

Abstract
Questions surround the mechanism of mutually exclusive expression by which Plasmodium falciparum mediates activation and silencing of var genes. These encode PfEMP1 proteins, which function as cytoadherent and immunomodulatory molecules at the surface of parasitized erythrocytes. Current evidence suggests that promoter silencing by var introns might play a key role in var gene regulation. To evaluate the impact of cis-acting regulatory regions on var silencing, we generated P. falciparum lines in which luciferase was placed under the control of an UpsA var promoter. By utilizing the Bxb1 integrase system, these reporter cassettes were targeted to a genomic region that was not in apposition to var sub-telomeric domains. This eliminated possible effects from surrounding telomeric elements and removed the variability inherent in episomal systems. Studies with highly synchronized parasites revealed that the UpsA element possessed minimal activity in comparison with a heterologous (hrp3) promoter. This may well result from the integrated UpsA promoter being largely silenced by the neighboring cg6 promoter. Our analyses also revealed that the DownsA 3’ untranslated region further decreased the luciferase activity from both cassettes, whereas the var A intron repressed the UpsA promoter specifically. By applying multivariate analysis over the entire cell cycle, we confirmed the significance of these cis-elements and found the parasite stage to be the major factor regulating UpsA promoter activity. Additionally, we observed that the UpsA promoter was capable of nucleating reversible silencing that spread to a downstream promoter. We believe these studies are the first to analyze promoter activity of Group A var genes which have been implicated in severe malaria, and support the model that var introns can further suppress var expression. These data also suggest an important suppressive role for the DownsA terminator. Our findings imply the existence of multiple levels of var gene regulation in addition to intrinsic promoter-dependent silencing.
Keywords: Plasmodium falciparum, var, Antigenic variation, Gene regulation, Multivariate analysis
1. Introduction
The Plasmodium falciparum Erythrocyte Membrane Protein 1 (PfEMP1) variant surface antigens encoded by the var gene family (Baruch et al., 1995; Smith et al., 1995; Su et al., 1995) have repeatedly been linked to the immuno-evasive strategies and cytoadherent phenotypes of parasitized erythrocytes (Biggs et al., 1992; Roberts et al., 1992; Bull and Marsh, 2002). The etiological agent of severe malaria, the P. falciparum parasite, activates the expression of only one PfEMP1 ligand at a time, thereby mediating binding to a variety of host endothelial cell surface receptors, including Cluster of Differentiation 36 (CD36) Inter-Cellular Adhesion Molecule 1 (ICAM-1) in the host microvasculature, or chondroitin sulfate A (CSA) in the placenta (Fried and Duffy, 1996; Andrews et al., 2003; Salanti et al., 2003). This sequesters the parasite, preventing immune-mediated clearance in the spleen (Barnwell, 1989).
Each var gene is defined by the possession of one or more combinations of multiple adhesive domains in a two-exon gene structure, with ~60 different var genes per haploid genome (Smith et al., 2000; Gardner et al., 2002). The entirety of the global var gene repertoire is vast, however, as different parasite isolates may possess a wholly unique array of var genes (Trimnell et al., 2006; Barry et al., 2007; Kraemer et al., 2007). Sequencing of clonal laboratory isolates revealed that var genes can be classified into several subfamilies, primarily Groups A, B and C. This is based on their chromosomal location, the nucleotide identity of their non-coding regions and the adhesive domains encoded within the protein – in particular the “head structure” made up of the paired amino-terminal Duffy-binding-like α (DBLα) and cysteine-rich interdomain region α (CIDRα) domains (Gardner et al., 2002; Kraemer and Smith, 2003; Lavstsen et al., 2003). Group B and C genes comprise the bulk of the var family in each genome, with Group B genes located subtelomerically and transcribed away from the telomere and Group C genes found in several tandem arrays in central chromosomal locations. Group A genes are also present in subtelomeric regions but in a tail-to-tail orientation with the Group B genes such that they are transcribed towards the telomere.
var groups also possess highly specific upstream non-coding regions, such that UpsA, UpsB, and UpsC promoters precede Group A, B, and C var coding sequences, respectively. The presence of a few transitional var genes represented by Group B/A (UpsB with a Group A head structure) and Group B/C (Ups B in a central chromosomal location) strengthens the hypothesis that these groupings are maintained by intragroup recombination with occasional intergroup mixing (Kraemer et al., 2007; Bull et al., 2008). The relevance of these classifications to clinical infection is apparent in the stratification of the different var gene subfamilies to different pathologies, such that clinical isolates obtained from children with severe malaria express more Group A and B var genes (Kaestli et al., 2006; Kyriacou et al., 2006; Rottmann et al., 2006), while placental isolates express a single var gene, var2csa (Tuikue Ndam et al., 2005; Magistrado et al., 2008).
To date, the var intron has been the only cis-acting sequence element implicated in the modulation of var promoter function. Transient transfection studies have documented the repression of a var promoter-driven reporter gene by a closely apposed var intron (Deitsch et al., 2001). This intron-mediated silencing was found to be dependent on a bidirectional promoter activity that originated from the central region of the intron (Calderwood et al., 2003; Gannoun-Zaki et al., 2005) and required a one-to-one pairing of one var promoter with one intron (Frank et al., 2006). However, this model of intron-mediated var silencing has been controversial, as the presence or absence of the var intron was reported to have no effect on the silencing of a var promoter driving a selectable marker (Voss et al., 2006, 2007). In those studies, parasites were able to recognize the intron-less var promoter as part of the var gene family, suggesting that all information necessary for silencing and mutually exclusive expression might reside within the var promoter sequence. These differing accounts were addressed by a subsequent report, demonstrating that various heterologous promoters had the same silencing effect on an upstream var promoter as did the var intron (Dzikowski et al., 2007). This implied that silencing of var expression only required a promoter activity within the vicinity of the var promoter.
Exactly how a second paired promoter might allow the parasite to recognize and “count” a var promoter remains unclear, but there appears to be a requirement for a specific nuclear element that can be titrated away by a highly active “uncounted” var promoter (Dzikowski and Deitsch, 2008). The intron has also been shown to insulate a downstream selectable marker from the spread of var promoter activation, which suggests that the intron is able to affect the epigenetic status of chromatin surrounding the var promoter (Voss et al., 2006).
While alignments of var promoter sequences stratify these genes into distinct and consistent functional classes, the downstream non-coding regions of the Group A and B genes are also highly conserved, and are termed DownsA and DownsB (Kraemer and Smith, 2003; Lavstsen et al., 2003). The potential regulatory effects of these elements on var gene promoters have not been explored. Additionally, although Group A var genes represent a clinically significant subpopulation, the specific effects of the Group A intron on UpsA expression have not been assessed. Here, we report our investigations into Group A var gene regulation using the firefly luciferase reporter gene. Our data show that the UpsA promoter is minimally active in this integrated context. These studies have not definitively determined whether, in this genomic context, UpsA is expressed at biologically relevant levels. Nevertheless, our multivariate analyses on tightly synchronized parasite lines provide evidence that the highly conserved Group A var 3’ untranslated region (UTR), referred to as DownsA, and the Group A intron, both decrease expression of a reporter produced from the UpsA var promoter, albeit with differing specificities. These findings, combined with previous studies of Group B and C var genes, suggest that the parasite possesses a consistent means of var family regulation, with additional control imposed by conserved subgroup-specific DNA cis-elements including the DownsA element.
2. Materials and methods
2.1. Parasite cultivation and transfection
Parasites were cultivated in human red blood cells at 3 to 4% hematocrit (Fidock et al., 1998). The previously described P. falciparum Dd2attB parasite line, which contains an attB site within the disrupted locus of the cg6 gene (Nkrumah et al., 2006), was grown continuously in the presence of 2.5 nM of the Plasmodium dihydrofolate reductase (dhfr)-specific antifolate inhibitor WR99210 (Jacobus Pharmaceuticals, Princeton, NJ). Predominantly ring-stage parasites at 4 to 8% parasitemia were electroporated with 50 μg of an attP-containing plasmid and 50 μg of the pInt plasmid, as previously described (Nkrumah et al., 2006). One hundred μg/mL of G418 sulfate (Cellgro) and 2.5 μg/mL blasticidin S hydrochloride (BSD; Invitrogen) were added on day 2 post-transfection to select for co-transfected parasites. These typically became microscopically visible 17-26 days post-transfection. At that time, G418 was removed to eliminate the pInt plasmid from the culture. Cloning was performed by limiting dilution at 0.7 parasites per well (Goodyer et al., 1997).
2.2. Plasmid preparation
To generate the control p∂LH plasmid that contains the luciferase gene and the hrp2 3’UTR but has no promoter driving luciferase expression, the hrp3 promoter of the luciferase expression plasmid pHLH (Wu et al., 1995) was removed by Asp718 and NsiI digestion, and replaced with an oligonucleotide linker prepared by annealing two primers: 620 (5’-GTACCAAAGTCGACAAATGCA) and 621 (5’-TTTGTCGACTTTG). The pALH plasmid was generated by inserting a 2.2 kb UpsA fragment that was PCR amplified from the var gene PFD1235w using primers 772 (5’-AAAggtaccGTATGTTATACC) and 773 (5’-GCAgaatgcattcTTATAACAAAGTATTTAAATAA). These primers introduced 5’ KpnI and 3’ XmnI sites (lowercase) that were used to subclone into the KpnI and BfrBI digested p∂LH plasmid. A 0.75 kb fragment of the PFD1235w intron, amplified with primers 636 (5’-TTTctgcagGTAAATGGAGTATATATATGTG) and 774 (5’-CGGctgcagCTATAATTAAAAAAAAAGGTATGTATG) that both contained a PstI site, was inserted into the PstI site of pALH and pHLH. This yielded the plasmids pALHi and pHLHi, respectively. The 1.4 kb DownsA region of PFD1235w was PCR amplified using primers 626 (5’-CGGATGTATGGAATATATaagcttAAAA) and 627 (5’- GATATctgcagTACTATTACATAATACAT TC), and cloned into HindIII-PstI-digested pALH and pHLH, yielding pALA and pHLA. All PFD1235w sequences were amplified from genomic DNA of the P. falciparum 3D7 strain. Firefly luciferase expression cassettes were isolated from the vectors pALHi, pALA, pHLH, pHLHi, pHLA and p∂LH with KpnI and BamHI digestion, and inserted into pCBD-P (Nkrumah et al., 2006; Supplementary Fig. S1) that had been digested with KpnI-BglII. This generated the plasmids pCBD-P-ALHi, pCBD-P-ALA, pCBD-P-HLH, pCBD-P-HLHi, pCBD-P-HLA and pCBD-P-∂LH, respectively. The ALH luciferase cassette was isolated from pALH by digestion with SalI and BamHI, and ligated into pCBD-P cut with SalI and BglII to yield the plasmid pCBD-P-ALH.
2.3. Detection of integration by PCR and Southern hybridization
To detect integration we utilized the following primers: 3’F1 (5’-CATTTGAATTATTGCTCAACGCT) and T3 (5’- ATTAACCCTCACTAAAGGGA) to detect the Dd2attB parental locus; 3’F1 and either 1089 (5’-CCTACAAATATTATATCAAGAAATTC), 1090 (5’- AATCCGAATGTTCTGAATACCAC) or 767 (5’-CCACCTCGAT ATGTGCATC) to detect the attL junction of the var-, hrp3-, or no promoter-driven cassettes, respectively; 594 (5’-CAACCTAGGTGATATATTTCTATTGGTATTTAT) and T3 to detect the attR junction; and 594 with 1089, 1090 or 767 to detect any var-, hrp3-or no promoter-driven attP vectors that were either replicating episomally or had integrated as concatamers. Southern blots were hybridized with either cg6 or luciferase [32P]-labeled probes, obtained by digestion of the plasmids pCG6-attB (Nkrumah et al., 2006) or pHLH (Wu et al., 1995), respectively.
2.4. Luciferase assays
Parasites lines were doubly synchronized in 5% D-sorbitol (w/v) for two or more successive growth cycles. Thin smears of the parasite cultures were then prepared and fixed with methanol to count developmental stages and percent parasitemias. The hematocrits of the cultures were determined immediately prior to release of the parasites by lysis of the infected red blood cells with 0.1% saponin. Following two washes in PBS, parasite pellets were frozen at -80°C until further processing. Parasites were lysed at room temperature in 1X Reporter Lysis Buffer (Promega) that was supplemented with protease inhibitor cocktail (Roche) and 0.5 mM phenylmethanesulfonyl fluoride (PMSF; Sigma). Supernatants were assayed for luciferase activity by detection of relative luciferase units (RLU) in a Bertholt auto-injection luminometer or in a Victor2 luminescence plate reader (Perkin Elmer), following the addition of 100 μL reconstituted Luciferase Assay Substrate (Promega). Luciferase activities were assessed as RLU per million parasites (RPM), calculated by dividing the background-subtracted RLU by the number of millions of parasites assayed.
2.5. RNA extraction, cDNA preparation and 5’RACE
Parasite RNA was prepared using TRIzol Reagent (Invitrogen), and stored at -80°C until further processing. After extraction with choloroform, the RNA was precipitated and resuspended in diethyl dicarbonate (DEPC)-treated water. cDNA synthesis was performed on 2.5 μg of DNase-treated RNA using SuperScript III First Strand Synthesis System (Invitrogen), with parallel reactions performed without reverse transcriptase (RT) to check for genomic DNA contamination. 5’ rapid amplification of 5’ complementary DNA ends (RACE) was performed on 10 μg of DNase-treated RNA using a FirstChoice RLM-RACE kit (Ambien) and primers 392 (5’- TACATATAAATGTATCTATCC) and 1799 (5’- CCTCATTGATATAAATATTGTAG) to the UpsA promoter region.
2.6. Real-time PCR and analysis
Real time PCR was performed in a DNA Engine Opticon 2 detector (Biorad) to quantify transcript levels using QuantiTect SYBR Green PCR Master Mix (Qiagen). Luciferase transcript was detected using primers 1791 (5’-GCTGGGCGTTAATCAGAGAG) and 1792 (5’-GTGTTCGTCTTCGTCCCAGT) as described (Frank and Deitsch, 2006). bsd transcript was detected using primers 1793 (5’-TTGTCTCAAGAAGAATCCAC) and 1794 (5’-TCCCCCAGTAAAATGATATAC) as described (Chookajorn et al., 2007). PCR was performed with 0.5 μM primers for 40 cycles, using cycling conditions of 94°C for 30 s, 53°C for 40 s and 68°C for 50 s, followed by melting curve analysis from 45° -100°C. The amounts of luciferase and bsd transcript were calculated using standard curves generated by a four-fold dilution series of genomic DNA harvested from the Dd2attB-ALHi parasite line. These were presented as copy number relative to that of actin. Transcript copy numbers of actin were determined as above, using primers A129 (5’-AGCAGCAGGAATCCACACA) and A130 (5’- TGATGGTGCAAGGGTTGTAA), as described (Salanti et al., 2003).
2.7. In vitro drug assays
Parasite susceptibilities to drug selection markers were measured using a 96-well version of a published 72-h SYBR Green I (Invitrogen) incorporation assay (Plouffe et al., 2008). These used ring-stage cultures seeded at 0.1% parasitemia and 1.6% hematocrit and serial two-fold dilutions of the transfection selection agents BSD or WR99210, prepared in complete medium. These agents were tested at final concentrations of 0.08 to 40 μg/mL or 1.95 to 1,000 nM, respectively.
2.8. Data analysis
Luciferase activities, obtained from single time-points of an intra-erythrocytic developmental cycle (IDC), were collated using Microsoft Excel 2004 and statistically analyzed using GraphPad Prism (Version 5). To assess the fold change in geometric means of RPM expression between the control lines ALH or HLH and their matched experimental lines, we calculated the difference between the log-transformed values of the control line and each matched line. This difference was equivalent to the log of the ratio (or fold difference) of the untransformed values. Calculation of the antilog of this ratio therefore represented the fold change in expression. To quantify the reduction in expression compared with the control lines, we converted the antilogs to their reciprocal values. Area under the curve (AUC) values were calculated by numerically integrating the associated activity data over time using the trapezoidal formula. This is a standard numerical technique that represents the individual integration intervals by trapezoids with width ΔX and heights Y1 and Y2, where ΔX refers to the change in time (h post invasion (pi) and Y refers to the activity values. These AUC values were summed up to the given time to arrive at the cumulative AUC value (CAUC). For example, CAUC at time point 3 was the sum of the AUC values obtained at time-points 1, 2 and 3. An explanation of the principal component analysis (PCA) and partial least squares (PLS) techniques used for multivariate modeling is included in Supplementary Data S1.
3. Results
3.1. Selection of PFD1235w as a representative of Group A var genes
In the 3D7 strain, the Group A var genes are generally considered to be comprised of 10 members (Supplementary Table S1; (Gardner et al., 2002; Kraemer and Smith, 2003; Trimnell et al., 2006)), seven of which have non-CD36 binding head structures and encode large multi-domain PfEMP1 proteins (Robinson et al., 2003). One of the seven large Group A members, PFD1235w, has been classified by some groups as a fourth strain-transcending var gene (var4) (Jensen et al., 2004). Here we refer to these seven large genes as var4-like. The three remaining Group A members have been defined as Type 3 var, and are termed “atypical” for their small size and lack of CIDR1 domains, yet their two-cysteine residue DBL1α1 domains, promoter sequences and telomere-directed transcriptional orientation still classify them as Group A family members. These Group A var genes encode PfEMP1 proteins that differ widely in size, yet their upstream gene flanking regions were observed to be remarkably similar (Supplementary Fig. S2A). Although the Group A var introns exhibited less homology, the DownsA sequence elements were also found to be highly conserved (Supplementary Fig. S2B-C).
Based on this analysis, we chose to analyze the effect of the intron and the DownsA region on the regulation of activity from the UpsA promoter, and selected the var gene PFD1235w (a var4 gene, see Supplementary Table S1) as representative of the Group A family members. By RT-PCR and 5’RACE, we mapped the transcription start site of PFD1235w to -1,367 bp upstream of the start codon (Supplementary Fig. S1A). We limited the size of the promoter region to 2.2 kb, which includes this putative site and avoids possible effects from the promoter of the rif gene PFD1230c located upstream of PFD1235w (Fig. 1A). We note that the transcriptional start site of rif genes has been documented to be within 300 bp upstream of the rif start codon (Kyes et al., 1999; Tham et al., 2007), a region excluded from this segment. Attempts to map the PFD1235w polyadenylation site by 3’RACE were hampered by the high homologies exhibited between var family members, but conventional RT-PCR mapping by specific primers showed that the site was greater than 500 bp downstream of the stop codon (data not shown). To maximize the likelihood of retaining the necessary regulatory elements within the DownsA terminator, we utilized 1.4 kb of sequence downstream of the PFD1235w stop codon, referred to here as the DownsA element. This region lacked the putative transcription start site of the rif gene PFD1240w, which starts 2.16 kb downstream.
Fig. 1.
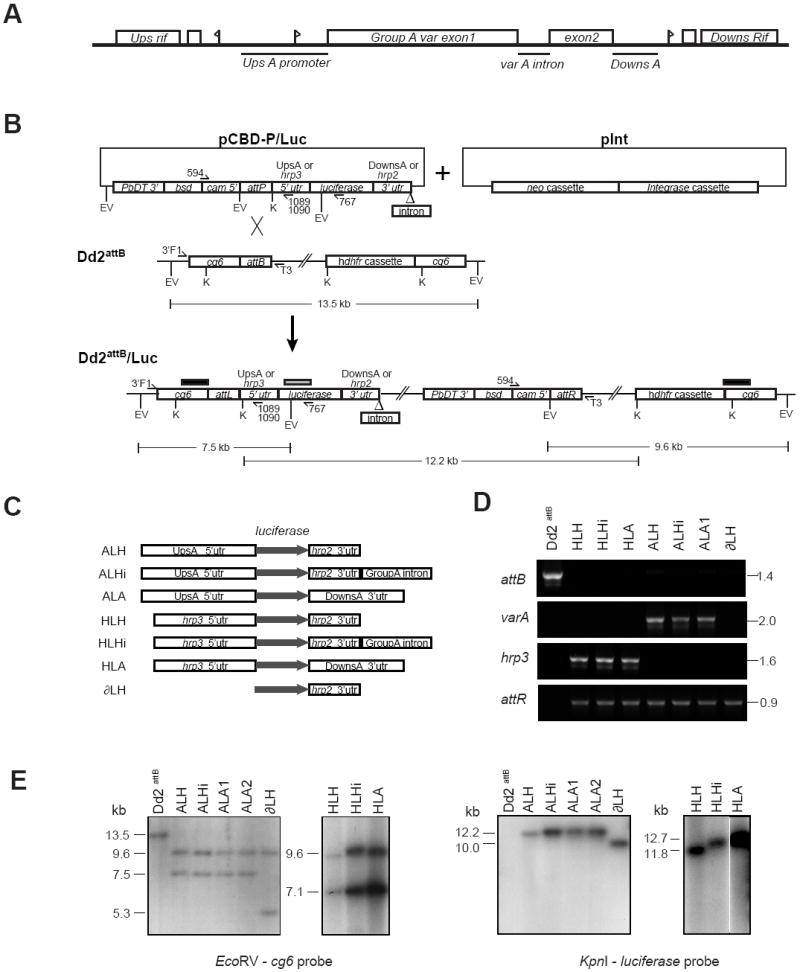
Generation and molecular characterization of luciferase reporter cassettes integrated into the Plasmodium falciparum Dd2attB parasite line. (A) Schematic of the genomic layout of the Group A var gene PFD1235w (previously named AL010226) in the P. falciparum 3D7 strain. Flags represent putative transcription start sites. Bars represent DNA regions chosen for analysis. The diagram is not drawn to scale. (B) Schematic of the integration of a generic luciferase reporter cassette-bearing pCBD-P plasmid (containing the attP element) into the attB locus of the Dd2attB parasite line. The attB docking site was engineered into the cg6 gene by homologous recombination (Nkrumah et al., 2006). Co-transfection of pCBD-P plasmids with the integrase-encoding plasmid pInt resulted in site-specific integration of the pCBD-P plasmid into the genome, flanked by attL and attR sequences. Upon recombination, the luciferase cassette resided downstream of the cg6 amino-terminal region, followed by the pBluescript plasmid backbone and the bsd selectable marker cassette placed in the opposite orientation to the reporter. Restriction digest fragment sizes refer to the integration of the pCBD-P-ALH plasmid. The positions of the probes used for Southern blot hybridization are indicated as bars above the Dd2attB/Luc locus (black bar: cg6 probe; gray bar: luciferase probe). EV, EcoRV; K, KpnI. (C) Experimental design of luciferase reporter assays, depicting the cassettes subcloned into pCBD-P and integrated into Dd2attB. The grey arrow indicates the luciferase open reading frame. UpsA, the 2.15 kb 5’ untranslated region (UTR) of PFD1235w; DownsA, the 1.4 kb 3’ UTR of PFD1235w; Group A intron, the 0.74 kb PFD1235w intron. (D) PCR reactions documenting the integration of luciferase reporters into Dd2attB. attB: Amplification of the Dd2attB parent strain and designated luciferase reporter lines with primers 3’F1 (cg6-specific) and T3 (pBS plasmid backbone). varA and hrp3: Results of PCR over the attL recombination site using the primer 3’F1 and either the UpsA-specific primer 1089 (varA) or the hrp3-specific primer 1090 (hrp3). attR: PCR amplification over the attR junction using primers 594 (specific to the cam promoter driving the bsd selectable marker in all pCBD-P plasmids) and T3. The parasite line ALA2 yielded similar results (data not shown). (E) Southern blot hybridization documented the insertion of single-copy luciferase cassettes into the attB site of the Dd2attB parasite. Genomic DNA was isolated from parasite lines and digested with EcoRV or KpnI. Hybridization with a cg6 probe revealed a 13.5 kb fragment in the EcoRV-digested Dd2attB genomic DNA that was absent in all second generation attP recombinants. This band was replaced in all lines by a 9.6 kb band corresponding to the attR junction and the human dhfr cassette, and a band of variable size (due to the different promoters tested) that corresponded to the attL junction and the 5’ portion of the luciferase cassette. The smaller band sizes are as follows: ALH, ALHi and ALA cassettes: 7.5 kb; HLH, HLHi and HLA cassettes: 7.1 kb; ∂LH cassette: 5.3 kb. Hybridization with a luciferase probe showed bands consisting of the entire inserted pCBD-P plasmid, the attR junction, and the plasmid backbone of the integrated attB locus. There was no evidence of plasmid retention or multi-copy insertion, which would have been apparent as bands ~3 kb smaller in size. Sizes were as follows: ALH: 12.2 kb; ALHi: 12.9 kb; ALA: 13.1 kb; ∂LH: 10.0 kb; HLH: 11.8 kb; HLHi: 12.6 kb; and HLA: 12.7 kb. The autoradiograph of the KpnI digested genomic DNA from the hrp3 containing lines HLH, HLHi and HLA was cropped to remove intervening lanes.
3.2. Generation of integrated single-copy luciferase cassettes within the Dd2attB parasite
To test the effect of Group A cis-elements on UpsA and hrp3 promoter expression, we took advantage of our recently published method of rapid site-specific integration (Nkrumah et al., 2006). This is based on mycobacteriophage Bxb1 integrase-mediated recombination between an attP element resident on a transfection plasmid and an attB site introduced into the P. falciparum genome. This method enabled us to efficiently generate isogenic parasite lines carrying integrated single-copy luciferase cassettes. This ensured that each parasite in a culture harbored the reporter cassette and produced a more reproducible luciferase signal compared with parasite lines that expressed luciferase from episomally replicating plasmids (data not shown).
To assess the ability of the UpsA promoter to drive expression of a luciferase reporter in the presence or absence of cis-acting elements, we co-transfected Dd2attB parasites with pCBD-P -based plasmids expressing various luciferase expression cassettes, together with the pInt plasmid. The pCBD-P plasmid contained a single attP element located between the blasticidin S-deaminase (bsd) selectable marker cassette and a multiple cloning site into which the luciferase cassettes were inserted (see Supplementary Fig. S1B). pInt expresses the Bxb1 mycobacteriophage serine integrase that catalyzes recombination between the episomal attP element and an attB site previously inserted into the P. falciparum cg6 locus of the Dd2attB parasite (Nkrumah et al., 2006). A representative schematic of pCBD-P plasmids and integrase-mediated recombination events is presented in Fig. 1B. In our pCBD-P plasmids, the luciferase reporter gene was under the control of the UpsA var promoter, the hrp3 promoter, or no promoter, in combination with either the hrp2 terminator (with or without the Group A intron) or the var DownsA terminator (Fig. 1C). Transfection of the various pCBD-P luciferase plasmids yielded the preclonal lines Dd2attB/ALH, Dd2attB/ALHi, Dd2attB/ALA1, Dd2attB/ALA2, Dd2attB/HLH, Dd2attB/HLHi, Dd2attB/HLA, and Dd2attB/∂LH (schematically represented in Fig. 1C).
Cloned lines (hereafter referred to only by their transgene cassette name) were obtained by limiting dilution and were confirmed by sequence-specific PCR screening (Fig. 1D). This documented the loss of the 1.4 kb Dd2attB parental product, and the acquisition of PCR bands specific for either the attL-UpsA or the attL-hrp3 promoter junctions in the UpsA or hrp3 promoter lines, respectively. All second-generation recombinant lines possessed the same 3’ attR junction, demonstrated by the 0.9 kb band that was PCR amplified from all lines. Southern blot hybridization of genomic DNA (Fig. 1E) confirmed the insertion into the attB-cg6 locus of a single copy of the plasmids harboring the luciferase cassettes. This insertion generated two smaller fragments upon EcoRV digestion, one 9.6 kb and the other variable in size depending upon the specific luciferase cassette (Fig. 1E). Given the presence of only one attB site in the Dd2 genome, any additional plasmid copies would retain an intact attP junction. PCR reactions to amplify the attP junction of pCBD-P plasmids did not yield any product, indicating the absence of plasmid concatamers or episomes. Southern blot banding patterns were consistent with single-copy integration and a lack of episomal plasmids, which would have been apparent as 7.2 to 10.2 kb bands (Fig. 1E). The sequence of experimental events involved in the generation of these clones and their culturing under different selection regimens is illustrated in Fig. 2A.
Fig. 2.
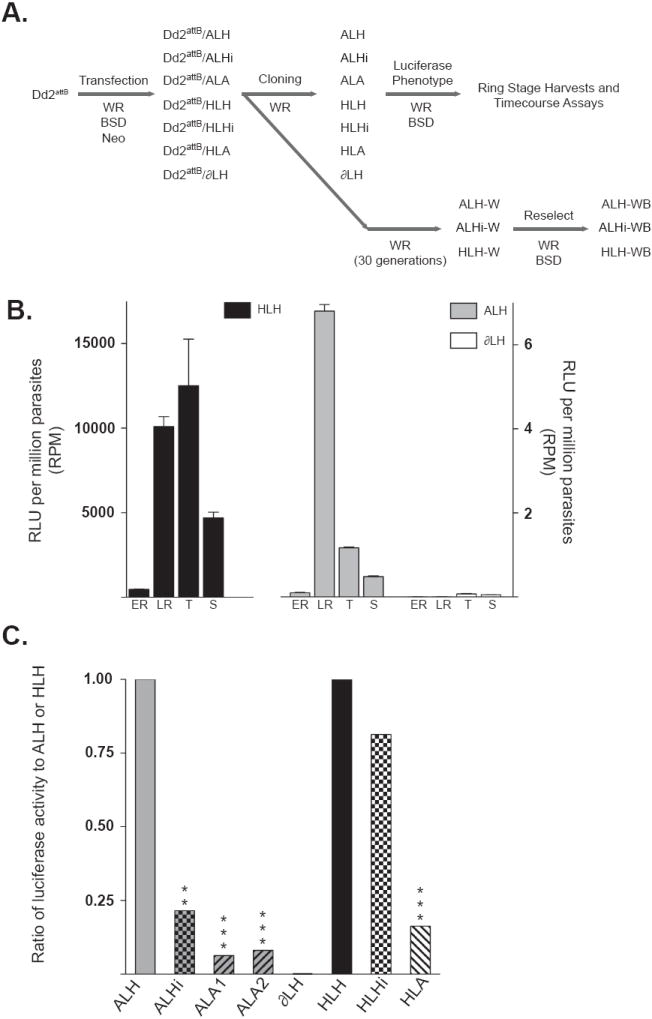
The DownsA terminator (corresponding to the downstream non-coding region from the Plasmodium falciparum Group A var gene PFD1235w) decreases luciferase expression of multiple promoters, while the intron from this same Group A gene represses the var promoter specifically. (A) Experimental design and order of experiments performed on reporter containing parasites. Preclonal parasite lines are denoted with the prefix “Dd2attB/”. (B) The stage-specific luciferase activity of integrated luciferase cassettes was assessed by harvesting parasites during early ring (eR), late ring (lR), trophozoite (T) and schizont (S) stages. Firefly luciferase activities of integrated reporter cassettes HLH (plotted on the left Y-axis), as well as ALH and ∂LH (plotted on the right Y-axis) are expressed as relative luciferase units (RLU) per million parasites (RPM). These data were obtained from a representative time course experiment performed in triplicate. (C) Ratios of expression from the luciferase cassettes, with significance derived from repeated measures ANOVA with Dunnett’s post-test (or a paired t-test in the case of ∂LH). ALH is defined as the control for UpsA promoter-containing lines and the promoter-less ∂LH line, HLH is defined as the control for the hrp3 promoter-containing lines. ALH and HLH are given ratios of one for comparative purposes. **, P value ≤ 0.01; ***, P value ≤ 0.001.
3.3. Integrated reporter cassettes are stage-specifically activated and exhibit promoter-dependent maximum activity levels
The stage-specificity of these promoters was confirmed by the analysis of luciferase activity levels at different developmental stages (Fig. 2B). These levels were calculated in RPM, to control for intra- and inter-assay differences in parasite densities. Assays showed that in the ALH line, the peak activity, ranging from < 1 to 10 RPM, occurred in the late ring stages, whereas in the HLH line the maximal luciferase activity, in the range of 1,000-10,000 RPM, was in the late ring and early trophozoite stages. Thus, while the HLH and ALH cassettes were consistently the most highly expressed during the ring stages, the UpsA-driven cassette had roughly 1,000–fold less activity. The ∂LH line exhibited little activity above background levels until late in the parasite life cycle, possibly as a result of read-through transcription from the upstream late-stage cg6 promoter. This activity was minimal during the periods of maximum ALH and HLH expression, yielding 0.1 RPM or less. This equated to background levels of luciferase activity. The ability of these promoters to control stage-specific timing of transcription within the cg6 locus confirmed the presence of the transcriptional motifs necessary for gene-specific transcriptional timing. The large discrepancy between the activities of the single-copy hrp3 and UpsA promoters suggests that the UpsA promoter is only minimally active in this context. This may be attributable to the silencing of this element by the upstream cg6 promoter. We note that these studies do not provide evidence of whether UpsA in this context is expressed at biological active levels (Fig. 1B).
3.4. The DownsA element significantly reduces luciferase activity, whereas the Group A var intron specifically represses the UpsA promoter element
Parasite lines generated in the Dd2attB background were doubly sorbitol synchronized and assayed for luciferase activity at late ring stages, approximately 16-20 hpi. While the DownsA element caused a marked decrease in UpsA activity in two separate clones (ALA1 and ALA2), the initial assessments of luciferase activity suggested that the Group A intron did not have a significant effect on UpsA promoter activity. This inference was obtained by comparison of the mean RPM values over nine individual assays (Table 1). Replacing the heterologous hrp2 3’ UTR with the DownsA terminator element significantly suppressed 94-95% of the ALH activity, from 4.7 RPM for ALH to 0.2 and 0.3 RPM for ALA1 and ALA2, respectively (P values <0.001). When the intron was placed downstream of the UpsA cassette, a comparison of mean values showed only a 20% decrease in luciferase activity, to 3.8 RPM (P value = 0.8). The promoter-less line ∂LH averaged 0.003 RPM (P value <0.001). The decrease in activity due to the DownsA terminator element was also evident when comparing lines expressing hrp3 promoter-driven cassettes with either the DownsA or hrp2 terminators (Table 1). In those lines, the HLH cassette was repressed 73% by the DownsA element (a mean RPM of 1,791 from HLA versus 6,601 from HLH, P value < 0.01), but was unaffected by the downstream placement of the Group A intron (a 6,601 RPM average value from the HLH cassette as compared with 7,636 RPM from the HLHi cassette, yielding 116% more activity; P value = 0.8).
Table 1.
Mean relative luciferase units per million Plasmodium falciparum parasites (RPM) and related statistics obtained from independent assays of parasite lines containing the integrated luciferase cassettes.
| Parasite linea | Mean RPM (n) | S.D. | Skewnessb | Kurtosisc | Geo. Meand | 95% CI of Geo. Mean | P valuee |
|---|---|---|---|---|---|---|---|
| ALH | 4.7 (9) | 5.86 | 0.865 | -0.972 | 2.07 | 0.687, 6.213 | n/a |
| ALHi | 3.8 (9) | 9.13 | 2.044 | 2.548 | 0.44 | 0.069, 2.831 | <0.05 |
| ALA1 | 0.2 (9) | 0.17 | 0.387 | -1.566 | 0.13 | 0.055, 0.311 | <0.01 |
| ALA2 | 0.3 (9) | 0.36 | 1.603 | 1.281 | 0.17 | 0.070, 0.401 | <0.001 |
| ∂LH | 0.003 (4) | 0.005 | 0.749 | -1.688 | 0.00004 | 3 × 10-8, 4 × 10-5 | 0.0145 |
| HLH | 6601 (5) | 5086 | 0.323 | -1.819 | 4917 | 1591, 15196 | n/a |
| HLHi | 7636 (5) | 8298 | 0.52 | -1.726 | 3993 | 720.5, 22133 | ns |
| HLA | 1791 (5) | 2036 | 0.413 | -1.975 | 802.3 | 112.7, 5713 | <0.01 |
All lines were derived from the P. falciparum Dd2attB strain.
Skewness describes the asymmetry of the distribution. Positive values have distributions asymmetrically skewed to the right (higher values), negative values are skewed to the left (lower values).
Kurtosis quantifies the shape of the distribution as Gaussian or non-Gaussian. Gaussian distributions have 0 kurtosis, distributions with more values in the center have negative kurtosis, and those with more values in the tails have positive kurtosis.
The geometric mean is the antilog of the mean of the log-transformed values, which allows comparison of the transformed mean values in the original units. The variance of the geometric mean is captured by the 95% confidence interval (CI).
P values were calculated using repeated measures ANOVA such that the UpsA promoter lines constituted one group (P value= 0.003) and the hrp3 promoter lines constituted a second group (P value= 0.0056). The Dunnett’s post-test was used for between-line comparisons. The P value for the ∂LH line was derived from a paired t test using matched ALH values. n/a, not applicable.
Closer inspection of the ALHi dataset, however, revealed that the intron did act to repress UpsA activity in the majority of the assays. Calculating the mean without log transformation had masked this effect. With the ALHi dataset, computation of the skewness (that quantifies the symmetry of the distribution, where perfect symmetry = 0) and kurtosis (that quantifies how closely the shape of the distribution is Gaussian and therefore = 0) revealed scores of 2.0 and 2.6, respectively, reflecting more values in the right-hand tail of the distribution (Table 1). To correct for non-Gaussian distributions, we log-transformed the values, and calculated the geometric means and 95% confidence intervals. After log10 transformation, the repressive effect of the Group A intron on the UpsA promoter was evident (Table 1).
Evaluation of the transformed data by repeated measures ANOVA showed significant differences in the means of the log-transformed RPM (log(RPM)) of both the UpsA promoter-containing lines (P value < 0.01) and the hrp3 promoter-containing lines (P value < 0.01). We defined the ALH and HLH lines as controls to allow for Dunnett’s post-test analysis and observed significant differences between the RPM obtained from ALH and either ALHi, ALA1 or ALA2. For the hrp3-driven group, we found significance only for the effect of the DownsA terminator element on the hrp3 promoter element. These data suggested that repression mediated by the intron was specific to the UpsA var promoter, whereas the DownsA element imposed a repressive effect that was less discriminatory.
Conversion of the ratio of the log values to the antilog yielded an expression ratio for ALHi/ALH of 0.22, documenting a 78% decrease in UpsA-driven luciferase expression when the Group A intron was placed downstream (Fig. 2C, see Table 2 for calculations and confidence interval values). The HLHi/HLH ratio of 0.81 showed that the Group A intron had a non-significant reduction in expression from the hrp3 promoter (less than 20%). Thus the Group A intron resulted in significantly decreased expression of the UpsA promoter within a chromosomal context. This effect was not seen when the intron was downstream of the hrp3 promoter and was therefore specific to the var promoter.
Table 2.
Means of log-transformed relative luciferase units per million Plasmodium falciparum parasites (log(RPM)) and related statistics.
| Parasite line | Mean of log(RPM)a | Difference of the mean log(RPM) valuesb | 95% C.I. of Differencec | Reciprocal ratio of expressiond | Reciprocal 95% C.I. of Ratiod |
|---|---|---|---|---|---|
| ALH | 0.314 | 0 | n/a | 1 | n/a |
| ALHi | -0.353 | 0.667 | 0.0315, 1.303 | 0.215 | 0.050, 0.93 |
| ALA1 | -0.884 | 1.198 | 0.563, 1.834 | 0.063 | 0.015, 0.27 |
| ALA2 | -0.775 | 1.089 | 0.454, 1.725 | 0.081 | 0.019, 0.35 |
| ∂LH | -4.426 | 4.633 | 1.750, 7.517 | 0.00002 | 3 × 10-8, 0.018 |
| HLH | 3.692 | 0 | n/a | 1 | n/a |
| HLHi | 3.601 | 0.090 | -0.410, 0.591 | 0.813 | 0.26, 2.56 |
| HLA | 2.904 | 0.787 | 0.287, 1.288 | 0.163 | 0.051, 0.51 |
Log10 transformation of the RPM values corrects for non-Gaussian distributions and permits evaluation of the fold-difference in expression.
Values were calculated by subtracting the mean of the log-transformed RPM values of each parasite line from the control line mean value. Mean ALHi, ALA, and ∂LH log(RPM) values were subtracted from the mean ALH log(RPM). Mean HLHi and HLA log(RPM) values were subtracted from the mean HLH log(RPM).
The 95% confidence interval (C.I.) of the difference in mean log(RPM) values were obtained from repeated measures ANOVA with Dunnett’s post tests performed separately on the UpsA-and the hrp3-promoter containing lines. ALH and HLH were defined as control lines for the respective groups. The 95% C.I. of the ∂LH line was obtained by a paired t test comparison of log-transformed ∂LH values with the matched log-transformed ALH values.
The ratio (or fold difference) of luciferase expression was the antilog of the difference in mean log(RPM). This was calculated per the formula: log(control) - log(treated) = log (control/treated). The reciprocal of the antilog was presented to show expression as the fold reduction from ALH or HLH.
The 95% C.I. of the ratio of expression versus ALH or HLH was calculated from the antilog of the 95% C.I. of the difference in the mean log(RPM) values. The reciprocal value was presented to illustrate the fold reduction in expression. n/a, not applicable.
In contrast, the repression effected by the DownsA element was not specific to the ALH cassette, as the element led to a significant reduction in activity produced by both the UpsA promoter and the hrp3 promoter elements. ALA/ALH ratios of 0.06 and 0.08 were obtained for ALA1 and ALA2, respectively (Fig. 2C). These ratios were very similar to those obtained prior to log transformation, which was as expected given the more Gaussian distribution of these datasets. The effect of the DownsA element on the hrp3 activity was not as severe, as the HLA/HLH ratio of expression was 0.16. Nevertheless, this resulted in a > 80% reduction in activity.
3.5. The DownsA region and the Group A var intron each reduce the total accumulated activity of UpsA luciferase and delay the time to maximal expression
The studies described above characterized the activity levels of luciferase enzyme cassettes at a single point during the IDC. The utility of this approach was hampered, however, by variability in relative activity levels between lines and within the same line in different assays. We postulated that this heterogeneity might be related to the stage specificity of the UpsA promoter. As a more comprehensive approach, we generated highly synchronized ALH, ALHi, ALA1 and ∂LH cultures and harvested time points at approximately 4, 10, 16, 21, 28, 33 and 38 h pi. These samples were subjected to luciferase assays to assess the impact of the Group A intron and DownsA terminator cis-elements on the level and timing of UpsA expression. Giemsa thin smears of each line were simultaneously prepared to monitor the developmental stages of the parasites at each time point (Fig. 3A). All lines consisted of > 95% ring stages during the first 24 h and the closely matched transitions into trophozoite and schizont stages during the second day indicated a high degree of synchronicity. Additionally, we followed the level and timing of the luciferase transcript to assess the relationship between transcript and activity levels.
Fig. 3.
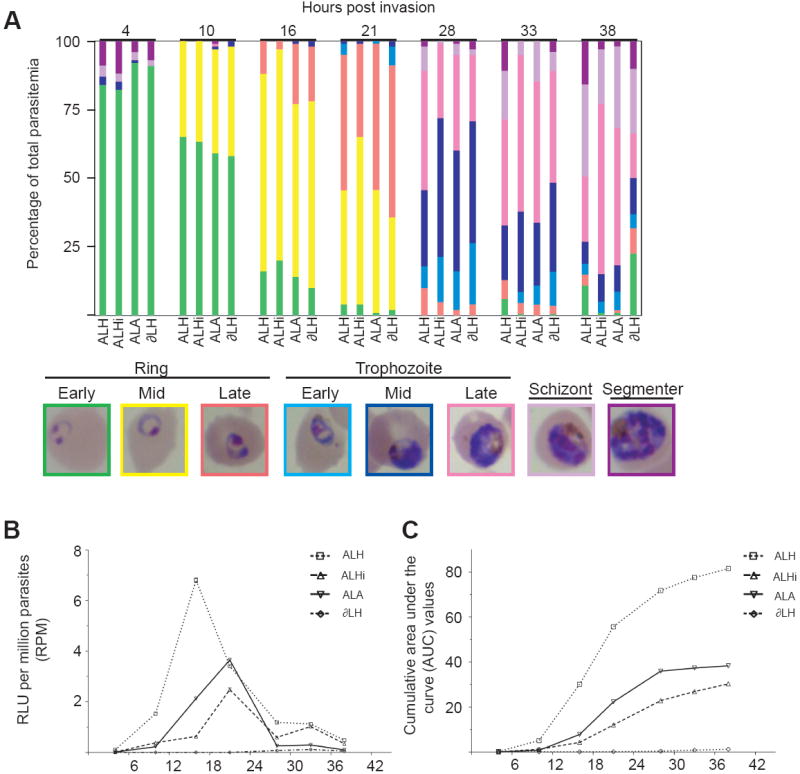
Regulatory elements from the Plasmodium falciparum Group A var gene PFD1235w repress cumulative luciferase activity from the UpsA promoter and affect the timing of maximal transcription. (A) Histogram of the percentages of each developmental stage obtained at the harvested time points. Representative Giemsa-stained images are presented, with the border color corresponding to the bar color on the histogram. Parasite lines were subjected to repeat rounds of double sorbitol synchronization, with the initial round performed just after blood cell invasion and a second one performed 12-15 h later. Replicate flasks were prepared to minimize parasite culture handling. (B) relative luciferase units (RLU) per million parasites (RPM) obtained from the time course experiment. (C) From this experiment, the cumulative area under the curve values were derived using a trapezoidal formula.
We observed that the luciferase activity produced by the UpsA var promoter in the ALH line was confined to 10-28 h pi, with the peak of RPM expression occurring at 16 h pi (Fig. 3B). Other groups have also documented peak var expression within the first 4-18 h pi (Kyes et al., 2003; Voss et al., 2003; Dahlback et al., 2007). At the 16 h pi time point, luciferase activity in the ALH line was found to be very high compared with the ALHi and ALA1 lines. The intronic and terminator cis-elements reduced UpsA activity by 90% and 70%, respectively, consistent with the reductions documented by the individual time point harvests. At 21 h pi, the activity of the ALH line dropped precipitously, while the ALHi and ALA1 lines reached their maximum intensity, approaching the activity of ALH (Fig. 3B).
The cumulative RPM of each line was then examined over the entire IDC by calculating the AUC, enabling an assessment of total UpsA expression levels (Fig. 3C). This led us to observe that the cumulative effects of the cis-elements on the UpsA promoter were more moderate than was apparent during maximum ALH expression, amounting to a 50% decrease in total accumulated luciferase activity when the DownsA terminator was utilized, and a 70% decrease in the total accumulated luciferase activity in the presence of the Group A intron. In all lines, luciferase expression tapered off after 28 h pi, but ALH and ALHi continued to see incremental cumulative increases up to 38 h pi.
The repressive effects of the Group A cis-elements appeared to be due primarily to the blunting of the maximal expression obtainable from the ALH luciferase cassette during 10-16 h pi, a critical stage-specific window of expression for the UpsA promoter. The developmental stages evident in these lines at these two time points appeared very similar, consisting of > 95% ring stages in all lines, and with similar counts of early-, mid- and late-ring forms (Fig. 3A). Despite this apparent similarity, close examination of the developmental stages and the luciferase activities over time showed that the actual IDC times (and the relative expression levels of the different luciferase cassettes) were notably different. Thus, assays performed on parasite IDCs outside the critical expression time-frame would misinterpret the effects of the cis-elements on UpsA expression, and would be under-representative of the true differences in activity levels. This would explain why some of the single time point assays failed to identify a repressive effect of the Group A intron (see above).
From our tightly synchronized time points, we used quantitative real-time PCR with cDNA preparations to assess the numbers of transcripts produced by the UpsA promoter and the downstream bsd selectable marker. This showed that each UpsA-containing line had a transcription profile similar to the one for enzyme activity. For ALH, UpsA transcription peaked at 16 h pi, whereas for ALHi and ALA1 maximum transcription was at 16-21 h pi (Fig. 4A). Again, these peaks of expression nearly overlapped with ALH at 21 h pi. These expression patterns appeared most similar to UpsC var promoter expression, which was maximally expressed 8-18 h pi but showed evidence of expression from 0-26 h pi (Voss et al., 2003).
Fig. 4.
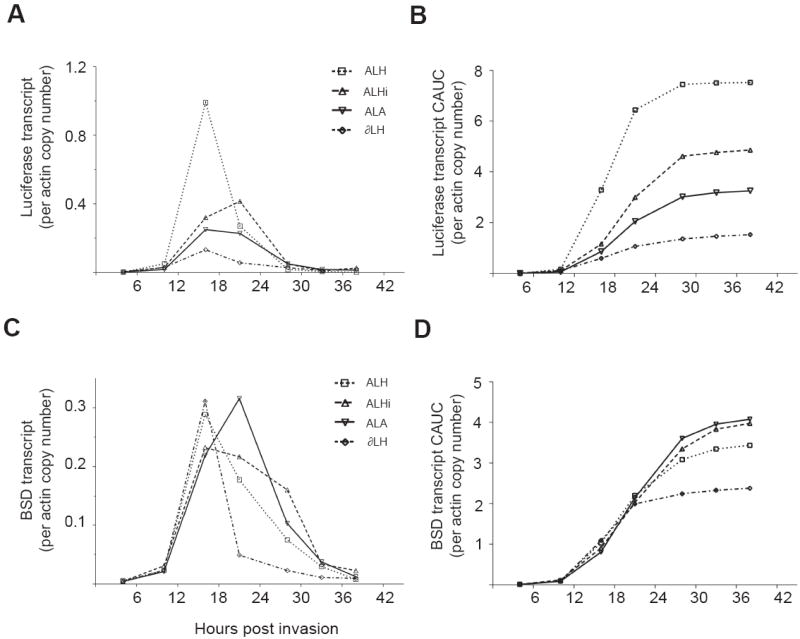
Regulatory elements from the Plasmodium falciparum Group A var gene PFD1235w repress cumulative UpsA–produced transcript levels. UpsA is the upstream non-coding region from the Group A var gene PFD1235w. (A-D) Luciferase (A) and bsd (C) transcript values assessed by quantitative real-time PCR with cDNA preparations. These values were normalized to the copy number of actin transcripts, a widely used reference gene. The cumulative area under the curve data are presented for luciferase in (B) and bsd in (D).
Our analysis showed that for ALH, the peak of transcription corresponded exactly to the peak in luciferase activity, with a sharper decrease in RNA levels than enzyme activity. For ALHi, transcripts accumulated to much greater levels before any notable change in luciferase activity occurred, so that there was a time delay between generation of the transcript and the luciferase activity. At 16 h pi there was a marked increase in luciferase transcript but the increase in luciferase activity was delayed until 21 h pi, at which time the ALHi luciferase transcript and associated activity attained maximum levels. The ALA1 luciferase transcript displayed a similar pattern of expression, but the activity was more closely correlated with the increase in transcript levels. Compared with ALH, we observed that accumulated UpsA-driven luciferase transcripts in the ALHi and ALA1 lines were reduced by 35% and 54% (Fig. 4B), a finding that recalled the 50–70% reduction in UpsA-driven luciferase activities in these lines (see above).
We then measured the level of bsd transcript to assess the impact of the UpsA promoter lines on the downstream marker (Fig. 4C). This revealed no significant difference in the overall level of transcript produced between the different UpsA lines when these were maintained under continuous BSD pressure, as assessed by AUC measurements (Fig. 4D). In this system, transcription of the bsd marker peaked at 16 to 21 h pi. This marker was driven by a 0.6 kb calmodulin promoter, shorter than the 1.0 kb calmodulin promoter previously documented to be active later in the IDC (Voss et al., 2006). This altered timing was reminiscent of similar findings that resulted from truncations of the hrp3 5’ UTR (Lopez-Estrano et al., 2007), and suggested that motifs necessary for stage-specific expression might be contained in the missing 0.4 kb calmodulin sequence. We note that our expression data were normalized against actin, a widely used reference gene that is highly active in later parasite stages (Bozdech et al., 2003). A control gene with a more uniform profile of transcriptional activity might not have produced such an apparent shift in calmodulin promoter timing of expression.
To further investigate the differences in activity and transcript levels between the lines, we applied PCA and PLS methods to generate statistical models of the multivariate data obtained from the time course experiment. Traditional methods of statistical analysis (such as ANOVA and Student’s t-test) treat each sample independently and assume statistical independence. For this reason they could not be applied, as there is both strong autocorrelation within each dependent variable (i.e. the current time value depended upon the previous time value) as well as possible correlation with the parasite line (i.e. between luciferase transcript and activity levels). PCA analysis (Supplementary Fig. S3A) summarized the related variation in the dataset and was used to screen for outliers, whereas PLS correlated the variation into related clusters by linear multivariate modeling, and statistically assessed the relative contributions of each variable to the model. These techniques yielded graphical depictions of the correlations between time course values and the variables of staging and reporter cassette type. These analyses are presented in Supplementary Data S1 and Supplementary Fig. S3B-D. A similar method of circular PCA has most recently been applied to the 48-h P. falciparum IDC transcriptome (Scholz and Fraunholz, 2008).
3.6. An expression cassette positioned in proximity to the UpsA var promoter can be reversibly repressed and repression correlates with the silencing of UpsA activity
As our experiments progressed, we observed that freshly thawed UpsA-promoter integrated cell lines, which had been frozen at the time of cloning, displayed a marked sensitivity to media supplemented with 2.0 μg/mL of BSD (a concentration typical of routine parasite culture). However, thawed lines containing hrp3-promoter driven luciferase cassettes were unaffected by the addition of BSD. Cell lines frozen during the cloning process had been grown for 30 generations without BSD drug pressure (Fig. 2A), in order to minimize the retention of non-integrated attP plasmids in the transgenic parasites. Continuous WR99210 pressure was maintained to prevent spontaneous excision of the parental attB locus by homologous recombination. We have previously documented that relieving drug pressure for 10 generations had no effect on the stability of the attB × attP locus, or on the growth rate of the parasites once drug pressure was reapplied (Nkrumah et al., 2006). The lag time exhibited in the growth profiles of these cultures suggested that the UpsA containing locus was subject to increased mechanisms of silencing specific to the var promoter. When the culture was released from selection pressure, silencing spread from the UpsA promoter and impacted expression from the bsd selectable marker cassette located downstream (see Fig. 1B for schematic). The spread of silencing from UpsB and UpsC var promoters has previously been documented when the parasites are grown in media permissive for var gene silencing (Voss et al., 2006, 2007). Here, the development of susceptibility to BSD pressure suggested that the UpsA promoter was capable of a similar spread of silencing.
To investigate this further, we assessed the growth curves of several clones of ALH, ALHi and HLH that had been grown in WR99210-containing medium and were reintroduced to BSD drug pressure (Fig. 5A). We found that the UpsA-containing cell lines ALH and ALHi required a minimum of eight generations to attain greater than 5% growth. In the case of one ALHi clone, parasitemia never surpassed > 5% even after 18 generations. The HLH cultures were unaffected by the addition of BSD pressure and showed growth profiles similar to the cultures maintained in WR99210 alone. All UpsA-containing parasite lines cultured in medium without BSD surpassed a parasitemia greater than 5% within two to four generations. Thus, cell lines containing the UpsA promoter required a greater length of time to attain optimal growth rates after the reapplication of BSD, a potential consequence of silencing mechanisms that spread from the var promoter to affect neighboring loci downstream.
Fig. 5.
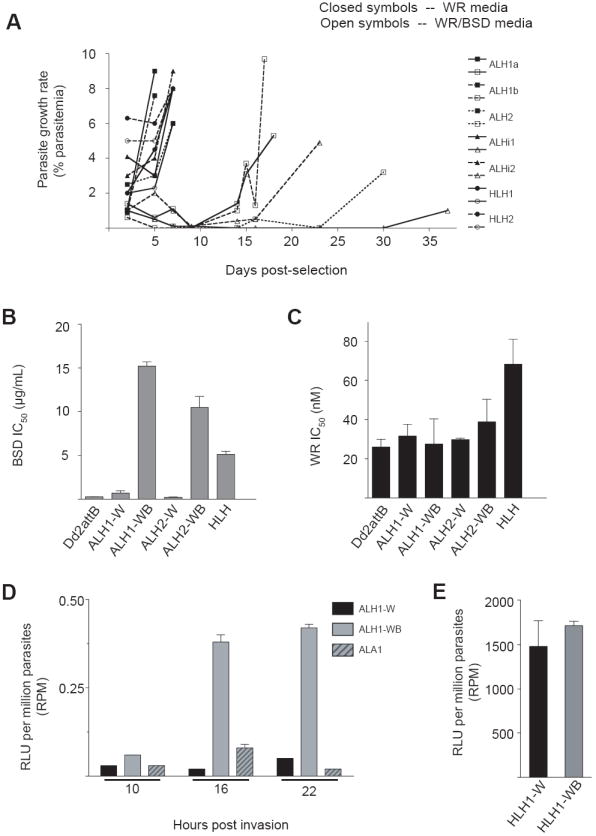
The Plasmodium falciparum UpsA promoter (from the PFD1235w var gene) is associated with epigenetic changes in the surrounding locus and affects the expression of surrounding elements. (A) Growth curves of lines cultured in the presence of the selection agents WR99210 and BSD (open symbols) or WR99210 alone (closed symbols). Parasite cultures were seeded into replicate wells at 1% parasitemia, all with 2.5 nM WR99210 but half with 2 μg/mL BSD. HLH transgenes containing the hrp3 promoter were not affected by the application of BSD pressure, while the growth of UpsA promoter-containing lines was delayed. Prior to application of drug, normal parasite growth was verified by attainment of parasitemia above 5% in complete medium. (B, C) In vitro SYBR Green I incorporation assays were performed against BSD (B) or WR99210 (C) using the ALH parasite lines selected in the presence of WR99210 and BSD (ALH-WB) or WR99210 alone (ALH-W). As a control, we included the HLH parasite line cultured in medium containing both WR99210 and BSD. IC50 values (shown as mean ± S.E.M., calculated from six independent experiments) were derived by regression analysis. (D-E) Luciferase activities of BSD pressured versus non-pressured cell populations, measured in relative luciferase units per million parasites. (D) Time-course of the luciferase activities of BSD sensitive and resistant populations of ALH parasites. As a control, we included the ALA1 parasite line that was cultured in the presence of WR99210 and BSD. (E) Luciferase activity of HLH cultures grown in the presence (HLH1-WB) or absence (ALA1-W) of BSD, harvested during late ring stages.
To quantify the level of BSD resistance in the sensitive (ALH-W) versus the resistant (ALH-WB) ALH clones, we performed 72-h drug assays using SYBR Green I on two separate thawed cultures of the ALH lines (ALH1 and ALH2), and compared this with the level of BSD resistance documented in BSD-resistant UpsA- and hrp3-containing lines. Cultures of ALH grown under continuous WR99210 pressure but without BSD (ALH1-W and ALH2-W) showed a reversion of their resistance to BSD, yielding IC50 values of 0.7 and 0.2 μg/mL, respectively, comparable to an IC50 value of 0.3 μg/mL obtained from the Dd2attB parental line (which lacks the bsd selectable marker; Fig. 5B). These IC50 values were greatly reduced in comparison with the UpsA-promoter integrant lines selected for growth in WR99210 plus BSD (ALH1-WB and ALH2-WB), which yielded IC50 values greater than 10 μg/mL for BSD. PCR analyses confirmed that the attP plasmid was still retained as an integrated element despite the absence of BSD drug pressure (data not shown). These data suggested that the UpsA promoter nucleated epigenetic silencing that spread downstream from the var promoter, reducing expression from the bsd selectable marker when the BSD selection pressure was removed
The repression of the BSD marker was completely reversed upon reacquisition of BSD resistance, however, as the IC50 values obtained from the ALH1-WB and ALH2-WB lines slightly exceeded the BSD resistance levels exhibited by the HLH line grown in WR99210-BSD media. All UpsA-promoter integrant lines showed similar levels of resistance once the culture reestablished typical growth under BSD pressure. The repression of the bsd selectable marker did not, however, extend to the human dhfr selectable marker located downstream within the attB × attP locus (see Fig. 1B). All ALH lines had similar profiles of resistance to WR99210, as shown by drug assays performed in parallel, whether or not they were resistant to BSD. These IC50 values were comparable to that obtained from the WR99210-resistant Dd2attB line (Fig. 5C), which was as expected given their continuous culture under WR99210 selection.
To address the activation status of the reporter gene in the absence of BSD pressure, parasite lines ALH1-W, ALH1-WB and ALA1 were synchronized and harvested for luciferase activity at approximately 10, 16 and 22 h pi as described above in the timecourse study (Fig. 5D). ALH parasites grown in the absence of BSD (ALH1-W) demonstrated a 95% reduction in luciferase expression at 16 h pi, and an 88% reduction in expression at 22 h pi, compared with activity obtained from ALH1-WB parasites. Activity from the ALA reporter construct exhibited levels of reduction from ALH1-WB similar to those obtained in the previous timecourse experiment (79% and 95% reductions at 16 and 22 h pi, respectively). HLH-W and HLH-WB parasites harvested during the same time period showed no demonstrable difference in activity levels over several sample time points (Fig. 5E).
Therefore UpsA promoter-containing parasites possessed a tendency towards progressively decreased expression, but were nonetheless responsive to neighboring loci. When the cell line was cultured in the presence of WR99210 and BSD drugs, parasites were under constant pressure to maintain both the dhfr and bsd selectable markers in an “on” state. When the parasite line was grown in the absence of BSD, the “on” activation status of the downstream bsd cassette was no longer a criterion for maintaining survival in the WR99210 selective medium and the locus became susceptible to inactivation by silencing from the UpsA promoter upstream. Those parasites that exhibited enhanced silencing of their reporter cassettes and their bsd selectable marker cassette were able to survive in the WR99210 media, and the activity levels of both the luciferase cassette and the selectable marker cassette were reduced. With the reapplication of BSD pressure and given sufficient time for the selection of a population of cells with an active selectable marker locus, however, these lines exhibited concomitant restoration of the upstream luciferase activity.
4. Discussion
In this study, we examined the transcriptional status of the highly conserved Group A var promoter and determined whether Group A cis-elements could regulate the expression of this P. falciparum var gene subfamily. Our initial studies of reporter activity documented minimal activity levels in association with single-copy UspA-driven cassettes, suggesting that the UpsA promoter in this context is largely inactive. Our multivariate analysis of highly synchronized, transfected lines provides evidence, however, that var4-like Group A family members are subject to regulation by the same genetic element, i.e. the intron, that has been shown to suppress the expression of Group B and C var genes (Deitsch et al., 2001; Dzikowski et al., 2006; Frank et al., 2006). We also report an additional and potent mechanism of down-regulation in the DownsA element that constitutes the Group A var 3’ UTR. Finally, the ability of transgenic UpsA-containing parasites to exhibit reversible repression is highly reminiscent of endogenous var gene silencing and activation mechanisms, and suggests that this subfamily is also capable of nucleating a suppressive chromatin environment that can impact surrounding loci.
The process by which the parasite comes to activate a singular var gene appears to proceed via a stochastic mechanism in which all var family members are capable of activation. However, studies have shown that the var gene subfamilies do not exhibit identical expression profiles. While studies of var gene silencing have largely referred to Group A, B, and C genes as a whole and assume their equivalence, in vitro surveys of var gene activation and silencing have found that Group A var genes are preferentially silenced in unselected cultures, with an apparently faster “off” rate and a slower “on” rate than other var groups (Frank et al., 2007; Mok et al., 2008). Those findings are corroborated in a study that examined changes in var gene transcription during the adaptation of patient isolates to in vitro culture. This study observed that Group A and E genes had the fastest decline in transcription (Peters et al., 2007). This was in marked contrast to assessments of var gene profiles in clinical isolates, in which Group A genes were frequently observed to be highly represented (Bull et al., 2005; Kyriacou et al., 2006), or in cultured parasites selected in vitro for particular cytoadherent phenotypes (Jensen et al., 2004; Duffy et al., 2005). Thus, there appears to be a strong pressure to maintain Group A genes in a silent state in the absence of phenotypic selection, due either to more layers of silencing being applied to this subfamily (a higher “off” rate) or to less frequent activation (a lower “on” rate).
Differences between var subfamilies are also apparent in their use of epigenetic markers of gene activation and silencing. Silent UpsB and UpsE var promoters have been found to physically associate with the NAD+ dependent histone deacetylase PfSir2p, whereas UpsC promoters do not (Freitas-Junior et al., 2005). In that study, UpsA promoters were not assessed. However, a concurrent study reported that these promoters could be preferentially activated and disregulated following disruption of PfSir2p (Duraisingh et al., 2005). Therefore, while the var subfamilies are not subject to identical mechanisms of silencing, they nonetheless seem to adhere to the same rules of mutual exclusion that allow singular var gene expression and ensure the production and transport of a single PfEMP1 antigen.
To address questions of UpsA-induced mutual exclusion, we attempted to express the bsd marker under the control of our UpsA element. Despite numerous transfections into different strains, we were unable to activate this marker or a separate selectable marker located elsewhere on the same plasmid. Additionally, single-copy integrants of the pCBD-P-ALH plasmid in the 3D7attB line were successfully obtained only when parasites were transfected and selected without BSD drug pressure, while pCBD-P-ALA integrated cells were never recovered in 3D7attB. This 3D7 strain is more sensitive than Dd2 to BSD (Mamoun et al., 1999; and data not shown), and seems to require more copies of the bsd selectable marker when the UpsA var silencing element is included on the plasmid. Taken together, these data provide evidence that the UpsA promoter is itself repressed and can nucleate the spread of repression to neighboring genes in an episomal or integrated context, as has been shown for UpsB and UpsC genes (Voss et al., 2006; Voss et al., 2007).
The low level of UpsA activity in the ALH line suggests that this promoter is silenced in the vast majority of the individual cells Nonetheless, our studies identified a clear repression by the intron on expression from the UpsA promoter in this chromosomal configuration. This might be explained, if a higher percentage of cells were exhibiting activation in the ALH culture than in the ALHi culture. Hence, in these two cultures, heterogeneous populations of UpsA activation might exist, which in the case of the ALHi culture might be prevented from attaining the same degree of activation as the ALH culture. This is reminiscent of the repression exhibited by the Group B intron on the UpsB and UpsC promoters, which were themselves largely inactive but became more repressed in the presence of the intron (Voss et al., 2006, 2007). In our experiments, the Group A intron repressed the UpsA promoter specifically, in a manner analogous to previous reports of Group B intron-mediated silencing of UpsB and UpsC var promoters (Dzikowski et al., 2006, 2007; Frank et al., 2006). This supports the “promoter-pairing” hypothesis of var gene regulation, in so far as the activity levels obtained from the UpsA promoter integrated at the cg6 locus were further reduced upon placement of the Group A intron downstream of the cassette. It is possible that the upstream cg6 promoter functioned as the paired promoter for the transgenic UpsA promoter and as such allowed the UpsA promoter to enter into a silent “paired” state. Yet it is abundantly clear that the intron further reduced the expression of this reporter cassette, suggesting that the UpsA promoter in the ALH culture is in some state of minimal activation. Whether this repressive effect transitions the UpsA promoter from an active to a silent state or imposes additional silencing on already reduced expression requires further testing, especially in light of the upstream cg6 promoter present in all Dd2attB transgenic parasites. In the absence of quantitative analysis of activity levels on a cell-by-cell basis, or a means to select a population of cells that is primarily “on” versus “off”, this theory remains speculative.
In contrast to the specific repression of activity exerted by the Group A intron, the DownsA sequence reduced the activity of either promoter tested, drawing into some question the relevance of this effect on Group A var gene regulation. Sequence comparisons of the DownsA elements reveal them to be highly conserved, despite minimal homology within the coding regions of Group A var genes. This conservation might be necessary to allow the correct alignment of Group A var open reading frames for protein domain shuffling during DNA recombination, and potentially to maintain sequence motifs that contribute to the regulation of Group A var gene expression. A separate mechanism of regulating malarial gene expression by 3’ UTRs has previously been documented in the sexual stages of the rodent parasite Plasmodium berghei (Mair et al., 2006). In these stages, translational repression occurs as a result of the interaction between mRNA species and the RNA helicase DOZI, thus preventing nascent polypeptide synthesis prior to zygote formation in the mosquito. In our system, the amounts of RNA produced by the ALA transgene correlated well with luciferase activity levels, suggesting a lack of retention of untranslated mRNA. Indeed, earlier studies suggest that var gene expression is regulated primarily at the level of transcription initiation (Scherf et al., 1998; Kyes et al., 2007; Schieck et al., 2007). One possibility to be tested is that the DownsA elements might regulate transcript levels by affecting mRNA stability, as has been found with conserved 3’ UTR elements in the Plasmodium knowlesi variant antigen SICAvar genes (Corredor et al., 2004).
The finding that DownsA non-specifically reduces protein expression suggests that this sequence destabilizes the mRNA independent of promoter context, but the striking homology exhibited amongst the 3’ flanking regions of Group A genes suggests a conservation of this repressive mechanism. RNA destabilization would result in decreased luciferase expression and transcript levels, as was found in this study. Intriguingly, comparison of the AT content of the 3’ UTRs of Group A, B, and C var genes reveals that the DownsA sequences consist of 89% AT, with T > A. The DownsB and several Group C 3’ UTRs consist of 70% AT with A > T. Additionally, while the DownsB and Group C 3’ UTRs possess a single putative poly(A) site within the 1,000 bp nearest the stop codon, the DownsA element utilized in this study possessed numerous putative poly(A) sites throughout the region of high homology, suggesting possible differences amongst these Groups in the degree of var mRNA stability.
Recent reports have documented the enrichment of a tri-methylation mark on the lysine at position 9 of histone H3 bound to silent var promoters. This mark extends from the var promoter into the coding region of the silenced gene and is present throughout the life cycle (Chookajorn et al., 2007; Lopez-Rubio et al., 2007). We performed preliminary chromatin immunoprecipitation experiments on parasite extracts of the ALHi and ALA1 cell lines, using antibodies to the H3K9Me3 molecular mark (Supplementary Fig. S4A-B). Both ALHi and ALA1 parasites showed a modest (two-fold) enrichment of the methylation mark at the luciferase gene compared to a non-specific control antibody, while a greater than two-fold enrichment of H3K9Me3 at the bsd gene was found within the ALHi parasite. Interestingly, the ALA1 parasites did not show enrichment of this marking at the downstream selectable marker. Furthermore, ALA parasites did not exhibit increased sensitivity to BSD when grown in the absence of this selection agent for 38 generations. An unanswered question in var gene research is the ability of the parasite to selectively activate one var gene while maintaining silencing on a second var gene located only several kilobases away. Although preliminary, our results suggest that the highly conserved DownsA region might contain an insulator that is capable of inhibiting the spread of H3K9Me3 and as such would restrict the silencing to one region.
The PfEMP1-encoding var genes play a key role in immune evasion, by permitting the presentation of different antigenic proteins with the potential for novel cytoadherent capabilities through mutually exclusive var gene activation. Our study illustrates that the var4-like Group A genes are minimally active in an integrated context, and are subject to intron-mediated repression, as has been shown for other var subfamilies. In addition, our studies suggest that the Group A terminator DownsA can act as a potent repressor of expression. These results implicate multiple layers of repression in the silencing of the Group A var subfamily, contributing to their highly silenced state. The ability of the UpsA promoter to nucleate silencing mirrors the abilities of other var gene Groups. Differences between var Group activation and switch rates though, mediated by subtly different repressive mechanisms, might in turn affect the duration and severity of infection and the development of immunity. Uncovering the mechanisms regulating var gene activation might assist the development of therapeutic modalities tailored to specific states of severe malaria.
Supplementary Material
Acknowledgments
We thank Dr. Marcus Lee for constructive comments on the manuscript. R.A.M. gratefully acknowledges support from the Cellular and Molecular Biology Training Grant (NIGMS T32 07491; PI. Dr. Pamela Stanley) and the Albert Einstein College of Medicine Medical Scientist Training Program (Director: Dr. Myles Akabas).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Andrews KT, Pirrit LA, Przyborski JM, Sanchez CP, Sterkers Y, Ricken S, Wickert H, Lepolard C, Avril M, Scherf A, Gysin J, Lanzer M. Recovery of adhesion to chondroitin-4-sulphate in Plasmodium falciparum varCSA disruption mutants by antigenically similar PfEMP1 variants. Mol Microbiol. 2003;49:655–669. doi: 10.1046/j.1365-2958.2003.03595.x. [DOI] [PubMed] [Google Scholar]
- Barnwell JW. Cytoadherence and sequestration in falciparum malaria. Exp Parasitol. 1989;69:407–412. doi: 10.1016/0014-4894(89)90190-2. [DOI] [PubMed] [Google Scholar]
- Barry AE, Leliwa-Sytek A, Tavul L, Imrie H, Migot-Nabias F, Brown SM, McVean GA, Day KP. Population genomics of the immune evasion (var) genes of Plasmodium falciparum. PLoS Pathog. 2007;3:e34. doi: 10.1371/journal.ppat.0030034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baruch DI, Pasloske BL, Singh HB, Bi X, Ma XC, Feldman M, Taraschi TF, Howard RJ. Cloning the P. falciparum gene encoding PfEMP1, a malarial variant antigen and adherence receptor on the surface of parasitized human erythrocytes. Cell. 1995;82:77–87. doi: 10.1016/0092-8674(95)90054-3. [DOI] [PubMed] [Google Scholar]
- Biggs BA, Anders RF, Dillon HE, Davern KM, Martin M, Petersen C, Brown GV. Adherence of infected erythrocytes to venular endothelium selects for antigenic variants of Plasmodium falciparum. J Immunol. 1992;149:2047–2054. [PubMed] [Google Scholar]
- Bozdech Z, Llinas M, Pulliam BL, Wong ED, Zhu J, DeRisi JL. The transcriptome of the intraerythrocytic developmental cycle of Plasmodium falciparum. PLoS Biol. 2003;1:E5. doi: 10.1371/journal.pbio.0000005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bull PC, Marsh K. The role of antibodies to Plasmodium falciparum-infected-erythrocyte surface antigens in naturally acquired immunity to malaria. Trends Microbiol. 2002;10:55–58. doi: 10.1016/s0966-842x(01)02278-8. [DOI] [PubMed] [Google Scholar]
- Bull PC, Berriman M, Kyes S, Quail MA, Hall N, Kortok MM, Marsh K, Newbold CI. Plasmodium falciparum variant surface antigen expression patterns during malaria. PLoS Pathog. 2005;1:e26. doi: 10.1371/journal.ppat.0010026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bull PC, Buckee CO, Kyes S, Kortok MM, Thathy V, Guyah B, Stoute JA, Newbold CI, Marsh K. Plasmodium falciparum antigenic variation Mapping mosaic var gene sequences onto a network of shared, highly polymorphic sequence blocks. Mol Microbiol. 2008;68:1519–1534. doi: 10.1111/j.1365-2958.2008.06248.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calderwood MS, Gannoun-Zaki L, Wellems TE, Deitsch KW. Plasmodium falciparum var genes are regulated by two regions with separate promoters, one upstream of the coding region and a second within the intron. J Biol Chem. 2003;278:34125–34132. doi: 10.1074/jbc.M213065200. [DOI] [PubMed] [Google Scholar]
- Chookajorn T, Dzikowski R, Frank M, Li F, Jiwani AZ, Hartl DL, Deitsch KW. Epigenetic memory at malaria virulence genes. Proc Natl Acad Sci USA. 2007;104:899–902. doi: 10.1073/pnas.0609084103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corredor V, Meyer EV, Lapp S, Corredor-Medina C, Huber CS, Evans AG, Barnwell JW, Galinski MR. A SICAvar switching event in Plasmodium knowlesi is associated with the DNA rearrangement of conserved 3’ non-coding sequences. Mol Biochem Parasitol. 2004;138:37–49. doi: 10.1016/j.molbiopara.2004.05.017. [DOI] [PubMed] [Google Scholar]
- Dahlback M, Lavstsen T, Salanti A, Hviid L, Arnot DE, Theander TG, Nielsen MA. Changes in var gene mRNA levels during erythrocytic development in two phenotypically distinct Plasmodium falciparum parasites. Malar J. 2007;6:78. doi: 10.1186/1475-2875-6-78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deitsch KW, Calderwood MS, Wellems TE. Malaria Cooperative silencing elements in var genes. Nature. 2001;412:875–876. doi: 10.1038/35091146. [DOI] [PubMed] [Google Scholar]
- Duffy MF, Byrne TJ, Elliott SR, Wilson DW, Rogerson SJ, Beeson JG, Noviyanti R, Brown GV. Broad analysis reveals a consistent pattern of var gene transcription in Plasmodium falciparum repeatedly selected for a defined adhesion phenotype. Mol Microbiol. 2005;56:774–788. doi: 10.1111/j.1365-2958.2005.04577.x. [DOI] [PubMed] [Google Scholar]
- Duraisingh MT, Voss TS, Marty AJ, Duffy MF, Good RT, Thompson JK, Freitas-Junior LH, Scherf A, Crabb BS, Cowman AF. Heterochromatin silencing and locus repositioning linked to regulation of virulence genes in Plasmodium falciparum. Cell. 2005;121:13–24. doi: 10.1016/j.cell.2005.01.036. [DOI] [PubMed] [Google Scholar]
- Dzikowski R, Frank M, Deitsch K. Mutually exclusive expression of virulence genes by malaria parasites is regulated independently of antigen production. PLoS Pathog. 2006;2:e22. doi: 10.1371/journal.ppat.0020022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dzikowski R, Li F, Amulic B, Eisberg A, Frank M, Patel S, Wellems TE, Deitsch KW. Mechanisms underlying mutually exclusive expression of virulence genes by malaria parasites. EMBO Rep. 2007;8:959–965. doi: 10.1038/sj.embor.7401063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dzikowski R, Deitsch KW. Active transcription is required for maintenance of epigenetic memory in the malaria parasite Plasmodium falciparum. J Mol Biol. 2008;382:288–297. doi: 10.1016/j.jmb.2008.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fidock DA, Nomura T, Wellems TE. Cycloguanil and its parent compound proguanil demonstrate distinct activities against Plasmodium falciparum malaria parasites transformed with human dihydrofolate reductase. Mol Pharmacol. 1998;54:1140–1147. doi: 10.1124/mol.54.6.1140. [DOI] [PubMed] [Google Scholar]
- Frank M, Deitsch K. Activation, silencing and mutually exclusive expression within the var gene family of Plasmodium falciparum. Int J Parasitol. 2006;36:975–985. doi: 10.1016/j.ijpara.2006.05.007. [DOI] [PubMed] [Google Scholar]
- Frank M, Dzikowski R, Costantini D, Amulic B, Berdougo E, Deitsch K. Strict pairing of var promoters and introns is required for var gene silencing in the malaria parasite Plasmodium falciparum. J Biol Chem. 2006;281:9942–9952. doi: 10.1074/jbc.M513067200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank M, Dzikowski R, Amulic B, Deitsch K. Variable switching rates of malaria virulence genes are associated with chromosomal position. Mol Microbiol. 2007;64:1486–1498. doi: 10.1111/j.1365-2958.2007.05736.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freitas-Junior LH, Hernandez-Rivas R, Ralph SA, Montiel-Condado D, Ruvalcaba-Salazar OK, Rojas-Meza AP, Mancio-Silva L, Leal-Silvestre RJ, Gontijo AM, Shorte S, Scherf A. Telomeric heterochromatin propagation and histone acetylation control mutually exclusive expression of antigenic variation genes in malaria parasites. Cell. 2005;121:25–36. doi: 10.1016/j.cell.2005.01.037. [DOI] [PubMed] [Google Scholar]
- Fried M, Duffy PE. Adherence of Plasmodium falciparum to chondroitin sulfate A in the human placenta. Science. 1996;272:1502–1504. doi: 10.1126/science.272.5267.1502. [DOI] [PubMed] [Google Scholar]
- Gannoun-Zaki L, Jost A, Mu J, Deitsch KW, Wellems TE. A silenced Plasmodium falciparum var promoter can be activated in vivo through spontaneous deletion of a silencing element in the intron Eukaryot. Cell. 2005;4:490–492. doi: 10.1128/EC.4.2.490-492.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardner MJ, Hall N, Fung E, White O, Berriman M, Hyman RW, Carlton JM, Pain A, Nelson KE, Bowman S, Paulsen IT, James K, Eisen JA, Rutherford K, Salzberg SL, Craig A, Kyes S, Chan MS, Nene V, Shallom SJ, Suh B, Peterson J, Angiuoli S, Pertea M, Allen J, Selengut J, Haft D, Mather MW, Vaidya AB, Martin DM, Fairlamb AH, Fraunholz MJ, Roos DS, Ralph SA, McFadden GI, Cummings LM, Subramanian GM, Mungall C, Venter JC, Carucci DJ, Hoffman SL, Newbold C, Davis RW, Fraser CM, Barrell B. Genome sequence of the human malaria parasite Plasmodium falciparum. Nature. 2002;419:498–511. doi: 10.1038/nature01097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodyer ID, Hayes DJ, Eisenthal R. Efflux of 6-deoxy-D-glucose from Plasmodium falciparum-infected erythrocytes via two saturable carriers. Mol Biochem Parasitol. 1997;84:229–239. doi: 10.1016/s0166-6851(96)02802-2. [DOI] [PubMed] [Google Scholar]
- Jackson JA. User’s Guide to Principal Components. John Wiley; New York: 1991. [Google Scholar]
- Jensen AT, Magistrado P, Sharp S, Joergensen L, Lavstsen T, Chiucchiuini A, Salanti A, Vestergaard LS, Lusingu JP, Hermsen R, Sauerwein R, Christensen J, Nielsen MA, Hviid L, Sutherland C, Staalsoe T, Theander TG. Plasmodium falciparum associated with severe childhood malaria preferentially expresses PfEMP1 encoded by group A var genes. J Exp Med. 2004;199:1179–1190. doi: 10.1084/jem.20040274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaestli M, Cockburn IA, Cortes A, Baea K, Rowe JA, Beck HP. Virulence of malaria is associated with differential expression of Plasmodium falciparum var gene subgroups in a case-control study. J Infect Dis. 2006;193:1567–1574. doi: 10.1086/503776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kraemer SM, Smith JD. Evidence for the importance of genetic structuring to the structural and functional specialization of the Plasmodium falciparum var gene family. Mol Microbiol. 2003;50:1527–1538. doi: 10.1046/j.1365-2958.2003.03814.x. [DOI] [PubMed] [Google Scholar]
- Kraemer SM, Kyes SA, Aggarwal G, Springer AL, Nelson SO, Christodoulou Z, Smith LM, Wang W, Levin E, Newbold CI, Myler PJ, Smith JD. Patterns of gene recombination shape var gene repertoires in Plasmodium falciparum: comparisons of geographically diverse isolates. BMC Genomics. 2007;8:45. doi: 10.1186/1471-2164-8-45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kyes S, Christodoulou Z, Pinches R, Kriek N, Horrocks P, Newbold C. Plasmodium falciparum var gene expression is developmentally controlled at the level of RNA polymerase II-mediated transcription initiation. Mol Microbiol. 2007;63:1237–1247. doi: 10.1111/j.1365-2958.2007.05587.x. [DOI] [PubMed] [Google Scholar]
- Kyes SA, Rowe JA, Kriek N, Newbold CI. Rifins: a second family of clonally variant proteins expressed on the surface of red cells infected with Plasmodium falciparum. Proc Natl Acad Sci USA. 1999;96:9333–9338. doi: 10.1073/pnas.96.16.9333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kyes SA, Christodoulou Z, Raza A, Horrocks P, Pinches R, Rowe JA, Newbold C. A well-conserved Plasmodium-falciparum var gene shows an unusual stage-specific pattern. Mol Microbiol. 2003;48:1339–1448. doi: 10.1046/j.1365-2958.2003.03505.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kyriacou HM, Stone GN, Challis RJ, Raza A, Lyke KE, Thera MA, Kone AK, Doumbo OK, Plowe CV, Rowe JA. Differential var gene transcription in Plasmodium falciparum isolates from patients with cerebral malaria compared to hyperparasitaemia. Mol Biochem Parasitol. 2006;150:211–218. doi: 10.1016/j.molbiopara.2006.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lavstsen T, Salanti A, Jensen AT, Arnot DE, Theander TG. Sub-grouping of Plasmodium falciparum 3D7 var genes based on sequence analysis of coding and non-coding regions. Malar J. 2003;2:27. doi: 10.1186/1475-2875-2-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez-Estrano C, Gopalakrishnan AM, Semblat JP, Fergus MR, Mazier D, Haldar K. An enhancer-like region regulates hrp3 promoter stage-specific gene expression in the human malaria parasite Plasmodium falciparum. Biochim Biophys Acta. 2007;1769:506–513. doi: 10.1016/j.bbaexp.2007.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez-Rubio JJ, Gontijo AM, Nunes MC, Issar N, Hernandez Rivas R, Scherf A. 5’ flanking region of var genes nucleate histone modification patterns linked to phenotypic inheritance of virulence traits in malaria parasites. Mol Microbiol. 2007;66:1296–1305. doi: 10.1111/j.1365-2958.2007.06009.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magistrado P, Salanti A, Tuikue Ndam NG, Mwakalinga SB, Resende M, Dahlback M, Hviid L, Lusingu J, Theander TG, Nielsen MA. VAR2CSA expression on the surface of placenta-derived Plasmodium falciparum-infected erythrocytes. J Infect Dis. 2008;198:1071–1074. doi: 10.1086/591502. [DOI] [PubMed] [Google Scholar]
- Mair GR, Braks JA, Garver LS, Wiegant JC, Hall N, Dirks RW, Khan SM, Dimopoulos G, Janse CJ, Waters AP. Regulation of sexual development of Plasmodium by translational repression. Science. 2006;313:667–669. doi: 10.1126/science.1125129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mamoun CB, Gluzman IY, Goyard S, Beverley SM, Goldberg DE. A set of independent selectable markers for transfection of the human malaria parasite Plasmodium falciparum. Proc Natl Acad Sci USA. 1999;96:8716–8720. doi: 10.1073/pnas.96.15.8716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mok BW, Ribacke U, Rasti N, Kironde F, Chen Q, Nilsson P, Wahlgren M. Default pathway of var2csa switching and translational repression in Plasmodium falciparum. PLoS ONE. 2008;3:e1982. doi: 10.1371/journal.pone.0001982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nkrumah LJ, Muhle RA, Moura PA, Ghosh P, Hatfull GF, Jacobs WR, Jr, Fidock DA. Efficient site-specific integration in Plasmodium falciparum chromosomes mediated by mycobacteriophage Bxb1 integrase. Nature Methods. 2006;3:615–621. doi: 10.1038/nmeth904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters JM, Fowler EV, Krause DR, Cheng Q, Gatton ML. Differential changes in Plasmodium falciparum var transcription during adaptation to culture. J Infect Dis. 2007;195:748–755. doi: 10.1086/511436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plouffe D, Brinker A, McNamara C, Henson K, Kato N, Kuhen K, Nagle A, Adrian F, Matzen JT, Anderson P, Nam TG, Gray NS, Chatterjee A, Janes J, Yan SF, Trager R, Caldwell JS, Schultz PG, Zhou Y, Winzeler EA. In silico activity profiling reveals the mechanism of action of antimalarials discovered in a high-throughput screen. Proc Natl Acad Sci USA. 2008;105:9059–9064. doi: 10.1073/pnas.0802982105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts DJ, Craig AG, Berendt AR, Pinches R, Nash G, Marsh K, Newbold CI. Rapid switching to multiple antigenic and adhesive phenotypes in malaria. Nature. 1992;357:689–692. doi: 10.1038/357689a0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson BA, Welch TL, Smith JD. Widespread functional specialization of Plasmodium falciparum erythrocyte membrane protein 1 family members to bind CD36 analysed across a parasite genome. Mol Microbiol. 2003;47:1265–1278. doi: 10.1046/j.1365-2958.2003.03378.x. [DOI] [PubMed] [Google Scholar]
- Rottmann M, Lavstsen T, Mugasa JP, Kaestli M, Jensen AT, Muller D, Theander T, Beck HP. Differential expression of var gene groups is associated with morbidity caused by Plasmodium falciparum infection in Tanzanian children. Infect Immun. 2006;74:3904–3911. doi: 10.1128/IAI.02073-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salanti A, Staalsoe T, Lavstsen T, Jensen AT, Sowa MP, Arnot DE, Hviid L, Theander TG. Selective upregulation of a single distinctly structured var gene in chondroitin sulphate A-adhering Plasmodium falciparum involved in pregnancy-associated malaria. Mol Microbiol. 2003;49:179–191. doi: 10.1046/j.1365-2958.2003.03570.x. [DOI] [PubMed] [Google Scholar]
- Scherf A, Hernandez-Rivas R, Buffet P, Bottius E, Benatar C, Pouvelle B, Gysin J, Lanzer M. Antigenic variation in malaria: in situ switching, relaxed and mutually exclusive transcription of var genes during intra-erythrocytic development in Plasmodium falciparum. Embo J. 1998;17:5418–5426. doi: 10.1093/emboj/17.18.5418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schieck E, Pfahler JM, Sanchez CP, Lanzer M. Nuclear run-on analysis of var gene expression in Plasmodium falciparum. Mol Biochem Parasitol. 2007;153:207–212. doi: 10.1016/j.molbiopara.2007.02.004. [DOI] [PubMed] [Google Scholar]
- Scholz M, Fraunholz MJ. A computational model of gene expression reveals early transcriptional events at the subtelomeric regions of the malaria parasite, Plasmodium falciparum. Genome Biol. 2008;9:R88. doi: 10.1186/gb-2008-9-5-r88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith JD, Chitnis CE, Craig AG, Roberts DJ, Hudson-Taylor DE, Peterson DS, Pinches R, Newbold CI, Miller LH. Switches in expression of Plasmodium falciparum var genes correlate with changes in antigenic and cytoadherent phenotypes of infected erythrocytes. Cell. 1995;82:101–110. doi: 10.1016/0092-8674(95)90056-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith JD, Subramanian G, Gamain B, Baruch DI, Miller LH. Classification of adhesive domains in the Plasmodium falciparum erythrocyte membrane protein 1 family. Mol Biochem Parasitol. 2000;110:293–310. doi: 10.1016/s0166-6851(00)00279-6. [DOI] [PubMed] [Google Scholar]
- Su XZ, Heatwole VM, Wertheimer SP, Guinet F, Herrfeldt JA, Peterson DS, Ravetch JA, Wellems TE. The large diverse gene family var encodes proteins involved in cytoadherence and antigenic variation of Plasmodium falciparum-infected erythrocytes. Cell. 1995;82:89–100. doi: 10.1016/0092-8674(95)90055-1. [DOI] [PubMed] [Google Scholar]
- Tham WH, Payne PD, Brown GV, Rogerson SJ. Identification of basic transcriptional elements required for rif gene transcription. Int J Parasitol. 2007;37:605–615. doi: 10.1016/j.ijpara.2006.11.006. [DOI] [PubMed] [Google Scholar]
- Trimnell AR, Kraemer SM, Mukherjee S, Phippard DJ, Janes JH, Flamoe E, Su XZ, Awadalla P, Smith JD. Global genetic diversity and evolution of var genes associated with placental and severe childhood malaria. Mol Biochem Parasitol. 2006;148:169–180. doi: 10.1016/j.molbiopara.2006.03.012. [DOI] [PubMed] [Google Scholar]
- Tuikue Ndam NG, Salanti A, Bertin G, Dahlback M, Fievet N, Turner L, Gaye A, Theander T, Deloron P. High level of var2csa transcription by Plasmodium falciparum isolated from the placenta. J Infect Dis. 2005;192:331–335. doi: 10.1086/430933. [DOI] [PubMed] [Google Scholar]
- Voss TS, Kaestli M, Vogel D, Bopp S, Beck HP. Identification of nuclear proteins that interact differentially with Plasmodium falciparum var gene promoters. Mol Microbiol. 2003;48:1593–1607. doi: 10.1046/j.1365-2958.2003.03528.x. [DOI] [PubMed] [Google Scholar]
- Voss TS, Healer J, Marty AJ, Duffy MF, Thompson JK, Beeson JG, Reeder JC, Crabb BS, Cowman AF. A var gene promoter controls allelic exclusion of virulence genes in Plasmodium falciparum malaria. Nature. 2006;439:1004–1008. doi: 10.1038/nature04407. [DOI] [PubMed] [Google Scholar]
- Voss TS, Tonkin CJ, Marty AJ, Thompson JK, Healer J, Crabb BS, Cowman AF. Alterations in local chromatin environment are involved in silencing and activation of subtelomeric var genes in Plasmodium falciparum. Mol Microbiol. 2007;66:139–150. doi: 10.1111/j.1365-2958.2007.05899.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Y, Sifri CD, Lei HH, Su XZ, Wellems TE. Transfection of Plasmodium falciparum within human red blood cells. Proc Natl Acad Sci USA. 1995;92:973–977. doi: 10.1073/pnas.92.4.973. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.