, Heumann D, Baumgartner JD, Glauser MP. High circulating levels of interleukin-6 in patients with septic shock: evolution during sepsis, prognostic value, and interplay with other cytokines. The Swiss-Dutch J5 Immunoglobulin Study Group. Am J Med. 1991;91:23–29. doi: 10.1016/0002-9343(91)90069-a. [DOI] [PubMed] [Google Scholar]
- 30.Iwamura H, Sato M, Wakitani K. Comparative study of glucocorticoids, cyclosporine A, and JTE-607 [(−)-Ethyl-N[3,5-dichloro-2-hydroxy-4-[2-(4-methylpiperazin-1-yl)ethoxy]be nzoyl]-L-phenylalaninate dihydrochloride] in a mouse septic shock model. J Pharmacol Exp Ther. 2004;311:1256–1263. doi: 10.1124/jpet.104.072421. [DOI] [PubMed] [Google Scholar]
- 31.Gao H, Neff TA, Guo RF, Speyer CL, Sarma JV, Tomlins S, Man Y, Riedemann NC, Hoesel LM, Younkin E, Zetoune FS, Ward PA. Evidence for a functional role of the second C5a receptor C5L2. FASEB J. 2005;19:1003–1005. doi: 10.1096/fj.04-3424fje. [DOI] [PubMed] [Google Scholar]
- 32.Remick DG, Bolgos GR, Siddiqui J, Shin J, Nemzek JA. Six at six: interleukin-6 measured 6 h after the initiation of sepsis predicts mortality over 3 days. Shock. 2002;17:463–467. doi: 10.1097/00024382-200206000-00004. [DOI] [PubMed] [Google Scholar]
- 33.Berclaz PY, Zsengeller Z, Shibata Y, Otake K, Strasbaugh S, Whitsett JA, Trapnell BC. Endocytic internalization of adenovirus, nonspecific phagocytosis, and cytoskeletal organization are coordinately regulated in alveolar macrophages by GM-CSF and PU.1. J Immunol. 2002;169:6332–6342. doi: 10.4049/jimmunol.169.11.6332. [DOI] [PubMed] [Google Scholar]
- 34.Remick DG, Newcomb DE, Bolgos GL, Call DR. Comparison of the mortality and inflammatory response of two models of sepsis: lipopolysaccharide vs. cecal ligation and puncture. Shock. 2000;13:110–116. doi: 10.1097/00024382-200013020-00004. [DOI] [PubMed] [Google Scholar]
- 35.Ebong SJ, Call DR, Bolgos G, Newcomb DE, Granger JI, O’Reilly M, Remick DG. Immunopathologic responses to non-lethal sepsis. Shock. 1999;12:118–126. doi: 10.1097/00024382-199908000-00005. [DOI] [PubMed] [Google Scholar]
- 36.Bonfield TL, Raychaudhuri B, Malur A, Abraham S, Trapnell BC, Kavuru MS, Thomassen MJ. PU.1 regulation of human alveolar macrophage differentiation requires granulocyte-macrophage colony-stimulating factor. Am J Physiol Lung Cell Mol Physiol. 2003;285:L1132–1136. doi: 10.1152/ajplung.00216.2003. [DOI] [PubMed] [Google Scholar]
- 37.del Fresno C, Soler-Rangel L, Soares-Schanoski A, Gomez-Pina V, Gonzalez-Leon MC, Gomez-Garcia L, Mendoza-Barbera E, Rodriguez-Rojas A, Garcia F, Fuentes-Prior P, Arnalich F, Lopez-Collazo E. Inflammatory responses associated with acute coronary syndrome up-regulate IRAK-M and induce endotoxin tolerance in circulating monocytes. J Endotoxin Res. 2007;13:39–52. doi: 10.1177/0968051907078623. [DOI] [PubMed] [Google Scholar]
- 38.Fan H, Cook JA. Molecular mechanisms of endotoxin tolerance. J Endotoxin Res. 2004;10:71–84. doi: 10.1179/096805104225003997. [DOI] [PubMed] [Google Scholar]
- 39.Kobayashi K, Hernandez LD, Galan JE, Janeway CA, Jr, Medzhitov R, Flavell RA. IRAK-M is a negative regulator of Toll-like receptor signaling. Cell. 2002;110:191–202. doi: 10.1016/s0092-8674(02)00827-9. [DOI] [PubMed] [Google Scholar]
- 40.Watson RS, Linde-Zwirble WT, Carcillo JA, Angus DC. Severe sepsis in children: A U.S. Epidemiologic Survey. Crit Care Med. 2001;28:A46. [Google Scholar]
- 41.Suri M, Harrison L, Van de Ven C, Cairo MS. Immunotherapy in the prophylaxis and treatment of neonatal sepsis. Curr Opin Pediatr. 2003;15:155–160. doi: 10.1097/00008480-200304000-00003. [DOI] [PubMed] [Google Scholar]
- 42.Banerjea MC, Speer CP. The current role of colony-stimulating factors in prevention and treatment of neonatal sepsis. Semin Neonatol. 2002;7:335–349. doi: 10.1016/s1084-2756(02)90116-8. [DOI] [PubMed] [Google Scholar]
- 43.Goldman S, Ellis R, Dhar V, Cairo MS. Rationale and potential use of cytokines in the prevention and treatment of neonatal sepsis. Clin Perinatol. 1998;25:699–710. [PubMed] [Google Scholar]
- 44.Perez EM, Weisman LE. Novel approaches to the prevention and therapy of neonatal bacterial sepsis. Clin Perinatol. 1997;24:213–229. [PubMed] [Google Scholar]
- 45.Bilgin K, Yaramis A, Haspolat K, Tas MA, Gunbey S, Derman O. A randomized trial of granulocyte-macrophage colony-stimulating factor in neonates with sepsis and neutropenia. Pediatrics. 2001;107:36–41. doi: 10.1542/peds.107.1.36. [DOI] [PubMed] [Google Scholar]
- 46.Miura E, Procianoy RS, Bittar C, Miura CS, Miura MS, Mello C, Christensen RD. A randomized, double-masked, placebo-controlled trial of recombinant granulocyte colony-stimulating factor administration to preterm infants with the clinical diagnosis of early-onset sepsis. Pediatrics. 2001;107:30–35. doi: 10.1542/peds.107.1.30. [DOI] [PubMed] [Google Scholar]
- 47.Carr R, Modi N, Dore C. G-CSF and GM-CSF for treating or preventing neonatal infections. Cochrane Database Syst Rev. 2003:CD003066. doi: 10.1002/14651858.CD003066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Rosenbloom AJ, Linden PK, Dorrance A, Penkosky N, Cohen-Melamed MH, Pinsky MR. Effect of granulocyte-monocyte colony-stimulating factor therapy on leukocyte function and clearance of serious infection in nonneutropenic patients. Chest. 2005;127:2139–2150. doi: 10.1378/chest.127.6.2139. [DOI] [PubMed] [Google Scholar]
- 49.Docke WD, Randow F, Syrbe U, Krausch D, Asadullah K, Reinke P, Volk HD, Kox W. Monocyte deactivation in septic patients: restoration by IFN-gamma treatment. Nat Med. 1997;3:678–681. doi: 10.1038/nm0697-678. [DOI] [PubMed] [Google Scholar]
- 50.Cioffi WG, Jr, Burleson DG, Jordan BS, Becker WK, McManus WF, Mason AD, Jr, Pruitt BA., Jr Effects of granulocyte-macrophage colony-stimulating factor in burn patients. Arch Surg. 1991;126:74–79. doi: 10.1001/archsurg.1991.01410250080013. [DOI] [PubMed] [Google Scholar]