

Abstract
Background
Time series gene expression data analysis is used widely to study the dynamics of various cell processes. Most of the time series data available today consist of few time points only, thus making the application of standard clustering techniques difficult.
Results
We developed two new algorithms that are capable of extracting biological patterns from short time point series gene expression data. The two algorithms, ASTRO and MiMeSR, are inspired by the rank order preserving framework and the minimum mean squared residue approach, respectively. However, ASTRO and MiMeSR differ from previous approaches in that they take advantage of the relatively few number of time points in order to reduce the problem from NP-hard to linear. Tested on well-defined short time expression data, we found that our approaches are robust to noise, as well as to random patterns, and that they can correctly detect the temporal expression profile of relevant functional categories. Evaluation of our methods was performed using Gene Ontology (GO) annotations and chromatin immunoprecipitation (ChIP-chip) data.
Conclusion
Our approaches generally outperform both standard clustering algorithms and algorithms designed specifically for clustering of short time series gene expression data. Both algorithms are available at http://www.benoslab.pitt.edu/astro/.
Background
Time series experiments have been widely used to study the dynamic behavior of the cells in a variety of biological processes, including cell proliferation [1], development [2], and response to extracellular stimuli [3,4]. Time series data can be broadly divided into two classes: the short-time series with few sampled time points (typically 3–8) and long-time series with more than 10 time points sampled. Most algorithms used to analyze time series datasets initially were based on general clustering methods like hierarchical clustering [5], k-means [6], Bayesian networks [7], and self-organizing maps [8]. Although these methods are capable of revealing some biological features, they are not taking into consideration the sequential nature of the time series data. More recently, some groups suggested methodologies specifically designed for clustering time series expression data, including the use of continuous representation of expression profiles [9], hidden Markov models [10], and others [11-14]. However, algorithms such as those developed by Bar-Joseph et al. [9], De Hoon et al. [12] and Peddada et al. [13] perform better on long time series datasets where the statistical power is higher. For short time series data, which represent about 80% of the time series gene expression datasets [15], they are expected to perform less optimal due to data overfitting caused by the small number of sampled time points.
In order to avoid that, some researchers have suggested the use of predefined patterns of expression profiles (either taken directly from the data or from prior biological observations) and matching the observed data to these profiles using some cost function [15-18]. Such approaches usually identify a large number of patterns, but many of them may arise randomly from noise due to the small number of sampled time points. The algorithm proposed by Ernst et al. [15] is capable of partially correcting for this problem with the implementation of heuristics: the user is required to select a set of potential profiles that are expected to represent better the real biological nature of such data. Last but not least, almost all of the approaches mentioned above use a cost function followed by a greedy algorithm to find clusters. As we will show later, such approaches may miss some biologically significant characteristics of the data.
In this paper, we present two new algorithms, ASTRO and MiMeSR, respectively, which are specifically designed to identify biologically relevant clusters of genes from short time series data. ASTRO and MiMeSR are inspired by the order preserving framework and the minimum mean squared residue approach, respectively. Other algorithms have used the same principles in the past, but in the biclustering context [19-21], which makes such algorithms NP hard [21]. We demonstrate the utility of ASTRO and MiMeSR using several well-defined short time datasets. We show that our approaches are robust to noise and random patterns and they can correctly detect the temporal expression profile of relevant functional categories in linear time. Comparative analysis also showed that our approaches outperform both general clustering algorithms and algorithms designed specifically for short time series gene expression data.
Results and Discussion
Robustness to noise
To test the robustness of ASTRO and MiMeSR to noise, we generated three sets of data, 1000 rows and 3, 5, and 7 time points respectively, with five order preserving submatrix which at the same time verify the minimum mean squared residue property embedded in it (domain knowledge). Then, we added 0%, 1%, 3%, and 5% level of noise into the simulated data. We ran each algorithm several times on each set of data and plot the average of the Adjusted Rand index (Figure 1). The Adjusted Rand index values lies between 0 and 1. Larger value means higher similarity between the clustering results. If the simulated result is perfectly consistent to the domain knowledge, the index value will be 1. If a clustering is no more than a random choice, the index will be zero [22]. The results in Figure 1 show that both algorithms perform equally well on the 5 time points dataset, while ASTRO is more robust on the 3 time points datasets and MiMeSR on the 7 time points dataset.
Figure 1.

ASTRO and MiMeSR performance on simulated data. Comparison of the two algorithms on simulated data with different noise levels.
Application on Saccharomyces cerevisiae amino acid starvation dataset
We tested the ability of ASTRO and MiMeSR to identify biologically relevant clusters from short time series data using the yeast amino acid (AA) starvation dataset [3]. Saccharomyces cerevisiae response to stress by AA starvation is measured at time points 0.5 h, 1 h, 2 h, 4 h, and 6 h and at the control (unstimulated) cells (time point 0 h). The data was filtered to remove genes with missing values and genes whose expression level did not change substantially between time points, filtering threshold ε < 2.0 for ASTRO and MiMeSR. The results show that both our approaches can correctly identify the temporal profiles of relevant functional groups. Statistical evaluation of our clusters was performed using external datasets, like the GO categories [23] and AA starvation ChIP-chip data [3,24]. Compared to general clustering algorithms (e.g., k-means) and algorithms designed specifically for clustering short time series gene expression data [17,25], our techniques were able to detect more significant patterns.
Evaluation using GO annotations
Figure 2A and 2B present the plot of the most significant clusters identified in this dataset by ASTRO (Z = 10-7 to 10-68) and MiMeSR (H < 2), respectively. The minimum number of genes per cluster in both cases was set to Kmin = 25. In principle, one might expect that the genes in biologically relevant clusters will also participate in the same biological processes. We used the on-line yeast GO Term Finder tool http://www.yeastgenome.org to assess GO membership of the genes in the identified clusters. We found that with the exception of the clusters with many genes of unknown function, the majority of the genes in the identified clusters belong to the same GO categories. The p-values for these clusters were ranging from 10-10 to 10-34 for ASTRO (Table 1) and from 10-34 to 10-68 for MiMeSR (Table 2.) The results also show that in general MiMeSR clusters are more homogenous than the ASTRO clusters regarding the GO pathways. For example, the percentage of the genes in the ASTRO clusters C1 and D1 that belong to the "ribosome biogenesis" category (Table 1) is smaller than MiMeSR clusters E2 and F2 (Table 2); consequently, their p-values are higher. The same is true for ASTRO cluster B1 and MiMeSR clusters D2 and G2.
Figure 2.

Clustering yeast time series data. The most statistically significant clusters as they were identified by (A) ASTRO and (B) MiMeSR.
Table 1.
Evaluation of the clusters identified by ASTRO using Gene Ontology data
| ASTRO (OPSM) | ||||
| Clusters | No. of genes | Top GO term | # of genes in category | p-value |
| A1 | 70 | Cellular biosynthetic process | 41 (58.6%) | 7.0e-16 |
| Biosynthetic process | 42 (60.0%) | 2.5e-13 | ||
| B1 | 86 | Translation | 55 (64.0%) | 1.5e-33 |
| Macromolecule biosynthetic process | 55 (64.0%) | 1.5e-27 | ||
| C1 | 25 | Ribosome biogenesis and assembly | 14 (56.0%) | 9.8e-15 |
| Ribonucleoprotein complex biogenesis and assembly | 14 (56.0%) | 9.6e-10 | ||
| D1 | 74 | Ribosome biogenesis and assembly | 44 (59.5%) | 2.6e-34 |
| Ribonucleoprotein complex biogenesis and assembly | 44 (59.9%) | 4.6e-31 | ||
| E1* | 33 | Sulfur metabolic process | 06 (18.2%) | 9.0e-05 |
| Sulfur amino acid metabolic process | 04 (12.1%) | 2.4e-03 | ||
| F1* | 26 | Amino acid transport | 04 (15.4%) | 1.4e-03 |
| Amine transport | 04 (15.4%) | 3.6e-03 | ||
* More than 20% genes of unknown function
Table 2.
Evaluation of the clusters identified by MiMeSR using Gene Ontology data
| MiMeSR (MMSR) | ||||
| Clusters | No. of genes | Top GO term | # of genes in category | p-value |
| A2 | 246 | Ribosome biogenesis and assembly | 103 (42.0%) | 2.0e-64 |
| Gene expression | 167 (68.0%) | 1.0e-49 | ||
| B2* | 66 | Sulfur metabolic process | 13 (20.0%) | 1.2e-12 |
| Sulfur amino acid metabolic process | 8 (12.1%) | 4.4e-08 | ||
| C2 | 133 | Ribosome biogenesis and assembly | 82 (62.0%) | 1.7e-68 |
| Ribonucleoprotein complex biogenesis and assembly | 84 (63.2%) | 5.6e-65 | ||
| D2 | 80 | Translation | 62 (77.5%) | 1.5e-46 |
| Macromolecule biosynthetic process | 66 (82.5%) | 1.9e-45 | ||
| E2 | 109 | Ribosome biogenesis and assembly | 73 (67.0%) | 1.7e-64 |
| Ribonucleoprotein complex biogenesis and assembly | 73 (67.0%) | 8.1e-59 | ||
| F2 | 60 | Ribosome biogenesis and assembly | 42 (70.0%) | 1.6e-37 |
| Ribonucleoprotein complex biogenesis and assembly | 42 (70.0%) | 2.2e-34 | ||
| G2 | 94 | Translation | 76 (81.0%) | 3.1e-60 |
| Macromolecule biosynthetic process | 77 (82.0%) | 5.2e-53 | ||
* More than 20% genes of unknown function
Evaluation using ChIP-chip data
Another reasonable assumption is that genes that belong to the same cluster (co-expressed genes) are more likely to be regulated by the same transcription factors (co-regulated genes). We evaluated the ASTRO and MiMeSR clusters using the published AA starvation ChIP-chip dataset on 34 transcription factors [24]. Each of the 34 transcription factors in the ChIP-chip dataset was tested for target over-representation in each of the clusters using the Fisher's exact test. The results are presented on Tables 3 (ASTRO) and 4 (MiMeSR). FHL1, and SFP1 appear to have overrepresented number of target genes in clusters A1, B1, D1 (Table 3), A2, C2, D2, and G2 (Table 4), which are also the most similar in their overall expression pattern (Figure 2A, 2B), especially in 0.5 hr and 1 hr time points. It is possible that these transcription factors act early on in the AA starvation response as it was previously suggested [26]. Consistent with our results, Jorgensen et al. [27] have found that FHL1 and SFP1 regulate many ribosome biosynthesis genes, which is the most significant GO process in cluster C1, D1, A2, C2, D2, E2, and F2 genes. Furthermore, MET31, MET32 and MET4 have been associated with regulation of sulfur metabolism genes [28], which is the most significant category for our cluster E1 genes.
Table 3.
Evaluation of the clusters identified by ASTRO using ChIP-chip data.
| ASTRO (OPSM) | ||||||
| TFs | A1 | B1 | C1 | D1 | E1 | F1 |
| ARO80 | 2/3% (5e-02) |
3/3% (7e-02) |
1/4% (4e-02) |
|||
| BAS1 | 1/3% (8e-02) |
|||||
| CBF1 | 5/15% (1e-02) |
|||||
| CHA4 |
4/4% (9e-03) |
|||||
| DAL81 |
1/4% (4e-03) |
|||||
| FHL1 |
25/36% (3e-20) |
43/50% (3e-42) |
3/10% (1e-02) |
19/25% (1e-11) |
||
| GCR2 | 3/4% (2e-02) |
|||||
| GCN4 | 3/9% (7e-02) |
|||||
| MET31 | 1/3% (7e-02) |
|||||
| MET32 |
4/12% (6e-12) |
|||||
| MET4 | 1/3% (7e-02) |
|||||
| SFP1 |
6/9% (1e-05) |
13/15% (2e-14) |
3/10% (1e-02) |
8/11% (5e-08) |
||
Bold letter boxes correspond to statistically overrepresented transcription factors in that cluster. Each box contains: (a) the number of genes, (b) the percent of genes in the cluster associated with this transcription factor, and (c) the p-value (Fisher's exact test).
Table 4.
Evaluation of the clusters identified by MiMeSR using ChIP-chip data.
| MiMeSR (MMSR) | |||||||
| TFs | A2 | B2 | C2 | D2 | E2 | F2 | G2 |
| ARO80 | 4/3% (3e-02) |
||||||
| BAS1 | |||||||
| CBF1 | 10/4% (1e-02) |
9/14% (3e-03) |
3/3% (1e-02) |
||||
| CHA4 | |||||||
| DAL81 | |||||||
| FHL1 |
86/35% (3e-20) |
27/20% (7e-14) |
48/60% (1e-52) |
5/5% (1e-02) |
3/5% (7e-02) |
62/65% (2e-70) |
|
| GCR2 |
4/5% (5e-03) |
||||||
| GCN4 | |||||||
| MET31 | |||||||
| MET32 |
4/5% (9e-03) |
3/3% (9e-02) |
|||||
| MET4 | |||||||
| SFP1 |
30/12% (1e-08) |
4/3% (1e-02) |
17/21% (1e-21) |
19/20% (1e-20) |
|||
Bold letter boxes correspond to statistically overrepresented transcription factors in that cluster. Each box contains: (a) the number of genes, (b) the percent of genes in the cluster associated with this transcription factor, and (c) the p-value (Fisher's exact test).
Comparison of the results in Tables 3 and 4 also shows that in general ASTRO finds more clusters that are statistically significantly enriched in genes bound by transcription factors. In other words, ASTRO performs better than MiMeSR in identifying co-regulated genes.
Comparison with other methods
We compared ASTRO and MiMeSR with the popular k-means general clustering algorithm and the recently published STEM [25] and FCV [17], both of which are designed specifically for short time series gene expression data. We used the Matlab 7.0.0 implementation of k-means with correlation distance. We ran the k-means algorithm for 10 clusters because most of our algorithm picked the same number. FCV was implemented according to the description provided in [17]. STEM ran over the web http://www.cs.cmu.edu/~jernst/stem/ with the following parameters: maximum unit of change in model profiles between time points = 4; number of model profiles = 50. For unbiased comparison, we ran these algorithms on the same dataset of 698 genes (filtering threshold ε < 2). We selected clusters with less than 50% genes of unknown function as depicted by the GO database. Figure 3A and 3B presents the comparative evaluation of these approaches using GO and ChIP data, respectively. A particular cluster was considered to be "significant" if the p-value of the top GO category or transcription factor-gene association was smaller than the threshold. A good clustering algorithm is expected to identify sets of genes that will participate in the same biological processes (GO annotation) and/or regulated by the same transcription factors. The more homogeneous these clusters are the more significant the annotation categories will become. ASTRO identified a higher number of significant clusters than the k-means and the FCV algorithms in all p-value thresholds (Figure 3A and 3B). Also, it performed equally well or better when compared to STEM. MiMeSR also performed equally well or better than all other algorithms with respect to the TF-gene association data; and it gave comparable results to the other algorithms in the GO category analysis. In particular, we found it to be more accurate than the other algorithms in predicting the more tight clusters (low p-values), whereas the other algorithms performed better in higher p-values. This shows the potential of our algorithms in identifying more tight, biologically relevant clusters. We note, however, that a thorough comparison of different methods is impossible when dealing with noisy datasets and algorithms with different reporting thresholds.
Figure 3.

Comparison of clustering approaches. Comparative analysis of clustering approaches using (A) GO data and (B) amino acid starvation ChIP-chip data. The y-axis represents the percent of clusters for which the p-value of their most significant category (GO or ChIP-chip) was lower than the given threshold.
Conclusion
In this study, we presented two new algorithms for analyzing short time series gene expression data: ASTRO that uses the order preserving matrix concept and MiMeSR that uses the minimum mean squared residue concept. Both these algorithms use linear algebra techniques to identify coherent gene clusters over all time points in linear time. This offers a significant advantage over existing methods that employ greedy approaches or heuristics, since ASTRO and MiMeSR avoid problems related to cost functions or the choice of predefined sets of expression profiles [19,21,25]. Also, the complexity of our methods is smaller than that of existing algorithms, even when it is compared to those that use a greedy approach to speed up their running time [9-15]. ASTRO identifies all gene clusters with coherent expression patterns, irrespectively of the magnitude of expression change. Genes belonging in such clusters are generally expected to participate in the same biological processes (see, also, Table 1). MiMeSR identifies all gene clusters with coherent expression change both in direction as well as in magnitude. Genes belonging in such clusters are expected to be regulated by the same set of transcription factors (although different transcription factors may act at different time points). In general, MiMeSR identifies clusters that are enriched for genes belonging to the same pathways, while ASTRO identifies clusters with genes that are regulated by the same transcription factors (Tables 1, 2, 3 and Figure 3). In terms of overall statistical significance of the identified clusters, MiMeSR outperforms ASTRO in most cases (Figure 3). In a direct comparison of the two algorithms in simulated data (Figure 1) we found that ASTRO outperforms MiMeSR when fewer data points are available, whereas MiMeSR performs better when more data points are available.
Testing ASTRO &MiMeSR in well-characterized short time series gene expression datasets showed that it is robust to noise and to random patterns, and that it can correctly predict the temporal expression profile of relevant functional categories as confirmed by statistical analysis of GO category membership over-representation and analysis of transcription factor occupancy in the promoters of the gene members of the various clusters. Our approaches are shown to outperform existing clustering algorithms, including the popular k-means, as well as FCV and STEM and they were able to distinguish between closely related but biologically distinct patterns. As expected, ASTRO finds more homogeneous clusters than MiMeSR, with respect to the percentage of genes associated with a given transcription factor. This is because it takes into consideration the magnitude of the change in gene expression, which is more closely related to the transcription factors involved.
In principle, ASTRO and MiMeSR can also be applied to long time series gene expression data (more than 10 time points) or gene expression data sampled over different conditions, but in this case the number of genes in each cluster is expected to be low. However, they can be adapted to identify local patterns, thus overcoming this problem.
Methods
General description of the algorithms
A time series gene expression dataset can be represented by an N × M matrix, A = [anm], with rows corresponding to the genes from G = {g1,..., gn,..., gN}, and columns corresponding to the time point measurements from T = {t1,..., tm,..., tM}. The entry anm is the expression level of gene n at time point tm or -simply- m. Given a short time-series gene expression dataset, our goal is to identify the sets of genes with coherent behavior, i.e., genes whose expression levels increase and/or decrease coherently across the time point experiments, by minimizing the effect of noise and random patterns. The input data are pre-processed to identify and remove from the matrix all genes whose expression level remains constant across time points. We consider the expression of a gene to be constant when the difference between the minimum and the maximum value (in log-scale) is less than a positive real number, ε. ε is a user-defined parameter and it can be based on prior knowledge on the expected level of noise on a given experiment.
ASTRO (Rank Order Preserving Matrix framework)
ASTRO seeks to identify sets of genes with similar expression profiles irrespectively of their expression fold-change. Therefore, only the direction of expression change is considered and not its magnitude. This reflects the biological fact that different gene products may be required at different quantities for a given cellular response or function. Under this assumption, the problem of finding a cluster of similarly expressed genes can be caste into a problem of finding an order preserving submatrix (OPSM) of the gene expression matrix A. A submatrix C of A is order preserving (OP) if there is a permutation of its columns under which the sequence of values in every row is strictly increasing or decreasing. In other words an OPSM is a set: C = {2-tuples (I,J), I ∈ G and J ∈ T}, such that each row induces the same rank order permutation on the columns. The problem of searching over all possible subsets of columns for identifying the most significant OPSM is NP hard [19]. However, taking advantage of the small number of sampled points in a short time series dataset, one may seek patterns of consistent gene expression over all time points. In such case the order will be required to be preserved in all columns (time-points) for the genes in a cluster (i.e. J = T). In fact, this assumption is now commonly used in analyzing short-time series experiments [9-15]. As we show below, this reduces the complexity of finding OPSMs, which offers an advantage over methods that use probabilistic models and greedy algorithms. ASTRO is guaranteed to find all OPSMs across all time points in O(NM) using linear algebra techniques.
Overview
In this part, we focus on genes with coherent behavior, i.e., genes whose expression levels increase and/or decrease coherently across all time points. ASTRO starts by filtering those genes with constant gene expression across all time points (using a threshold ε). It then proceeds by constructing the rank matrix of the time series gene expression data. Next, it identifies all distinct coherent patterns in the ranked matrix. Finally, it assigns each gene to its corresponding cluster by performing a row comparison between the set of distinct rows of the ranked matrix and the ranked matrix itself. See Additional file 1 for ASTRO pseudo codes.
Rank Matrix Construction
A rank matrix is an N × M matrix, R = [rnm], in which every row (gene) is a vector of the ranks of the corresponding expression values in A in increasing order. For example, if the expression levels of gene gi are Ai* = (5, 10, 15, 8) then the corresponding row in the rank matrix would be Ri* = (1, 3, 4, 2). The ranking is performed in increasing order. If more than two entries have the same value, the user can decide to give them the same ranking or rank them in the order they appear. In this study, we choose the former. By replacing each entry of the gene expression matrix with their rank along the rows, we are no longer considering the expression level of a given gene per se, but its dynamics over all time points. The advantage of this method is its speed and that it avoids the use of probabilistic models, greedy algorithms, or costly column permutations. Also, one will notice that for any k > 1 rows of the ranked matrix that are similar under any permutation of the columns of the gene expression matrix; they will always belong to the same cluster. Finally, the fact that the rank is conserved under any permutation of the columns of the gene expression matrix further reduces the chance that a random pattern might be picked up in a cluster (see, also, the Statistical Significance and Complexity Analysis section).
Pattern Identification
Given a gene expression data matrix, A, the exact number of distinct OP expression profiles that can be found in the dataset (time points t = 1,..., M) is the number of distinct rows, NU, of its corresponding ranked matrix R. The set of distinct OP patterns, U, can thus be identified by considering the rank matrix R as a set of rows and identify all subsets of identical rows in it. ASTRO is guaranteed to identify the exact number of distinct OP patterns in a given matrix in O(NM).
Identification of Order Preserving Clusters
Once the exact number of distinct OP patterns has been identified, ASTRO assigns each gene to one of the NU groups by comparing each distinct row Uk* of the ranked matrix to the rows Rn* of the ranked matrix itself, and assign gene n to cluster G{k} each time Uk* = Rn*. This approach is guaranteed to identify all OP clusters of size K × M, with Kmin ≤ K ≤ N, and Kmin is the minimum number of genes in a cluster.
Statistical Significance and Complexity Analysis of ASTRO
The statistical significance of each identified cluster with K genes is assessed by computing the tail probability that a random dataset of size N × M will contain a cluster with K or more genes in it [19]. In principle, the probabilistic description of the reference random matrix would be that of the observed noise in the microarray experiment [29,30]. Since this distribution is difficult to calculate in closed form, we calculate the upper bound of this tail probability following the approach described below. Let's assume we have a dataset with M time points that are independent, identically distributed according to the uniform distribution. Then the probability that a random row (gene) supports a given cluster is equal to the number of possible column permutations or 1/M!. Since the rows are assumed to be independent, the probability of having at least K rows in the cluster is the k-tail of the (N,(1/M!)) binomial distribution, i.e.:
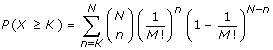 |
As there are Ms = M! ways to choose an OP cluster of size M, the following expression Z(M,K) is an upper bound on the probability of having a cluster of size M with K or more genes:
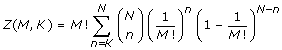 |
We use this bound to estimate the significance of any given cluster of size M with K members. The best cluster is the one with the largest statistical significance, i.e., the one with the smallest Z(M,K). Therefore, as long as that upper bound probability is smaller than any desired significance level, the identified cluster in the real gene expression matrix will be statistically significant.
The overall complexity of ASTRO is ~O(NM). Recall that the time series gene expression A is an N × M matrix. The rank matrix can be identified with an O(NM) complexity. The number of distinct OP patterns and the set of distinct OP patterns can be identified with a complexity less than O(N). Finally, clusters can be identified with a complexity less than O(N). In all, the complexity of ASTRO is O(NM) + O(N) + O(N), which is ~O(NM), less that the complexity of existing approaches.
MiMeSR (Minimum Mean Squared Residue)
MiMeSR seeks to solve the same problem, but unlike ASTRO, it takes into consideration the magnitude of the expression change in the analysis. This is based on the (biological) assumption that if a set of genes is regulated by the same transcription factors across all time points (even if different transcription factors are active at different time points,) then the expression pattern of these genes will not only be the same in terms of direction, but also in magnitude. In other words, MiMeSR aims to identify more coherent clusters of co-regulated genes rather than simply genes with similar expression patterns under a given set of conditions. Under this hypothesis, the problem of finding a cluster of similarly expressed genes is a problem of finding submatrices of the gene expression matrix, A, with minimum mean squared residue or coherent values [20,21]. A cluster here is defined as a submatrix C = [cij] of A (with i and j correspond to the gene and time point, respectively), such that its mean squared residue H(C) < δ. The mean squared residue H(C) of C is computed using the following formula:
where ciT is the mean of the ith row (expression of gene i over all time points), cGj is the mean of the jth column (expression of all genes at the j time point) and cGT is the mean of all the elements of C. When C = [c(i,j)] = [ai + bj] = [ai] + [bj], where [ai] a matrix with constant values on rows, and [bj] a matrix with constant values on columns, then it can be shown that the mean squared residue, H(C), of C is zero.
Proof.
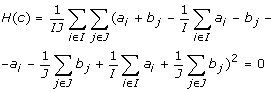 |
The MiMeSR algorithm that we develop in this study uses this concept to search for submatrices with mean squared residue smaller that a given threshold, δ → 0.
Cheng and Church have shown that when the search extends over all possible subsets of columns, then the solution is NP hard [20]. As we will show below, looking for patterns consistent over all time points (i.e. J = T) reduces the algorithmic complexity to O(NMK), and produces biologically relevant results. MiMeSR uses linear algebra and arithmetic tools to solve the problem, which is advantageous over greedy algorithms or the use of heuristics that were used in the past.
Overview
MiMeSR starts by filtering those genes whose expression levels do not change significantly during the time course (threshold ε). Then, it writes the gene expression matrix A as the sum of matrix Z1, with constant values on columns, and Z2 = A - Z1. Finally, it identifies submatrices with constant values on rows in Z2, which correspond to the minimum mean squared residue clusters in the gene expression matrix A. See Additional file 1 for MiMeSR pseudo codes.
Identification of minimum mean squared residue clusters
MiMeSR extracts minimum mean squared residue submatrices from the gene expression matrix using the following approach. For a given row i of matrix A, a new matrix Z1 is constructed with constant values in the columns. All rows in Z1 are identical to row A[i]. Then Z2 is calculated as Z2 = A - Z1. Then, MiMeSR identifies the submatrix with constant values on rows across the whole time points in Z2. This step is easily performed by identifying the set of rows of Z2 such that max(Z2(n,:) - min(Z2(n,:)) <ε, with ε → 0. The submatrices with constant values on rows in Z2 correspond to submatrices with minimum mean squared residue (coherent values) in A. For simplicity and without loss of generality, let us consider an example of the synthetic gene expression matrix A, with coherent values cluster in it, corresponding to rows r1, r3 and r4 (Figure 4.) By subtracting from A matrix Z1 (constructed using the first row of A, (2 4 6 3)), a new matrix Z2 is generated whose rows r1, r3, and r4 correspond to the submatrix with constant values on rows. Note that, the same cluster will be constructed by using any of the rows r1, r3, or r4. Therefore, after a cluster has been identified, its rows are not further considered in the construction of new Z1 matrices. This approach is guaranteed to identify all submatrices with minimum mean squared residue across all time point experiments. Note that, since the operation Z2 = A - Z1 is performed using all the rows of A during each iteration, and since we are seeking for the set of rows of Z2 such that max(Z2(n,:) - min(Z2(n,:)) < ε, MiMeSR can allow rows (genes) to belong to more than one cluster. The biological equivalent of this notion is that genes may be involved in more than one genetic pathway or to be regulated by more than one transcription factors.
Figure 4.

Example of the minimum mean squared residue method.
Statistical Significance and Complexity Analysis of MiMeSR
For practical reasons, it is important to assess the effects of the ε parameter on the clusters that are identified by MiMeSR. This can be done by sensitivity analysis in which the parameter ε is perturbed and the results are compared. For this analysis, it is usually sufficient to consider one or two values above and below the originally selected value of ε. Only clusters that are consistently identified by MiMeSR as ε varies should be retained for further examination. Note that the number of genes in these clusters may also change. The user therefore needs to determine a rule for dealing with genes that may be dropped from the clusters as ε changes. The most conservative approach would be to retain only the genes that remain in the clusters for all values of ε around its selected value. It can be easily shown that the overall complexity of MiMeSR is ~O(NMK), where K is the number of minimum mean squared residue clusters in A. Note that K corresponds to the maximum number of constant columns matrices that can be constructed using the rows of A without identifying redundant clusters.
Authors' contributions
PVB and ABT designed the study, analyzed the results and wrote the paper. KVB and TM designed the web server and contributed to the writing of the paper.
Supplementary Material
Supplementary materials. The following additional data are available with the online version of this paper. Additional file 1 contains the pseudo codes for ASTRO and MiMeSR.
Acknowledgments
Acknowledgements
This work was supported by NIH grants 1R01LM009657-01, and 2R24RR014214-05 and by NIH-NIAID contract no. NO1 AI-50018. P.V.B. was also supported by NIH grant 1R01LM007994-01.
Contributor Information
Alain B Tchagang, Email: abt10@pitt.edu.
Kevin V Bui, Email: kvb2@pitt.edu.
Thomas McGinnis, Email: tfm3@pitt.edu.
Panayiotis V Benos, Email: benos@pitt.edu.
References
- Spellman PT, Sherlock G, Zhang MQ, Iyer VR, Anders K, Eisen MB, Brown PO, Botstein D, Futcher B. Comprehensive identification of cell cycle-regulated genes of the yeast Saccharomyces cerevisiae by microarray hybridization. Mol Biol Cell. 1998;9:3273–3297. doi: 10.1091/mbc.9.12.3273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arbeitman MN, Furlong EE, Imam F, Johnson E, Null BH, Baker BS, Krasnow MA, Scott MP, Davis RW, White KP. Gene expression during the life cycle of Drosophila melanogaster. Science. 2002;297:2270–2275. doi: 10.1126/science.1072152. [DOI] [PubMed] [Google Scholar]
- Gasch AP, Spellman PT, Kao CM, Carmel-Harel O, Eisen MB, Storz G, Botstein D, Brown PO. Genomic expression programs in the response of yeast cells to environmental changes. Mol Biol Cell. 2000;11:4241–4257. doi: 10.1091/mbc.11.12.4241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guillemin K, Salama NR, Tompkins LS, Falkow S. Cag pathogenicity island-specific responses of gastric epithelial cells to Helicobacter pylori infection. Proc Natl Acad Sci USA. 2002;99:15136–15141. doi: 10.1073/pnas.182558799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisen MB, Spellman PT, Brown PO, Botstein D. Cluster analysis and display of genome-wide expression patterns. Proc Natl Acad Sci USA. 1998;95:14863–14868. doi: 10.1073/pnas.95.25.14863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tavazoie S, Hughes JD, Campbell MJ, Cho RJ, Church GM. Systematic determination of genetic network architecture. Nat Genet. 1999;22:281–285. doi: 10.1038/10343. [DOI] [PubMed] [Google Scholar]
- Friedman N, Linial M, Nachman I, Pe'er D. Using Bayesian networks to analyze expression data. J Comput Biol. 2000;7:601–620. doi: 10.1089/106652700750050961. [DOI] [PubMed] [Google Scholar]
- Tamayo P, Slonim D, Mesirov J, Zhu Q, Kitareewan S, Dmitrovsky E, Lander ES, Golub TR. Interpreting patterns of gene expression with self-organizing maps: methods and application to hematopoietic differentiation. Proc Natl Acad Sci USA. 1999;96:2907–2912. doi: 10.1073/pnas.96.6.2907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bar-Joseph Z, Gerber GK, Gifford DK, Jaakkola TS, Simon I. Continuous representations of time-series gene expression data. J Comput Biol. 2003;10:341–356. doi: 10.1089/10665270360688057. [DOI] [PubMed] [Google Scholar]
- Schliep A, Schonhuth A, Steinhoff C. Using hidden Markov models to analyze gene expression time course data. Bioinformatics. 2003;19:i255–263. doi: 10.1093/bioinformatics/btg1036. [DOI] [PubMed] [Google Scholar]
- Ramoni MF, Sebastiani P, Kohane IS. Cluster analysis of gene expression dynamics. Proc Natl Acad Sci USA. 2002;99:9121–9126. doi: 10.1073/pnas.132656399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Hoon MJ, Imoto S, Miyano S. Statistical analysis of a small set of time-ordered gene expression data using linear splines. Bioinformatics. 2002;18:1477–1485. doi: 10.1093/bioinformatics/18.11.1477. [DOI] [PubMed] [Google Scholar]
- Peddada SD, Lobenhofer EK, Li L, Afshari CA, Weinberg CR, Umbach DM. Gene selection and clustering for time-course and dose-response microarray experiments using order-restricted inference. Bioinformatics. 2003;19:834–841. doi: 10.1093/bioinformatics/btg093. [DOI] [PubMed] [Google Scholar]
- Sharan R, Elkon R, Shamir R. Cluster analysis and its applications to gene expression data. Ernst Schering Res Found Workshop. 2002:83–108. doi: 10.1007/978-3-662-04747-7_5. [DOI] [PubMed] [Google Scholar]
- Ernst J, Nau GJ, Bar-Joseph Z. Clustering short time series gene expression data. Bioinformatics. 2005;21:i159–168. doi: 10.1093/bioinformatics/bti1022. [DOI] [PubMed] [Google Scholar]
- Lu X, Zhang W, Qin ZS, Kwast KE, Liu JS. Statistical resynchronization and Bayesian detection of periodically expressed genes. Nucleic Acids Res. 2004;32:447–455. doi: 10.1093/nar/gkh205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moller-Levet CS, Cho KH, Wolkenhauer O. Microarray data clustering based on temporal variation: FCV with TSD preclustering. Appl Bioinformatics. 2003;2:35–45. [PubMed] [Google Scholar]
- Zhao LP, Prentice R, Breeden L. Statistical modeling of large microarray data sets to identify stimulus-response profiles. Proc Natl Acad Sci USA. 2001;98:5631–5636. doi: 10.1073/pnas.101013198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben-Dor A, Chor B, Karp R, Yakhini Z. Discovering local structure in gene expression data: the order-preserving submatrix problem. J Comput Biol. 2003;10:373–384. doi: 10.1089/10665270360688075. [DOI] [PubMed] [Google Scholar]
- Cheng Y, Church GM. Biclustering of expression data. Proc Int Conf Intell Syst Mol Biol. 2000;8:93–103. [PubMed] [Google Scholar]
- Madeira SC, Oliveira AL. Biclustering Algorithms for Biological Data Analysis: A Survey. IEEE/ACM Transactions on Computational Biology and Bioinformatics. 2004;1:24–45. doi: 10.1109/TCBB.2004.2. [DOI] [PubMed] [Google Scholar]
- Yeung KY, Ruzzo WL. Principal component analysis for clustering gene expression data. Bioinformatics. 2001;17:763–774. doi: 10.1093/bioinformatics/17.9.763. [DOI] [PubMed] [Google Scholar]
- Harris MA, Clark J, Ireland A, Lomax J, Ashburner M, Foulger R, Eilbeck K, Lewis S, Marshall B, Mungall C, et al. The Gene Ontology (GO) database and informatics resource. Nucleic Acids Res. 2004:D258–261. doi: 10.1093/nar/gkh036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harbison CT, Gordon DB, Lee TI, Rinaldi NJ, Macisaac KD, Danford TW, Hannett NM, Tagne JB, Reynolds DB, Yoo J, et al. Transcriptional regulatory code of a eukaryotic genome. Nature. 2004;431:99–104. doi: 10.1038/nature02800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ernst J, Bar-Joseph Z. STEM: a tool for the analysis of short time series gene expression data. BMC Bioinformatics. 2006;7:191. doi: 10.1186/1471-2105-7-191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ernst J, Vainas O, Harbison CT, Simon I, Bar-Joseph Z. Reconstructing dynamic regulatory maps. Mol Syst Biol. 2007;3:74. doi: 10.1038/msb4100115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jorgensen P, Rupes I, Sharom JR, Schneper L, Broach JR, Tyers M. A dynamic transcriptional network communicates growth potential to ribosome synthesis and critical cell size. Genes Dev. 2004;18:2491–2505. doi: 10.1101/gad.1228804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blaiseau PL, Isnard AD, Surdin-Kerjan Y, Thomas D. Met31p and Met32p, two related zinc finger proteins, are involved in transcriptional regulation of yeast sulfur amino acid metabolism. Mol Cell Biol. 1997;17:3640–3648. doi: 10.1128/mcb.17.7.3640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dudoit S, Yee Hwa Yang YH, Callow MJ, Speed TP. Statistical methods for identifying differentially expressed genes in replicated cDNA microarray experiments. Statistica sinica. 2002;12:111–139. [Google Scholar]
- Hassibi A, Vikalo H. IEEE International Workshop On Genomic Signal Processing and Statistics: 2005. New Port, Rhode Island, USA; 2005. A probabilistic model for inherent noise and systematic errors of microarrays. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary materials. The following additional data are available with the online version of this paper. Additional file 1 contains the pseudo codes for ASTRO and MiMeSR.