

Abstract
A statistical modeling approach is proposed for use in searching large microarray data sets for genes that have a transcriptional response to a stimulus. The approach is unrestricted with respect to the timing, magnitude or duration of the response, or the overall abundance of the transcript. The statistical model makes an accommodation for systematic heterogeneity in expression levels. Corresponding data analyses provide gene-specific information, and the approach provides a means for evaluating the statistical significance of such information. To illustrate this strategy we have derived a model to depict the profile expected for a periodically transcribed gene and used it to look for budding yeast transcripts that adhere to this profile. Using objective criteria, this method identifies 81% of the known periodic transcripts and 1,088 genes, which show significant periodicity in at least one of the three data sets analyzed. However, only one-quarter of these genes show significant oscillations in at least two data sets and can be classified as periodic with high confidence. The method provides estimates of the mean activation and deactivation times, induced and basal expression levels, and statistical measures of the precision of these estimates for each periodic transcript.
Advances in microarray technologies (1–5) have enabled investigators to explore the dynamics of transcription on a genomewide scale. The current challenge is to extract useful and reliable information out of these large data sets. A common, first approach is cluster analysis. The primary objective of cluster analysis is to group genes that have comparable patterns of variation. This method is valuable for reducing the complexity of large data sets and for identifying predominant patterns within the data (6). However, additional methods are needed to extract information about individual genes from these large data sets.
Toward these goals we consider a statistical method to identify genes whose transcript profiles respond to a stimulus. In general terms, this approach involves modeling the association of a generic response with a specific experimental variable, for example, timing, cell type, temperature, or drug dosage, using a set of interpretable parameters. One objective is to estimate pertinent parameters for individual transcripts, with the goal of testing specific hypotheses concerning transcript response to the stimulus. If the statistical model provides an adequate representation of the expression data for a specific gene, then the corresponding model parameter estimates can provide certain response characteristics for that gene. For example, model parameters may describe the magnitude, duration, or timing of the response. This modeling strategy can be used for two group comparisons, where the objective may be to identify genes that are differentially expressed between normal and abnormal tissues, or in drug discovery studies, where the objective may be to identify transcripts affected by drug dosage.
To demonstrate the utility of this approach, we formulated a model for identifying periodically transcribed genes of the budding yeast Saccharomyces cerevisiae. In this case, the stimulus is synchronous resumption of the cell cycle by releasing the cells from a fixed arrest point. The response is a pulse of transcription, and the key experimental variable is cell cycle timing (7–9).
Four synchronized cell cycle data sets have been generated and made available for general exploration (8, 9). These large data sets have been analyzed by visual inspection (8), Fourier transform and hierarchical clustering (9), k-means (10) and QT clustering (11), self-organizing maps (12), and singular value decomposition (13, 14). Fourier transform analysis of three data sets, where the threshold for periodicity was based on the behavior of known periodic genes, led to a report that there are 800 periodically transcribed genes (9). Later, k-means clustering was applied to one data set, and five periodic clusters with 524 members were identified (10). However, only 330 genes were identified by both approaches. For comparison, we have used statistical modeling to look for regularly oscillating profiles within these large data sets. This approach complements clustering methods in that rather than seeking to group together genes having similar expression patterns it aims to directly identify transcripts affected by a given stimulus and to provide specific information regarding individual response patterns. As amplified below, the method also allows for heterogeneity in response patterns among samples, with expected robustness of inferences on response parameters to certain types of experimental variations.
Methods
A Modeling Framework.
Let Yjk denote the expression level for the jth gene in the kth sample in a stimulus experiment. The number, J, of genes studied often will be of high dimension, typically in the thousands, while the number of samples, K, may be comparatively few. A standard statistical approach would relate the mean of the vector response, Y′k = (Y1k, … , YJk) for the kth sample to a corresponding vector of p covariates xk = (x1k, … , xpk) that codes the stimulus categories and possible other characteristics of the kth sample using a regression function, say Δ(xk, θ)′ = {Δ1k(xk, θ), … , ΔJk(xk, θ)}, where θ′ = (θ1, … , θJ) may include gene-specific and other parameters and is to be estimated. Under such a regression model the elements of the vector of differences Yk − Δk(xk, θ) have mean zero, but can be expected to be correlated due, for example, to variations in mRNA extraction, amplification, and assessment among samples. Such variations can be acknowledged by introducing additional parameters, which we refer to as heterogeneity parameters into the model for the mean of Yk. In fact, for sample k, one can introduce both an additive heterogeneity parameter δk and a multiplicative heterogeneity parameter λk giving a model, δk + λkΔjk(xk, θ) for the expectation of Yjk. The average of the δks and λks are restricted to be zero and one, respectively, to avoid possible identifiability problems relative to the regression parameters θ of primary interest. The high dimension of Yk will allow those heterogeneity parameters to be precisely estimated in many applications. The inclusion of these parameters may make plausible an assumption that Yk given xk are nearly independent, especially for in vitro experiments. Under such an assumption, one can simplify the modeling and numerical procedure for the estimation of θ.
Following the approach described in the seminal statistical paper by
Liang and Zeger (15), estimation of the mean parameter vector
η′ = {δ1, … , δK,
λ1, … , λK, θ} can proceed
by specifying a “working” covariance matrix for
Yk, which under the above independence
assumption will be approximated by a diagonal matrix, written as
Vk = diag(v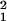 ,
… , v
,
… , v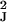 ), so that the expression level for
each of the J genes is allowed to have a distinct variance.
), so that the expression level for
each of the J genes is allowed to have a distinct variance.
Estimates of the vector of mean parameters η can now be estimated as η̂′ = {δ̂1, … , δ̂K, λ̂1, … , λ̂K, θ̂}, a solution to the estimating equation,
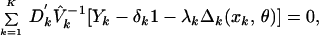 |
1 |
where Dk is the matrix of partial
derivatives of the mean of Yk with respect to
the parameter η, V̂k denotes
Vk with each v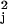 replaced by a consistent estimate v̂
replaced by a consistent estimate v̂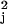 ,
and 1 denotes a column vector of ones of length J. Under the
above modeling assumptions, η̂ will be approximately jointly
normally distributed provided both J and K are
large, and the variance of η̂ can be consistently estimated (as
J and K become large) by a standard sandwich
formula (15, 16).
,
and 1 denotes a column vector of ones of length J. Under the
above modeling assumptions, η̂ will be approximately jointly
normally distributed provided both J and K are
large, and the variance of η̂ can be consistently estimated (as
J and K become large) by a standard sandwich
formula (15, 16).
The mean parameter estimation procedure just outlined is expected to be useful in various types of microarray data sets. It will allow the estimation of meaningful gene-specific parameters to characterize expression levels in response to a stimulus, and, in that sense, is complementary to cluster analysis that seeks to group genes having similar expression patterns, with less emphasis on pattern characteristics. For example, in the context of comparing the expression patterns between diseased and nondiseased tissues, one may define a binary indicator xk that takes value zero for nondiseased tissue samples and one for diseased tissue samples, and specify a regression function, Δjk(xk, θ) = θj0 + θj1xk, under which the jth gene would be differentially expressed between normal and abnormal tissues, whenever θj1 ≠ 0. The regression variable xk also could be expanded to allow the regression function to depend on other measured characteristics of the kth sample (or the kth study subject). Similarly, in a study of variations of expression over time one would define xk = tk, the timing of the kth sample to be gathered, and one can choose a linear or other functional form to model the regression function Δjk(xk, θ).
In the remainder of this paper we focus on the specific setting of transcript response patterns when cells are released from cell cycle arrest. Our particular interest will be the identification of genes having periodic expression level changes over multiple cell cycles. A key data analysis challenge relates to the possibility of a single nonoscillating pulse of transcription for some genes, after the resumption of cell cycling.
Modeling Periodic Transcription.
Consider a synchronized experiment to identify mRNAs that are transcribed once per cell cycle. Suppose that when activated, the jth mRNA reaches an elevated value (αj + βj) and when deactivated it falls to a basal expression level (αj) (Fig. 1). Naturally, βj is interpreted as the difference between averaged peak and trough expression levels. Considering multiple copies of the jth mRNA, transcribed and dissipated at consecutive times in multiple cells with imperfect synchronization, the mean expression level of the jth transcript at the time tk may be modeled as:
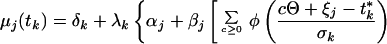 |
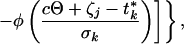 |
in which j = 1, 2, … , J and k = 1, 2, … , K for all J transcripts at all K time points, where ζj, ξj are the activation and deactivation times for the jth gene, respectively, t*k = tk + τ, where τ denotes the difference of actual cell cycle timing and observed timing and is typically known as phase, Θ is the cell cycle span, and the summation is over multiple cell cycles, c = 0, 1, 2, … . The standard deviation, σk, depicts the variation of “true” cell-specific timings around tk, which we assume to follow a normal distribution with mean tk, resulting in the cumulative normal distribution function φ(.) in the mean model. Also, δk, λk are the additive and multiplicative heterogeneity parameters for the kth sample as described above, and here xk = tk. The above single-pulse model (SPM) specifies a model for the mean expression of each gene as the cell cycle proceeds. Gene-specific activation and deactivation times as well as the background and elevated expression levels are estimated for each gene. SPM also allows for variation between samples, for the fact that the synchrony is imperfect and, as described below, for the synchrony to deteriorate over time (Fig. 1). Further detail on the development of the SPM is given in the Appendix, which is published as supplemental material on the PNAS web site, www.pnas.org. The resulting mean expression model has been shown visually to reproduce the profiles observed for periodic transcripts measured by conventional means (17).
Figure 1.

The basic assumption of the SPM is that a cell cycle-regulated transcript will be transcribed at one invariant time and will be dissipated at a subsequent time during the cell cycle. (A) For example, we consider a single transcript that is activated at (ζ = 10′) and deactivated at (ξ = 55′) during two consecutive cell cycles of length (Θ = 80′) from a basal (α = 0) to an induced (α + β = 1) level of expression. (B) In a typical synchrony experiment, multiple transcripts are made per cell and RNA is harvested from many cells. These cells are not perfectly synchronized and the synchrony deteriorates with time, leading to attenuation of simple pulses (dashed line) into smooth peaks (dotted line) that dampen out with time (solid line). In the example shown, the ages of cells vary from a standard deviation of 3 to 19 min. (C) The expression values obtained (dots) are subject to both additive and multiplicative heterogeneity as well as additional variability beyond what has been modeled, and the differences of which are known as residuals. Using these residuals, we estimate their standard deviation and evaluate the significance of the pulse height in relation to this standard deviation via the Z scores.
The SPM described above can be applied by using the mean model estimation procedure outlined above. To simplify numerical aspects we have used a multistage procedure: (i) heterogeneity parameters, (δk, λk), k = 1, … , K, are estimated by using all genes when the pulse heights are set to zero, (ii) the cell cycle span, Θ, is estimated by using a group of known cell cycle genes under a pulse model, (iii) the synchronization variability, σk, k = 1, … , K, is estimated by using the same group of known genes, and (iv) gene-specific parameters (αj, βj, ζj, ξj), j = 1, … , J, are estimated while other estimated parameters are treated as fixed at their estimated values. Although a simultaneous estimation approach using the estimating Eq. 1 would be preferable, the impact on estimation of the gene-specific parameters of their variance estimates is likely to be minimal as gene-specific parameters are weakly correlated with other parameters. Fixing the cell cycle span and sample-specific parameters allows a separate simple calculation of the gene-specific parameter estimates, and of their variance estimates, for each of the J genes. Further detail on these calculations is given in the Appendix.
To test the fit of the SPM we introduced additional polynomial functions of time in the mean model and tested the hypothesis that the polynomial coefficients were identically zero. Specifically, the SPM is augmented and written as
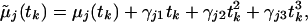 |
allowing a departure from the SPM. A score-type test statistic for
(γj1, γj2, γj3) =
(0, 0, 0) is then constructed using the asymptotic normal theory
described above. This score statistic,
χ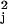 , will have an approximate chi-square
distribution with three degrees of freedom under the SPM model, for
sufficiently large J and K. We choose 11.3, the
1% upper percentage of this chi-square distribution to identify genes
with patterns that depart significantly from the SPM. For the
cdc28 data set, for example, only 262 genes give test
statistics that exceeded the critical value. Note that other deviations
than those polynomial terms could be specified but are not pursued
here.
, will have an approximate chi-square
distribution with three degrees of freedom under the SPM model, for
sufficiently large J and K. We choose 11.3, the
1% upper percentage of this chi-square distribution to identify genes
with patterns that depart significantly from the SPM. For the
cdc28 data set, for example, only 262 genes give test
statistics that exceeded the critical value. Note that other deviations
than those polynomial terms could be specified but are not pursued
here.
For those genes for which the expression pattern does not depart significantly from SPM, we estimate activation time (ζj), deactivation time (ξj), basal expression level (αj), and elevation in expression level during the interval (βj), along with their estimated standard deviations. Under the SPM, expression levels are cell cycle regulated if and only if βj ≠ 0. We choose a critical value of 5 for the absolute value of each Zj, the ratio of the estimate of βj to its estimated standard deviation, to reject the null hypothesis. This value, far in the tail of normal distribution, is expected to preserve a genomewide significance level of about 0.3% (two-sided) even with as many as 6,000 genes under study. Some of genes that showed evidence of departure from SPM also may have expression patterns that vary with the cell cycle. One could test βj = 0 also for those genes in the context of the augmented mean model, μ̃j(tk), described above, though the interpretation of such a test would be conditional on the adequacy of the augmented model.
Nature of the Data.
Three data sets were used in this analysis. The cdc28 data set was generated by Cho et al. (8), and synchrony was established by using a temperature-sensitive cdc28 mutation to reversibly arrest cells in G1. Oligonucleotide arrays (Affymetrix, Santa Clara, CA) were hybridized to fluorescently labeled cDNAs made from each sample, and the absolute fluorescence intensity values are assumed to be proportional to the amounts of each transcript in each target sample (7). These data were downloaded from http://genomics.stanford.edu. The two other sets of data (alpha factor and cdc15) were generated by Spellman et al. (9) using an alpha factor-mediated G1 arrest and a temperature-sensitive cdc15 mutation to induce a reversible M-phase arrest, respectively. In this case, fluorescently labeled cDNAs were made from RNA from each time point and a second fluorescent dye was used to label cDNA made from an asynchronous control culture. Control and test cDNAs were mixed and hybridized to arrays of PCR-amplified yeast ORFs. Fluorescence intensity values of both dyes were measured, and logarithmic ratios of test versus control values were generated. Obtained ratios are assumed to approximate the corresponding true ratios of test versus control mRNA levels (9). These data and the cdc28 data, rescaled to mimic the ratio data, were accessed from the public domain site http://cellcycle-www.stanford.edu. Our results are based on the analysis of these data sets and as such will be influenced by all sources of variation involved in the preparation and processing of these arrayed samples.
Results and Discussion
The primary assumptions of SPM are that cell cycle-regulated transcripts will peak only once per cycle and that these pulses occur at invariant times in consecutive cycles. SPM includes terms that enable additive and multiplicative heterogeneity across samples to be accommodated. Fig. 2 shows additive heterogeneity estimates for each data set. Additive heterogeneity is minimal when logarithmic ratios are used. When the absolute intensity is considered for the cdc28 data set, the additive heterogeneity is most evident at the 90-min time point. This confirms the concern over this particular time point (8) and provides a means of correcting for its heterogeneity.
Figure 2.

Parameters estimated for the data sets from synchronization by alpha factor (A), cdc15 (B), and cdc28 (C for ratio data and D for absolute intensity) data sets. The left column reflects the estimated additive heterogeneity for each time point. The middle column indicates the estimated cell cycle span for each synchrony as the profiled, weighted least square on a probability scale. To facilitate visual inspection, we transformed this sum of squares to a probability scale using exp[−L(Θ)]/∫exp[−L(Θ)]. This would be a posterior probability, if expression values followed the normal distribution. The right column shows estimated standard deviations associated with deteriorating synchrony.
We estimated cell cycle span for each data set by using a set of 104
known cell cycle-regulated genes and profiling over a range of possible
cell cycle spans (see Appendix). As expected, the cycle span
differs for each synchrony method. Cycle spans for the alpha factor and
cdc15 data sets show bimodal distributions (Fig. 2). These
may be due to recovery artifacts that differentially affect the first
cycle and alter the timing of a subset of the transcripts. We have used
the estimated cell cycle span that minimizes a certain weighted sum of
squares, giving a value of 58 min for the alpha factor synchrony, 115
for the cdc15 cells, and 85 for the cdc28
culture. Fig. 2 also shows the estimated standard deviations associated
with loss of synchrony over time. Once these values have been obtained,
the χ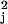 values are calculated for the
jth gene for j = 1, … , J, and
gene-specific parameters are estimated for all genes having
transcription patterns consistent with the SPM (i.e.,
χ
values are calculated for the
jth gene for j = 1, … , J, and
gene-specific parameters are estimated for all genes having
transcription patterns consistent with the SPM (i.e.,
χ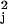 values less than 11.3). Gene-specific
parameters include the mean activation and deactivation times and the
basal and elevated levels.
values less than 11.3). Gene-specific
parameters include the mean activation and deactivation times and the
basal and elevated levels.
Fig. 3 shows the microarray data (solid lines) for five periodic genes and the fitted SPM to these profiles (dotted lines). Clearly, the model closely approximates the profile of the data and provides mean activation and deactivation times (in brackets) that are consistent with the patterns observed. The Z values for these oscillations vary from about 18 for RFA1 in the cdc15 data set to about 3.5 for MCM3 in the alpha factor data set. The fact that the periodic behavior of MCM3 is still evident gives us confidence that we have set a sufficiently conservative threshold for each Zj. The top three transcripts have been classified as G1-specific, MCB-regulated genes (9). However, the PDS1 pulse is delayed compared to the other two. RFA1 and CLB6 are activated at about the same time, but the pulse of CLB6 mRNA is shorter lived. These differences are reflected in the activation and deactivation times calculated for each gene by SPM and can be used to identify coordinately regulated transcripts.
Figure 3.

The fit of the SPM (dotted lines) to the microarray data (solid lines) from three different synchronized cell cycles for five periodically transcribed genes. The logarithmic ratio data versus time is plotted for the alpha factor (Right), cdc15 (Center), and cdc28 (Left) synchronies. Beneath each plot, the times of activation and deactivation for each transcript are shown in brackets, followed by Z score and χ2 statistic calculated under SPM, which indicate the significance of the pulse height and deviation from SPM, respectively.
Identification of Cell Cycle-Regulated Transcripts Using SPM on the cdc28 Data Set.
A total of 607 genes met SPM thresholds for periodicity (i.e., |Zj| value of 5 or greater) using absolute fluorescence intensity measurements directly from the cdc28 data (8). We obtained about the same number of genes by using either the logarithm of the intensity or the logarithmic ratios of intensities as generated by Spellman et al. (1, 2, 9). However, only about 500 genes were identified in all three analyses. Thus, any single data transformation may miss about 20% of the potential positives, due to Z values that are close to our threshold. In all subsequent analysis, we have used logarithmic ratios of the cdc28 data to be consistent with the alpha factor and cdc15 data.
Lists of cell cycle-regulated genes in the cdc28 data set have been compiled by visual inspection (8) and k-means clustering (10). SPM analysis confirms a majority of these assignments and identifies many more candidate oscillating transcripts. The application of the k-means approach provided by Tavezoie et al. (10) used an initial filtering strategy to select the 3,000 yeast genes, which showed the highest coefficient of variation over the time course. Then, the iterative k-means procedure was used to partition all 3,000 profiles into 30 clusters. The requirement that all 3,000 profiles fit into one of 30 clusters necessitated the assembly of large clusters with loosely correlated patterns of expression. Five of these clusters had mean temporal profiles that were clearly periodic over two cell cycles. However, only about half of the profiles of the 524 cluster members exceeded the thresholds for periodicity in SPM.
To see whether SPM could identify a tight cluster of periodic genes, we computed the χ2 and Z values for a cluster of G1-specific transcripts which was assembled at three different thresholds by Heyer et al. (11), using the QT_Clust algorithm. In this case, we find that all the tightest cluster members either exceed or come very close to the threshold for periodicity set in SPM (Fig. 4 Top). Inspection of the borderline cases indicates that they are likely to be periodic and thus our Z value threshold is conservative. When the cluster threshold is set lower, membership doubles and again nearly all profiles are at the SPM threshold or well above it (Fig. 4 Middle). However, as noted by Heyer et al. (11) further relaxation of the cluster threshold to include 272 profiles leads to the inclusion of many poorly matching patterns that also have low Z values by SPM (Fig. 4 Bottom). This finding indicates the efficiency of both approaches in identifying the most periodic transcripts. It also illustrates the value of having two completely different methods of analyzing the data to establish meaningful thresholds and characterize the less robust response patterns.
Figure 4.

Periodic transcripts that peak in the G1 phase of the cell cycle were identified by Heyer et al. (11) using the QT_Clust algorithm and varying the cluster diameter threshold from <0.3 (top 41 genes), to <0.5 (83 genes), to <1.2 (272 genes). The transcript profiles for members of these successively larger G1 clusters were analyzed by SPM, and their Z and χ2 values are plotted (Left). The Z score and χ2 thresholds of SPM are superimposed on these plots to show that the proportion of these profiles that would be classified as periodic (lower right quadrant of each plot). (Right) the distribution of mean activation and deactivation times is plotted for each group. These parameter estimates were calculated by SPM only for those profiles that exceed SPM thresholds.
Another feature of SPM is its estimation of gene-specific parameters. Fig. 4 also shows how the distributions of activation and deactivation times broaden as cluster membership increases. This broadening indicates that in addition to containing nonperiodic profiles, this group contains genes with different kinetics of expression. Thus, SPM enables these clusters of similar expression patterns to be further subdivided, depending on the question of interest.
Using All the Data Sets to Estimate the Number of Periodic Genes.
One limitation of these cell cycle data sets is the small number of samples and the lack of multiple measurements at any time point, which makes the identification of false positives and false negatives problematic. To mitigate this problem, we have used SPM to identify periodic transcripts from the cdc28, cdc15, and alpha factor data sets separately and then compared the results. SPM identifies about twice as many periodic genes in the cdc28 data set as in either of the other two synchronies (Fig. 5), and overall there are 1,088 genes that show significant oscillations in at least one data set. Included among the 1,088 candidate periodic genes identified by SPM are 81% of the 104 known periodic genes. A total of 254 genes oscillate significantly in at least two databases, which represents 4% of all genes, but includes 46% of the known periodic genes. Thus, SPM identifies the known periodic transcripts well above the level expected by chance. Only one-quarter of the known periodic genes are among the 71 genes that score as periodic in all three data sets. A total of 834 genes appear periodic in only one data set and as such further data collection will be required before this large group of genes can be unambiguously classified. Complete lists of the periodic transcripts identified by SPM are available at our web site (www.fhcrc.org/labs/breeden/SPM).
Figure 5.

Periodic transcripts identified by SPM with thresholds of |Z| >5 and χ2 < 11.3 are depicted to show the extent of agreement between the three data sets. Logarithmic ratio data provided by Spellman et al. (9) for each of the three data sets was analyzed by SPM. The total number of periodic genes identified in each data set is shown and is represented by a circle. Agreement between data sets is indicated by the intersections of the circles. A total of 71 genes meet the SPM threshold for periodicity in all three data sets; 254 score as periodic in at least two databases; 834 appear periodic in only one data set; and 1,088 meet the SPM threshold in only one database. If we use an additional criterion of R2 > 0.6 to identify the profiles among these 1,088 for which the model provides an explanation for 60% or more of the expression data variation, we find 473 profiles. This list is available at our website (www.fhcrc.org/labs/breeden/SPM).
Spellman et al. (9) used Fourier analysis of the combined data from the same three data sets to identify periodic transcripts. Using the known periodic genes as a guide for setting their threshold, they estimated that 799 genes are periodic. Only 65% of these genes also are picked up by SPM as being periodic in at least one data set. This difference may be explained, in part, by our conservative threshold for Z because reducing the threshold value for Z to 4.0 enables 79% of these genes to be classified as periodic in at least one data set.
Nearly all of the genes that exceed the threshold for periodicity by SPM in at least two data sets also are recognized by the method of Spellman et al. (9). Here again, as with clustering, the most robust periodic patterns are identified by both methods. However, there are 571 genes that appear to be periodic by SPM criteria in at least one data set but are not classified as such by Spellman et al. (9). As noted above, these cannot be unambiguously classified as periodic without further corroborating data. They are either false negatives in two data sets or false positives in one. Experimental variation is much more likely to result in a nonperiodic pattern than it is to produce a smoothly oscillating profile. With SPM, the peaks also must occur at the same time in consecutive cell cycles and peaks and troughs are not recognized if they are represented by a single point in the profile (see Appendix). These restrictions should reduce the impact of noise and result in a lower false positive error rate. However, we cannot eliminate the impact of noise in the data and, with so few data points to base these assignments upon, many remain ambiguous. The 254 genes that score as periodic in two data sets can be considered periodic with reasonably high confidence, but they include only about half of the known periodic genes and as such clearly underestimate the number. Unless more data are generated, classification of the other transcripts will remain ambiguous. In other words, in spite of the accumulation of nearly one half million data points, we can identify only about half of the periodic transcripts of budding yeast with high confidence. These ambiguities, combined with the fact that statistical methods are most reliable when there is a large number of independent samples, leads us to conclude that another data set, traversing two full cell cycles and having closer time points will be required to more completely identify and order the periodic transcripts of this important model organism.
If even half of these 1,088 genes are actually periodic (see Fig. 5), they would comprise about 10% of all budding yeast genes, which could be viewed as an enormous regulatory burden to the cells, especially if there are many different ways in which this regulation is accomplished. On the other hand, if there are only 20 different circuits that achieve this regulation and gene products have evolved into these limited expression patterns based on the cell's need for them, one could view it as a highly parsimonious strategy for limiting the biosynthetic load on the cell.
Conclusion
In this report we use a statistical model (SPM) to identify and characterize single pulses of transcription that occur at invariant times in consecutive cell cycles. SPM is a specific application of statistical modeling, but the basic strategy can be applied to any large data set to identify genes undergoing a transcriptional response to a stimulus. Due to its relative simplicity, statistical modeling can be used to interrogate large data sets without using additional filters to reduce the number of genes to be analyzed. It also includes heterogeneity parameters that will tend to reduce the impact of noise in the data sets. SPM identifies regularly oscillating transcripts without regard to the abundance of the transcript or the height or timing of the peak and provides estimates of the mean time of activation and deactivation. These values are only estimates, but they are unbiased under the assumed SPM and can be considered defining characteristics of individual genes. SPM also provides statistical measures of the precision of the parameter estimates so that optimal groupings can be made and subjected to further analysis. These features of statistical modeling complement and augment the other methods used to analyze microarray data.
Supplementary Material
Acknowledgments
We thank Drs. M. Campbell, J. Cooper, and J. Sidorova for helpful discussions about the data. We also thank the investigators who contributed their yeast expression data to the public domain, and L. Heyer, S. Kruglyak and G. Church for providing cluster information for comparison. This research is supported by grants from the National Institutes of Health (GM41073 to L.B. and PO1 CA53996 to L.P.Z. and R.P.).
Abbreviation
- SPM
single-pulse model
References
- 1.Schena M, Shalon D, Davis R W, Brown P O. Science. 1995;270:467–470. doi: 10.1126/science.270.5235.467. [DOI] [PubMed] [Google Scholar]
- 2.Schena M, Shalon D, Heller R, Chai A, Brown P O, Davis R W. Proc Natl Acad Sci USA. 1996;93:10614–10619. doi: 10.1073/pnas.93.20.10614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lander E S. Nat Genet Suppl. 1999;21:3–4. doi: 10.1038/4427. [DOI] [PubMed] [Google Scholar]
- 4.DeRisi J L, Iyer V R, Brown P O. Science. 1997;278:680–686. doi: 10.1126/science.278.5338.680. [DOI] [PubMed] [Google Scholar]
- 5.Fodor S P A, Read J L, Pirrung M C, Stryer L, Lu A T, Solas D. Science. 1991;251:767–773. doi: 10.1126/science.1990438. [DOI] [PubMed] [Google Scholar]
- 6.Sherlock G. Curr Opin Immunol. 2000;12:201–205. doi: 10.1016/s0952-7915(99)00074-6. [DOI] [PubMed] [Google Scholar]
- 7.Cho R J, Fromont-Racine M, Wodicka L, Feierbach B, Stearns T, Legrain P, Lockhart D J, Davis R W. Proc Natl Acad Sci USA. 1998;95:3752–3757. doi: 10.1073/pnas.95.7.3752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cho R J, Campbell M J, Winzeler E A, Steinmetz L, Conway A, Wodicka L, Wolfsberg T G, Gabrielian A E, Landsman D, Lockhart D J, et al. Mol Cell. 1998;2:65–73. doi: 10.1016/s1097-2765(00)80114-8. [DOI] [PubMed] [Google Scholar]
- 9.Spellman P T, Sherlock G, Zhang M Q, Iyer V R, Anders K, Eisen M B, Brown P O, Botstein D, Futcher B. Mol Biol Cell. 1998;9:3273–3297. doi: 10.1091/mbc.9.12.3273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tavazoie S, Hughes J D, Campbell M J, Cho R J, Church G M. Nat Genet. 1999;22:281–285. doi: 10.1038/10343. [DOI] [PubMed] [Google Scholar]
- 11.Heyer L J, Kruglyak S, Yooseph S. Genome Res. 1999;9:1106–1115. doi: 10.1101/gr.9.11.1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tamayo P, Slonim D, Mesirov J, Zhu Q, Kitareewan S, Dmitrovsky E, Lander E S, Golub T R. Proc Natl Acad Sci USA. 1999;96:2907–2912. doi: 10.1073/pnas.96.6.2907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Holter N S, Mitra M, Maritan A, Cieplak M, Banavar J R, Fedoroff N V. Proc Natl Acad Sci USA. 2000;97:8409–8414. doi: 10.1073/pnas.150242097. . (First Published July 11, 2000; 10.1073/pnas.150242097) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Alter O, Brown P O, Botstein D. Proc Natl Acad Sci USA. 2000;97:10101–10106. doi: 10.1073/pnas.97.18.10101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Liang K Y, Zeger S L. Biometrika. 1986;73:13–22. [Google Scholar]
- 16.Prentice R L, Zhao L P. Biometrics. 1991;47:825–839. [PubMed] [Google Scholar]
- 17.Breeden L L. Methods Enzymol. 1997;283:332–341. doi: 10.1016/s0076-6879(97)83027-3. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.