

Abstract
Elimination of pathogens is the basis of host resistance to infections; however, relationship between persisting pathogens and disease has not been clarified. Leishmania major infection in mice is an important model of host–pathogen relationship. Infected BALB/c mice exhibit high parasite numbers in lymph nodes and spleens, and a chronic disease with skin lesions, splenomegaly, and hepatomegaly, increased serum IgE levels and cytokine imbalance. Although numerous gene loci affecting these disease symptoms have been reported, genes controlling parasites’ elimination or dissemination have never been mapped. We therefore compared genetics of the clinical and immunologic symptomatology with parasite load in (BALB/c × CcS-11) F2 hybrids and mapped five loci, two of which control parasite elimination or dissemination. Lmr5 influences parasite loads in spleens (and skin lesions, splenomegaly, and serum IgE, IL-4, and IFNγ levels), and Lmr20 determines parasite numbers in draining lymph nodes (and serum levels of IgE and IFNγ), but no skin or visceral pathology. Three additional loci do not affect parasite numbers but influence significantly the disease phenotype—Lmr21: skin lesions and IFNγ levels, Lmr22: IL-4 levels, Lmr23: IFNγ levels, indicating that development of L. major-caused disease includes critical regulations additional to control of parasite spread.
Keywords: Leishmania major, Mouse, Genetics of parasite elimination, Susceptibility to leishmaniasis, Functional heterogeneity
Parasite infection induces in the host both the responses that limit parasite growth and multiplication and responses that are either neutral or can lead to damage of host’s tissues. Interaction between the infecting parasite and the host’s immune system is especially important for understanding of pathogenesis of diseases caused by chronic infections.
One of the important unresolved questions in pathogenesis of chronic infections is the relationship between the pathogen load and clinical symptoms. Whereas early elimination of infectious agents generally precludes development of chronic disease, the relationship between load and spread of persisting pathogens and manifestations and course of ensuing disease has not been clarified. Although the number of studies addressing this problem is limited, they provided examples of unexpected genetic and functional complexity in infections with various pathogens such as Borrelia burgdorferi (Ma et al. 1998), Toxoplasma gondii (Johnson et al. 2002), and Trypanosoma cruzi (Marinho et al. 2004). This involved strain differences in both the relationship between pathogen load and intensity of symptoms of disease and in dissemination to different organs. However, the relationship between genetic control of parasite load and parasite-induced pathology has never been analyzed simultaneously in segregating populations (Råberg et al. 2007).
Leishmania infection in mice has served as a paradigm of interaction of pathogen and host. Leishmania parasites are transmitted to vertebrate hosts by infected phlebotomine sandflies and infect professional phagocytes (macrophages, monocytes, and neutrophils) as well as dendritic cells and fibroblasts. The major host cell is macrophage, in which parasites multiply, eventually rupturing the cell and spreading to uninfected cells. They affect skin, mucous membranes, lymph nodes, spleen, bone marrow, liver, lungs, and, in rare cases, even the brain. The expansion of the parasites is determined by multiple factors, including both parasite characteristics (reviewed in McMahon-Pratt and Alexander 2004) and host genetics (reviewed in Lipoldová and Demant 2006). Leishmania parasites can evade the first-line defense systems, inhibit several functions of host’s immune cells (Olivier et al. 2005), and prime the development of an immune state favorable for the parasite. In the mouse model, distinct clinical manifestation of cutaneous and visceral leishmaniasis observed in humans can be reproduced in some mouse strains. Mice from several strains (BALB/c, SWR/J, P/J) develop severe systemic disease affecting skin and visceral organs (Sacks and Noben-Trauth 2002), whereas mice of the majority of inbred strains (e.g., C3H, CBA, C57BL/6, B10.D2, 129/Sv, and STS/A) after infection with L. major develop no pathological changes or only small spontaneously healing cutaneous lesions restricted to the infection site. The wide spectrum of clinical symptoms and immunological reactions in susceptible strains provides an opportunity to define control of individual components of disease phenotype. However, there are no data about the relationship of parasite load to manifestations of disease.
As the non-healer phenotype of BALB/c mice is controlled by multiple genes, we used a special tool for genetic analysis of multigenic traits, the recombinant congenic (RC) strains. Each of the 20 CcS/Dem RC mouse strains carries a different random subset of 12.5% genes of the resistant donor strain STS/A (STS) on the background of the susceptible strain BALB/cHeA (BALB/c). As a consequence, the individual STS genes contributing to the resistance to infection L. major were distributed among different RC strains. Previously, we studied three CcS/Dem strains: the most resistant strain CcS-5, the strain CcS-20 that exhibits intermediate susceptibility, and the susceptible strain CcS-16. This resulted in mapping of 17 loci, Lmr3–Lmr19 (Leishmania major response 3–19), which are associated with 13 different combinations of pathological symptoms and immunological reactions (Lipoldová et al. 2000; Badalová et al. 2002; Vladimirov et al. 2003; Havelková et al. 2006).
In the present study, in addition to the disease symptoms and immunological parameters, we also measured load of parasites in draining lymph nodes and spleen. This allowed us to analyze for the first time the genetic relationship between the presence of parasites and manifestations of disease. We mapped genes controlling these parameters in the strain CcS-11, which after infection presents smaller skin lesions, lower splenomegaly and hepatomegaly, fewer parasites in spleen, and lower concentrations of IgE and IL-4 in serum than the susceptible strain BALB/c (Lipoldová et al. 2002 and this paper). We show here that the degree of elimination of parasites is genetically controlled and that this control is organ specific. Different genes control parasite load predominantly in lymph nodes or in spleen. Moreover, we show that additional genes that do not affect parasites’ presence in these two organs also significantly affect the course of disease.
Materials and methods
Mice Males and females of strains BALB/c and CcS-11 were 8 to 24 weeks old at the time of infection. When used for these experiments, strain CcS-11 was in more than 38 generations of inbreeding. The parts of its genome inherited from the BALB/c or STS parents were defined (Stassen et al. 1996). Two hundred and ninety-nine F2 hybrids between CcS-11 and BALB/c (age 9 to 13 weeks at the time of infection, mean age 11 weeks, median 12 weeks) were produced at the Institute of Molecular Genetics. They comprised 155 females and 144 males. During the experiment, male and female mice were placed into separate rooms and males were caged individually. They were tested in three successive experimental groups. The research had complied with all relevant European Union guidelines for work with animals.
Parasites Leishmania major LV 561 (MHOM/IL/67/LRC-L137 JERICHO II) was maintained in rump lesions of BALB/c females. Amastigotes were transformed to promastigotes using SNB-9 (Diamond and Herman 1954). Promastigotes (107) from 6-day-old subculture 2 were inoculated in 50 μl sterile saline s.c. into mouse rump.
Disease phenotype The size of the primary skin lesions was measured weekly using a Vernier caliper gauge. The mice were killed 8 weeks after infection. Blood, spleen, liver, and inguinal lymph nodes were collected for further analysis.
Quantification of parasite load in spleens and inguinal lymph nodes by PCR–ELISA Total DNA from spleens and inguinal lymph nodes was prepared using a standard proteinase procedure. To detect Leishmania DNA by PCR, two primers (digoxigenin-labeled F 5′-ATT TTA CAC CAA CCC CCA GTT-3′ and biotin-labeled R 5′-GTG GGG AGG GGG CGT TCT-3′; Rodgers et al. 1990; VBC-GENOMICS Biosciences Research, Vienna, Austria) were used for amplification of the 120-bp conservative region of the minicircle kinetoplast of Leishmania parasites. Briefly, 50 ng of extracted DNA with 5 pmol of each kinetoplast primer, 0.5 mM concentration of each dNTP, 2 mM MgCl2, 50 mM KCl, 10 mM Tris–HCl (pH 8.3), and 2 U of Taq polymerase (Invitrogen Brazil Ltda., São Paulo, Brazil) was amplified in a total volume of PCR reaction of 20 μl. As a positive control, we used 20 ng of L. major DNA per reaction (0.3 fg of L. major DNA approximately corresponds to one parasite). After an initial hot start at 94°C for 90 s, the conditions used for amplification were: 94°C for 30 s, 53°C for 45 s, 72°C for 60 s (26 cycles for DNA from lymph nodes and 33 cycles for DNA from spleens), followed by the final extension 72°C for 10 min, using the I-Cycler (Bio-Rad Laboratories, Hercules, CA, USA). Concentration of the amplified 120-bp region of Leishmania DNA in PCR product was determined using the modified ELISA protocol (Pharmingen, San Diego, CA, USA). Plates were coated with 100 μl per well streptavidin solution (0.5 μg/ml in 0.1 M NaHCO3 pH 7.0) at 4°C overnight. PCR samples were diluted to 2% concentration in 2% FCS/PBS. The positive PCR control was used as a standard for ELISA. Plates were incubated at room temperature for 2 h and then incubated at room temperature for 45 min with the anti-digoxigenin detecting antibody, Fab fragments (Roche Diagnostic GmbH, Mannheim, Germany) 0.15 U/ml, diluted in 2% FCS/PBS. As a substrate for color reaction development, we used 2,2′-azino-bis(3-ethylbenzo-thiazoline-6-sulfonic acid) (ABTS) solution (150 mg ABTS in 500 ml 0.1 M citric acid, pH 4.35) (Sigma-Aldrich Co., St. Louis, MO, USA) with 0.03% H2O2. The color reaction was stopped after 10 min by adding 50 μl 0.5 M oxalic acid per well. The Leishmania loads were determined using the ELISA Reader Tecan and the curve fitter program KIM-E (Schoeller Pharma, Prague, Czech Republic) using least squares-based linear regression analysis.
Cytokine and IgE levels IFNγ, IL-4, IL-12, and IgE levels in serum were determined using the primary and secondary monoclonal antibodies (IFNγ = R4-6A2, XMG1.2; IL-4 = BVD4-1D11, BVD6-24G2; IL-12 = Red-T and G297-298; IgE = R35-72, R3592) and standards from Pharmingen, San Diego, CA (recombinant mIFNγ, mIL-4, mIL-12 p70 heterodimer, and purified mIgE = C38-2). The ELISA measurements of IFNγ, IL-4, IL-12, and IgE levels were performed as described in the previous section.
Genotyping of F2 mice DNA was isolated from tails using a standard proteinase procedure. The strain CcS-11 differs from BALB/c at STS-derived segments on eight chromosomes, respectively (Stassen et al. 1996). These differential segments were typed in the F2 hybrid mice between CcS-11 and BALB/c using 16 microsatellite markers (Research Genetics, Huntsville, FL, USA): D1Mit403, D3Mit45, D7Mit54, D7Mit18, D7Nds1, D7Mit282, D7Mit67, D7Mit259, D8Mit85, D10Mit12, D10Mit46, D12Mit37, D16Mit73, D19Mit51, D19Mit60, and D19Mit46. The maximal distance between any two markers in the chromosomal segments derived from the strain STS or from the nearest BALB/c derived markers was 19 cM.The PCR genotyping of polymorphic simple sequence length markers with fragment length difference of more than 10 bp was performed using unlabeled primers (D1Mit403, D7Mit18, D7Nds1, D7Mit282, D7Mit67, D7Mit259, D10Mit12, D10Mit46, D12Mit37, D16Mit73, D19Mit51, D19Mit60, and D19Mit46). DNA was amplified in a 20-µl PCR reaction with 0.11 μM of forward and reverse primer, 0.2 mM concentration of each dNTP, 1.0–3.0 mM MgCl2 (the optimal concentration for the most primers was 1.5 mM), 50 mM KCl, 10 mM Tris–HCl (pH 8.3), 0.5 U of Taq polymerase (GIBCO, Grand Island, NY, USA), and approximately 40 ng of tail DNA. PCR reaction was performed using the I-Cycler (Bio-Rad Laboratories), according to the following scheme: an initial hot start 3 min at 94°C, followed by 40 cycles of 94°C for 30 s for denaturing, 55° for 60 s for annealing (except markers D7Mit18 and D7Mit259, for which optimal Ta = 57°), 72° for 60 s for elongation, and finally 3 min at 72° for final extension. Each PCR product was mixed with 5 μl of loading buffer and electrophoresed in 2–3% agarose gel for 20 min to 1.5 h at 150 V. The PCR genotyping for simple sequence length polymorphic markers with fragment length difference of less than 10 bp (D3Mit45, D7Mit54, and D8Mit85) was performed using [γ-32P]ATP end-labeled primers as described elsewhere (Krulová et al. 1997).
Statistical analysis The linkage of genes that control parasite spread, organ pathology, and immune parameters with microsatellite markers was examined with analysis of variance (NCSS, Kaysville, UT, USA) and PROC GLM statement of SAS 8.2 for Windows (SAS Institute, Gary, NC, USA). Genotype, sex, and age were fixed factors and the experiment was random factor. In order to obtain normal distribution, the obtained values of dependent variables were transformed as shown in the legends of the tables. We tested individual effects of all markers as well as of all their pairwise combinations using the backward elimination procedure (Tripodis et al. 2001). The phenotypes of compound genotypes that significantly deviated from simple additive effects of the alleles indicated interactions. Markers and interactions with P <0.05 were combined in a single comparison.The time course of skin lesion development was evaluated on the basis of weekly measurements of lesion size in each mouse in weeks 4 to 8 after infection. Mixed model of analysis of variance (Proc Mixed, SAS) with marker as the fixed variable, the week of observation as the covariate, and mouse as random variable have been used to evaluate the linkage.The percentage of variance explained by an independent variable, the partial R2, was computed in the standard way by subtracting the regression sums of squares of the model without the variable of interest (SS(β1, β2, β3, β4/β0)) from the regression sums of squares of the full model (SS(β1, β2, β3, β4, β5/β0)); this difference divided by the residual sums of squares (RSS(β1, β2, β3, β4, β5/β0)) algebraically expressed as
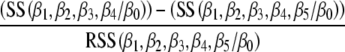 |
indicated the contribution of the independent variable.To obtain genome-wide significance values (corrected P), the observed P values (αT) were adjusted according to Lander and Schork (1994) using the formula:
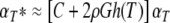 |
where G = 1.75 M (the length of the segregating part of the genome = 12.5% of 14 M), C = 8 (number of chromosomes segregating in cross between CcS-11 and BALB/c, respectively); ρ = 1.5 for F2 hybrids; h(T) = the observed statistic (F ratio). Direct comparisons of this method with permutation analyses in establishing genome-wide significance thresholds found them comparable (Singer et al. 2005).Power computation for detection of sex difference in marker effects on parasite load were carried forth. Across genotypes (D1Mit403, D10Mit12), the sample size was balanced between males (n = 144) and females (n = 144) for D1Mit403 and males (n = 143) and females (n = 146) for D10Mit12. The marker frequencies for each genotype ranged for CC from 59 to 65, for CS from 149 to 157, and for SS from 66 to 79. Under these sample sizes, we were able to detect on the log scale at 0.80 power and α = 0.05 (two-sided) effect sizes for sex by marker interaction in ranges from 0.75 to 0.86 standard deviation units or larger, depending upon the specific differential effect.
Results
Differences in organ pathology and immune response between strains CcS-11 and BALB/c The parasite load, disease symptoms, and immunological parameters in BALB/c and CcS-11 mice are given in Table 1. BALB/c mice develop larger skin lesions during the course of infection (weeks 5, 6, 7, and 8) and larger splenomegaly and hepatomegaly compared to the strain CcS-11, with no significant sex difference in size of skin lesions, splenomegaly, and hepatomegaly (data not shown). Larger numbers of parasites in spleen were also observed in strain BALB/c, but there were no significant differences in parasite load in draining lymph nodes. Parasite numbers in spleens of strain CcS-11 and in draining lymph nodes of both BALB/c and CcS-11 were significantly higher in males (Table 1, Fig. 1). Serum IgE and IL-4 levels were higher in strain BALB/c than in strain CcS-11, but there were no strain differences in serum concentrations of IFNγ and IL-12. Serum IgE and cytokine levels were not influenced by sex (Table 1).
Table 1.
Comparison of immunopathological parameters in L. major infected mice of the strains BALB/c and CcS-11
| Phenotype | Strain | P value | |||
|---|---|---|---|---|---|
| BALB/c | CcS-11 | Strain difference | |||
| Lesion size 5 weeks (mm2) | 60.79 ± 6.33 (n = 19) | 29.55 ± 6.33 (n = 19) | 1.34 × 10−3 | ||
| Lesion size 6 weeks (mm2) | 91.67 ± 7.78 (n = 19) | 41.33 ± 7.78 (n = 19) | 5.88 × 10−5 | ||
| Lesion size 7 weeks (mm2) | 107.42 ± 9.52 (n = 19) | 46.54 ± 9.52 (n = 19) | 6.91 × 10−5 | ||
| Lesion size 8 weeks (mm2) | 129.31 ± 11.73 (n = 19) | 55.05 ± 11.73 (n = 19) | 7.86 × 10−5 | ||
| Splenomegaly (spleen-to-body weight ratio × 1,000) | 17.84 ± 1.13 (n = 19) | 12.26 ± 1.13 (n = 19) | 1.51 × 10−3 | ||
| Hepatomegaly (liver-to-body weight ratio × 1,000) | 68.06 ± 1.55 (n = 19) | 60.32 ± 1.55 (n = 19) | 2.75 × 10−3 | ||
| IgE in serum (μg/ml) | 10.22 | 3.54 ± 0.29 (n = 16) | 3.97 | 1.62 ± 0.28 (n = 18) | 6.79 × 10−5 |
| IFNγ in serum (ng/ml) | 5.57 | 1.31 ± 0.05 (n = 18) | 4.45 | 1.22 ± 0.06 (n = 14) | 0.257 |
| IL-12 in serum (ng/ml) | 2.83 | 1.04 ± 0.20 (n = 16) | 1.74 | 0.55 ± 0.20 (n = 15) | 0.119 |
| IL-4 in serum (ng/ml) | 4.65 | 1.61 ± 0.23 (n = 12) | 2.33 | 0.83 ± 0.26 (n = 9) | 3.75 × 10−2 |
| Parasites in draining lymph nodes, total group (arbitrary units) | 1.61 | 5.08 ± 0.20 (n = 17) | 2.30 | 5.44 ± 0.19 (n = 19) | 0.211 |
| Parasites in draining lymph nodes, females | 0.57 | 4.04 ± 0.23a (n = 9) | 0.46 | 3.83 ± 0.22b (n = 10) | 0.643 |
| Parasites in draining lymph nodes, males | 9.28 | 6.83 ± 0.37a (n = 8) | 8.54 | 6.75 ± 0.35b (n = 9) | 0.931 |
| Parasites in spleen, total group (arbitrary units) | 1.58 | 5.06 ± 0.29 (n = 18) | 0.31 | 3.43 ± 0.28 (n = 19) | 4.02 × 10−4 |
| Parasites in spleen, females | 1.10 | 4.70 ± 0.45c (n = 8) | 0.18 | 2.89 ± 0.40d (n = 10) | 8.36 × 10−3 |
| Parasites in spleen, males | 3.08 | 5.73 ± 0.34c (n = 10) | 0.58 | 4.06 ± 0.36d (n = 9) | 6.01 × 10−3 |
Means and standard errors of means and P values were computed by analysis of variance. Lesion size (mm2), splenomegaly and hepatomegaly (spleen and liver-to-body weight ratio × 1,000, respectively) exhibited normal distribution without transformation. The following transformations were used to obtain normal distribution: IgE levels in serum: IgE (μg/ml)—natural logarithm of value to the power of 1.5; IFNγ (ng/ml)—natural logarithm of the value to the power of 0.5; IL-4 (ng/ml)—natural logarithm to the power of 1.1; IL-12 (ng/ml)—natural logarithm; parasite load detected as parasite DNA content (arbitrary units) in spleen and in lymph nodes—natural logarithm of (value × 100). Number of tested mice is shown in brackets. The numbers in bold give the average non-transformed values.
aSex difference in BALB/c, P = 9.26 × 10−5
bSex difference in CcS-11, P = 1.14 × 10−5
cSex difference in BALB/c, P = 0.133
dSex difference in CcS-11, P = 8.93 × 10−3
Fig. 1.

Parasites in hematoxylin–eosin-stained inguinal lymph node sections of female and male mice. a Uninfected BALB/c male, bL. major infected BALB/c male, c uninfected BALB/c female, dL. major infected BALB/c female, e uninfected CcS-11 male, fL. major infected CcS-11 male, g uninfected CcS-11 female, hL. major infected CcS-11 female. Arrows show groups of Leishmania parasites (with a dark nucleus, smaller kinetoplast, and light cytoplasm) among normal lymph node cells (b, d, f) and parasites inside a macrophage (h)
Loci that influence parasite burden, organ pathology, and immunological parameters in the strain CcS-11 The summary of the detected loci and their phenotypic effects is given in Table 2. Linkage analysis of F2 hybrids between BALB/c and CcS-11 indicated four novel loci Lmr20, Lmr21, Lmr22, and Lmr23 on chromosomes 1, 7, 12, and 16, respectively. For the previously detected locus Lmr5 on chromosome 10 (Lipoldová et al. 2000), we detected an additional effect on parasite load in spleen and on IL-4 level in serum in interaction with Lmr22. These data are given in Tables 3, 4, and 5 (single gene effects) and Tables 6, 7, and 8 (gene interactions). These tables describing detected linkages give besides the statistical significance of linkage also two other quantitative characteristics (Lander and Botstein 1989) that describe the size of their effect: (a) a magnitude of effect of each locus that describes how large the phenotypic difference is (e.g., in IgE level) between mice with different alleles at this locus and (b) the explained variance (% of variance) that is the proportion of phenotypic variation in the tested population that is caused by the effects of these alleles. The first parameter in our data varies between 1.4 and 2.5 (i.e., difference between the extreme phenotypes caused by alleles at a locus is between 40% and 150%) and the percent of explained variance 1–14. These value ranges are common for majority of quantitative trait loci (QTLs) in the mouse (Flint et al. 2005).
Table 2.
Summary of Lmr loci that control response to L. major in the strain CcS-11
| Locus | Chr. | Marker (position in cM) | Trait controlled |
|---|---|---|---|
| Lmr20 | 1 | D1Mit403 (100) | Parasite load in draining LNs, serum IgE, serum IFNγ (in interaction with Lmr23) |
| Lmr21 | 7 | D7Mit67 (63.5) | Lesion development, serum IFNγ (in interaction with Lmr5) |
| Lmr5 | 10 | D10Mit12 (56); D10Mit46 (63) | Lesion size, parasite load in spleen, splenomegaly, serum IgE, serum IFNγ (in interaction with Lmr21), serum IL-4 (in interaction with Lmr22) |
| Lmr22 | 12 | D12Mit37 (1) | Serum IL-4 (in interaction with Lmr5) |
| Lmr23 | 16 | D16Mit73 (8.6) | Serum IFNγ (in interaction with Lmr20) |
Only the QTLs with corrected P < 0.05 (genome-wide significance) are shown.
Table 3.
Loci that control parasite load in draining lymph nodes and spleens and splenomegaly in L. major infected F2 hybrids between CcS-11 and BALB/c
| Phenotype | Locus | Group | Marker | Genotype | P value | Corrected P value | Percent of explained variance | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| CC | CS | SS | ||||||||||
| Parasites in draining lymph nodes (arbitrary units) | Lmr20 | Both sexes | D1Mit403 | 7.74 ± 0.51 (n = 66) | 6.26 ± 0.33 (n = 152) | 4.35 ± 0.51 (n = 66) | 8.35 × 10−5 | 5.54 × 10−3 | 4.25 | |||
| Females | D1Mit403 | 4.96 ± 0.55 (n = 37) | 3.34 ± 0.40 (n = 70) | 1.90 ± 0.55 (n = 37) | 1.16 × 10−3 | 5.89 × 10−2 | 8.39 | |||||
| Males | D1Mit403 | 10.87 ± 0.89 (n = 29) | 9.34 ± 0.53 (n = 82) | 7.31 ± 0.89 (n = 29) | 0.301 | NS | – | |||||
| Parasites in spleen (arbitrary units) | Lmr5 | Both sexes | D10Mit12 | 2.09 | 5.34 ± 0.14 (n = 60) | 0.98 | 4.58 ± 0.09 (n = 150) | 0.96 | 4.56 ± 0.12 (n = 79) | 8.09 × 10−5 | 5.39 × 10−3 | 4.54 |
| Females | D10Mit12 | 1.60 | 5.07 ± 0.20 (n = 31) | 0.60 | 4.10 ± 0.14 (n = 69) | 0.52 | 3.96 ± 0.17 (n = 46) | 8.57 × 10−5 | 5.76 × 10−3 | 11.74 | ||
| Males | D10Mit12 | 2.57 | 5.55 ± 0.20 (n = 29) | 1.70 | 5.14 ± 0.12 (n = 81) | 2.15 | 5.37 ± 0.18 (n = 33) | 0.173 | NS | – | ||
| Splenomegaly (spleen-to-body weight ratio × 1,000) | Lmr5 | Both sexes | D10Mit12 | 21.43 ± 0.61 (n = 62) | 18.82 ± 0.39 (n = 151) | 17.49 ± 0.53 (n = 82) | 8.63 × 10−6 | 6.98 × 10−4 | 11.60 | |||
| D10Mit46 | 20.64 ± 0.63 (n = 61) | 18.76 ± 0.40 (n = 151) | 17.52 ± 0.56 (n = 76) | 1.27 × 10−3 | 6.21 × 10−2 | 4.32 | ||||||
| Females | D10Mit12 | 22.59 ± 0.86 (n = 33) | 19.06 ± 0.59 (n = 70) | 16.91 ± 0.71 (n = 48) | 6.48 × 10−6 | 5.57 × 10−4 | 14.47 | |||||
| D10Mit46 | 22.56 ± 0.96 (n = 28) | 19.42 ± 0.59 (n = 73) | 17.27 ± 0.72 (n = 49) | 1.06 × 10−4 | 7.08 × 10−3 | 11.50 | ||||||
| Males | D10Mit12 | 19.42 ± 0.84 (n = 29) | 18.29 ± 0.50 (n = 81) | 17.71 ± 0.77 (n = 34) | 0.318 | NS | – | |||||
| D10Mit46 | 18.78 ± 0.78 (n = 33) | 18.07 ± 0.51 (n = 78) | 18.34 ± 0.87 (n = 27) | 0.750 | NS | – | ||||||
Mean and SE values were obtained by analysis of variance. Only linkages significant at whole genome level in the total group or in at least in one sex are given. The parasite loads were estimated by PCR–ELISA (see Materials and methods). In order to obtain normal distribution required for analysis of variance, value of parasites in spleens was transformed to the natural logarithm of (value × 100). Splenomegaly (spleen-to-body weight ratio × 1,000) and parasite load in draining lymph nodes exhibited normal distribution without transformation. The numbers in bold give the average non-transformed values. C and S indicate the presence of BALB/c and STS allele, respectively; n indicates number of mice.
Table 4.
Loci controlling skin lesion development after infection with L. major in both sexes and separately in female and male F2 hybrids between CcS-11 and BALB/c
| Locus | Group | Marker | Genotype | P value | Corrected P value | ||
|---|---|---|---|---|---|---|---|
| CC | CS | SS | |||||
| Lmr5 | Both sexes | D10Mit12 | 79.09 ± 1.61 (n = 62) | 71.46 ± 1.03 (n = 151) | 59.18 ± 1.40 (n = 82) | 1.16 × 10−6 | 5.27 × 10−5 |
| D10Mit46 | 79.84 ± 1.64 (n = 61) | 70.94 ± 1.04 (n = 151) | 58.90 ± 1.47 (n = 76) | 1.05 × 10−6 | 4.83 × 10−5 | ||
| Females | D10Mit12 | 79.06 ± 2.29 (n = 33) | 66.16 ± 1.58 (n = 70) | 52.01 ± 1.91 (n = 48) | 6.34 × 10−6 | 2.67 × 10−4 | |
| D10Mit46 | 79.19 ± 2.52 (n = 28) | 66.60 ± 1.56 (n = 73) | 52.26 ± 1.91 (n = 49) | 1.88 × 10−5 | 7.28 × 10−4 | ||
| Males | D10Mit12 | 79.12 ± 2.15 (n = 29) | 76.03 ± 1.29 (n = 81) | 69.23 ± 1.98 (n = 34) | 0.147 | NS | |
| D10Mit46 | 80.39 ± 2.03 (n = 33) | 75.00 ± 1.32 (n = 78) | 70.89 ± 2.24 (n = 27) | 0.207 | NS | ||
| Lmr21 | Both sexes | D7Mit282 | 109.53 ± 3.63 (n = 77) | 102.90 ± 2.64 (n = 140) | 86.35 ± 3.98 (n = 63) | 4.3 × 10−3 | 0.0972 (suggestive) |
| D7Mit67 | 113.18 ± 3.58 (n = 79) | 101.54 ± 2.69 (n = 132) | 85.39 ± 3.53 (n = 77) | 4.25 × 10−4 | 0.0116 | ||
| Females | D7Mit282 | 103.20 ± 4.92 (n = 45) | 93.89 ± 3.63 (n = 80) | 81.06 ± 6.57 (n = 25) | 0.1012 | NS | |
| D7Mit67 | 110.14 ± 4.62 (n = 46) | 91.70 ± 3.62 (n = 71) | 76.65 ± 5.44 (n = 32) | 1.8 × 10−3 | 0.0458 | ||
| Males | D7Mit282 | 109.67 ± 5.18 (n = 32) | 112.75 ± 3.79 (n = 60) | 98.87 ± 4.76 (n = 38) | 0.0895 | NS | |
| D7Mit67 | 113.89 ± 5.39 (n = 33) | 112.31 ± 3.84 (n = 61) | 99.95 ± 4.45 (n = 45) | 0.172 | NS | ||
Mean and SE values were obtained by analysis of variance. Lesion size (mm2) exhibited normal distribution without transformation. C and S indicate the presence of BALB/c and STS allele, respectively; n indicates number of mice. The values for Lmr5 were calculated by analysis of covariance from all five time points (from 4 to 8 weeks of infection) as described in Materials and methods; the values for Lmr21 by analysis of variance for week 8.
Table 5.
Loci controlling IgE level in L. major infected F2 hybrids between CcS-11 and BALB/c
| Locus | Group | Marker | Genotype | P value | Corrected P value | Percent of explained variance | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| CC | CS | SS | |||||||||
| Lmr20 | Both sexes | D1Mit403 | 21.63 | 5.39 ± 0.20 (n = 69) | 16.86 | 4.75 ± 0.13 (n = 157) | 12.69 | 4.05 ± 0.20 (n = 69) | 4.35 × 10−5 | 3.07 × 10−3 | 6.09 |
| Females | D1Mit403 | 19.87 | 5.17 ± 0.25 (n = 39) | 13.25 | 4.15 ± 0.18 (n = 73) | 10.00 | 3.49 ± 0.24 (n = 41) | 2.36 × 10−5 | 1.81 × 10−3 | 11.76 | |
| Males | D1Mit403 | 18.54 | 4.99 ± 0.33 (n = 30) | 17.44 | 4.83 ± 0.20 (n = 84) | 13.86 | 4.26 ± 0.34 (n = 28) | 0.235 | NS | – | |
| Lmr5 | Both sexes | D10Mit12 | 18.03 | 4.92 ± 0.22 (n = 62) | 16.19 | 4.65 ± 0.14 (n = 150) | 11.82 | 3.88 ± 0.19 (n = 81) | 5.58 × 10−4 | 3.02 × 10−2 | 4.70 |
| Females | D10Mit12 | 17.25 | 4.81 ± 0.28 (n = 33) | 13.90 | 4.27 ± 0.19 (n = 70) | 11.49 | 3.81 ± 0.24 (n = 47) | 2.98 × 10−2 | NS | – | |
| Males | D10Mit12 | 18.38 | 4.97 ± 0.33 (n = 29) | 18.78 | 5.02 ± 0.20 (n = 80) | 11.92 | 3.90 ± 0.30 (n = 34) | 8.64 × 10−3 | NS | – | |
Mean and SE values were obtained by analysis of variance. In order to obtain normal distribution required for analysis of variance value of IgE in serum (μg/ml) was transformed by natural logarithm of value to the power of 1.5. The numbers in bold give the average non-transformed values. C and S indicate the presence of BALB/c and STS allele, respectively; n indicates number of mice.
Table 6.
Interactions between loci (Lmr20 and Lmr23) that control level of IFNγ in serum in L. major infected F2 hybrids between CcS-11 and BALB/c
| Expl. var. = 6.93% | P = 5.15 × 10−4 | Corr. P = 2.01 × 10−2 | |||||
|---|---|---|---|---|---|---|---|
| D1Mit403 (Lmr20) | |||||||
| D16Mit73 (Lmr23) | CC | CS | SS | ||||
| CC | 14.03 | 1.63 ± 0.11 (n = 11) | 12.05 | 1.58 ± 0.06 (n = 37) | 7.48 | 1.42 ± 0.10 (n = 16) | |
| CS | 15.68 | 1.66 ± 0.06 (n = 35) | 7.89 | 1.44 ± 0.05 (n = 69) | 6.34 | 1.36 ± 0.09 (n = 20) | |
| SS | 4.36 | 1.21 ± 0.11 (n = 12) | 6.75 | 1.38 ± 0.07 (n = 33) | 16.95 | 1.68 ± 0.10 (n = 16) | |
Mean and SE values were obtained by analysis of variance. The linkage significant at whole genome level is given. In order to obtain normal distribution, we used natural logarithm of (IFNγ in serum [ng/ml] to the power of 0.5) and natural logarithm (IL-4 in serum [ng/ml] to the power of 1.1). The numbers in bold give the average non-transformed values. C and S indicate the presence of BALB/c and STS allele, respectively; n indicates number of mice.
Table 7.
Interactions between loci (Lmr21 and Lmr5) that control level of IFNγ in serum in L. major infected F2 hybrids between CcS-11 and BALB/c
| Expl. var. = 7.18% | P = 4.96 × 10−4 | Corr. P = 1.95 × 10−2 | |||||
|---|---|---|---|---|---|---|---|
| D7Mit67 (Lmr21) | |||||||
| D10Mit12 (Lmr5) | CC | CS | SS | ||||
| CC | 4.86 | 1.26 ± 0.09 (n = 18) | 13.66 | 1.62 ± 0.07 (n = 25) | 21.93 | 1.76 ± 0.11 (n = 11) | |
| CS | 11.81 | 1.57 ± 0.06 (n = 34) | 8.86 | 1.48 ± 0.05 (n = 57) | 11.71 | 1.57 ± 0.07 (n = 30) | |
| SS | 11.59 | 1.57 ± 0.09 (n = 16) | 6.71 | 1.38 ± 0.07 (n = 30) | 4.91 | 1.26 ± 0.09 (n = 17) | |
Mean and SE values were obtained by analysis of variance. The linkage significant at whole genome level is given. In order to obtain normal distribution, we used natural logarithm of (IFNγ in serum [ng/ml] to the power of 0.5) and natural logarithm (IL-4 in serum [ng/ml] to the power of 1.1). The numbers in bold give the average non-transformed values. C and S indicate the presence of BALB/c and STS allele, respectively; n indicates number of mice.
Table 8.
Interactions between loci that control level of IL-4 in serum in L. major infected F2 hybrids between CcS-11 and BALB/c
| Expl. var. = 7.71% | P = 7.04 × 10−4 | Corr. P = 2.69 × 10−2 | |||||
|---|---|---|---|---|---|---|---|
| D10Mit12 (Lmr5) | |||||||
| D12Mit37 (Lmr22) | CC | CS | SS | ||||
| CC | 9.04 | 2.38 ± 0.38 (n = 6) | 3.57 | 1.30 ± 0.19 (n = 24) | 5.90 | 1.88 ± 0.23 (n = 17) | |
| CS | 4.57 | 1.58 ± 0.20 (n = 23) | 7.77 | 2.20 ± 0.13 (n = 55) | 4.15 | 1.48 ± 0.21 (n = 20) | |
| SS | 5.10 | 1.71 ± 0.23 (n = 17) | 7.09 | 2.09 ± 0.18 (n = 26) | 3.42 | 1.26 ± 0.24 (n = 15) | |
Mean and SE values were obtained by analysis of variance. The linkage significant at whole genome level is given. In order to obtain normal distribution, we used natural logarithm of (IFNγ in serum [ng/ml] to the power of 0.5) and natural logarithm (IL-4 in serum [ng/ml] to the power of 1.1). The numbers in bold give the average non-transformed values. C and S indicate the presence of BALB/c and STS allele, respectively; n indicates number of mice.
Parasite presence in secondary immune organs is controlled by two loci, Lmr20 and Lmr5 (Table 3). Lmr20 on chromosome 1 is linked to D1Mit403 and it controls parasite load in draining lymph nodes (corrected P = 5.54 × 10−3). Its BALB/c allele is associated with higher parasite numbers. The effects of Lmr20 account for 4.25% of variance in parasite load in the lymph nodes. In addition, there was a pronounced sex effect (P = 10−9) on parasite numbers in draining lymph nodes and spleen. However, there was no interaction between sex and the marker and no sufficient power to establish whether the effects of Lmr20 are sex specific. This qualification concerns the effects of the individual Lmr loci; the sex difference within the two strains is highly significant (Table 1).Lmr5 on chromosome 10 is linked to D10Mit12 and it influences parasite load in spleens and splenomegaly (corrected P = 5.39 × 10−3 and 6.98 × 10−4, respectively). The effects of Lmr5 account for 4.54% of variance in parasite number in spleens and 11.60% of variance in splenomegaly. Higher parasite numbers and a more severe splenomegaly are associated with BALB/c allele. Separate analysis of females and males revealed that Lmr5 determines parasite numbers in spleen and splenomegaly in females (corrected P = 5.76 × 10−3 and 5.57 × 10−4, respectively). It accounts in females for 11.74% and 14.47% of variance in parasite numbers and splenomegaly, respectively, whereas differences in parasite numbers in spleens and splenomegaly in males were not significant (Table 3). However, absence of interaction between sex and marker and power computations indicated that the present experiments did not have sufficient power to establish whether Lmr5 has a sex-specific effect. The present data are compatible with additive effects of sex and genotype at Lmr5.
Skin lesions are controlled by two loci: Lmr5 and Lmr21 (Table 4 and Fig. 2). Lmr5 on chromosome 10 is linked to D10Mit12 and D10Mit46 (corrected P = 5.27 × 10−5 and 4.83 × 10−5, respectively). Lmr21 on chromosome 7 is linked to D7Mit67 and D7Mit282 (lesion size on week 8; corrected P = 0.0116 and 0.0972). BALB/c allele of both Lmr5 and Lmr21 is associated with larger lesions. The impact of Lmr5 and Lmr21 on phenotype is higher in females than in males. In females, Lmr5 on chromosome 10 is linked with D10Mit12 and D10Mit46 (corrected P = 2.67 × 10−4 and 7.28 × 10−4, respectively), and Lmr21 on chromosome 7 is linked with D7Mit67 (corrected P = 0.046). In males, we did not detect Lmr5 or Lmr21.
Fig. 2.

Effects of genotype and sex on size of cutaneous lesion at Lmr5 (a, b) and Lmr21 (c, d) weeks 4 to 8 after infection. The means and standard errors of means (error bars) for each of the three genotypes at different time points are given. The numbers of mice for D10Mit12 were 151 females and 144 males; for D7Mit67 149 females and 139 males. C and S indicate the presence of BALB/c and STS allele, respectively
Components of the systemic immune response: Of the four tested components (serum levels of IgE, IFNγ, IL-4, and IL-12) we did not detect any linkage of IL-12 levels. The linkages of the other three components involve five loci, Lmr20, Lmr21, Lmr5, Lmr22 and Lmr23 (Table 5 single gene effects and Tables 6, 7, and 8 gene interactions). Serum IgE concentrations are influenced by Lmr20 and Lmr5. Lmr20 on chromosome 1 is linked to D1Mit403 (corrected P = 3.07 × 10−3, 6.09% of variance) and its BALB/c allele is associated with higher serum levels of IgE. Influence of this locus on IgE level is observed only in females (corrected P = 1.81 × 10−3, 11.76% of variance). Lmr5 on chromosome 10 is linked to D10Mit12 (corrected P value = 3.02 × 10−2, 4.70% of variance). When analyzed separately, females and males exhibit allelic differences of the same size and direction, but do not reach significant P values due to insufficient number of mice in the separate sexes. Serum IFNγ concentrations are influenced by two inter-locus interactions. Lmr20 marked by D1Mit403 interacts with Lmr23 linked to D16Mit73 (Table 6). The highest level of IFNγ is observed in STS homozygotes in both loci (corrected P = 2.01 × 10−2). This interaction accounts for 6.93% of variance.Serum IFNγ level is also controlled by the interaction of Lmr21 (linked to D7Mit67) with Lmr5 (linked to D10Mit12) (corrected P = 1.95 × 10−2, 7.18% of variance). The lowest IFNγ levels are observed either in STS homozygotes at both loci or in BALB/c homozygotes in both loci (Table 7). A separate statistical evaluation of these interactions in females and males was not possible due to the low numbers of mice in some genotypes, but the interaction effects were similar in both sexes (data not shown).Serum IL-4 concentration is influenced by one inter-locus interaction. Lmr22 marked by D12Mit37 interacts with Lmr5 linked to D10Mit12. The highest and lowest level of IL-4 is observed in BALB/c and STS homozygotes, respectively, at both loci (corr. P = 2.69 × 10−2). This interaction accounts for 7.71% of variance (Table 8).
Sex differences in genetic control of lesion size, splenomegaly, and serum level of IgE were revealed when analyzing the male and female F2 hybrids separately. Influence of sex was high for skin lesions during the whole course of infection (P < 1.68 × 10−5) and IgE level in serum (P = 1.41 × 10−2). The impact of the Lmr loci controlling these parameters appears to be stronger in females than in males. However, this influence of sex was independent of the segregating genes, as there was no significant interaction between sex and any of the tested markers.
Discussion
There is extensive literature about genetic control of L. major-induced disease (Demant et al. 1996; Beebe et al. 1997; Roberts et al. 1997, 1999; Lipoldová et al. 2000; Badalová et al. 2002; Vladimirov et al. 2003; Havelková et al. 2006), but until now there are no reports on the genetics of parasite elimination. However, the genetics of host–pathogen interactions has two important parts: genetics of infection that describes genes that participate in the control of the spread of the infectious agent within the host and genetics of disease development that analyzes the immune and pathological responses of the host. We used genetic dissection to elucidate the functional relationship between the two components of the disease. This study represents the first genome-wide search for loci controlling parasite elimination and dissemination after L. major infection. Our results show that these processes correlate only partly.
Controlling loci and their function We have detected five loci that in the strain CcS-11 control host–parasite interaction (Table 2, Fig. 3). We have found that parasite numbers in lymph nodes and in spleen are controlled independently: the locus Lmr20 on chromosome 1 influences parasite load in draining lymph nodes, whereas parasite load in spleen is determined by the locus Lmr5 on chromosome 10. Lmr20 is not involved in regulation of skin lesion size, splenomegaly, or hepatomegaly, but it influences systemic immunological changes: it controls serum IgE levels in females, and it interacts with Lmr23 on chromosome 16 in regulation of IFNγ level in serum. The control of spread of parasites to spleen is exercised by another locus—Lmr5. In females, Lmr5 controls parasite numbers in spleen and splenomegaly. This locus also influences pathology of another organ (the skin lesions). It has an impact on the systemic immune response by influencing serum IgE level, and in interactions with Lmr21 on chromosome 7 and Lmr22 on chromosome 12, also serum levels of IFNγ and IL-4, respectively. The BALB/c allele of Lmr5 is associated with a larger parasite load, splenomegaly, and skin lesion size as well as higher IgE levels.The second locus that controls development of the skin lesions, Lmr21 on chromosome 7, is not involved in control of parasite load in the lymph nodes nor in the spleen. Loci Lmr22 (chromosome 12) and Lmr23 (chromosome 16) control only systemic immune responses—serum IL-4 and IFNγ level, respectively. They do not affect parasite load, splenomegaly, or skin lesions.It is important to understand that, as in any QTL study, failure to find a linkage between a phenotype and a marker does not rule out that such linkage may exist, although its phenotypic effect are likely smaller than in the detected linkages. So for a QTL, which affects several but not all parameters of a complex disease, this indicates that it has predominant effects on some parameters, although it might modify to a lesser extent other parameters as well.Most loci we describe here affect several phenotypic traits. It is possible that some of these loci contain several linked genes, each affecting a different trait or a different subset of these traits. This is an important issue that needs to be resolved by recombinational analysis, as postulation of separate but linked loci merely on basis of a segregation study could be erroneous because of likely confounding random variations.
Fig. 3.

Position of the loci that control response to L. major in strain CcS-11. The regions of STS and BALB/c origin are represented as dark and white, respectively; the boundary regions of undetermined origin are shaded. Only the markers defining the boundaries the STS-derived segment and the markers that were tested for linkage are shown. The markers that exhibit significant P values (corrected for genome-wide search) are shown in boxes. The arrows indicate the regions of significant linkage
Control of parasite elimination differs between organs The loci Lmr20 and Lmr5 control differentially parasite numbers in lymph nodes and spleen, respectively, suggesting a predominantly organ-specific control of parasite load. This hypothesis is compatible with the mechanistic studies of others. The enzymes inducible nitric oxide synthase and phagocyte NADPH oxidase, which are required for the control of L. major, display organ- and stage-specific anti-Leishmania effects (Stenger et al. 1996; Blos et al. 2003). Inducible nitric oxide synthase has been shown to control resistance to parasites in skin and draining lymph nodes, but not in spleen of resistant strain C57BL/6 (Stenger et al. 1996). On the other hand, activity of phagocyte NADPH oxidase is essential for the clearance of L. major in the spleen, but it is dispensable for the resolution of the acute skin lesions and it exerted only a limited effect on the containment of the parasites in the draining lymph node (Blos et al. 2003). Similarly, the genes Lmr20 and Lmr5 might operate in different pathways of parasite killing or their function might be exerted by mediators produced by cells that influence these pathways.
Control of cytokine levels and its relationship to organ pathology and parasite numbers Several Lmr loci control IFNγ levels in combination with organ or skin pathology and parasite numbers. Different loci control different groups of phenotypes: Lmr5—organ pathology, parasite numbers in spleen, IFNγ level, and IL-4 levels; Lmr20—parasite numbers in lymph nodes and IFNγ level; Lmr21—organ pathology and IFNγ level; Lmr23—IFNγ only (For interaction of these loci see Table 2). This suggests that pathology-controlling and/or parasite-controlling genes Lmr5, Lmr21, and Lmr20 might operate via pathways that involve IFNγ. Some genes, such as Lmr22 and Lmr23, are involved only in control of systemic immune response. These observations are in agreement with our previous detailed data on relationship of levels of IFNγ, IL-12, TNFα, IL-4 and IL-6, and organ pathology (Havelková et al. 2006) that indicated that there is no simple correlation between production of cytokines and development of disease. The influence of individual Lmr loci on several phenotypes can be due either to multiple effects of a single gene or these loci may comprise two or more closely linked genes with different effects.
Sex influence on parasite elimination, organ pathology, and systemic disease Our data show a sex influence on parasite numbers in secondary immune organs. Although strains BALB/c and CcS-11 did not exhibit any sex influence on lesion size, males of strain CcS-11 contained more parasites in spleens than females and males of both strains had much higher parasite load in lymph nodes. Previously, Mock and Nacy (1988) also reported higher parasite numbers in livers of males of BALB/cAnPt, DBA/2N, CDF1 and DBA/2J mice intravenously injected by L. major. These data collectively suggest that some genes controlling susceptibility to L. major might operate differently in the two sexes; however, we did not find instances of significant marker–sex interactions. Lmr20 influences IgE level in serum only in females. Genes controlling infections that appear to be sex dependent have been observed also with other pathogens. For example, Rmp4 (resistance to mouse pox 4) controls susceptibility to ectromelia virus in female mice only (Brownstein and Gras 1995) and Hrl (herpes resistance locus) exhibits higher influence on susceptibility to Herpes simplex virus in male than in female mice (Lundberg et al. 2003). Sex-specific QTLs influence also susceptibility to Theiler’s murine encephalomyelitis virus-induced demyelination: loci Tmved7 and 8 affect male mice only, whereas locus Tmved9 controls susceptibility only in females. Locus Tmved6 operates both in females and males, but it has an opposite effect on disease susceptibility in males and females (Butterfield et al. 2003).
Potential candidate genes The Lmr loci described here are mapped with a precision of 6–30 cM. The regions of the most probable linkage (estimated as the distance of markers that exhibit no significant linkage but are the closest to the markers with linkage) were between 4 and 19 cM long (Fig. 3), which is customary for the newly detected loci (Flint et al. 2005). These broad loci contain each several genes that have been shown previously to influence infection with L. major or that also influence responses to other pathogens as well as some immunological traits (for references, see Tables 9 and 10); however, the effects of many of Lmr loci might be caused by genes that are presently not suspected candidates. Presently, we are producing mice with recombinant haplotypes that carry each Lmr locus in a very short segment on chromosome. The testing of these strains will restrict the present number of the candidate genes to the most likely ones. In summary, we show that L. major parasite load and disease manifestations are controlled by distinct sets of genes. Other genes also determine important immunological and pathological phenotypes of infected mice, but they do not appreciably affect parasite burden. By indicating which of the loci that control immune response and/or organ pathology are involved also in elimination or suppression of the parasite and which are not, these results represent an important step in a progress towards integration of genetic data and results from functional immunological studies.An important corollary of these findings is that in humans there also likely exist polymorphic genes that control numbers of parasites and their organ distribution as well as other genes that affect some components of immune response and/or development or organ pathology but not parasite load. These polymorphisms will result in important individual differences in the “functional architecture” of the disease, its progression being in different individuals driven by different pathways. An apparently identical clinical phenotype in different individuals therefore can result from different functional pathways, and hence could differ in prognosis and response to therapy.
Table 9.
Potential candidate genes for Lmr loci controlling response to L. major in strain CcS-11
| Chr. | Locus | Marker | Position in cM | Potential candidate gene | Position in cM |
|---|---|---|---|---|---|
| Fcer1a, Fc receptor, IgE, high affinity I, alpha polypeptide (Lewis et al. 2004) | 94.2 | ||||
| Fcer1g, Fc receptor, IgE, high affinity I, gamma polypeptide (Lewis et al. 2004) | 93.3 | ||||
| Fcgr2b, Fc receptor, IgG, low affinity Iib (Padigel and Farrell 2005; Woelbing et al. 2006) | 92.3 | ||||
| Cluster of IFN activated genes (Ifi201, Ifi202a, Ifi203, Ifi204, Ifi505) (Blos et al. 2003) | 95.2–95.3 | ||||
| 1 | Lmr20 | D1Mit403 | 100 | Tgfb2 transforming growth factor, beta 2 (Padigel and Farrell 2005) | 101.5 |
| 7 | Lmr21 | D7Mit67 | 63.5 | Il4r, IL-4 receptor alpha chain (CD124) (Gessner et al. 1994) | 62.0 |
| 10 | Lmr5 | D10Mit12 | 56.0 | ||
| D10Mit46 | 63.0 | Ifng, interferon gamma (Nacy et al. 1985) | 67.0 | ||
| 12 | Lmr22 | D12Mit37 | 1.0 | Adam17 (a disintegrin and metalloproteinase domain 17) (Moss et al. 1997) | 3.0 |
| 16 | Lmr23 | D16Mit73 | 8.6 | Two clusters of genes that affect B-cell development (Igl-C5, Igll1, Vpreb1, Vpreb2; Igl-C1, Igl-C2, Igl-C3, Igl-V1, Igl-V2, Igl1-r) (Hoerauf et al. 1996; DeKrey et al. 2003) | 9.8 |
| 13.0 | |||||
Table 10.
Co-mapping with loci controlling other infections and other possibly related QTLs
| Chr. | Locus | Marker | Position in cM | Co-localization with other possibly related QTLs | Position in cM |
|---|---|---|---|---|---|
| Tir3c, trypanosomiasis infection response 3c (Iraqi et al. 2000) | 93.0 | ||||
| Ssta2, susceptibility to Salmonella typhimurium antigens 2 (Trezena et al. 2002) | 95.8 | ||||
| 1 | Lmr20 | D1Mit403 | 100 | Bbaa12, Borrelia burgdorferi-associated arthritis 12 (Roper et al. 2001) | 96.0 |
| berr1, Berghei resistance locus 1 (Bagot et al. 2002) | 102.0 | ||||
| Tria4, T-cell receptor induced activation 4 (Havelková et al. 1999) | 46.0 | ||||
| Heal6, wound healing/regeneration 6 (Mcbrearty et al. 1998) | 52.4 | ||||
| Tsiq1, T-cell secretion of IL-4 QTL1 (Choi et al. 2005) | 59.0 | ||||
| 7 | Lmr21 | D7Mit67 | 63.5 | Dice2, determination of interleukin 4 commitment 2 (Bix et al. 1998) | 63.5 |
| 12 | Lmr22 | D12Mit37 | 1.0 | Cinda2, cytokine-induced activation (Krulová et al. 1997) | 1.0 |
| Cd4ts5, CD4 T-cell subset 5 (Jackson et al. 2003) | 6.0 | ||||
| 16 | Lmr23 | D16Mit73 | 8.6 | Dice1, determination of interleukin 4 commitment 1 (Bix et al. 1998; Baguet et al. 2004) | 8.0 |
Acknowledgement
Support for this work was provided by the Grant Agency of the Czech Republic (grant 310/06/1745), the European Commission (INTAS Genomics 05-1000004-7761), Academy of Sciences of the Czech Republic (project grant AVOZ50520514), and Ministry of Education of the Czech Republic (project grant LC06009). The work of P. D., L. Q., and A. H. was supported by Roswell Park Cancer Institute’s Institutional Funds.
Open Access This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
References
- Badalová J, Svobodová M, Havelková H, Vladimirov V, Vojtíšková J et al (2002) Separation and mapping of multiple genes that control IgE level in Leishmania major infected mice. Genes Immun 3:187–195 [DOI] [PubMed]
- Bagot S, Campino S, Penha-Goncalves C, Pied S, Cazenave PA et al (2002) Identification of two cerebral malaria resistance loci using an inbred wild-derived mouse strain. Proc Natl Acad Sci U S A 99:9919–9923 [DOI] [PMC free article] [PubMed]
- Baguet A, Epler J, Wen Kwun W, Bix M (2004) A Leishmania major response locus identified by interval-specific congenic mapping of a T helper type 2 cell bias-controlling quantitative trait locus. J Exp Med 200:1605–1612 [DOI] [PMC free article] [PubMed]
- Beebe AM, Mauze S, Schork NJ, Coffman RL (1997) Serial backcross mapping of multiple loci associated with resistance to Leishmania major in mice. Immunity 6:551–557 [DOI] [PubMed]
- Bix M, Wang ZE, Thiel B, Schork NJ, Locksley RM (1998) Genetic regulation of commitment to interleukin 4 production by a CD4(+) T cell-intrinsic mechanism. J Exp Med 188:2289–2299 [DOI] [PMC free article] [PubMed]
- Blos M, Schleicher U, Soares Rocha FJ, Meissner U, Rollinghoff M et al (2003) Organ-specific and stage-dependent control of Leishmania major infection by inducible nitric oxide synthase and phagocyte NADPH oxidase. Eur J Immunol 33:1224–1234 [DOI] [PubMed]
- Brownstein DG, Gras L (1995) Chromosome mapping of Rmp-4, a gonad-dependent gene encoding host resistance to mousepox. J Virol 69:6958–6964 [DOI] [PMC free article] [PubMed]
- Butterfield RJ, Roper RJ, Rhein DM, Melvold RW, Haynes L et al (2003) Sex-specific quantitative trait loci govern susceptibility to Theiler's murine encephalomyelitis virus-induced demyelination. Genetics 163:1041–1046 [DOI] [PMC free article] [PubMed]
- Choi P, Xanthaki D, Rose SJ, Haywood M, Reiser H et al (2005) Linkage analysis of the genetic determinants of T-cell IL-4 secretion, and identification of Flj20274 as a putative candidate gene. Genes Immun 6:290–297 [DOI] [PubMed]
- DeKrey GK, Jones JJ, Mbow ML, Brodskyn CI, Titus RG (2003) Short report: Requirement of B cells for delayed type hypersensitivity-like pathology after secondary infection with Leishmania major in resistant C57BL/6 mice. Am J Trop Med Hyg 69:481–483 [PubMed]
- Demant P, Lipoldová M, Svobodová M (1996) Resistance to Leishmania major in mice. Science 274: 1392–1393 [DOI] [PubMed]
- Diamond LS, Herman CM (1954) Incidence of trypanosomes in the Canada goose as revealed by bone marrow culture. J Parasitol 40:195–202 [DOI]
- Flint J, Valdar W, Shifman S, Mott R (2005) Strategies for mapping and cloning quantitative trait genes in rodents. Nat Rev Genet 6:271–286 [DOI] [PubMed]
- Gessner A, Schroppel K, Will A, Enssle KH, Lauffer L et al (1994) Recombinant soluble interleukin-4 (IL-4) receptor acts as an antagonist of IL-4 in murine cutaneous leishmaniasis. Infect Immun 62:4112–4117 [DOI] [PMC free article] [PubMed]
- Havelková H, Kosařová M, Krulová M, Demant P, Lipoldová M (1999) T-cell proliferative response is controlled by loci Tria4 and Tria5 on mouse chromosomes 7 and 9. Mamm Genome 10:670–674 [DOI] [PubMed]
- Havelková H, Badalová J, Svobodová M, Vojtíšková J, Kurey I et al (2006) Genetics of susceptibility to leishmaniasis in mice: four novel loci and functional heterogeneity of gene effects. Genes Immun 7:220–233 [DOI] [PubMed]
- Hoerauf A, Rollinghoff M, Solbach W (1996) Co-transfer of B cells converts resistance into susceptibility in T cell-reconstituted, Leishmania major-resistant C.B-17 scid mice by a non-cognate mechanism. Int Immunol 8:1569–1575 [DOI] [PubMed]
- Iraqi F, Clapcott SJ, Kumari P, Haley CS, Kemp SJ et al (2000) Fine mapping of trypanosomiasis resistance loci in murine advanced intercross lines. Mamm Genome 11:645–648 [DOI] [PubMed]
- Jackson AU, Galecki AT, Burke DT, Miller RA (2003) Genetic polymorphisms in mouse genes regulating age-sensitive and age-stable T cell subsets. Genes Immun 4:30–39 [DOI] [PMC free article] [PubMed]
- Johnson J, Suzuki Y, Mack D, Mui E, Estes R et al (2002) Genetic analysis of influences on survival following Toxoplasma gondii infection. Int J Parasitol 32:179–185 [DOI] [PubMed]
- Krulová M, Havelková H, Kosařová M, Holáň V, Hart AAM et al (1997) IL-2-induced proliferative response is controlled by loci Cinda1 and Cinda2 on mouse chromosomes 11 and 12: a distinct control of the response induced by different IL-2 concentrations. Genomic 42:11–15 [DOI] [PubMed]
- Lander E, Botstein D (1989) Mapping Mendelian factors underlying quantitative traits using RFLP linkage maps. Genetics 121:185–199 [DOI] [PMC free article] [PubMed]
- Lander ES, Schork NJ (1994) Genetic dissection of complex traits. Science 265:2037–2048 [DOI] [PubMed]
- Lewis G, Rapsomaniki E, Bouriez T, Crockford T, Ferry H et al (2004) Hyper IgE in New Zealand black mice due to a dominant-negative CD23 mutation. Immunogenetics 56:564–571 [DOI] [PubMed]
- Lipoldová M, Demant P (2006) Genetic susceptibility to infectious disease: lessons from mouse models of leishmaniasis. Nat Rev Genet 7:294–305 [DOI] [PubMed]
- Lipoldová M, Svobodová M, Krulová M, Havelková H, Badalová J et al (2000) Susceptibility to Leishmania major infection in mice: multiple loci and heterogeneity of immunopathological phenotypes. Genes Immun 1:200–206 [DOI] [PubMed]
- Lipoldová M, Svobodová M, Havelková H, Krulová M, Badalová J et al (2002) Mouse genetic model for clinical and immunological heterogeneity of leishmaniasis. Immunogenetics 54:174–183 [DOI] [PubMed]
- Lundberg P, Welander P, Openshaw H, Nalbandian C, Edwards C et al (2003) A locus on mouse chromosome 6 that determines resistance to herpes simplex virus also influences reactivation, while an unlinked locus augments resistance of female mice. J Virol 77:11661–11673 [DOI] [PMC free article] [PubMed]
- Ma Y, Seiler KP, Eichwald EJ, Weis JH, Teuscher C et al (1998) Distinct characteristics of resistance to Borrelia burgdorferi-induced arthritis in C57BL/6N mice. Infect Immun 66:161–168 [DOI] [PMC free article] [PubMed]
- Marinho CR, Bucci DZ, Dagli ML, Bastos KR, Grisotto MG et al (2004) Pathology affects different organs in two mouse strains chronically infected by a Trypanosoma cruzi clone: a model for genetic studies of Chagas' disease. Infect Immun 72:2350–2357 [DOI] [PMC free article] [PubMed]
- McBrearty BA, Clark LD, Zhang XM, Blankenhorn EP, Heber-Katz E (1998) Genetic analysis of a mammalian wound-healing trait. Proc Natl Acad Sci U S A 95:11792–11797 [DOI] [PMC free article] [PubMed]
- McMahon-Pratt D, Alexander J (2004) Does the Leishmania major paradigm of pathogenesis and protection hold for New World cutaneous leishmaniases or the visceral disease? Immunol Rev 201:206–224 [DOI] [PubMed]
- Mock BA, Nacy CA (1988) Hormonal modulation of sex differences in resistance to Leishmania major systemic infections. Infect Immun 56:3316–3319 [DOI] [PMC free article] [PubMed]
- Moss ML, Jin SL, Milla ME, Bickett DM, Burkhart W et al (1997) Cloning of a disintegrin metalloproteinase that processes precursor tumour-necrosis factor-alpha. Nature 385:733–736 [DOI] [PubMed]
- Nacy CA, Fortier AH, Meltzer MS, Buchmeier NA, Schreiber RD (1985) Macrophage activation to kill Leishmania major: activation of macrophage for intracellular destruction of amastigotes can be induced by both recombinant interferon- and non-interferon lymphokines. J Immunol 135:3505–3511 [PubMed]
- Olivier M, Gregory DJ, Forget G (2005) Subversion mechanisms by which Leishmania parasites can escape the host immune response: a signaling point of view. Clin Microbiol Rev 18:293–305 [DOI] [PMC free article] [PubMed]
- Padigel UM, Farrell JP (2005) Control of infection with Leishmania major in susceptible BALB/c mice lacking the common (gamma)-chain for FcR is associated with reduced production of IL-10 and TGF-beta by parasitized cells. J Immunol 174:6340–6345 [DOI] [PubMed]
- Råberg L, Sim D, Read AF (2007) Disentangling genetic variation for resistance and tolerance to infectious diseases in animals. Science 318:812–814 [DOI] [PubMed]
- Roberts LJ, Baldwin TM, Curtis JM, Handman E, Foote SJ (1997) Resistance to Leishmania major is linked to H2 region on chromosome 17 and to chromosome 9. J Exp Med 9:1705–1710 [DOI] [PMC free article] [PubMed]
- Roberts LJ, Baldwin TM, Speed TP, Handman E, Foote SJ (1999) Chromosomes X, 9, and the H2 locus interact epistatically to control Leishmania major infection. Eur J Immunol 29:3047–3050 [DOI] [PubMed]
- Rodgers MR, Popper SJ, Wirth DF (1990) Amplification of kinetoplast DNA as a tool in the detection and diagnosis of Leishmania. Exp Parasitol 71:267–275 [DOI] [PubMed]
- Roper RJ, Weis JJ, McCracken BA, Green CB, Ma Y et al (2001) Genetic control of susceptibility to experimental Lyme arthritis is polygenic and exhibits consistent linkage to multiple loci on chromosome 5 in four independent mouse crosses. Genes Immun 2:388–397 [DOI] [PubMed]
- Sacks D, Noben-Trauth N (2002) The immunology of susceptibility and resistance to Leishmania major in mice. Nat Rev Immunol 2:845–858 [DOI] [PubMed]
- Singer JB, Hill AE, Nadeau JH, Lander ES (2005) Mapping quantitative trait loci for anxiety in chromosome substitution strains of mice. Genetics 169:855–862 [DOI] [PMC free article] [PubMed]
- Stassen APM, Groot PC, Eppig JT, Demant P (1996) Genetic composition of the recombinant congenic strains. Mamm Genome 7:55–58 [DOI] [PubMed]
- Stenger S, Donhauser N, Thuring H, Rollinghoff M, Bogdan C (1996) Reactivation of latent leishmaniasis by inhibition of inducible nitric oxide synthase. J Exp Med 183:1501–1514 [DOI] [PMC free article] [PubMed]
- Trezena AG, Souza CM, Borrego A, Massa S, Siqueira M et al (2002) Co-localization of quantitative trait loci regulating resistance to Salmonella typhimurium infection and specific antibody production phenotypes. Microbes Infect 4:1409–1415 [DOI] [PubMed]
- Tripodis N, Hart AA, Fijneman RJ, Demant P (2001) Complexity of lung tumor modifiers: mapping of thirty genes and twenty five interactions in half of the mouse genome. J Natl Cancer Inst 93:1484–1489 [DOI] [PubMed]
- Vladimirov V, Badalová J, Svobodová M, Havelková H, Hart AAM et al (2003) Different genetic control of cutaneous and visceral disease after Leishmania major infection in mouse. Infect Immun 71:2041–2046 [DOI] [PMC free article] [PubMed]
- Woelbing F, Kostka SL, Moelle K, Belkaid Y, Sunderkoetter C et al (2006) Uptake of Leishmania major by dendritic cells is mediated by Fc(gamma) receptors and facilitates acquisition of protective immunity. J Exp Med 203:177–188 [DOI] [PMC free article] [PubMed]