

Abstract
Lifetime imaging microscopy is a powerful tool to probe biological phenomena independent of luminescence intensity and fluorophore concentration. We describe time-resolved imaging of long-lifetime luminescence with an unmodified commercial laser scanning confocal/multiphoton microscope. The principle of the measurement is displacement of the detection pinhole to collect delayed luminescence from a position lagging the rasting laser beam. As proof of principle, luminescence from microspheres containing europium (Eu3+), a red emitting probe, was compared to that of short-lifetime green-fluorescing microspheres and/or fluorescein and rhodamine in solution. Using 720-nm two-photon excitation and a pinhole diameter of 1 Airy unit, the short-lifetime fluorescence of fluorescein, rhodamine and green microspheres disappeared much more rapidly than the long-lifetime phosphorescence of Eu3+ microspheres as the pinhole was repositioned in the lagging direction. In contrast, repositioning of the pinhole in the leading and orthogonal directions caused equal loss of short- and long-lifetime luminescence. From measurements at different lag pinhole positions, a lifetime of 270 μs was estimated for the Eu3+ microspheres, consistent with independent measurements. This simple adaptation is the basis for quantitative 3-D lifetime imaging microscopy.
Keywords: confocal microscope, europium, lifetime imaging, luminescence, two-photon excitation
1 Introduction
Conventional fluorescence microscopy creates images based on the intensity and distribution of extrinsically added fluorophores, engineered proteins, and specimen autofluorescence. Fluorescence images depend on various factors, including fluorophore concentration, intensity of excitation, photobleaching, and detector sensitivity. In quantitative fluorescence microscopy, fluorescence intensity can change independently of the phenomenon being investigated. For example, leakage of fluorophores, photobleaching, and changes in cell shape can alter fluorescence measurements independently of the biological parameter under study.1 Ratio imaging overcomes signal variations due to dye redistribution and photobleaching and is used effectively to image calcium, pH, and other cellular parameters.2–4 However, the spectral characteristics of many fluorophores are not amenable to ratio imaging.
An alternative approach is fluorescence lifetime imaging microscopy (FLIM) that uses the lifetime of luminescence decay to visualize biological parameters of interest. Lifetime represents the average time a fluorophore spends in the excited state prior to emitting a photon and returning to ground state.5 Fluorescence lifetime is independent of the concentration of luminophores (fluorophores and phosphors), excitation intensity, and other factors that introduce artifacts into intensity measurements.6 For luminophores of sufficiently different lifetimes, a single excitation source and detector can be used to discriminate multiple probes of differing lifetimes but similar spectral characteristics.7
Lifetimes of luminescence probes range from pico- to microsecond durations.8 Because of the frequency of use of shorter-lifetime probes, most commercial FLIM systems are geared toward measuring lifetimes on a nanosecond scale.9 Other applications, however, require long lifetime (phosphorescence).
In FLIM, lifetime is assessed by both time and frequency domain techniques. In the time domain technique, the specimen is exposed to brief pulses of light. Fluorescence decay is then measured either from emission intensity at different time intervals after the pulses (time-gated detection) or by time-resolved photon counting.6,10,11 By fitting measured fluorescence with an exponential decay function, lifetimes are estimated. In the frequency-based technique, modulated light of defined amplitude and phase excites the specimen. Because of the delay between emission and excitation, lifetime can be calculated from phase shift of the emitted luminescence relative to the excitation. Both time and frequency domain techniques have been adapted to wide-field fluorescence microscopy and to laser scanning confocal and multiphoton microscopy.9,12–15 The principal advantage of confocal and mulitphoton microscopy is 3-D resolving power.9,16 However, confocal and multiphoton FLIM systems are generally configured to measure short lifetimes in the pico- and nanosecond range. Adaptation of a confocal and multiphoton FLIM system for long-lifetime imaging in the microsecond range requires additional equipment and modifications.9,17
Here, we describe a novel and cost-effective technique for submicron lifetime imaging of long-lifetime luminescence based on shifting the pinhole of a laser scanning confocal microscope. This technique can be applied using an unmodified commercial laser scanning confocal microscope. We call this technique pinhole shifting lifetime imaging microscopy (PSLIM).
2 Methods and Materials
2.1 Imaging of Long-Lifetime Europium Microspheres and Short-Lifetime Luminophores
Imaging was performed with a Zeiss LSM 510 META NLO confocal microscope (Thornwood, New York) equipped with a Coherent Mira 900 femtosecond pulsed Ti:Sapphire laser for multiphoton excitation (Santa Clara, California). Images were collected as 512×512 pixel scans at 8-bit intensity resolution using a 63× 1.4 NA planapochromat oil immersion lens at pinhole diameters calibrated in Airy units appropriate for the wavelength of fluoresced light. Pinhole shifting was accomplished with Zeiss LSM software, which permits displacement of detector pinholes in both parallel and orthogonal directions with respect to the rasting laser spot. The software provides a pinhole adjustment menu. This menu can be accessed from the “maintain” button on the LSM software. The menu specifies pinhole physical displacement in microns or scaling in Airy units appropriate for the NA and magnification of the objective lens.
Two-photon excitation of long-lifetime europium microspheres, short-lifetime green microspheres, and rhodamine and fluorescein in solution was accomplished using 720-nm light from the Ti:Sapphire laser. Green and red luminescence was divided by a 545-nm long-pass dichroic mirror and directed to photomultipliers through 525-nm (50-nm bandpass) and 590-nm (50-nm bandpass) barrier filters, respectively. Images were acquired at room temperature. Acousto-optical tunable filters (AOTFs) were set for 29% laser intensity at the specimen plane for all experiments unless indicated otherwise. All microspheres were 1 μm in diameter.
2.2 Imaging of Europium and Blue Microspheres
In some experiments, long-lifetime europium microspheres were imaged simultaneously with short-lifetime blue microspheres. Excitation was performed at 720 nm from the multiphoton Ti:Sapphire laser. Red and blue luminescences were separated by a 545-nm long-pass dichroic mirror and directed to photomultipliers through 590-nm (50-nm bandpass) and 500-nm (20-nm bandpass) barrier filters, respectively. Images were acquired at room temperature. All microspheres were 1 μm in diameter.
2.3 Slide Preparation
Solutions (5 μl) of europium (1 × 107 microspheres/μL) and green or blue microspheres (3.6 × 107 microspheres/μL) and/or fluorescein or rhodamine (16 mM) were added to 200 μl glycomethacrylate. The microsphere solutions were sonicated for 30 min before addition to glycomethacrylate. Aliquots (50 μl) were then pipetted on a glass slide and covered with a 0.17-mm-thick glass coverslip. The glass slides were placed under UV light at 4 °C for 30 min. At this point, the slides were stored at room temperature for experimentation.
2.4 Software
Intensity for images was determined using Zeiss LSM software (Carl Zeiss, GmBH) and Photoshop (Adobe Systems, San Jose, California). A region inside microspheres was selected, and the average intensity from this region was determined. Plotting of graphs was performed using SigmaPlot (Systat Software, Inc., San Jose, California). All illustrations were made in Corel Draw (Corel Corporation, Eden Prairie, Minnesota).
2.5 Luminophores and Chemicals
Europium, green, and blue luminescent microspheres (1-μm-diam size) were obtained from Invitrogen (Carlsbad, California). Fluorescein and rhodamine were obtained from Sigma Corp. (St. Louis, Missouri). Glycomethacrylate was purchased from Electron Microscopy Sciences (Hatfield, Pennsylvania).
3 Results
3.1 Principle of Luminescence Lifetime Imaging Microscopy by Pinhole Shifting
In confocal microscopy, the detection pinhole is positioned to collect light exactly from the position within the specimen over which the laser beam is being scanned (Fig. 1). Indeed, when the pinhole is misaligned in the leading, lagging, or orthogonal directions, collection of reflected light and short-lifetime luminescence drops precipitously. However, as shown in Fig. 1, when the pinhole is shifted in the lagging direction, delayed luminescence—namely, long-lifetime luminescence—should be selectively transmitted through the pinhole with rejection of short-lifetime fluorescence. The lifetimes collected depend on the distance of pinhole shifting and the speed of the rasting laser beam across the specimen. Speed is inversely proportional to dwell time, which is defined as the amount of time the laser beam resides over each spot in the specimen. This principle leads us to hypothesize that delayed luminescence of long-lifetime probes can be selectively detected by shifting the detection pinhole in the lagging direction in relation to the rasting laser spot. We call this technique pinhole shifting lifetime imaging microscopy (PSLIM).
Fig. 1.

Principle of long-lifetime imaging microscopy by confocal pinhole shifting. When the detection pinhole is aligned to the crossover of the rasting laser spot, short-lifetime luminescence passes the pinhole to the photodetector. When the pinhole is shifted in the lagging direction with respect to the rasting laser spot, short-lifetime luminescence is rejected and long-lifetime luminescence is collected instead. The lifetimes collected depend on the speed of the rasting laser spot and the pinhole shift distance.
The lagging pinhole collects decaying luminescence from a position on the specimen that follows the crossover of the rasting laser beam (Fig. 1). The lifetimes collected depend on the pinhole shift distance and the dwell time of the rasting laser spot. For a confocal microscope operating at the diffraction limit, lifetimes collected for different pinhole shifts (τcollected) are given by
| (1) |
where m is pinhole shift in Airy units corresponding to the diameter of the Airy disk projected on the pinhole plane, and Δt is dwell time of the laser spot.
In confocal microscopy, the diameter of the smallest resolvable point in the specimen plane (dl) is:
| (2) |
where λ is the wavelength of fluorescence, and NA is the numerical aperture of the objective lens.11 The diameter of this Airy disk as projected on the pinhole (D1) is
| (3) |
where M is magnification at the pinhole image plane. Dl defines the physical size of 1 Airy unit in the pinhole plane. To maximize axial resolution, pinhole diameters in confocal microscopy are typically set to 1 Airy unit. Here, we will use Airy units to describe pinhole shifting distances. Accordingly, lateral shifts of the pinhole by 1, 2, and 3 Airy units correspond to physical shifting distances of D1,2D1,3D1, and so forth.
3.2 Images of Long-Lifetime Europium and Short-Lifetime Probes after Pinhole Shifting
We hypothesized that shifting the detection pinhole of a confocal microscope in the lagging direction to the rasting laser spot will enable selective imaging of long-lifetime luminescence. To test this hypothesis, we imaged long-lifetime europium microspheres in comparison to short-lifetime green microspheres and two diffusely distributed short-lifetime fluorophores, fluorescein and rhodamine. Europium is a luminescent lanthanide metal characterized by a lifetime of several hundred microseconds and a large Stoke's shift between excitation and emission wavelengths.18–21 Accordingly, we used europium microspheres as a long-lifetime specimen. Green microspheres, rhodamine, and fluorescein are short-lifetime fluorophores (τ≤20 ns) and were imaged simultaneously with europium.
Images of europium and green 1-μm microspheres were acquired with a laser dwell time of 102 μs per pixel using 720-nm multiphoton excitation from a pulsed Ti:Sapphire laser. Pinholes in the red and green channels were first aligned to the rasting laser spot in the normal fashion for confocal imaging, and an image was acquired [Fig. 2(a), center column]. The image shows europium microspheres in the red channel and green microspheres in both the green and red channels (yellow in the overlay because of bleed-through of green fluorescence into the red channel). The pinholes in each channel were then shifted by 1 Airy unit in the lagging direction in relation to the rasting laser spot, and another image was acquired [Fig. 2(a), left column]. In the new, pinhole-shifted image, red-luminescing europium microspheres remained visible, but the green microspheres disappeared. The disappearance of the green fluorescence was even more striking because the original, unshifted green-fluorescence image was oversaturated. In contrast, when the pinhole was shifted 1 Airy unit in the leading direction relative to the rasting laser spot, both red and green luminescence were lost entirely [Fig. 2(a), right column].
Fig. 2.
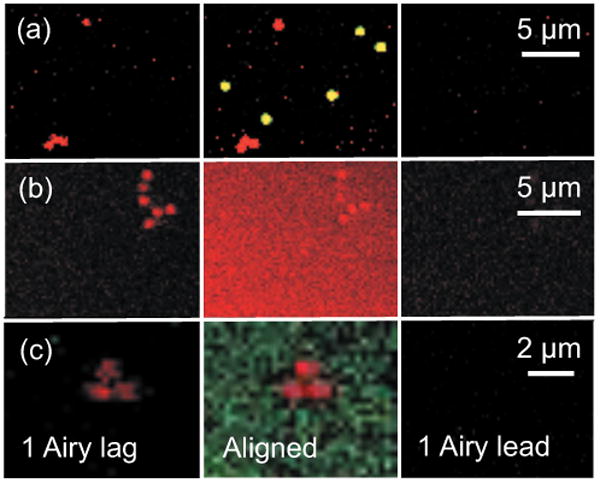
Confocal images of long-lifetime europium microspheres and short-lifetime green microspheres, rhodamine, and fluorescein. In (a), 1-μm europium microspheres were imaged with 1-μm green microspheres. In (b), europium microspheres were imaged with rhodamine (400 μM) in solution. In (c), europium microspheres were imaged with fluorescein (400 μM) in solution. The center column shows images obtained with the pinholes aligned to the laser spot. The left column shows images obtained with pinholes shifted in the lagging direction relative the rasting laser spot by 1 Airy unit. The right column shows images obtained with pinholes shifted by 1 Airy unit in the leading direction. Laser power was 13% in (a) and (c) and 20% in (b). Dwell time was 102 μs for (a) and (b) and 204 μs for (c).
As another test of the pinhole shifting hypothesis, europium microspheres were embedded in methacrylate with 400 μM rhodamine in solution. Images were then acquired using 720-nm multiphoton excitation and a laser dwell time of 102 μs per pixel. Since emissions from europium and rhodamine are both red, only images from the red channel were collected. The first image acquired was with the pinhole aligned to the rasting laser spot [Fig. 2(b), center column]. This unshifted pinhole image showed the red luminescence of the europium microspheres against a diffuse background of red rhodamine fluorescence. The pinhole was then shifted by 1 Airy unit in the lagging direction, and another image was acquired [Fig. 2(b), left column]. In the lagging image, the long-lifetime red luminescence of the europium microspheres remained, but the short-lifetime luminescence of rhodamine disappeared. Importantly, the unshifted and shifted images were collected at the same instrumental settings of gain, offset, and laser intensity. In contrast, when the pinholes were shifted 1 Airy unit distance in the leading direction, both europium and rhodamine luminescence disappeared [Fig. 2(b), right column].
In a last series of experiments, europium microspheres were embedded with 400 μM fluorescein in solution and imaged as described earlier with a dwell time of 204 μs per pixel. With normal alignment of the pinholes in the red and green channels, the europium microspheres were red spots on a diffuse green background of fluorescein fluorescence [Fig. 2(c), center column]. When the pinholes were then shifted 1 Airy unit in the lagging direction, imaging showed retention of the long-lifetime red europium luminescence, but short-lifetime green fluorescein fluorescence was virtually completely lost [Fig. 2(c), left column]. In contrast, when the pinholes were shifted 1 Airy unit distance in the leading direction, both europium and fluorescein luminescence disappeared [Fig. 2(c), right column]. Overall, these experiments support the theory that shifting the pinhole in the lagging direction of the rasting laser spot by 1 Airy unit selectively images long-lifetime luminescence over the short-lifetime fluorescence.
3.3 Selection of Laser Dwell Time for Long-Lifetime Imaging
To evaluate experimentally the importance of dwell time in detection of long-lifetime probes by confocal multiphoton microscopy, 1-μm europium and blue microspheres on glass slides were imaged at dwell times of 3, 12, 51, and 102 μs using 720-nm multiphoton excitation. The photomultiplier gain, laser power, and AOTF settings were the same for each dwell time. At dwell times of 3 and 12 μs, fluorescence of the short-lifetime blue microspheres was imaged easily, but long-lifetime europium beads were barely discernable at 3 μs and quite grainy at 12 μs [Fig. 3(a)]. Europium luminescence at short dwell times was dim because the dwell times were simply too short to collect a sufficiently large fraction of the long-lifetime luminescence. In contrast, when we used longer dwell times of 51 and 102 μs, both short-lifetime blue microspheres and long-lifetime europium microspheres were imaged clearly.
Fig. 3.
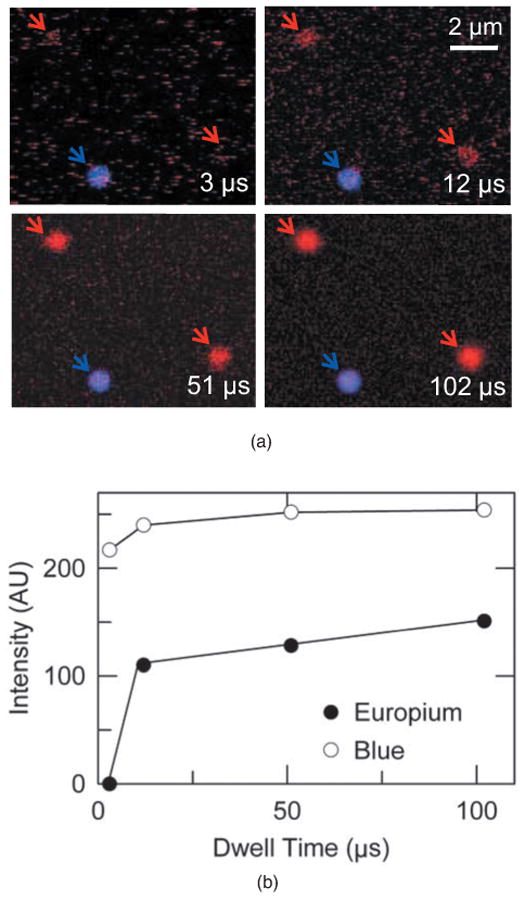
Confocal overlay images and intensities of europium and blue microspheres at different laser dwell times. In (a), images of europium (red arrows) and blue (blue arrow) microspheres were acquired at dwell times of 3, 12, 51, and 102 μs. In (b), intensities were calculated from pixel values of images of a europium microsphere (lower-right red arrow) and the blue microsphere in (a) and plotted against dwell time. Laser power was 29%.
The mean intensity of blue and europium microspheres, indicated by arrows in Fig. 4(a) for different dwell times, was determined. The intensity of the blue microsphere remained between 217 and 254 (AU) for all dwell times [open circles, Fig. 3(b)]. In contrast, the intensity of long-lifetime europium microspheres (lower-right corner) could not be reliably measured at a dwell time of 3 μs. At dwell times of 12, 51, and 102 μs, average pixel intensities became 110, 128, and 151 (AU), respectively [closed circles, Fig. 3(b)].
Fig. 4.

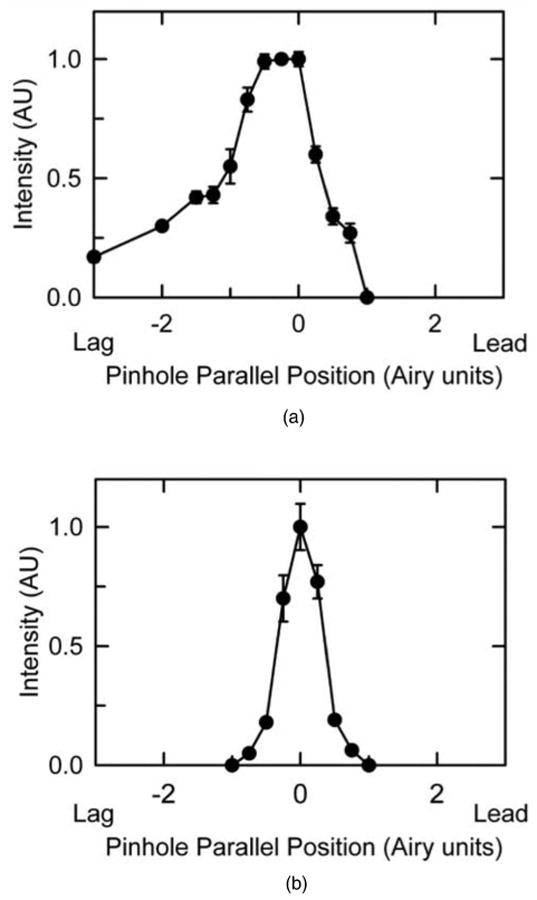
Confocal images and intensities of europium microspheres for different pinhole positions. In (a), images of europium microspheres were obtained for different aligned and lagging pinhole positions, as indicated. In (b), mean intensities of three different europium microspheres measured at lagging pinhole positions of 1, 2, and 3 Airy units were plotted and fitted to an exponential equation of the form I=e−t/τ, where the measured intensity at different pinhole positions represented different time points of measurement. Fitting yielded an average lifetime of 270±2.8 μs (SEM). Laser intensity was 14%, and dwell time was 204 μs.
3.4 Measurement of Long-Lifetime Europium
Our results to this point showed the ability of using the shifted pinhole to image long-lifetime luminescence selectively. Theory predicts that luminescence lifetimes can be measured by shifting the pinhole in the lagging direction and measuring the intensity of luminescence at different pinhole positions. The dwell time (Δt) of the rasting laser spot defines the duration of measurement, whereas the pinhole shift defines when Δt begins and stops in relation to luminescence decay curve. The following equations show these relationships:
| (4) |
| (5) |
where tstart and tstop are start and stop times representing a window in the luminescence decay curve, and Δt and m are as defined in Eq. (1). In order to measure a lifetime, at least two pinhole shifts—namely, two windows of measurement—are required.
Each pinhole shift corresponds to a time window of intensity measurement. The average lifetime of light traversing a pinhole of 1 Airy unit (τ0) can be approximated as
| (6) |
where m is pinhole shift in Airy units and Δt is laser dwell time. The measured intensity at different pinhole shifts or time points can fitted to an exponential equation of the form I=e−tτ, where I is intensity measured for a time window (pinhole shift). From this fitting, the lifetime τ of the probe can be estimated. For two measurements made at two different pinhole positions, lifetime can be also calculated by the following equation:
| (7) |
where τ is the lifetime of the fluorophore or phosphor, Io and I1 are intensities measured at the two pinhole shift positions, and Δt is as defined in Eq. (1).22–24
The validity and utility of these equations was evaluated using europium microspheres. Europium microspheres were imaged using 720-nm multiphoton excitation, a dwell time of 204 μs, and lagging detection pinhole positions relative to the rasting laser spot of 1, 2, and 3 Airy unit distances (m=1, 2, and 3).
As the pinhole position was shifted in the lagging direction, luminescence intensity of individual europium beads decreased gradually and progressively [Fig. 4(a)]. From images of three different europium microspheres imaged at three different lagging pinhole positions, intensities of luminescence were calculated from pixel values. By fitting a plot of intensity versus pinhole position to a two-parameter single exponential decay function, an average luminescence lifetime for europium from three independent measurements was estimated to be 270±2.8 μs [Fig. 4(b)]. For comparison, the estimate based on Eq. (7) from two measurements at pinhole positions of 1 and 2 Airy unit distances was 290 μs.
3.5 Intensity of Europium and Green Microspheres for Different Pinhole Positions
The lifetime of collected luminescence depends on the distance of pinhole shifting and the dwell time of the rasting laser beam across the specimen, as shown by Eq. (1). To test the effects of pinhole position on lifetimes collected, europium and green microspheres on glass slides were imaged at different pinhole positions in the leading and lagging directions using 720-nm multiphoton excitation and a dwell time of 102 μs. The loss of luminescence was very similar for the green microspheres in the leading and lagging directions of pinhole shift. A pinhole shift of 1 Airy unit in either the lagging or leading directions caused a virtually complete loss of fluorescence of green microspheres [Fig. 5(b)]. A nearly identical decrease of luminescence occurred when europium microspheres were imaged at 1 Airy unit distance in the leading direction, but loss of luminescence was much less when the pinhole was shifted 1 Airy unit in the lagging direction [Fig. 5(a)]. In the leading direction, a pinhole shift of 1 Airy unit distance caused all luminescence to be lost, while in the lagging direction, a pinhole shift of 1 Airy unit decreased the luminescence but did not make it disappear (55% loss of intensity). Even a pinhole shift of 3 Airy units [physical movement of pinhole by 330 μm or a time delay of 306 μs after excitation calculated using Eq. (4)] still did not extinguish europium luminescence in the lagging direction (84% loss of intensity). For the short-lifetime green microsphere, even the smallest shift of 0.25 Airy unit (∼22 μm) caused luminescence to decrease, while all luminescence was lost at a pinhole shift of 1 Airy unit distance in both the lagging and leading directions.
Fig. 5.
Intensity of luminescence as the pinhole was shifted in lagging and leading directions. Intensities were measured from three different europium (a) and green (b) microspheres at different leading and lagging pinhole positions. Data are meanmeans ± SEM.
Overall, short-lifetime luminescence decreased sharply and symmetrically as the pinhole was shifted in both the lagging and leading directions. For long-lifetime luminescence, in contrast, luminescence decreased much more slowly as the pinhole was shifted in the lagging direction compared to the leading direction [Fig. 5(a)]. For shifts in the leading direction, luminescence of the fluorophore and the luminophore were lost equally. These results support the theoretical prediction that pinhole shifting in the lagging direction selectively images long-lifetime luminescence.
3.6 Effect of Orthogonal Pinhole Shifts on Short- and Long-Lifetime Luminescence Measurements
The results shown so far examined the effects of shifting the pinhole parallel to the rasting laser spot. We also considered the effects of orthogonal shifts. For orthogonal pinhole shifts, the change of lifetime collected after a 1-pixel orthogonal shift corresponds to the time required to scan an entire row of pixels, where each row is typically 512 pixels or greater. Thus, both long- and short-lifetime luminescence are rapidly lost as the detection pinhole is shifted in either orthogonal direction unless dwell times are exceptionally short and lifetimes are particularly long.
To test this expectation, we evaluated the effect of shifting pinholes in the orthogonal directions with respect to the rasting laser spot on luminescence of europium and green microspheres using 720-nm multiphoton excitation and a dwell time of 102 μs. By convention, confocal/multiphoton images are collected by beginning scans at the top (north) and collecting successive left-to-right line scans to reach the bottom (south). As the pinholes were shifted orthogonally in either direction (i.e., north versus south), luminescence intensity of both europium and green microspheres decreased rapidly to virtually zero at 1 Airy unit distance (∼110 μm) (Fig. 6). These results illustrate that shifting the detection pinholes in orthogonal directions does not distinguish long- from short-lifetime luminescence and that luminescence of both short- and long-lifetime probes drops rapidly with as little as a 1 Airy unit pinhole shift.
Fig. 6.
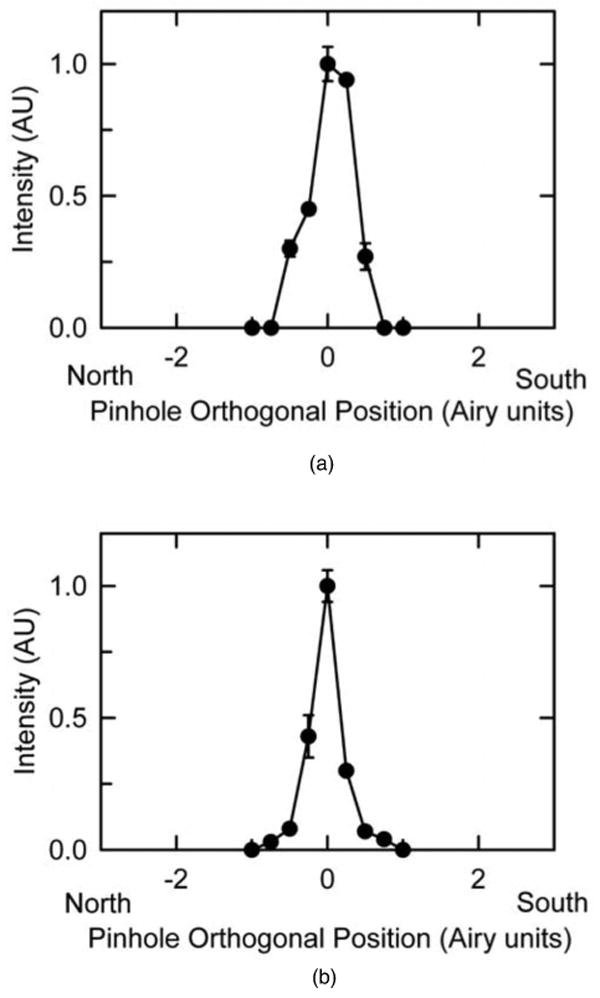
Intensity of luminescence as the pinhole was shifted in orthogonal directions. Intensities were measured from three different europium (a) and green (b) microspheres for different pinhole positions orthogonal to the rasting laser spot in the north and south directions. Data are means ± SEM.
3.7 Effect of Pinhole Diameter on Long-Lifetime Imaging
In PSLIM, delayed luminescence lagging the rasting laser spot is selectively collected by shifting the pinhole in the lagging direction. In the preceding examples, pinholes were set at 1 Airy unit—namely, the diameter of the Airy disk that is projected onto the pinhole from the specimen plane.
In order to test the effects of pinhole diameters larger than 1 Airy unit, europium and blue microspheres on glass slides were imaged at 1, 3, and 5 Airy unit pinhole diameters using 720-nm multiphoton excitation and a dwell time of 102 μs. At a pinhole diameter of 1 Airy unit, europium microspheres were imaged to their true diameter of 1 μm (Fig. 7). However, as the pinhole diameter was increased to 3 and 5 Airy units, the europium microspheres developed a comet-shaped tail in the leading direction relative to the rasting laser spot that increased apparent microsphere diameter in the x direction to 1.5 and 2.1 μm, respectively. The europium microspheres were resolved to their true diameter of 1 μm in the y direction at all pinhole diameters.
Fig. 7.

Confocal images of 1-μm-diameter europium and blue microspheres for different pinhole diameters. Europium (left column) and blue (right column) microspheres were imaged with the pinhole aligned to laser the spot and pinhole diameters of 1 to 5 Airy units, as indicated. The laser was rasted from left to right (x axis) and from top to bottom (y axis).
In contrast, the diameter of short-lifetime blue microspheres in confocal images remained 1 μm at all pinhole diameters. The blue microspheres were saturated, which caused blooming of the images and should have exaggerated any distortion, but the true shape of the blue microspheres was maintained at all pinhole diameters.
4 Discussion
4.1 Principle of Long-Lifetime Luminescence Imaging by Confocal Pinhole Shifting
Long-lifetime confocal/multiphoton imaging requires modifications and potentially expensive add-on equipment to existing microscopes. Here, we developed a technique of long-lifetime imaging microscopy by confocal pinhole shifting to perform lifetime imaging that requires no special modification of a standard commercial laser scanning confocal/multiphoton microscope. The principle for PSLIM is that when the pinhole of a confocal microscope is shifted in the lagging direction, long-lifetime luminescence becomes selectively transmitted through the pinhole, whereas collection of short-lifetime luminescence drops sharply (Fig. 1). This expectation was tested by experiments with europium, a long-lifetime luminophore, and different short-lifetime luminophores. While other techniques for measuring long-lifetime luminophores like europium chelates have been published previously by Connally et al.,19–21 this is the first report of imaging of long-lifetime europium using the pinhole shifting of a confocal microscope with multiphoton excitation for lifetime imaging of luminophores with 3-D resolution. Shifting the pinhole in the lagging direction of the rasting laser spot by 1 Airy unit distance or more caused the short-lifetime luminescence of green microspheres, rhodamine, and fluorescein to disappear, with retention of long-lifetime europium luminescence (Fig. 2, left columns). Shifting the pinhole in the leading direction caused both the short- and long-lifetime luminescence to disappear virtually equally (Fig. 2, right columns). These results validate the principle of selective long-lifetime luminescence imaging by shifting the pinhole of a confocal microscope in the lagging direction of the rasting laser spot.
In all these images (Fig. 2), a lateral shift in the apparent position of the long-lifetime europium microspheres occurred as the pinhole was displaced in the lagging direction. When the pinhole was shifted by 1 Airy unit distance in the lagging direction, the image of the europium microsphere was shifted in the leading direction by a corresponding distance (about 0.2 μm) with respect to the image obtained for the pinhole aligned to the rasting laser spot. The amount of pixel shift of the image depends on the pinhole position and the pixel size selected and generally could not be recognized except at very high magnification.
4.2 Effect of Dwell Time on Lifetime Imaging
Imaging of long-lifetime luminophores requires selection of appropriate dwell times. Accordingly, for europium with a lifetime of several hundred microseconds,18 dwell times of at least tens of microseconds were needed to collect a significant fraction of total luminescence. In contrast, for short-lifetime fluorescence probes, even the smallest dwell times of our scanning microscope (∼1 μs) collected all emitted luminescence.
Our results show that long-lifetime europium microspheres could be imaged and clearly resolved at dwell times of 51 and 102 μs [Fig. 3(a)], but at shorter dwell times of 3 and 12 μs, images were grainy with the inability to clearly resolve the europium microspheres. The graininess of the images was due to a low signal and correspondingly low signal-to-noise ratio (SNRs). SNRs in the images improved with increasing dwell times, as expected due to collection of more photons. While the signal was proportional to the number of photons, noise was proportional to the square root of the number of photons collected. Thus, as the signal increases with longer dwell times, SNRs improved.
For short-lifetime microspheres, in contrast, mean intensity did not change as dwell time increased [Fig. 3(b)]. Thus, fluorescence of short-lifetime blue microspheres was independent of dwell times and could be resolved even at the shortest dwell time of 3 μs. For short-lifetime microspheres going from dwell times of 3 to 102 μs, many more photons will be collected, and an increase in pixel intensity might be expected to occur. However, the results for blue microspheres showed that intensity increased from 217 to 254 (AU) a factor of only 1.2. The lack of difference is due to normalization of pixel intensities for dwell time by the microscope software. Thus, pixel intensities are proportional to photon/μs that are collected instead of the total number of photons collected during the dwell time. Because lifetimes are much shorter than dwell times for typical short-lifetime fluorophores, the efficiency of capture of emitted fluorescent photons does not increase with increasing dwell times, and pixel intensity (photons/μs) for short-lifetime luminescence is essentially independent of laser dwell times. Longer dwell times improve SNRs but also increase the probability of photobleaching. In our experiments, we did not observe photobleaching even at the longest dwell times. In general, when using longer dwell times, laser intensity should be decreased to minimize photobleaching while maintaining an acceptable SNR.
4.3 Measuring Lifetimes
Using PSLIM and shifting the pinhole by different distances, we could measure luminescence during different windows of decay. Images of luminescence were acquired for three different lagging pinhole positions for europium microspheres [Fig. 4(a)]. Intensity measured from the pixel values was plotted against the time of measurement, and lifetime was estimated by fitting the measurement with a single exponential decay function. Using this technique, lifetime of europium microspheres was measured as an average of 270±2.8 (SEM) μs from three independent measurements [Fig. 4(b)], which is comparable with published values.18 Also, the lifetime of europium encapsulated in a bead may differ from that in other physical environments, and lifetimes we measured at different dwell times ranged between 270 and 312 μs, in close agreement. At least two pinhole positions are needed to estimate a lifetime by PSLIM in a procedure analogous to time-gated lifetime measurement.10,23 Although gating techniques are faster, time-correlated photon counting techniques are in general more accurate.8
Our results showed that lifetimes as long as several hundred microseconds and possibly longer can be measured by PSLIM, but the shortest lifetime that can be measured using pinhole shifting is limited by the speed/dwell time of the rasting laser spot. Estimation of lifetime requires at least two windows of measurement—namely, two shifted pinhole positions. In PSLIM, the width of the windows of measurement equals the dwell time of the rasting laser spot. Since the shortest dwell time of our microscope was ∼1 μs, the shortest lifetime measurable by PSLIM will be about 1 μs. Future development of faster scanning technologies, such as acousto-optic scanning, would decrease the dwell times and hence allow shorter lifetimes to be measured by PSLIM.
4.4 Quantification of Pinhole Shifting
In the lagging direction, pinhole shifting caused europium microspheres to lose their luminescence much more slowly than in the leading direction. Even a pinhole shift of 3 Airy units (physical shift distance of ∼330 μm or a time delay of 306 μs) in the lagging direction retained 16% of the luminescence, whereas for short-lifetime green microspheres, all luminescence was lost at a pinhole shift of 1 Airy unit distance in both the lagging and leading directions (Fig. 5). Precise pinhole positioning is essential both for confocal microscopy and for accurate lifetime imaging. For short-lifetime green microspheres, a pinhole shift as little as 0.25 Airy unit decreased the brightness of confocal images (Fig. 5). Thus, confocal imaging must begin with precise alignment of the pinhole with respect to the rasting laser spot.
4.5 Distortions Associated with Large Pinhole Diameters
Collection of long-lifetime luminescence for pinhole diameters larger than 1 Airy unit caused asymmetric distortions in the images of long-lifetime europium microspheres (Fig. 7). Increasing the pinhole diameter allowed collection of delayed luminescence simultaneously with the short-lifetime luminescence without shifting the pinhole. However, the short- and long-lifetime luminescence that simultaneously passes through the pinhole comes from different parts of the specimen. Thus, collection of delayed luminescence with pinhole diameters greater than 1 Airy unit caused distortions in the images of long-lifetime specimens. Microspheres displayed a diameter greater than their true diameter of 1 μm in the x direction, with the diameter increasing to 1.5 and 2.1 μm with 3 and 5 Airy unit pinhole diameters. Nonetheless, at a pinhole diameter of 1 Airy unit, long-lifetime europium microspheres were resolved to their true diameter. Ultimately, the presence of these distortions and their magnitude will depend on the diameter of the pinhole, dwell time of the rasting laser spot, and lifetime of the specimen.
4.6 Multiple Pinholes for PSLIM
By using multiple pinholes—namely, a pinhole array—one could in principle record different time points of the luminescence decay simultaneously during a single scan. Use of a pinhole array would reduce the number of scans and hence the degree of photobleaching and phototoxicity. To detect luminescence, each pinhole in the array would need to be coupled to a separate light detector. Alternatively, detection in the pinhole plane might be accomplished with a photodiode array. Software development will also be needed to correct for the small pixel shifts associated with detection of delayed luminescence by pinhole shifting. Such software could analyze the acquired data and create lifetime maps of the images acquired. Light detected simultaneously from multiple pinholes can also be used to ratio signals acquired at different pinhole positions as a measure of the amount of delayed luminescence. Advantages of ratio imaging include an automatic correction for fluctuations in laser intensity and variations in probe concentrations.
4.7 Comparison with Other Lifetime Techniques
Compared to lifetime imaging by wide field microscopy, lifetime imaging using confocal and multiphoton microscopy provides much improved 3-D resolution of submicron structures.25 Previously, adaptation of confocal/multiphoton microscopes to lifetime imaging has required add-on equipment that potentially can reduce functionality and ease of use for more routine applications.15,17,26,27 Most often, such systems are adapted to measure lifetimes in the pico- and nanosecond time scale, the range of lifetimes exhibited by most dyes used for biological application.9,17 However, lifetime applications such as oxygen sensing require measurements of lifetimes on microsecond time scales.17,26 Compared to previous approaches for measuring long lifetimes, PSLIM using a confocal microscope system requires no add-on equipment, does not decrease functionality or ease of use for other applications, can be used with probes of long lifetimes from one to several hundred microseconds, and can be applied immediately with most laser scanning confocal microscopes. Due to limitations in scanning speeds, PSLIM is not currently applicable to measuring pico- and nanosecond lifetimes.
4.8 Detection Efficiency
The shifted pinhole collects a fraction of the decaying long-lifetime luminescence. The fraction collected decreases with increasing pinhole shift distances due to the decaying nature of the luminescence. This technique is less efficient in terms of the total photons collected compared to techniques such as streak cameras, where the entire luminescence decay is collected after excitation.28
5 Conclusions
We have shown here the application of a standard commercial confocal multiphoton microscope to perform lifetime imaging. By shifting the detection pinhole of the confocal microscope in the lagging direction parallel to the rasting laser spot, long-lifetime luminescence was selectively imaged. For biomedical studies, PSLIM may become useful to image oxygen-sensing phosphorescent probes whose lifetime is proportional to oxygen concentration. Imaging other biological parameters may also become possible by PSLIM and needs to be explored.
Acknowledgments
This work was supported, in part, by Grant Nos. DK37043 and DK073336 from the National Institutes of Health. Portions of this work were in partial fulfillment of the requirements for a Ph.D. to V.K.R. from the Department of Biomedical Engineering at the University of North Carolina at Chapel Hill.
Contributor Information
Venkat K. Ramshesh, Medical University of South Carolina, Center for Cell Death, Injury and Regeneration and Hollings Cancer Center, Charleston, South Carolina 29425
John J. Lemasters, Medical University of South Carolina, Center for Cell Death, Injury and Regeneration and Departments of Pharmaceutical Sciences and Biochemistry & Molecular Biology and Hollings Cancer Center, Charleston, South Carolina 29425
References
- 1.Ramanujan VK, Zhang JH, Biener E, Herman B. Multi-photon fluorescence lifetime contrast in deep tissue imaging: prospects in redox imaging and disease diagnosis. J Biomed Opt. 2005;10:1–11. doi: 10.1117/1.2098753. [DOI] [PubMed] [Google Scholar]
- 2.Hyatt CJ, Lemasters JJ, Muller-Borer BJ, Johnson TA, Cascio WE. A superfusion system to study border zones in confluent cultures of neonatal rat heart cells. Am J Physiol. 1998;274:H2001–H2008. doi: 10.1152/ajpheart.1998.274.6.H2001. [DOI] [PubMed] [Google Scholar]
- 3.Launikonis BS, Zhou J, Royer L, Shannon TR, Brum G, Ríos E. Confocal imaging of [Ca2+] in cellular organelles by SEER, shifted excitation and emission ratioing of fluorescence. J Physiology. 2005;567:523–543. doi: 10.1113/jphysiol.2005.087973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Smiley S, Reers M, Mottola-Hartshorn C, Lin M, Chen A, Smith T, Steele G, Jr, Chen L. Intracellualr heterogentiy in mitochondrial membrane potentials revealed by a J-aggregate-forming liophilic cation JC-1. Proc Natl Acad Sci U S A. 1991;88:3671–3675. doi: 10.1073/pnas.88.9.3671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lakowicz JR. Principles of Fluorescence Spectroscopy. Springer; New York: 1986. [Google Scholar]
- 6.Periasamy A, Wodnicki P, Wang XF, Kwon S, Gordon GW, Herman B. Time-resolved fluorescence lifetime imaging microscopy using a picosecond tunable dye laser system. Rev Sci Instrum. 1996;67:3722–3731. [Google Scholar]
- 7.Draaijer A, Sanders R, Gerritsen HC. Fluorescence lifetime imaging, a new tool in confocal microscopy. In: Pawley JB, editor. Handbook of Biological Confocal Microscopy. 2nd. Plenum Press; New York: 1995. pp. 491–505. [Google Scholar]
- 8.Suhling K, French PMW, Phillips D. Time-resolved fluorescence microscopy. Photochem Photobiol. 2004;4:13–22. doi: 10.1039/b412924p. [DOI] [PubMed] [Google Scholar]
- 9.Elson D, Requejo-Isidro J, Munro I, Reavell F, Siegel J, Suhling K, Tadrous P, French P. Time-domain fluorescence lifetime imaging applied to biological tissue. Photochem Photobiol. 2004;3:795–801. doi: 10.1039/b316456j. [DOI] [PubMed] [Google Scholar]
- 10.Gerritsen HC, Asselbergs MAH, Agronskaia AV, Van Sark WG. Fluorescence lifetime imaging in scanning microscopes: acquisition speed, photon economy and lifetime resolution. J Microsc. 2002;206:218–224. doi: 10.1046/j.1365-2818.2002.01031.x. [DOI] [PubMed] [Google Scholar]
- 11.Becker W, Bergmann A, Hink MA, Konig K, Benndorf K, Biskup C. Fluorescence lifetime imaging by time-correlated single-photon counting. Microsc Res Tech. 2004;63:58–66. doi: 10.1002/jemt.10421. [DOI] [PubMed] [Google Scholar]
- 12.Lakowicz J, Berndt K. Lifetime-sensitive fluorescence imaging using an rf phase sensitive camera. Rev Sci Instrum. 1991;62:1727–1734. [Google Scholar]
- 13.Gratton E, Limkeman M. A continuosly variable frequency cross-correlation phase fluorometer with picosecond resolution. Biophys J. 1983;44:315–324. doi: 10.1016/S0006-3495(83)84305-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gratton E, Breusegmen S, Sutin J, Ruan Q, Barry N. Fluorescence lifetime imaging for two-photon microscope: time-domain and frequency-domain methods. J Biomed Opt. 2003;8:381–390. doi: 10.1117/1.1586704. [DOI] [PubMed] [Google Scholar]
- 15.Buranachai C, Kamiyama D, Chiba A, Williams BD, Clegg RM. Rapid frequency-domain FLIM spinning disk confocal microscope: lifetime resolution, image improvement and wavelet analysis. J Fluoresc. 2008;18(5):929–942. doi: 10.1007/s10895-008-0332-3. [DOI] [PubMed] [Google Scholar]
- 16.Buurman EP, Sanders P, Draaijer A, Gerritsen HC, van Venn JJF, Houpt PM, Levine YK. Fluorescence lifetime imaging using a confocal microscope. Scanning. 1992;14:155–159. [Google Scholar]
- 17.Zhong W, Urayama P, Mycek MA. Lifetime modulation of a ruthenium-based dye in living cells: the potential for oxygen sensing. J Phys D. 2003;36:1689–1695. [Google Scholar]
- 18.Haugland RP. Handbook of Fluorescent Probes and Research Products. 9th. Molecular Probes, Inc.; Eugene, OR: 2002. pp. 543–595. [Google Scholar]
- 19.Connally R, Veal D, Piper J. High resolution detection of fluorescently labeled microorganisms in environmental samples using time-resolved fluorescence microscopy. FEMS Microbiol Ecol. 2002;41:239–245. doi: 10.1111/j.1574-6941.2002.tb00985.x. [DOI] [PubMed] [Google Scholar]
- 20.Connally R, Veal D, Piper J. Flash lamp-excited time-resolved fluorescence microscope suppresses autofluorescence in water concentrates to deliver an 11-fold increase in signal-to-noise ratio. J Biomed Opt. 2004;9:725–734. doi: 10.1117/1.1756594. [DOI] [PubMed] [Google Scholar]
- 21.Connally R, Veal D, Piper J. Time-resolved fluorescence microscopy using an improved europium chelate BHHST for the in-situ detection of cryptosporidium and giardia. Microsc Res Tech. 2004;64:312–322. doi: 10.1002/jemt.20087. [DOI] [PubMed] [Google Scholar]
- 22.Ballew RM, Demas JN. An error analysis of the rapid lifetime determination method for the evaluation of single exponential decays. Anal Chem. 1989;61:30–33. doi: 10.1021/ac981050d. [DOI] [PubMed] [Google Scholar]
- 23.Sharman KK, Periasamy A, Ashworth H, Demas JN, Snow NH. Error analysis of the rapid lifetime determination method for double-exponential decays and new windowing schemes. Anal Chem. 1999;71:947–952. doi: 10.1021/ac981050d. [DOI] [PubMed] [Google Scholar]
- 24.Periasamy A, Wang XF, Wodnick P, Gordon GW, Kwon S, Diliberto PA, Herman B. High-speed fluorescence microscopy: lifetime imaging in the biomedical sciences. J Microsc Soc Am. 1995;1:13–23. [Google Scholar]
- 25.Grauw CJD, Gerritsen HC. Multiple time-gated module for fluorescence lifetime imaging. Appl Spectrosc. 2001;55:670–678. [Google Scholar]
- 26.Gerritsen HC, Sanders R, Draaijer A, Ince C, Levine YK. Fluorescence lifetime imaging of oxygen in living cells. J Fluoresc. 1996;7:11–15. doi: 10.1007/BF01881442. [DOI] [PubMed] [Google Scholar]
- 27.Van Munster EB, Goedhart J, Kremers GJ, Manders EMM, Gadella TWJ., Jr Combination of a spinning disc confocal unit with frequency-domain fluorescence lifetime imaging microscopy. Cytometry, Part A. 2007;71A:207–214. doi: 10.1002/cyto.a.20379. [DOI] [PubMed] [Google Scholar]
- 28.Krishnan R, Masuda A, Centonze V, Herman B. Quantitative imaging of protein-protein interactions by multiphoton fluorescence lifetime imaging microscopy using a streak camera. J Biomed Opt. 2003;8:362–367. doi: 10.1117/1.1577574. [DOI] [PubMed] [Google Scholar]