

Abstract
Dopaminergic neurodegeneration during Parkinson disease (PD) involves several pathways including proteasome inhibition, α-synuclein (α-syn) aggregation, mitochondrial dysfunction, and glutathione (GSH) depletion. We have utilized a systems biology approach and built a dynamic model to understand and link the various events related to PD pathophysiology. We have corroborated the modeling data by examining the effects of α-syn expression in the absence and presence of proteasome inhibition on GSH metabolism in dopaminergic neuronal cultures. We report here that the expression of the mutant A53T form of α-syn is neurotoxic and causes GSH depletion in cells after proteasome inhibition, compared to wild-type α-syn-expressing cells and vector control. Modeling data predicted that GSH depletion in these cells was due to ATP loss associated with mitochondrial dysfunction. ATP depletion elicited by combined A53T expression and proteasome inhibition results in decreased de novo synthesis of GSH via the rate-limiting enzyme γ-glutamyl cysteine ligase. Based on these data and other recent reports, we propose a novel dynamic model to explain how the presence of mutated α-syn protein or proteasome inhibition may individually impact on mitochondrial function and in combination result in alterations in GSH metabolism via enhanced mitochondrial dysfunction.
Keywords: Parkinson's disease, Neurodegeneration, α-Synuclein, Protein aggregation, Proteasome inhibition, Mitochondrial dysfunction, Glutathione, Systems biology, Dynamic model, In silico, Free radicals
Introduction
Parkinson disease (PD) is an age-associated neurodegenerative disease clinically defined as a progressive movement disorder. PD is manifested as motor impairment with characteristic symptoms including resting tremor, rigidity, postural instability and bradykinesia. In advanced PD, patients also exhibit cognitive deficits and depression. Pathologically, PD is exemplified by selective loss of dopaminergic neurons in the substantia nigra pars compacta (SN) region of the ventral midbrain [1].
Research data have clearly indicated that during PD, SN dopaminergic neurons are subject to oxidative and nitrosative stress [1–5]. The earliest reported detectable event related to oxidative/nitrosative stress is depletion of the thiol antioxidant glutathione (GSH+ GSSG) [6]. GSH depletion during PD precedes mitochondrial damage and dopamine loss and the degree of its loss has been observed to correlate with disease severity [7,8]. During PD, there is a selective inhibition of mitochondrial complex I (CI) resulting in mitochondrial dysfunction [9]. PD toxins such as 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) and rotenone act on dopaminergic neurons via selective CI inhibition. The selective CI inhibition during sporadic PD might involve specific oxidative/nitrosative modifications of different CI subunits [7–13]. The CI is considered to be one of the respiratory complexes most severely affected by age-related oxidative stress, resulting in mitochondrial dysfunction [14].
Proteasome inhibition is another key phenomenon affecting neuronal viability in PD. Systemic administration of proteasome inhibitors to adult rats resulted in most of the biochemical symptoms of progressive parkinsonism with selective lesioning of SN neurons [15]. Further, proteasome inhibition has also recently been demonstrated to alter overall mitochondrial homeostasis [16]. During PD, SN neurons accumulate proteins, leading to formation of intraneuronal deposits termed “Lewy bodies” (LBs) [17]. α-Synuclein (α-syn; MW 18 kDa), the major component of LBs, is a presynaptic protein with a probable role in synaptic function. α-Syn is lipid associated under normal physiological conditions but tends to aggregate in the lipid-free state into higher molecular weight oligomers [18]. Formation of these aggregates has been directly linked to neurodegeneration in PD. Two mutations in the α-syn gene (A30P and A53T) have been associated with familial parkinsonism, but the biochemical consequences of these mutations in disease pathology are not completely understood [19].
It is therefore evident that neurodegeneration in PD involves an interplay between GSH depletion, oxidative/nitrosative stress, mitochondrial dysfunction, proteasome inhibition and protein aggregation. But the precise relationship among these pathways has not been completely defined. Moreover, the temporal order of biochemical events in neurodegeneration and how the relative contributions of each of the different pathways influence the others are not clear. Because PD pathology simultaneously involves more than one process, it would be interesting to evaluate the synergistic effect of simultaneously occurring processes. Systems biology can answer such questions by recapitulating the entire set of PD pathways simultaneously. Such a dynamic model utilizes the kinetics of the pathway intermediates and therefore permits a better appreciation of the relative contributions made by different components than do current static pathway diagrams.
Using dynamic modeling, we have previously reported the analysis of α-syn aggregation and tyrosine hydroxylase-mediated regulation of dopamine synthesis in terms of their relevance to PD [20,21]. With experimental validation, we have also integrated GSH metabolism and mitochondrial dysfunction in silico in association with PD [22] and demonstrated that curcumin could be a neuroprotective agent against oxidative/nitrosative stress and GSH depletion in this condition [23]. In the current study, we have generated a dynamic model of PD linking proteasome inhibition and α-syn expression with GSH homeostasis, mitochondrial physiology, and apoptosis. We have validated the modeling data using dopaminergic neuronal cell lines that express either wild-type (WT) or A53T mutant α-syn protein and have assessed the effects on GSH metabolism during proteasome inhibition. Based on our data, we propose a novel dynamic model for the functional relationship among these various PD-related cellular pathways.
Overview of dynamic in silico modeling of α-syn expression, proteasome inhibition, mitochondrial physiology, and GSH metabolism
We have recently reported dynamic modeling of α-syn aggregation with relevance to PD [20]. We have also reported in silico models (a) linking GSH metabolism and mitochondrial dysfunction associated with PD [22] and (b) exploring the neuroprotective effects of curcumin in PD [23]. In the current model, we have linked the modules of α-syn expression, aggregation, and proteasome inhibition with mitochondrial physiology and looked into their effects on GSH metabolism.
Model description
The integrated model comprises three primary modules:
Mitochondrial bioenergetics: This model includes oxidative phosphorylation, ATP synthesis, and generation of RS [reactive species, which includes reactive oxygen species (ROS) and reactive nitrogen species (RNS)]. This module has been described earlier [22,23].
GSH synthesis and metabolism: This model includes transcription of γ-glutamyl cysteine ligase (GCL) mRNA by Nrf2 (nuclear erythroid 2 p45-related factor 2, or NF-E2-related factor 2), JunD, and cFos-Jun transcription factors. Under normal conditions, Nrf2 remains complexed with Keap1 protein in the cytoplasm, which directs it to proteasomal degradation. This complex (Nrf2–Keap1) dissociates under the influence of ROS and proteasome inhibition to provide free Nrf2 in the cytoplasm, which then translocates to the nucleus and upregulates GCL gene expression, thereby increasing GCL enzyme activity and GSH synthesis [23,24]. These two interlinked models evaluate the generation of ATP, RS, and GSH. ATP is required for GSH synthesis and GSH levels influence ATP synthesis via their effects on oxidative stress. This module has been described earlier [22,23].
α-Syn aggregation and proteasomal degradation: This includes aggregation of α-syn under the influence of oxidative stress, Fe+2, and the NO2 free radical that also results in increased truncation of α-syn in the system. Aggregation of α-syn is modeled as a nucleation-dependent process in which “seeds” are initially formed that are converted to oligomers, then protofibrils, and finally to LBs. The model also shows proteasomal degradation of various substrate proteins, including α-syn, synphilin, and Pael-R, which is mediated by K-48-dependent ubiquitination of the substrate proteins [20].
The aggregated form of proteins (which includes soluble intermediates of the protein aggregation process but not LBs) has been modeled to cause inhibition of the proteasomal degradation of various substrate proteins. This module was integrated with modules 1 and 2. The nodes included in each of the individual modules are qualitatively described in this section, with the equations and kinetic parameters for each node tabulated in Supplementary Table 1.
-
Mitochondrial bioenergetics and dysfunction
We have recently described this module [22,23], which includes the following:
mitochondrial oxidative phosphorylation system and its role in ATP production and ROS generation;
generation of RNS in the mitochondria;
redox homeostasis via GSH, glutathione peroxidase, and glutathione reductase;
oxidative stress and mitochondrial dysfunction due to GSH depletion.
-
GSH synthesis and metabolism
The model incorporating GSH synthesis and its cellular distribution, antioxidant function, and interrelationship with mitochondrial dysfunction has been described earlier [22,23].
There are two transcription factors responsible for GCL mRNA transcription, cFos–Jun and Nrf2–Jun-D [46–51]. Nrf2–Jun-D binds with the antioxidant response element of the GCL promoter, resulting in transcription of GCL mRNA. Nrf2-mediated GCL transcription is upregulated under oxidative/electrophile stress [24–29].
-
Protein aggregation and proteasomal dysfunction
This block includes expression of α-syn control (rat α-syn), WT α-syn, and A53T α-syn as explained under Model description. It includes α-syn aggregation and formation of oligomers and then protofibrils. Both represent the soluble toxic species that are able to produce RS by altering dopamine homeostasis in the system [30,31]. Protofibrils further aggregate to form larger more aggregated species (called syn-agg in the model), which leads to the formation of LBs. LBs are considered to be neuroprotective [32] because they can be cleared by macroautophagy. In our system, expression of Pael-R and synphilin is also shown. Synphilin is responsible for K-63-mediated ubiquitination and driving α-syn toward formation of LBs directly instead of its sequential integration to form toxic oligomers and protofibrils, thereby playing a neuroprotective role [20].
Proteasomal degradation of substrate proteins and its inhibition
α-Syn, Pael-R, synphilin, and other cellular proteins are degraded by the proteasome. When cellular proteins are either damaged or misfolded because of oxidative damage, they are ubiquitinated, marking them for proteasomal degradation. Ubiquitination and protein targeting are facilitated by E1, E2, and E3 ligases (parkin) and other proteins of the ubiquitination–proteasomal machinery [20]. The degradation activity of the proteasome (predominantly the chymotrypsin activity of the 20S proteasome catalytic subunit) is inhibited by toxic protein aggregates (soluble toxic species) and this process is exacerbated during oxidative stress [33,34]. Under the influence of oxidative stress when more toxic aggregates accumulate in the system, this results in further inhibition of proteasomal degradation, setting up a vicious cycle of toxic aggregation and RS production. These substrate proteins, which are not degraded by the proteasome after its inhibition, form LB-like inclusions [35]. To demonstrate the effect of proteasomal inhibitors (PIs), one generic PI species is used to demonstrate the impact of proteasomal inhibition (Ki = 1 μM).
Interrelationship between mitochondrial dysfunction and protein aggregation
Recently, it has been demonstrated that α-syn aggregates lead to inhibition of CI activity [36]. In our model, α-syn aggregates have also been shown to cause direct inhibition of CI activity, resulting in ROS generation and a decrease in ATP production. CI has been shown to be inhibited by the α-syn aggregates in a noncompetitive manner by decreasing the Vmax of the reaction. ATP has been set up as a substrate for GSH synthesis to mimic the energy requirement for GSH synthesis.
In our dynamic virtual PD system, the three interrelated phenomena, (a) oxidative stress and protein aggregation, (b) accumulation of toxic protein aggregates and inhibition of proteasomal machinery, and (c) CI inhibition and subsequent mitochondrial dysfunction due to toxic aggregates resulting in GSH depletion, have been modeled.
Model construction
The model is a dynamic representation of mitochondrial bioenergetics, GSH synthesis/metabolism modulated by oxidative stress, and aggregation of α-syn (and other substrate proteins) and includes inhibition of proteasomal degradation by toxic aggregates/PI. A bottom–up approach is adopted for building the complete dynamic system. The various phenomena are first built as base modules and progressively integrated. As described earlier, the base modules developed and integrated include (a) mitochondrial bioenergetics and production of ROS/RNS, (b) GSH synthesis and metabolism, and (c) protein aggregation (including α-syn) and proteasomal machinery.
The process of model development involves a detailed study of the individual phenomenon and all the species involved therein. Interactions between the species, regulatory mechanisms, feedback controls, etc., are put together to get static interaction maps. This map is then made dynamic by incorporating physiological concentrations of the species, rate, and mechanism of reaction in flux equations that drive the reactions. Most of these data are obtained from published scientific literature, in which experimental and structural biology techniques have been used. Data unavailable in literature are optimized based on explicit assumptions. Once individual modules have been built and validated against published experimental data, the cross talk between these phenomena is worked out. The output from a module could either generate the input for another module or provide regulatory control. These interactions are used to cross-link and integrate the base modules to form the complete integrated system that can be used for analysis. The emergent behavior of such a complex network would be very different from the results seen in isolated experiments studying these phenomena. Moreover, such a system enables a higher magnification and transparency for profile analysis of relevant intermediates apart from key endpoint markers with manipulation of different triggers, inhibitors, and activators. A key requirement for “molecular target”-based drug discovery research is a thorough understanding of disease physiology in human at the molecular level. Because the dynamic in silico model offers a distinct ability to manipulate and assay any of the molecular players, studies like percentage knockout, inhibition, dose–response, overexpression, and mutational analysis could be performed with virtually any relevant biomarker.
In the current study, time-dependent potential differences, proton pump, ion flux, and other metabolic reactions have been modeled utilizing modified ordinary differential equations (ODEs) and mass action kinetics as described earlier [22,23]. Cell Works proprietary platform icPHYS software was used to create and integrate the pathways with ODEs solved by the Radau method.
The reaction kinetics for all nodes in the dynamic integrated model is summarized in Supplementary Table 1 with corresponding references. The model construction and its assumptions regarding GSH metabolism and mitochondrial dysfunction have been described earlier [22]. Following are the other key assumptions in the current version of our model:
The expression levels of WT and A53T α-syn are kept equal at 13 times higher than control. Control represents the rat α-syn already present in the cell type, whereas the WT and A53T represent the overexpressed human α-syn forms in the experimental system.
A53T α-syn is considered to have twofold higher aggregation kinetics than WT α-syn [37].
Nrf2–Keap complex dissociation is shown to be activated by oxidative stress and proteasome inhibition in the system. The effect of proteasome is considered to be indirect. Once Nrf2 is unbound from Keap1, it is not targeted for degradation by proteasome, resulting in increases in its concentration and translocation to the nucleus. In the presence of proteasomal inhibition, Nrf2 will not be degraded and the probability of dissociation of the complex will increase, resulting in liberation of Nrf2.
PI is taken as a generic inhibitor and not as any specific inhibitor. PI is introduced into the model as an arbitrary species representative of various known inhibitors. It has been modeled to inhibit the degradation flux for various substrates of the proteasome. In this study, the concentrations of PI for which system is tested are in the range of 0–5 μM. The Ki of PI for proteasomal degradation fluxes is 1 μM.
Simulation protocol
Initial conditions
The initial conditions of the model representing normal mitochondrial bioenergetics with the free radicals generated being scavenged by the GSH antioxidant system have been described earlier [22,23]. Regarding α-syn, initial conditions represent expression of control α-syn (rat α-syn) only.
α-Syn expression
For control experiments (representing cells containing rat α-syn and transfected with vector alone), the WT and A53T α-syn expression flux is kept off and the control expression flux is switched on. For WT or A53T α-syn experiments, WT or A53T α-syn expression flux is switched on along with control α-syn expression to mimic the condition under which rat α-syn is also expressed along with human α-syn.
Proteasome inhibition
In our model, PI has been introduced as a generic species with concentrations of 0, 0.5, 1.25, 2.5, and 5 μM. We have tested these concentrations of the PI in control, WT, and A53T α-syn expression systems.
Materials and methods
All tissue culture materials were procured from Life Technologies/Invitrogen (Carlsbad, CA, USA) or Cellgro (Kansas City, MO, USA). Anti-α-syn antibody was obtained from Chemicon (Temecula, CA, USA). Materials related to protein chemistry were obtained from Bio-Rad Laboratories (Hercules, CA, USA) and Sigma (St. Louis, MO, USA). Effectene transfection reagent was obtained from Qiagen (Valencia, CA, USA).
Cell culture
The 1RB3AN27 (N27) rat dopaminergic neuronal cell line was used throughout this study. N27 cells were grown as described previously [22,23].
Generation of stable α-syn cell lines
WT and A53T mutant α-synuclein cDNAs were separately sub-cloned into a pTR-UF12d mammalian expression vector at HindIII/SpeI sites upstream of a green fluorescence protein (GFP) marker. For generation of stable cell lines, N27 cells were plated in six-well plates and transfected with constructs (WT or A53T α-syn vs vector alone) using Effectene reagent (Qiagen). For selection of clones, a hygromycin-resistance vector was cotransfected along with all the expression vectors. Clones growing in selection medium containing hygromycin and exhibiting GFP fluorescence were selected and screened for α-syn expression. WT and A53T clones showing comparably equal expression via Western blot were chosen for further experiments.
Western blot
Cell extracts for anti-α-syn Western blots were prepared as described earlier [38]. Briefly, cells were harvested, resuspended in 1×PBS, and sonicated (25 s) on ice. The extract was solubilized in lysis buffer [10 mM Tris–Cl, pH 7.8, 10 mM NaCl, 0.5% Nonidet P-40, 0.1% SDS, complete protease inhibitor cocktail tablets (1×concentration; Roche Applied Sciences)] and fractionated into soluble and insoluble fractions by centrifuging at 12,000 g. The soluble fraction was resolved on SDS–PAGE followed by anti-α-syn Western blot. For anti-GSH Westerns, gels were run without DTT or β-mercaptoethanol in the SDS–PAGE loading buffer.
Cell viability assay
Cells seeded in 96-well plates at a density of 10×103/well were treated with PI for 18 h followed by quantitation of viable cells using the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay as described previously [22].
Estimation of total glutathione (GSH+ GSSG) levels
Total cellular glutathione estimation was carried out using a kit from Oxis Research (Portland, OR, USA) as outlined earlier [39]. All estimations were conducted in triplicate and normalized per protein.
γ-Glutamyl cysteine ligase activity
GCL assays were carried out as described earlier [40]. Briefly, cells obtained after various treatments were washed in 1×PBS and resuspended in 0.1 M Tris–Cl, pH 8.0. The cell suspension was sonicated and used for GCL enzyme assay. Enzyme activity was determined at 37°C by adding cell lysates to a reaction mixture containing 0.1 M Tris–HCl buffer, pH 8.0, 150 mM KCl, 5 mM sodium ATP, 2 mM phosphoenol pyruvate, 10 mM l-glutamate, 10 mM α-aminobutyrate, 20 mM MgCl2, 2 mM EDTA, 0.2 mM NADH, 17 U pyruvate kinase, and 17 U lactate dehydrogenase. Absorbance at 340 nm was monitored as a measure of enzyme activity. Assays run in the absence of α-aminobutyrate served as a blank. GCL activity values were normalized per protein. Control GCL assays were conducted on all samples in the absence of exogenous ATP in the reaction mixture to assess the contribution of endogenous ATP. For this experiment, up to 75 μg total protein sample was used.
γ-Glutamyl transpeptidase (GGT) assay
GGT activity was measured according to the method described earlier [41] with slight modifications. Briefly, cells were washed with 1×PBS and sonicated in ice-cold 0.1% Triton X-100 in 1×PBS and then centrifuged at 4000 g for 5 min at 4°C. The supernatant (50 μg of protein) was added to a reaction mixture containing 0.1 M ammediol/HCl buffer, pH 8.6, 20 mM glycylglycine, 200 μM γ-Glu-AMC, and 0.1% Triton X-100. The activity was kinetically assayed at 37°C with an excitation wavelength of 370 nm and an emission wavelength of 440 nm. GGT activity was expressed as nanomoles AMC (7-amino-4-methylcoumarin) produced per milligram protein per minute.
Preparation of mitochondria
Mitochondria from N27 cells were prepared as previously described [42].
Estimation of ATP content
The ApoGlow Rapid Apoptosis Screening Kit (Cambrex, Rockland, ME, USA) was used to measure ATP levels based on the luciferase assay according to the manufacturer's instructions. Relative luminescence units were measured on a Turner T-20E luminometer.
Statistical analysis
Data are expressed as means ± SD and significance testing was performed using ANOVA.
Results
Dopaminergic neurotoxicity cell model involving α-syn expression and proteasome inhibition
In the current study, we have utilized stable N27 neuronal cell lines expressing either WT human α-syn or the A53T mutant. The expression of α-syn in N27 cell clones was confirmed by Western blot analysis (Fig. 1A). The clones selected were mild expressers such that α-syn expression by itself did not result in significant cell death. When α-syn clones were incubated with increasing concentrations of the PI lactacystin or MG132 for 18 h, the A53T-expressing cells were more susceptible to cell death than either WT α-syn cells or vector control (Fig. 1B). This suggests that proteasome inhibition has neurotoxic effects predominantly in the presence of the A53T mutant protein, thus representing a cell model of α-syn-mediated neurotoxicity. The A53T clones showed ∼50% cell death at 2.5 μM concentration of PI and these conditions were employed for subsequent experiments. Although the same concentration (2.5 μM) was required for both the inhibitors to cause 50% cell death, lactacystin exhibited greater neurotoxicity at 3.75 μM.
Fig. 1.
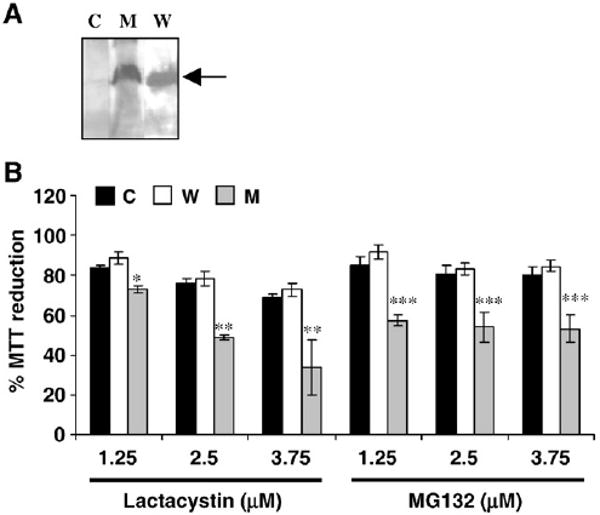
Generation of neuronal cell model of α-syn toxicity (A and B are in vitro data). (A) Western blot analysis of α-syn expression in N27 clones expressing either WT α-syn (W) or A53T mutant (M) compared to vector-transfected control cells (C). Arrow indicates α-syn protein (MW ∼18 kDa). (B) Cell viability of N27 α-syn clones in the presence of increasing concentrations of the PIs lactacystin and MG132 (18 h). All values are represented as a percentage compared to the respective untreated cells. *p< 0.005, **p< 0.001 and ***p< 0.001 compared to control cells.
In silico model linking α-syn, proteasome inhibition, and apoptosis
To recapitulate the same events in silico, we built a dynamic model of α-syn-mediated neurotoxicity utilizing the assumptions and simulation protocol outlined above. The α-syn model was linked to the proteasome machinery and the integrated GSH–mitochondrial model [22]. The system was challenged with increasing concentrations of a generic PI to analyze the synergistic effect of α-syn expression and proteasome inhibition on cell viability. To plot cell viability, we took the inverse of 8-hydroxy guanosine (8-OHdG) generation as a marker of apoptosis and multiplied it with a scaling factor to have comparable levels.
Similar to the cell culture data, A53T expression showed increased cell death (∼40%) compared to WT (30%) and control (∼15%) in the presence of PI (2.5 μM), thus generating a kinetic model that could be subsequently analyzed in terms of mitochondrial function and GSH dynamics (Fig. 2A). Further, A53T clones showed more DNA damage (∼50%) in the presence of PI in silico compared to WT (∼35%) and control (∼15%) (Fig. 2B).
Fig. 2.
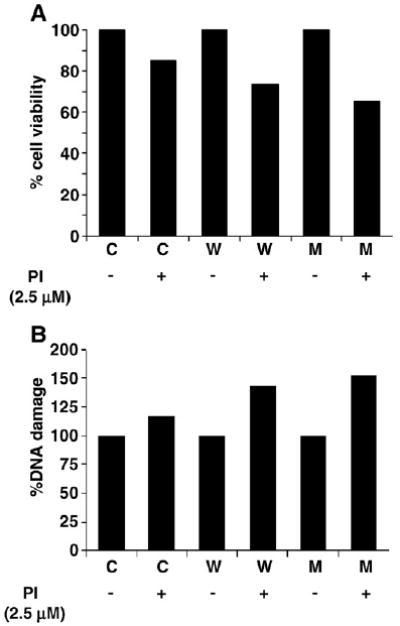
Generation of dynamic model of α-syn toxicity (A and B are in silico data). (A) Dose-dependent effect of PI (2.5 μM) in silico on cell viability (with 8-OHdG levels as marker of apoptosis) in cells expressing either WT (W) or A53T α-syn (M) compared to control (C). (B) Percentage DNA damage without and with PI (2.5 μM) in control, WT, and A53T α-syn cells measured by the amount of 8-OHdG, a biomarker for DNA damage and apoptosis.
In vitro and in silico analyses of GSH metabolism in α-syn clones
It has earlier been reported that the combination of α-syn expression and proteasome inhibition results in depletion of cellular GSH levels [43,44]. We therefore analyzed the effects of α-syn expression in the absence and presence of PI on GSH metabolism in our cells. It has been previously reported that lactacystin by itself can induce GSH synthesis in cells [45,46]. Hence, for experiments involving GSH metabolism, MG132 was utilized. In the absence of PI, total GSH levels were higher in WT α-syn cells (22.9 ng/mg protein) than in controls (15.2 ng/mg protein) and highest in A53T α-syn clones (42.7 ng/mg protein) (Fig. 3A). Upon incubation with the MG132 (1.25 to 5 μM), total GSH levels in control cells increased and then leveled off, demonstrating a final increase of over 100% (34.3 ng/mg protein) in control cells. WT cells also demonstrated a similar trend but the elevation in total GSH levels was only ∼45% (32 ng/mg protein). In contrast, in the A53T-expressing cells, GSH levels decreased by ∼40% from 42.7 to 26.5 ng/mg in a dose-dependent manner (Fig. 3A) such that there was ∼25% less GSH compared to control at 5 μM MG132. Our modeling studies also inferred that the levels of GSH after A53T expression declined with the introduction of PI (Figs. 3B and C). The trends of the GSH levels in WT and control ±PI showed a pattern similar to that seen in cell culture but only the control showed an elevation in GSH levels upon exposure to PI.
Fig. 3.
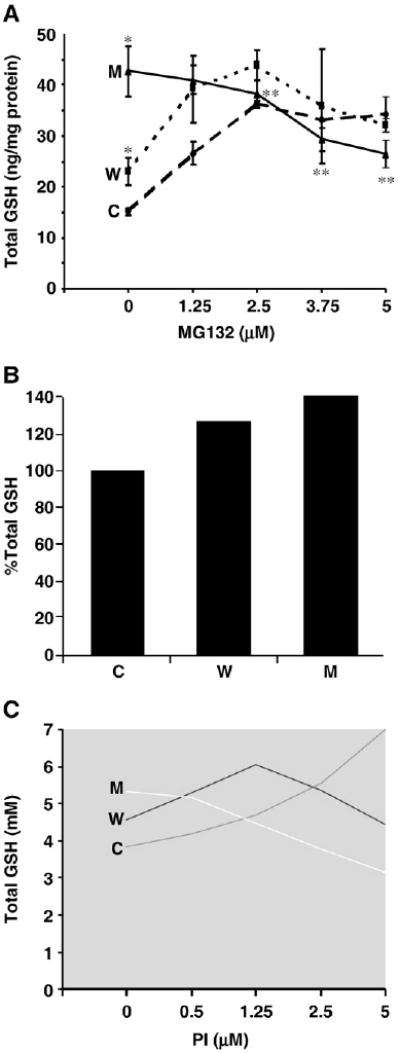
Effects of α-syn expression (±PI) on cellular total GSH levels in α-syn N27 clones vs vector control (A is in vitro data, B and C are in silico data). (A) Total GSH levels in α-syn clones in the absence and presence of MG132. *p< 0.01 compared to control cells; **p< 0.01 compared to untreated mutant cells (C, control; W, wild type; M, A53T mutant). In silico data on the effects of α-syn expression on GSH (total) levels in the (B) absence and (C) presence of PI (2.5 μM) (C) are shown.
In the absence of PI, an elevation in GSH levels might be attributed to increased synthesis via elevated activity of the GSH synthesizing enzyme GCL. It has been demonstrated that the expression of GCL mRNA is dependent on the transcription factor Nrf2, which in turn is activated by toxic insults such as oxidative stress [25–29] (see Materials and methods). Recently, it has been shown that Nrf2 levels are increased by proteasome inhibition [32]. Because α-syn expression has been demonstrated to contribute to increased oxidative stress [30,31] and proteasome inhibition [47,48], we surmised that this in turn might induce GCL mRNA expression via the Nrf2 pathway. Figs. 4A–D show the simulation data indicating α-syn (WT and A53T)-dependent induction of the Nrf2 pathway and increased expression of GCL mRNA and protein (both by ∼15% in WT and ∼25% in A53T compared to control), ultimately leading to increased GCL activity. However, in our model, we did not observe a significant difference in Nrf2 degradation by α-syn aggregation-mediated proteasome inhibition. The difference was significant when different concentrations of PI were used (data not shown). Alternatively, Nrf2 translocation into the nucleus was significantly increased (by ∼15% in WT and ∼25% in A53T compared to control) owing to increased dissociation of Nrf2–Keap1 complex mediated by increased ROS (Fig. 4A). This results in upregulation of GCL in control, WT, and A53T α-syn cells, which contributes differently to the production of ROS, such that the Nrf2 upregulation can be correlated with different α-syn forms (Fig. 4). In vitro enzyme assays carried out on α-syn clones showed a significant increase in GCL activity by ∼30 and ∼45% in WT and A53T cells, respectively, compared to control (Fig. 5A), thus corroborating the in silico data.
Fig. 4.
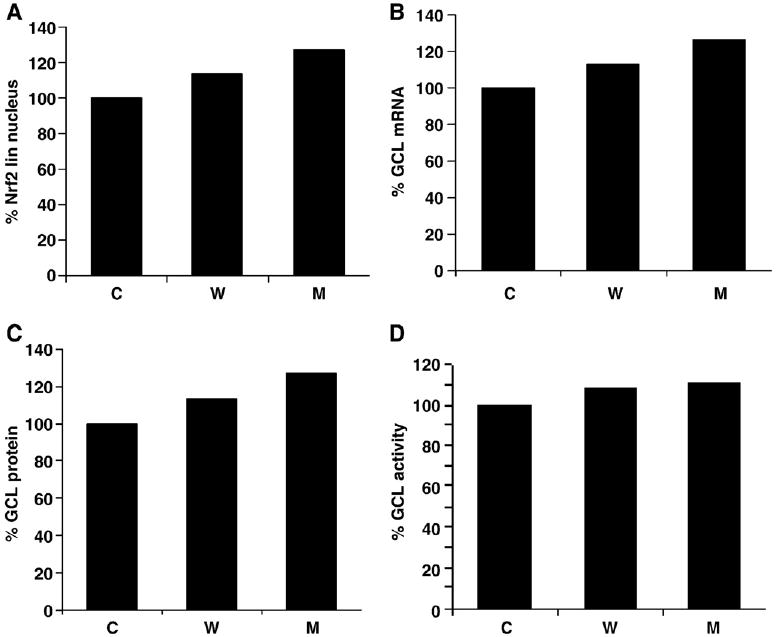
Simulation data showing the effects of α-syn expression and proteasome inhibition on GCL expression (A-D are in silico data). (A) Increased nuclear accumulation of Nrf2 transcription factor in the presence of α-syn (WT and mutant) compared to control. Nrf2-dependent elevation in (B) GCL mRNA expression, (C) GCL protein levels and (D) GCL enzyme activity in α-syn clones compared to control cells is shown.
Fig. 5.
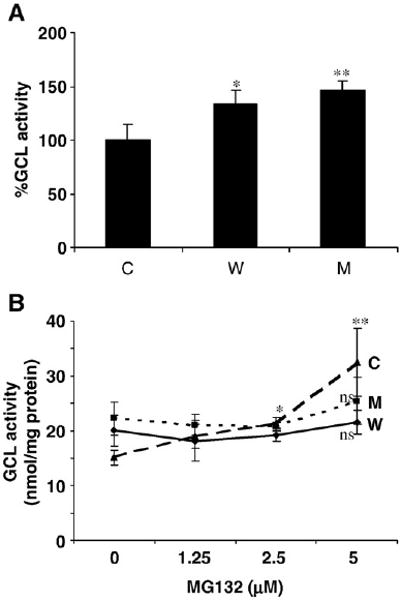
Effects of α-syn expression (±PI) on GCL activity in α-syn-expressing vs control cells (A and B are in vitro data). (A) GCL activity in the absence of proteasome inhibition. *p> 0.05 compared to control cells; **p< 0.05 compared to control cells. (B) GCL activity in the presence of MG132 addition. *p< 0.005 compared to untreated cells; **p< 0.05 compared to A53T cells; ns, not significant compared to untreated cells.
GCL assays in α-syn clones incubated with MG132, however, showed increased activity only in control cells (by ∼200%) and not in WT and A53T cells (Fig. 5B). These data suggested that the increase in GSH levels in the control cells exposed to PI could be explained by increased GCL activity; however, this did not explain the increase in the WT α-syn cells, which showed no elevation in GCL activity with proteasome inhibition. However, decreased GSH levels in A53T cells (+PI) might be due to some other factor affecting GCL activity. Because GCL activity is energy dependent, it is possible that ATP depletion might lead to decreased GCL activity.
Recently, Devi et al. [36] demonstrated that α-syn could be imported and accumulated in the mitochondria in neuronal cultures, leading to mitochondrial dysfunction via inhibition of CI. This inhibition was significantly more in A53T-expressing cells compared to WT. Dynamic modeling using these data showed that α-syn expression and proteasome inhibition caused increased accumulation of ROS (Fig. 6A) and CI inhibition at least in A53T cells versus controls (Fig. 6B). Simulation data also indicated that CI inhibition by α-syn aggregates is less than that of rotenone (data not shown). Quantitative estimation of ATP in silico confirmed that ATP levels were reduced in the A53T-expressing system incubated with PI compared to WT or control (Fig. 6C). Therefore, based on the in silico studies, because GCL activity requires ATP, the decreased GSH in the A53T-expressing cells exposed to PI could be due to a decrease in ATP levels as a consequence of mitochondrial dysfunction. Furthermore, GCL activity measured in silico in the absence of excess addition of ATP to the reaction resulted in decreased activity in A53T cells treated with PI compared to WT or control (Fig. 6D).
Fig. 6.
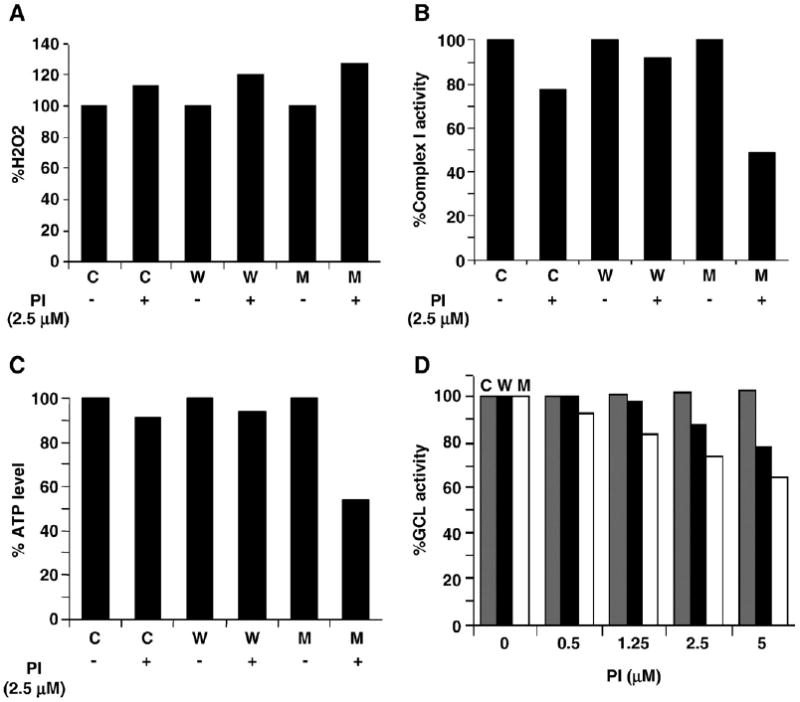
In silico simulations representing the effects of α-syn expression on mitochondrial function and GCL activity (A-D are in silico data). The effects of α-syn expression on (A) H2O2 levels, (B) CI activity, and (C) ATP levels in the absence and absence of PI (2.5 μM) in control (C), WT (W), and A53T mutant (M) α-syn expression systems are shown. (D) α-Syn expression and PI treatment leads to decreased GCL activity owing to decreased mitochondrial ATP levels in A53T α-syn (M) compared to WT and control.
To corroborate the in silico data, we assessed GCL activities in vitro in total cell extracts from A53T (vs WT and control cells) in the absence of exogenous ATP (Fig. 7A). With increasing concentrations of MG132, GCL activity was significantly decreased in A53T α-syn cells (by ∼40% at 5 μM MG132) compared to WT and control, suggesting that mitochondrial dysfunction and ATP depletion may be more affected in the A53T-expressing cells. Furthermore, ATP content was significantly more depleted by proteasomal inhibition in the A53T cells (by ∼50% at 5 μM MG132) than in either WT α-syn cells or controls (Fig. 7B).
Fig. 7.
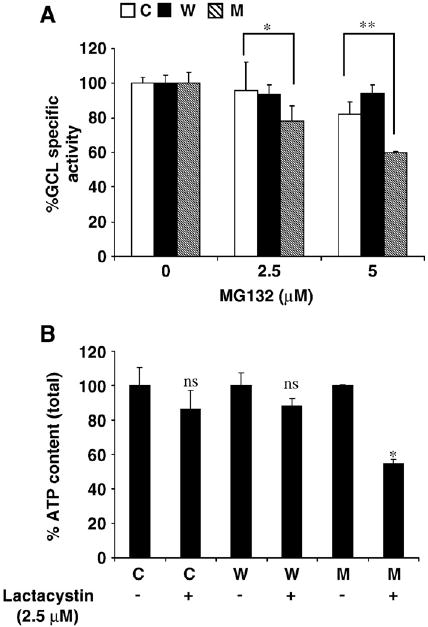
Role of ATP in GSH metabolism in α-syn N27 clones (A and B are in vitro data). (A) Effect of MG132 on GCL activity in the absence of exogenous ATP in α-syn-expressing vs untreated cells (C, control; W, wild type; M, A53T mutant). *p< 0.5 A53T compared to control cells; **p< 0.005 A53T compared to control cells. (B) Total ATP content in α-syn cells compared to control in the presence and absence of lactacystin (2.5 μM). *p< 0.001 compared to untreated A53T cells, ns, not significant compared to untreated cells.
Other mechanisms that might contribute to GSH depletion
GGT is a membrane-bound enzyme that cleaves GSH [49]. To analyze whether GGT activity might contribute to GSH depletion in our α-syn clones, we performed enzyme assays on cell extracts from α-syn clones (WT and A53T) (±PI). Fig. 8A shows that the GGT activity is increased in WT and A53T cells by ∼35 and ∼80%, respectively, in the absence of PI compared to control. But these values did not further alter significantly in the presence of PI compared to untreated cells. These data suggest that GGT activity might not contribute to GSH depletion during exposure to PI. Simulation data from our in silico model showed that although there was an increase in GGT activity in the absence of PI, with PI administration, there was a further increase in the A53T cells compared to WT and control cells (Figs. 8B and 8C).
Fig. 8.
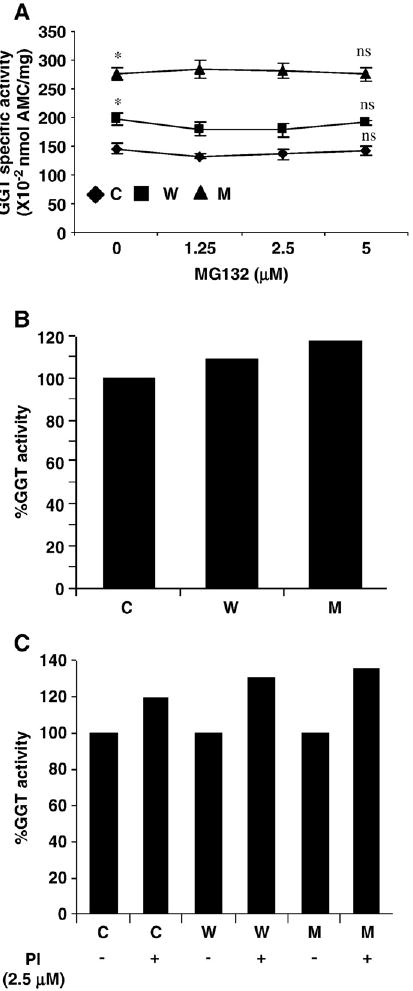
Effects of α-syn expression (±PI) on GGT activity (A is in vitro data; B and C are in silico data). (A) GGT activity in α-syn clones in the absence and presence of MG132 (C, control; W, wild type; M, A53T mutant). *p< 0.001 compared to control cells; ns, not significant. In silico data regarding the effects of α-syn expression on GGT activity in the (B) absence and (C) presence of PI (2.5 μM) are shown.
Titration of free GSH by glutathionylation of proteins might be another mechanism that could contribute to GSH depletion in A3T α-syn cells exposed to PI. We subjected soluble and insoluble fractions of α-syn clones and controls (±PI) to anti-GSH Western blots followed by quantitation of the GSH/β-actin signal. Fig. 9 shows that there is no significant change in the glutathionylation status in A53T cells compared to vector controls, suggesting that glutathionylation might not play a role in GSH depletion.
Fig. 9.
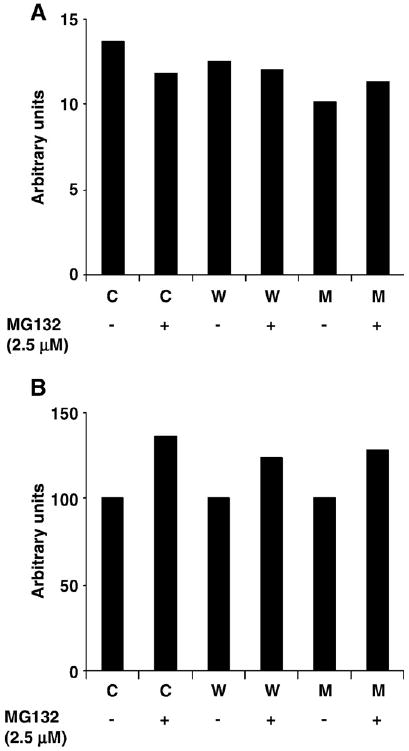
Effects of proteasome inhibition on glutathionylation levels in α-syn clones (A and B are in vitro data). Soluble and insoluble fractions from all three clones (C, control; W, wild type; M, A53T mutant) ± PI were run on SDS-PAGE followed by anti-GSH Western analysis. (A and B) The densitometric scanning ratio of anti-GSH/anti-β-actin signal in soluble and insoluble fractions of α-syn clones, respectively, is shown.
Therefore, a decrease in both mitochondrial function and GSH levels in the A53T and not any other cell types investigated in our study suggests that the combined detrimental effects of A53T expression and proteasome inhibition might ultimately result in neurodegeneration with relevance to PD.
Discussion
α-Syn protein has been directly linked to PD-associated neurodegeneration. Although there are studies that imply the toxic role of α-syn, there are others that have indicated a neuroprotective effect, thus making its role in PD controversial.
Several reports suggest that expression of WT α-syn at normal physiological levels provides neuroprotection [50]. This has been corroborated by da Costa et al. [51], who demonstrated that WT but not PD-related A53T mutant α-syn protects neuronal cells from apoptotic stimuli. But, exposure to the toxin 6-hydroxydopamine (6-OHDA) abolished this antiapoptotic phenotype by triggering α-syn aggregation, thereby contributing to PD pathology [51,52]. Similarly, expression of WT α-syn was neuroprotective against the neurotoxicity induced by serum deprivation and excitotoxicity. Furthermore, α-syn restored the GSH levels depleted during serum deprivation [53]. α-Syn probably regulates neuronal survival via Bcl-2 family expression and the PI3/Akt kinase pathway and inactivation of proapoptotic proteins [54]. Furthermore, α-syn expression protected neurons against oxidative stress via inactivation of the proapoptotic JNK signaling pathway in neuronal cells [55]. Recently, Quilty et al. [56] demonstrated that α-syn expression is upregulated in neurons in response to chronic oxidative stress and is associated with neuroprotection.
Similarly, α-syn overexpression in vivo was demonstrated to protect against paraquat-induced neurodegeneration and this neuroprotection was mediated by elevated expression of HSP70, a chaperone protein that has been shown to counteract paraquat toxicity in other models [57]. Jensen et al. [58] have shown that WT α-syn can protect naïve but not differentiated dopaminergic cells against 1-methyl-4-phenylpyridinium (MPP+)-mediated neurotoxicity. Therefore, depending on the status of cellular differentiation α-syn could be either neuroprotective or neurotoxic. Furthermore, exposure toMPP+caused an elevation in the expression of α-syn in human neuroblastoma cells, thus linking it to neuroprotection in experimental models [59,60]. Neuroprotection in these neurons was associated with phosphorylation events that in turn were linked to cell proliferation [59]. Upregulation of α-syn mRNA and protein was observed in a rodent model of apoptosis of dopaminergic neurons induced by injury [61]. However, α-syn in this model was expressed only in normal neurons rather than in dying neurons. But α-syn expression was decreased in rat SN after induction of apoptosis by intrastriatal 6-OHDA, thus supporting the neuroprotective capacity of α-syn [62]. These observations were strengthened by the observation that α-syn mRNA was significantly decreased in the SN of PD patients compared to the controls [63]. In a related study, Tanaka et al. [64] showed that expression of α-syn and its interacting partner synphilin-1 caused formation of aggresomes that were positive for phosphorylated α-syn. Interestingly, such aggresomes were found to a large extent in nonapoptotic cells compared to apoptotic neurons, suggesting a disconnection between aggresome formation and apoptosis.
Alternatively, the neurotoxic role (highlighted in our study) indicates that pathophysiological alterations such as overexpression, genetic mutations, and exposure to toxins cause the toxic gain of function of α-syn [18]. Accordingly, overexpression of human α-syn was sufficient to cause a familial PD-like condition, thus emphasizing the neurotoxicity of α-syn [65]. α-Syn overexpression leading to toxic aggregation in various in vivo models has resulted in neurotoxicity [66]. Familial mutations in α-syn were linked to sporadic PD when WT α-syn was identified as part of LBs [67,68]. Transgenic mice over-expressing mutant α-syn in the brain display cellular dysfunction and neurodegeneration [69,70]. α-Syn aggregation has been associated with adverse cellular effects such as proteasome dysfunction [71], altered macroautophagy [71], or defective chaperone-mediated autophagy [72] and impaired ER–Golgi vesicular traffic [73].
Apart from these effects, α-syn toxicity in PD models is anticipated to link with other pathways such as mitochondrial dysfunction, proteasome inhibition, and oxidative stress. Accordingly, the presence of α-syn-containing intraneuronal inclusions has been associated with PD itself and in animal models of PD exposed to either mitochondrial or proteasomal inhibitors [15,74]. There are reports linking α-syn expression and mitochondrial dysfunction connected with PD. Increased expression or mutation of α-syn has been suggested to result in mitochondrial dysfunction [75–77]. Expression of WT α-syn in neurons results in abnormal mitochondrial structures that were restored by treatment with the antioxidant vitamin E [78]. This suggests that α-syn expression might cause mitochondrial dysfunction via oxidative stress. Increased SN mitochondrial pathology with abnormal structures in α-syn transgenic mice was observed after MPTP treatment [79]. Exposure to pesticides such as paraquat and maneb has been reported to lead to the presence of disorganized and degenerative mitochondria in A53T human α-syn-expressing but not WT α-syn-expressing transgenic mice, suggesting the involvement of mutant α-syn in mitochondrial dysfunction [80]. A recent report suggested that a portion of α-syn might be localized to the mitochondrial membrane within normal SN dopaminergic neurons [81].
Cuervo et al. [72] demonstrated that WT α-syn protein is degraded by the lysosome, but the A53T is not, thereby increasing its cellular levels. Similar to bacterial toxins, the A53T protein can form annular pores leading to inappropriate membrane permeabilization of vesicles [82,83] and tighter interaction with brain mitochondrial membranes than monomeric or fibrillar α-syn [84]. In a related study, Anandatheerthavarada et al. [85] demonstrated that the Alzheimer amyloid precursor protein exhibits mitochondrial targeting and transmembrane arrest, thus impairing mitochondrial function in neurons. The first ever evidence that directly links α-syn expression and mitochondrial dysfunction with relevance to PD has recently been reported by Devi et al. [36]. In this study, it was demonstrated that human α-syn protein exhibits mitochondrial targeting that allows its accumulation within the mitochondrial inner membrane in neurons. Furthermore, the accumulated α-syn directly interacts and inhibits CI and this effect is more profound with A53T than with WT α-syn. Similar observations were also made within SN and striatal mitochondria from postmortem human PD brains.
Likewise, proteasome inhibition has also been suggested to affect mitochondrial function and GSH homeostasis associated with PD. Systemic injections of the PI epoxomicin into adult rats resulted not only in striatal dopamine depletion and dopaminergic SN neurodegeneration but also in the presence of intracytoplasmic inclusions [15]. Striatal microinfusion of lactacystin resulted in neuronal inclusions that stain for components of the ubiquitin–proteasome pathway [86]. Treatment of mesencephalic neurons with PI induced neurotoxicity accompanied by mitochondrial dysfunction and GSH depletion [87]. Proteasomal inhibition might also result in decreased mitochondrial turnover by the lysosomes [16], resulting in many defective A53T-containing mitochondria and leading to oligomerization within mitochondrial membranes.
In the current study, we have constructed a dynamic model for PD incorporating α-syn, proteasome inhibition, and mitochondrial dysfunction and analyzed their impact on GSH metabolism. Using stable α-syn-expressing N27 cell lines, we first developed a PI-mediated neurotoxic model of PD and tested it in terms of cell viability and GSH metabolism. Along the same lines, we built a dynamic in silico model and analyzed the mechanism of PI-mediated GSH depletion with “what-if” analysis. We have again validated these conclusions by carrying out cell culture experiments and, based on these data, we have reinforced each module. Based on these data, we have come up with a comprehensive model linking GSH metabolism, mitochondrial dysfunction, α-syn expression, and proteasome inhibition (Fig. 10). The majority of the data obtained from the in silico model are in line with in vitro experimental data.
Fig. 10.
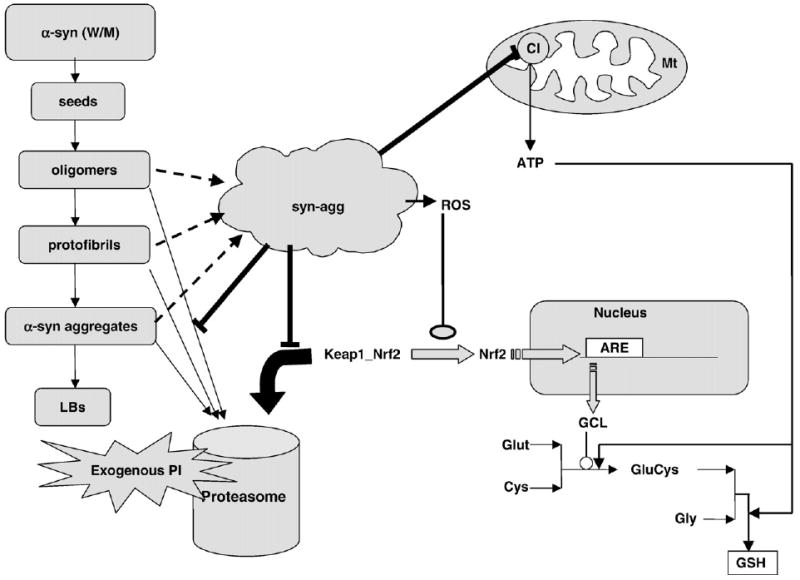
Schematic overview of the integrated model linking α-syn, the proteasome machinery, mitochondrial dysfunction, and GSH metabolism. In the presence of exogenous PI, the degradation of Nrf2 by the proteasome machinery is less. Consequently, the accumulated Nrf2 moves to the nucleus and upregulates GCL transcription. However, inhibition of proteasome leads to lowered degradation of aggregation intermediates, leading to accumulation of toxic α-syn aggregates in cell. These aggregates target the mitochondria leading to significant inhibition of CI, thus lowering ATP synthesis in the cell. ATP depletion ultimately results in decreased GSH synthesis in the cell, although the synthesis of the rate-limiting enzyme GCL has been upregulated by Nrf2. Abbreviations used: W/M, wild type or mutant (A53T); Gly, glycine; Glut, glutamate; GluCys, glutamate-cysteine; ARE, antioxidant response element; LBs, Lewy bodies; syn-agg, toxic α-syn aggregates.
α-Syn expression in N27 cells resulted in elevated GSH levels due to increased GCL (Figs. 4D and 5A) and GGT activities (Figs. 8A and 8B). Both of these genes have been previously demonstrated to be stress induced as a compensatory mechanism by various agents [88,89]. In the presence of further stress imposed by PI, GSH levels were increased in control cells owing to further increases in GCL activity (Fig. 5B). In WT-expressing cells, the addition of proteasomal inhibition also resulted in increased GSH levels (Fig. 3A) although the mechanism is unknown but clearly does not involve increases in either GCL or GGT activities. Conversely, challenging the A53T α-syn cells with PI resulted in significant GSH depletion (Fig. 3A). Our data demonstrate that GCL activity in A53T α-syn cells (+PI) measured in the absence of exogenous ATP was decreased (Figs. 6D and 7A), suggesting that ATP is probably the rate-limiting factor for GCL activity, which subsequently impinges on GSH levels. Whereas neither α-syn expression nor proteasomal inhibition alone resulted in significant ATP depletion, in combination these effects induced mitochondrial dysfunction leading to subsequent GSH depletion.
Based on previous data and our current data, we propose that A53T α-syn, which is not easily degradable, can accumulate and disrupt mitochondrial function. In the presence of PI, mitochondrial turnover is reduced further, contributing to the neurotoxicity of A53T, resulting in decreased ATP synthesis and in turn decreasing de novo synthesis of GSH. This constitutes a novel comprehensive PD model that serves to explain how the presence of mutant α-syn or proteasome inhibition may individually impact on mitochondrial function and in combination may alter the GSH metabolism via mitochondrial dysfunction. As such, this model serves to reconcile several previously disparate features of the disease in a sequential manner and to help explain the progression of the disease based on the concerted molecular actions of these components.
Supplementary Material
Acknowledgments
This work was supported by NIH R01 NS045615, RL1 NS062415, and P01 AG025901 (J.K.A.) and by DST, India, and Cell Works Group, Inc., India (both to M.M.S.B.). Cell Works Group is a commercial entity involved in creating in silico models of various pathways related to disease physiology.
Abbreviations
- PD
Parkinson disease
- SN
substantia nigra
- α-syn
α-synuclein
- CI
mitochondrial complex I
- RS
reactive species
- ROS
reactive oxygen species
- RNS
reactive nitrogen species
- GSH
glutathione reduced
- GSSG
glutathione oxidized
- GCL
γ-glutamyl cysteine ligase
- GGT
γ-glutamyl transpeptidase
- MPTP
1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine
- PI
proteasome inhibitor
- WT
wild type
- Nrf2
nuclear erythroid 2 p45-related factor 2 or NF-E2-related factor-2
Footnotes
Appendix A. Supplementary data: Supplementary data associated with this article can be found, in the online version, at doi:10.1016/j.freeradbiomed.2008.08.002.
References
- 1.Beal MF. Does impairment of energy metabolism result in excitotoxic neuronal death in neurodegenerative illnesses? Ann Neurol. 1992;31:119–130. doi: 10.1002/ana.410310202. [DOI] [PubMed] [Google Scholar]
- 2.Burke RE. Parkinson's disease. In: Koliatsos VE, Ratan RR, editors. Cell Death and Disease of the Nervous System. Totowa, NJ: Humana Press; 1998. pp. 459–475. [Google Scholar]
- 3.Adams JD, Jr, Chang ML, Klaidman L. Parkinson's disease—redox mechanisms. Curr Med Chem. 2001;8:809–814. doi: 10.2174/0929867013372995. [DOI] [PubMed] [Google Scholar]
- 4.Sayre LM, Smith MA, Perry G. Chemistry and biochemistry of oxidative stress in neurodegenerative diseases. Curr Med Chem. 2001;8:721–738. doi: 10.2174/0929867013372922. [DOI] [PubMed] [Google Scholar]
- 5.Danielson SR, Andersen JK. Oxidative and nitrative protein modifications in Parkinson's disease. Free Radic Biol Med. 2008;44:1787–1794. doi: 10.1016/j.freeradbiomed.2008.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Perry TL, Yong VW. Idiopathic Parkinson's disease, progressive supranuclear palsy and glutathione metabolism in the substantia nigra of patients. Neurosci Lett. 1986;67:269–274. doi: 10.1016/0304-3940(86)90320-4. [DOI] [PubMed] [Google Scholar]
- 7.Sriram K, Shankar SK, Boyd MR, Ravindranath V. Thiol oxidation and loss of mitochondrial complex I precede excitatory amino acid mediated neurodegeneration. J Neurosci. 1998;18:10287–10296. doi: 10.1523/JNEUROSCI.18-24-10287.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jha N, Jurma O, Lalli G, Liu Y, Pettusi EH, Greenamyre JT, Liu RM, Forman HJ, Andersen JK. Glutathione depletion in PC12 results in selective inhibition of mitochondrial complex I activity: implications for Parkinson's disease. J Biol Chem. 2000;275:26096–27001. doi: 10.1074/jbc.M000120200. [DOI] [PubMed] [Google Scholar]
- 9.Schapira AH, Cooper JM, Dexter D, Clark JB, Jenner P, Marsden CD. Mitochondrial complex I deficiency in Parkinson's disease. J Neurochem. 1990;54:823–827. doi: 10.1111/j.1471-4159.1990.tb02325.x. [DOI] [PubMed] [Google Scholar]
- 10.Lin TK, Hughes G, Muratovska A, Blaikie FH, Brookes PS, Darley-Usmar V, Smith RA, Murphy MP. Specific modification of mitochondrial protein thiols in response to oxidative stress: a proteomics approach. J Biol Chem. 2002;277:17048–17056. doi: 10.1074/jbc.M110797200. [DOI] [PubMed] [Google Scholar]
- 11.Murray J, Taylor SW, Zhang B, Ghosh SS, Capaldi RA. Oxidative damage to mitochondrial complex I due to peroxynitrite: identification of reactive tyrosines by mass spectrometry. J Biol Chem. 2003;278:37223–37230. doi: 10.1074/jbc.M305694200. [DOI] [PubMed] [Google Scholar]
- 12.Keeney PM, Xie J, Capaldi RA, Bennett JP., Jr Parkinson's disease brain mitochondrial complex I has oxidatively damaged subunits and is functionally impaired and misassembled. J Neurosci. 2006;26:5256–5264. doi: 10.1523/JNEUROSCI.0984-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mythri RB, Jagatha B, Pradhan N, Andersen J, Bharath MM. Mitochondrial complex I inhibition in Parkinson's disease: how can curcumin protect mitochondria? Antioxid Redox Signaling. 2007;9:399–408. doi: 10.1089/ars.2006.1479. [DOI] [PubMed] [Google Scholar]
- 14.Lenaz G, Bovina C, Castelluccio C, Fato R, Formiggini G, Genova ML, Marchetti M, Pich MM, Pallotti F, Parenti Castelli G, Biagini G. Mitochondrial complex I defects in aging. Mol Cell Biochem. 1997;178:329–333. [PubMed] [Google Scholar]
- 15.McNaught KS, Perl DP, Brownell AL, Olanow CW. Systemic exposure to proteasome inhibitors causes a progressive model of Parkinson's disease. Ann Neurol. 2004;56:149–162. doi: 10.1002/ana.20186. [DOI] [PubMed] [Google Scholar]
- 16.Sullivan PG, Dragicevic NB, Deng JH, Bai Y, Dimayuga E, Ding Q, Chen Q, Bruce-Keller AJ, Keller JN. Proteasome inhibition alters neural mitochondrial homeostasis and mitochondria turnover. J Biol Chem. 2004;279:20699–20707. doi: 10.1074/jbc.M313579200. [DOI] [PubMed] [Google Scholar]
- 17.Forno LS. Neuropathology of Parkinson's disease. J Neuropathol Exp Neurol. 1996;55:259–272. doi: 10.1097/00005072-199603000-00001. [DOI] [PubMed] [Google Scholar]
- 18.Rajagopalan S, Andersen JK. Alpha synuclein aggregation: is it the toxic gain of function responsible for neurodegeneration in Parkinson's disease? Mech Ageing Dev. 2001;122:1499–1510. doi: 10.1016/s0047-6374(01)00283-4. [DOI] [PubMed] [Google Scholar]
- 19.Abeliovich A, Beal MF. Parkinsonism genes: culprits and clues. J Neurochem. 2006;99:1062–1072. doi: 10.1111/j.1471-4159.2006.04102.x. [DOI] [PubMed] [Google Scholar]
- 20.Raichur A, Vali S, Gorin F. Dynamic modeling of alpha-synuclein aggregation for the sporadic and genetic forms of Parkinson's disease. Neuroscience. 2006;142:859–870. doi: 10.1016/j.neuroscience.2006.06.052. [DOI] [PubMed] [Google Scholar]
- 21.Kaushik P, Gorin F, Vali S. Dynamics of tyrosine hydroxylase mediated regulation of dopamine synthesis. J Comput Neurosci. 2007;22:147–160. doi: 10.1007/s10827-006-0004-8. [DOI] [PubMed] [Google Scholar]
- 22.Vali S, Mythri R, Jagatha B, Padiadpu J, Ramanujan KS, Andersen JK, Gorin F, Bharath MMS. Integrating glutathione metabolism and mitochondrial dysfunction with implications for Parkinson's disease: a dynamic model. Neuroscience. 2007;149:904–917. doi: 10.1016/j.neuroscience.2007.08.028. [DOI] [PubMed] [Google Scholar]
- 23.Jagatha B, Mythri RB, Vali S, Bharath MM. Curcumin treatment alleviates the effects of glutathione depletion in vitro and in vivo: therapeutic implications for Parkinson's disease explained via in silico studies. Free Radic Biol Med. 2008;44:907–917. doi: 10.1016/j.freeradbiomed.2007.11.011. [DOI] [PubMed] [Google Scholar]
- 24.Dickinson DA, Iles KE, Zhang H, Blank V, Forman HJ. Curcumin alters EpRE and AP-1 binding complexes and elevates glutamate–cysteine ligase gene expression. FASEB J. 2003;17:473–475. doi: 10.1096/fj.02-0566fje. [DOI] [PubMed] [Google Scholar]
- 25.Dickinson DA, Levonen AL, Moellering DR, Arnold EK, Zhang H, Darley-Usmar VM, Forman HJ. Human glutamate cysteine ligase gene regulation through the electrophile response element. Free Radic Biol Med. 2004;37:1152–1159. doi: 10.1016/j.freeradbiomed.2004.06.011. [DOI] [PubMed] [Google Scholar]
- 26.Zhang DD. Mechanistic studies of the Nrf2–Keap1 signaling pathway. Drug Metab Rev. 2006;38:769–789. doi: 10.1080/03602530600971974. [DOI] [PubMed] [Google Scholar]
- 27.Kobayashi A, Kang MI, Watai Y, Tong KI, Shibata T, Uchida K, Yamamoto M. Oxidative and electrophilic stresses activate Nrf2 through inhibition of ubiquitination activity of Keap1. Mol Cell Biol. 2006;26:221–229. doi: 10.1128/MCB.26.1.221-229.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lee JM, Shih AY, Murphy TH, Johnson JA. NF-E2-related factor-2 mediates neuroprotection against mitochondrial complex I inhibitors and increased concentrations of intracellular calcium in primary cortical neurons. J Biol Chem. 2003;278:37948–37956. doi: 10.1074/jbc.M305204200. [DOI] [PubMed] [Google Scholar]
- 29.D'Autréaux B, Toledano MB. ROS as signalling molecules: mechanisms that generate specificity in ROS homeostasis. Nat Rev Mol Cell Biol. 2007;8:813–824. doi: 10.1038/nrm2256. [DOI] [PubMed] [Google Scholar]
- 30.Lotharius J, Barg S, Wiekop P, Lundberg C, Raymon HK, Brundin P. Effect of mutant alpha-synuclein on dopamine homeostasis in a new human mesencephalic cell line. J Biol Chem. 2002;277:38884–38894. doi: 10.1074/jbc.M205518200. [DOI] [PubMed] [Google Scholar]
- 31.Sidhu A, Wersinger C, Vernier P. Does alpha-synuclein modulate dopaminergic synaptic content and tone at the synapse? FASEB J. 2004;18:637–647. doi: 10.1096/fj.03-1112rev. [DOI] [PubMed] [Google Scholar]
- 32.Tanaka M, Kim YM, Lee G, Junn E, Iwatsubo T, Mouradian MM. Aggresomes formed by alpha-synuclein and synphilin-1 are cytoprotective. J Biol Chem. 2004;279:4625–4631. doi: 10.1074/jbc.M310994200. [DOI] [PubMed] [Google Scholar]
- 33.Lindersson E, Beedholm R, Højrup P, Moos T, Gai W, Hendil KB, Jensen PH. Proteasomal inhibition by alpha-synuclein filaments and oligomers. J Biol Chem. 2004;279:12924–12934. doi: 10.1074/jbc.M306390200. [DOI] [PubMed] [Google Scholar]
- 34.Snyder H, Mensah K, Theisler C, Lee J, Matouschek A, Wolozin B. Aggregated and monomeric alpha-synuclein bind to the S6′ proteasomal protein and inhibit proteasomal function. J Biol Chem. 2003;278:11753–11759. doi: 10.1074/jbc.M208641200. [DOI] [PubMed] [Google Scholar]
- 35.McNaught KS, Shashidharan P, Perl DP, Jenner P, Olanow CW. Aggresome-related biogenesis of Lewy bodies. Eur J Neurosci. 2002;16:2136–2148. doi: 10.1046/j.1460-9568.2002.02301.x. [DOI] [PubMed] [Google Scholar]
- 36.Devi L, Raghavendran V, Prabhu BM, Avadhani NG, Anandatheerthavarada HK. Mitochondrial import and accumulation of alpha-synuclein impairs complex I in human dopaminergic neuronal cultures and Parkinson's disease brain. J Biol Chem. 2008;283:9089–9100. doi: 10.1074/jbc.M710012200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Narhi L, Wood SJ, Steavenson S, Jiang Y, Wu GM, Anafi D, Kaufman SA, Martin F, Sitney K, Denis P, Louis JC, Wypych J, Biere AL, Citron M. Both familial Parkinson's disease mutations accelerate alpha-synuclein aggregation. J Biol Chem. 1999;274:9843–9846. doi: 10.1074/jbc.274.14.9843. [DOI] [PubMed] [Google Scholar]
- 38.Zhou W, Hurlbert MS, Schaack J, Prasad KN, Freed CR. Overexpression of human alpha-synuclein causes dopamine neuron death in rat primary culture and immortalized mesencephalon-derived cells. Brain Res. 2000;866:33–43. doi: 10.1016/s0006-8993(00)02215-0. [DOI] [PubMed] [Google Scholar]
- 39.Bharath S, Cochran BC, Hsu M, Liu J, Ames BN, Andersen JK. Pre-treatment with R-lipoic acid alleviates the effects of GSH depletion in PC12 cells: implications for Parkinson's disease therapy. Neurotoxicology. 2002;23:479–486. doi: 10.1016/s0161-813x(02)00035-9. [DOI] [PubMed] [Google Scholar]
- 40.Seelig GF, Meister A. γ-Glutamylcysteine synthetase from erythrocytes. Methods Enzymol. 1985;113:390–392. doi: 10.1016/s0076-6879(85)13051-x. [DOI] [PubMed] [Google Scholar]
- 41.Forman HJ, Shi MM, Iwamoto T, Liu RM, Robison TW. Measurement of γ-glutamyl transpeptidase and γ-glutamylcysteine synthetase activities in cells. Methods Enzymol. 1995;252:66–71. doi: 10.1016/0076-6879(95)52009-0. [DOI] [PubMed] [Google Scholar]
- 42.Trounce IA, Kim YL, Jun AS, Wallace DC. Assessment of mitochondrial oxidative phosphorylation in patient muscle biopsies, lymphoblasts, and transmitochondrial cell lines. Methods Enzymol. 1996;264:484–509. doi: 10.1016/s0076-6879(96)64044-0. [DOI] [PubMed] [Google Scholar]
- 43.Tanaka Y, Engelender S, Igarashi S, Rao RK, Wanner T, Tanzi RE, Sawa AL, Dawson V, Dawson TM, Ross CA. Inducible expression of mutant alpha-synuclein decreases proteasome activity and increases sensitivity to mitochondria-dependent apoptosis. Hum Mol Genet. 2001;10:919–926. doi: 10.1093/hmg/10.9.919. [DOI] [PubMed] [Google Scholar]
- 44.Lee M, Hyun D, Halliwell B, Jenner P. Effect of the overexpression of wild-type or mutant alpha-synuclein on cell susceptibility to insult. J Neurochem. 2001;76:998–1009. doi: 10.1046/j.1471-4159.2001.00149.x. [DOI] [PubMed] [Google Scholar]
- 45.Dick LR, Cruikshank AA, Destree AT, Grenier L, McCormack TA, Melandri FD, Nunes SL, Palombella VJ, Parent LA, Plamondon L, Stein RL. Mechanistic studies on the inactivation of the proteasome by lactacystin in cultured cells. J Biol Chem. 1997;272:182–188. doi: 10.1074/jbc.272.1.182. [DOI] [PubMed] [Google Scholar]
- 46.Sekhar KR, Soltaninassab SR, Borrelli MJ, Xu ZQ, Meredith MJ, Domann FE, Freeman ML. Inhibition of the 26S proteasome induces expression of GLCLC, the catalytic subunit for γ-glutamylcysteine synthetase. Biochem Biophys Res Commun. 2000;270:311–317. doi: 10.1006/bbrc.2000.2419. [DOI] [PubMed] [Google Scholar]
- 47.Lindersson E, Beedholm R, Højrup P, Moos T, Gai W, Hendil KB, Jensen PH. Proteasomal inhibition by alpha-synuclein filaments and oligomers. J Biol Chem. 2004;279:12924–12934. doi: 10.1074/jbc.M306390200. [DOI] [PubMed] [Google Scholar]
- 48.Snyder H, Mensah K, Theisler C, Lee J, Matouschek A, Wolozin B. Aggregated and monomeric alpha-synuclein bind to the S6′ proteasomal protein and inhibit proteasomal function. J Biol Chem. 2003;278:11753–11759. doi: 10.1074/jbc.M208641200. [DOI] [PubMed] [Google Scholar]
- 49.Zhang H, Forman HJ, Choi J. γ-Glutamyl transpeptidase in glutathione biosynthesis. Methods Enzymol. 2005;401:468–483. doi: 10.1016/S0076-6879(05)01028-1. [DOI] [PubMed] [Google Scholar]
- 50.Sidhu A, Wersinger C, Moussa CEH, Vernier P. The role of α-synuclein in both neuroprotection and neurodegeneration. Ann N Y Acad Sci. 2004;1035:250–270. doi: 10.1196/annals.1332.016. [DOI] [PubMed] [Google Scholar]
- 51.da Costa CA, Ancolio K, Checler F. Wild-type but not Parkinson's disease-related Ala-53→Thr mutant alpha-synuclein protects neuronal cells from apoptotic stimuli. J Biol Chem. 2000;275:24065–24069. doi: 10.1074/jbc.M002413200. [DOI] [PubMed] [Google Scholar]
- 52.Alves Da Costa C, Paitel E, Vincent B, Checler F. Alpha-synuclein lowers p53-dependent apoptotic response of neuronal cells: abolishment by 6-hydroxydopamine and implications for Parkinson's disease. J Biol Chem. 2002;277:50980–50984. doi: 10.1074/jbc.M207825200. [DOI] [PubMed] [Google Scholar]
- 53.Lee M, Hyun D, Halliwell B, Jenner P. Effect of the overexpression of wild-type or mutant alpha-synuclein on cell susceptibility to insult. J Neurochem. 2001;76:998–1009. doi: 10.1046/j.1471-4159.2001.00149.x. [DOI] [PubMed] [Google Scholar]
- 54.Seo JH, Rah JC, Choi SH, Shin JK, Min K, Kim HS, Park CH, Kim S, Kim EM, Lee SH, Lee S, Suh SW, Suh YH. Alpha-synuclein regulates neuronal survival via Bcl-2 family expression and PI3/Akt kinase pathway. FASEB J. 2002;16:1826–1828. doi: 10.1096/fj.02-0041fje. [DOI] [PubMed] [Google Scholar]
- 55.Hashimoto M, Hsu LJ, Rockenstein E, Takenouchi T, Mallory M, Masliah E. Alpha-synuclein protects against oxidative stress via inactivation of the c-Jun N-terminal kinase stress-signaling pathway in neuronal cells. J Biol Chem. 2002;277:11465–11472. doi: 10.1074/jbc.M111428200. [DOI] [PubMed] [Google Scholar]
- 56.Quilty MC, King AE, Gai WP, Pountney DL, West AK, Vickers JC, Dickson TC. Alpha-synuclein is upregulated in neurons in response to chronic oxidative stress and is associated with neuroprotection. Exp Neurol. 2006;199:249–256. doi: 10.1016/j.expneurol.2005.10.018. [DOI] [PubMed] [Google Scholar]
- 57.Manning-Bog AB, McCormack AL, Purisai MG, Bolin LM, Di Monte DA. Alpha-synuclein overexpression protects against paraquat-induced neurodegeneration. J Neurosci. 2003;23:3095–3099. doi: 10.1523/JNEUROSCI.23-08-03095.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Jensen PJ, Alter BJ, O'Malley KL. Alpha-synuclein protects naïve but not dbcAMP-treated dopaminergic cell types from 1-methyl-4-phenylpyridinium toxicity. J Neurochem. 2003;86:196–209. doi: 10.1046/j.1471-4159.2003.01835.x. [DOI] [PubMed] [Google Scholar]
- 59.Gómez-Santos C, Ferrer I, Reiriz J, Viñals F, Barrachina M, Ambrosio S. MPP increases alpha-synuclein expression and ERK/MAP-kinase phosphorylation in human neuroblastoma SH-SY5Y cells. Brain Res. 2002;935:32–39. doi: 10.1016/s0006-8993(02)02422-8. [DOI] [PubMed] [Google Scholar]
- 60.Kalivendi SV, Cunningham S, Kotamraju S, Joseph J, Hillard CJ, Kalyanaraman B. Alpha-synuclein up-regulation and aggregation during MPP+-induced apoptosis in neuroblastoma cells: intermediacy of transferring receptor iron and hydrogen peroxide. J Biol Chem. 2004;279:15240–15247. doi: 10.1074/jbc.M312497200. [DOI] [PubMed] [Google Scholar]
- 61.Kholodilov NG, Oo TF, Burke RE. Synuclein expression is decreased in rat substantia nigra following induction of apoptosis by intrastriatal 6-hydroxydopamine. Neurosci Lett. 1999;275:105–108. doi: 10.1016/s0304-3940(99)00740-5. [DOI] [PubMed] [Google Scholar]
- 62.Kholodilov NG, Neystat M, Oo TF, Lo SE, Larsen KE, Sulzer D, Burke RE. Increased expression of α-synuclein in the substantia nigra pars compacta identified by mRNA differential display in a model of developmental target injury. J Neurochem. 1999;73:2586–2599. doi: 10.1046/j.1471-4159.1999.0732586.x. [DOI] [PubMed] [Google Scholar]
- 63.Neystat M, Lynch T, Przedborski S, Kholodilov N, Rzhetskaya M, Burke RE. Alpha synuclein expression in substantia nigra and cortex in Parkinson's disease. Mov Disord. 1999;14:417–422. doi: 10.1002/1531-8257(199905)14:3<417::aid-mds1005>3.0.co;2-x. [DOI] [PubMed] [Google Scholar]
- 64.Tanaka M, Kim YM, Lee G, Junn E, Iwatsubo T, Mouradian MM. Aggresomes formed by alpha-synuclein and synphilin-1 are cytoprotective. J Biol Chem. 2004;279:4625–4631. doi: 10.1074/jbc.M310994200. [DOI] [PubMed] [Google Scholar]
- 65.Singleton AB, Farrer M, Johnson J, Singleton A, Hague S, Kachergus J, Hulihan M, Peuralinna T, Dutra A, Nussbaum R, Lincoln S, Crawley A, Hanson M, Maraganore D, Adler C, Cookson MR, Muenter M, Baptista M, Miller D, Blancato J, Hardy J, Gwinn-Hardy K. alpha-Synuclein locus triplication causes Parkinson's disease. Science. 2003;302:841. doi: 10.1126/science.1090278. [DOI] [PubMed] [Google Scholar]
- 66.Abeliovich A, Beal MF. Parkinsonism genes: culprits and clues. J Neurochem. 2006;99:1062–1072. doi: 10.1111/j.1471-4159.2006.04102.x. [DOI] [PubMed] [Google Scholar]
- 67.Polymeropoulos MH. Genetics of Parkinson's disease. Ann N Y Acad Sci. 2000;920:28–32. doi: 10.1111/j.1749-6632.2000.tb06901.x. [DOI] [PubMed] [Google Scholar]
- 68.Spillantini MG, Schmidt ML, Lee VM, Trojanowski JQ, Jakes R, Goedert M. Alpha-synuclein in Lewy bodies. Nature. 1997;388:839–840. doi: 10.1038/42166. [DOI] [PubMed] [Google Scholar]
- 69.Gispert S, Del Turco D, Garrett L, Chen A, Bernard DJ, Hamm-Clement J, Korf HW, Deller T, Braak H, Auburger G, Nussbaum RL. Transgenic mice expressing mutant A53T human alpha-synuclein show neuronal dysfunction in the absence of aggregate formation. Mol Cell Neurosci. 2003;24:419–429. doi: 10.1016/s1044-7431(03)00198-2. [DOI] [PubMed] [Google Scholar]
- 70.Lee MK, Stirling W, Xu Y, Xu X, Qui D, Mandir AS, Dawson TM, Copeland NG, Jenkins NA, Price DL. Human alpha-synuclein-harboring familial Parkinson's disease linked Ala-53 → Thr mutation causes neurodegenerative disease with alpha-synuclein aggregation in transgenic mice. Proc Natl Acad Sci U S A. 2002;99:8968–8973. doi: 10.1073/pnas.132197599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Stefanis L, Larsen KE, Rideout HJ, Sulzer D, Greene LA. Expression of A53T mutant but not wild-type alpha-synuclein in PC12 cells induces alterations of the ubiquitin-dependent degradation system, loss of dopamine release, and autophagic cell death. J Neurosci. 2001;21:9549–9560. doi: 10.1523/JNEUROSCI.21-24-09549.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Cuervo AM, Stefanis L, Fredenburg R, Lansbury PT, Sulzer D. Impaired degradation of mutant alpha-synuclein by chaperone-mediated autophagy. Science. 2004;305:1292–1295. doi: 10.1126/science.1101738. [DOI] [PubMed] [Google Scholar]
- 73.Cooper AA, Gitler AD, Cashikar A, Haynes CM, Hill KJ, Bhullar B, Liu K, Xu K, Strathearn KE, Liu F, Cao S, Caldwell KA, Caldwell GA, Marsischky G, Kolodner RD, Labaer J, Rochet JC, Bonini NM, Lindquist S. Alpha-synuclein blocks ER–Golgi traffic and Rab1 rescues neuron loss in Parkinson's models. Science. 2006;313:324–328. doi: 10.1126/science.1129462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Fornai F, Schluter OM, Lenzi P, Gesi M, Ruffoli R, Ferrucci M, Lazzeri G, Busceti CL, Pontarelli F, Battaglia G, Pellegrini A, Nicoletti F, Ruggieri S, Paparelli A, Sudhof TC. Parkinson-like syndrome induced by continuous MPTP infusion: convergent roles of the ubiquitin–proteasome system and alpha-synuclein. Proc Natl Acad Sci USA. 2005;102:3413–3418. doi: 10.1073/pnas.0409713102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Beal MF. Mitochondria take center stage in aging and neurodegeneration. Ann Neurol. 2005;58:495–505. doi: 10.1002/ana.20624. [DOI] [PubMed] [Google Scholar]
- 76.Abou-Sleiman PM, Muqit MM, Wood NW. Expanding insights of mitochondrial dysfunction in Parkinson's disease. Nat Rev Neurosci. 2006;7:207–219. doi: 10.1038/nrn1868. [DOI] [PubMed] [Google Scholar]
- 77.Smith WW, Jiang H, Pei Z, Tanaka Y, Morita H, Sawa A, Dawson VL, Dawson TM, Ross CA. Endoplasmic reticulum stress and mitochondrial cell death pathways mediate A53T mutant alpha-synuclein-induced toxicity. Hum Mol Genet. 2005;14:3801–3811. doi: 10.1093/hmg/ddi396. [DOI] [PubMed] [Google Scholar]
- 78.Hsu LJ, Sagara Y, Arroyo A, Rockenstein E, Sisk A, Mallory M, Wong J, Takenouchi T, Hashimoto M, Masliah E. alpha-Synuclein promotes mitochondrial deficit and oxidative stress. Am J Pathol. 2000;157:401–410. doi: 10.1016/s0002-9440(10)64553-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Song DD, Shults CW, Sisk A, Rockenstein E, Masliah E. Enhanced substantia nigra mitochondrial pathology in human alpha-synuclein transgenic mice after treatment with MPTP. Exp Neurol. 2004;186:158–172. doi: 10.1016/S0014-4886(03)00342-X. [DOI] [PubMed] [Google Scholar]
- 80.Norris EH, Uryu K, Leight S, Giasson BI, Trojanowski JQ, Lee VM. Pesticide exposure exacerbates alpha-synucleinopathy in an A53T transgenic mouse model. Am J Pathol. 2007;170:658–666. doi: 10.2353/ajpath.2007.060359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Li WW, Yang R, Guo JC, Ren HM, Zha XL, Cheng JS, Cai DF. Localization of alpha-synuclein to mitochondria within midbrain of mice. Neuroreport. 2007;18:1543–1546. doi: 10.1097/WNR.0b013e3282f03db4. [DOI] [PubMed] [Google Scholar]
- 82.Lashuel HA, Hartley D, Petre BM, Walz T, Lansbury PT., Jr Neurodegenerative disease: amyloid pores from pathogenic mutations. Nature. 2002;418:291. doi: 10.1038/418291a. [DOI] [PubMed] [Google Scholar]
- 83.Volles MJ, Lansbury PT., Jr Zeroing in on the pathogenic form of alpha-synuclein and its mechanism of neurotoxicity in Parkinson's disease. Biochemistry. 2003;42:7871–7878. doi: 10.1021/bi030086j. [DOI] [PubMed] [Google Scholar]
- 84.Ding TT, Lee SJ, Rochet JC, Lansbury PT., Jr Annular alpha-synuclein protofibrils are produced when spherical protofibrils are incubated in solution or bound to brain-derived membranes. Biochemistry. 2002;41:10209–10217. doi: 10.1021/bi020139h. [DOI] [PubMed] [Google Scholar]
- 85.Anandatheerthavarada HK, Biswas G, Robin MA, Avadhani NG. Mitochondrial targeting and a novel transmembrane arrest of Alzheimer's amyloid precursor protein impairs mitochondrial function in neuronal cells. J Cell Biol. 2003;161:41–54. doi: 10.1083/jcb.200207030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Fornai F, Lenzi P, Gesi M, Ferrucci M, Lazzeri G, Busceti CL, Ruffoli R, Soldani P, Ruggieri S, Alessandri MG, Paparelli A. Fine structure and biochemical mechanisms underlying nigrostriatal inclusions and cell death after proteasome inhibition. J Neurosci. 2003;23:8955–8966. doi: 10.1523/JNEUROSCI.23-26-08955.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Kikuchi S, Shinpo K, Tsuji S, Takeuchi M, Yamagishi S, Makita Z, Niino M, Yabe I, Tashiro K. Effect of proteasome inhibitor on cultured mesencephalic dopaminergic neurons. Brain Res. 2003;964:228–236. doi: 10.1016/s0006-8993(02)04030-1. [DOI] [PubMed] [Google Scholar]
- 88.Zhang H, Dickinson DA, Liu RM, Forman HJ. 4-Hydroxynonenal increases γ-glutamyl transpeptidase gene expression through mitogen-activated protein kinase pathways. Free Radic Biol Med. 2005;38:463–471. doi: 10.1016/j.freeradbiomed.2004.10.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Dickinson DA, Iles KE, Watanabe N, Iwamoto T, Zhang H, Krzywanski DM, Forman HJ. 4-Hydroxynonenal induces glutamate cysteine ligase through JNK in HBE1 cells. Free Radic Biol Med. 2002;33:974. doi: 10.1016/s0891-5849(02)00991-7. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.