

Abstract
Post-translational histone modifications, such as acetylation, phosphorylation, ubiquitination, and methylation, have been correlated with regulation of gene expression. In Saccharomyces cerevisiae, Set1 has been identified as the sole histone methyltransferase required for histone H3 lysine 4 (Lys4) methylation. Yeast cells that do not express Set1 have several apparent phenotypes, including slow growth and defects in telomere, HML, and rDNA silencing. However, the mechanism by which the Set1 methyltransferase mediates differential histone H3 methylation (mono-, di-, and tri-) is still not understood, and the involvement of domains or regions in Set1 contributing to H3 Lys4 methylation has not been well characterized. In this study, the N terminus of Set1 was shown to be important for global and gene specific histone H3 trimethylation. We show that Set1 trimethyl-defective mutants can rescue a set1Δ slow growth defect. In contrast, Set1 trimethyl mutants were defective in telomere, rDNA, HML, and HMR silencing. Taken together, these data suggest that histone H3 Lys4 trimethylation is required for proper silencing, while mono- and/or dimethylation is sufficient for cell growth.
The existence of histone methylation has been known for over 30 years (1) and recently site specific histone methylation and the corresponding methyltransferases have been identified (2, 3). The catalytic core for most histone lysine methyltransferases resides in the SET domain (2, 4). The SET domain is an evolutionarily conserved motif, with homologues present in organisms ranging from yeast to humans (5). In Saccharomyces cerevisiae, Set1 is the sole methyltransferase responsible for histone H3 lysine 4 (H3 Lys4) methylation and catalyzes the addition of up to three methyl groups to its substrate resulting in mono-, di-, or trimethylated lysine 4 (6-10). Cells that do not express Set1 have several apparent phenotypes, including slow growth and defects in telomere, HML, and rDNA silencing (6, 10-13).
Purification of Set1 indicates that Set1 is in a high molecular weight complex with seven other proteins (Swd1, Swd2, Swd3, Bre2, Sdc1, Spp1, and Shg1) and together with Set1 is referred to as COMPASS or SET1C (8, 9, 14). Several of the Set1-associated proteins are also critical for Set1-mediated histone H3 Lys4 methylation suggesting that their interactions with Set1 regulate enzymatic activity or targeting of Set1 to chromatin (8-10). More recently, human homologues of Set1 (MLL1, MLL2, and KIAA0339) have also been purified as protein complexes and have been shown to have histone H3 Lys4 methyltransferase activity (15-18). Furthermore, several of their complex members have been identified as the human homologues of the proteins associated with Set1 (14, 16, 17).
In budding yeast, H3 Lys4 trimethylation and Set1 are highly concentrated at chromatin near the 5′ ends of open reading frames and is associated with transcriptionally active chromatin (7, 19, 20). In contrast, H3 Lys4 dimethylation can be located at intergenic regions, promoters, and open reading frames (13, 19, 21). Therefore, it is thought that H3 Lys4 dimethylation is more global and is located at regions of chromatin that are transcriptionally active, competent for transcription, or transcriptionally repressed (7, 13, 19-21). These results indicate that the different forms of H3 Lys4 methylation may have distinct and/or overlapping biological roles. However, the mechanism by which Set1 methyltransferase differentially methylates H3 (mono-, di-, and tri-) is still not understood, and potential domains within Set1 that regulate H3 Lys4 methylation have not been well characterized.
Set1 and COMPASS subunits have also been found to interact with the CTD of RNA polymerase II when it is phosphorylated at serine 5, and this interaction is dependent on the Paf1 elongation complex (19, 21). In addition, yeast deletion strains of Paf1 complex members lack both H3 Lys4 and Lys79 methylation (19, 21, 22). Based on these observations and chromatin localization studies of Set1 and Lys4 methylation, it has been proposed that H3 Lys4 methylation may play a role in transcription activation and/or elongation (19, 21). However, the exact mechanism by which Set1 and H3 Lys4 methylation functions in these processes is still poorly understood.
In this study, we generated N-terminal Set1 deletion mutants that lack global and gene specific H3 Lys4 trimethylation while maintaining normal levels of mono- and dimethylated H3 Lys4. We determined that the putative RNA recognition motif (RRM) domain in the N terminus of Set1 is needed for histone H3 Lys4 trimethylation. Using Set1 trimethyl-defective mutants, we showed that histone H3 Lys4 trimethylation alone is required for proper silencing of telomeres, rDNA, HML, and HMR loci, while only mono- and dimethylation were sufficient for proper cell growth suggesting that the different forms of histone H3 Lys4 methylation are playing distinct biological roles.
MATERIALS AND METHODS
Yeast Plasmids and Strains
Construction of the plasmid expressing FLAG-tagged full-length Set1 (1–1080) and Set1 (780–1080) has been described previously (6). The N-terminal truncation of Set1 (829–1080) was made by PCR amplification of the desired region of Set1 using the full-length SET1 plasmid as template. All constructs were engineered with a single FLAG epitope at the N terminus and subcloned into a yeast expression plasmid under the control of the ADH1 promoter and containing a URA3 or TRP1 selectable marker. The RRM deletion mutant (1–1080 ΔRRM) was made via site-directed deletion using the Stratagene Quikchange site-directed mutagenesis kit. Briefly, complementary oligonucleotides were designed that had homology both to the regions immediately upstream and downstream of the region to be deleted. PCR amplification of the template (Set1 1–1080) and subsequent steps were performed as described by the manufacturer. The following strains were used: MBY1198 (MATα his3Δ200 ade2∷hisG leu2Δ0 ura2Δ0 met15Δ0 trp1Δ63 Ty1his3AI-236r, Ty1ade2AI515), MBY1217 (MATα his3Δ200 ade2∷hisG leu2Δ0 ura2Δ0 met15Δ0 trp1Δ63 Ty1his3AI-236r, Ty1ade2AI515 set1Δ∷TRP1) (6), MSY421 (MATα ura3–52 leu2–3,112 trp1 his3 Δ[HHT1-HHF1 ] Δ[HHT2-HHF2 ] pMS329 copy I (URA3, HHT1-HHF-1) (23), MBY1587 (MATα ura3–52 leu2–3,112 trp1 his3 Δ[HHT1-HHF1] Δ[HHT2-HHF2] pMS329 copy I (URA3, HHT1-HHF-1) set1Δ∷KanMX4) (6), UCC506 (MATa ade2–101 his3-Δ200 leu2-Δ1 lys2–801 trp1-Δ1 ura3–52 URA∷Tel-V-R) (24) UCC506 set1Δ (isogenic to UCC506, containing set1Δ∷KanMX4; this study). UCC7262 (MATa ade2 his3 leu2 lys2 ura3 hhf1-hht1∷LEU2 hhf2-hht2∷MET15 ADE2-TEL-VR hmr∷URA3,pMP9) (25), UCC7262 set1Δ (isogenic to UCC7262, containing set1Δ∷KanMX4; this study). UCC7266 (MATa ade2 his3 leu2 lys2 ura3 hhf1-hht1∷LEU2 hhf2-hht2∷MET15 ADE2-TEL-VR hml∷URA3,pMP9) (25), UCC7266 set1Δ (isogenic to UCC7262, containing set1Δ∷KanMX4; this study). UCC1188 (MATα leu2-Δ1 lys2–801 trp1 ura3 hhf1-hht1∷LEU2 hhf2-hht2∷HIS3 RDN1∷URA3,pMP9) (25), UCC1188 set1Δ (isogenic to UCC1188, containing set1Δ∷KanMX4; this study).
Preparation of Yeast Whole Cell Extracts and Immunoprecipitations
For analysis of yeast histones, yeast whole cell extracts were prepared as follows: 5-ml cultures of yeast were grown to mid-log phase (A600 = 1.0). Cells were harvested, washed with water and resuspended in 250 μl of 2 m NaOH with 8% β-mercaptoethanol. Cells were incubated on ice for 5 min and then pelleted at 13,000 rpm for 2 min at 4 °C. Cell pellets were resuspended gently in 250 μl of Buffer A (40 mm HEPES-KOH, pH 7.5, 350 mm NaCl, 0.1% Tween 20, 10% glycerol, 1 μg/ml leupeptin, aprotinin, and pepstatin A, 1 mm phenylmethylsulfonyl fluoride) and pelleted as described above. Cell pellets were resuspended in 180 μl of 2× SDS-sample buffer. Five μl or 10 μl of each sample were loaded per lane for Western blotting. For immunoprecipitation of yeast Set1, 50-ml cultures of MBY1198 and MBY1217 expressing blank vector or the indicated FLAG-Set1 (1–1080) or FLAG-tagged Set1 mutants (1–1080 ΔRRM, 780–1080 and 829–1080) were grown to mid-log phase and harvested. Cells were washed with water, resuspended in Buffer A (see above) and lysed with glass beads using a mini-bead beater (Biospec Products). Lysates were clarified by centrifugation and removed to a new tube. M2 resin (Sigma) was added (10 μl) to each lysate and rotated at 4 °C for 2 h. M2 resin was pelleted by spinning at 3000 rpm in a microcentrifuge and washed two times for 5 min each with 1 ml of Buffer A. Immunoprecipitated Set1 was eluted from the M2-resin by the addition of 10 μl of 2× SDS-Laemmli sample buffer.
Electrophoresis and Western Blotting
Western blot analysis to detect methylated histones was performed as described previously (6). To detect FLAG-Set1, immunoprecipitates were resolved on 8% or 10% SDS-PAGE gels, transferred to PVDF1 membrane, and immunoblotted with monoclonal anti-FLAG antibody. Western blots directed against glyceraldehyde-3-phosphate dehydrogenase were also performed on the IP input to control for protein levels (data not shown).
Chromatin Immunoprecipitations
For analysis of PYK1, 100-ml cultures were grown as described (19). Briefly, cultures were grown in YP-ethanol (1% yeast extract, 2% peptone, 2% ethanol) to mid-log phase (A600 = 1.0) and split. Cells were washed and resuspended in either 50 ml of YP-ethanol or YPD (1% yeast extract, 2% peptone, 2% glucose) to induce expression of PYK1. Cells were induced for 1.5 h, harvested, and cross-linked with 1% formaldehyde. For analysis of GAL10, cultures were grown as described previously (26). Briefly, 100-ml cultures were grown in SC media (0.67% (w/v) yeast nitrogen base supplemented with amino acids, 2% glucose) lacking uracil overnight and split. Cells were harvested and resuspended in either SC-Ura or SC-Ura containing 0.5% glucose and 2% galactose. Cells were induced for 4 h, harvested, and cross-linked with 1% formaldehyde.
Chromatin immunoprecipitations were performed as described previously using H3 Lys4 dimethyl- and H3 Lys4 trimethyl-specific antibodies (Upstate Biotechnology) (27). Immunoprecipitated DNA was analyzed by PCR using primers that have been described previously to analyze the promoter, 5′ and 3′ regions of PYK1 and GAL10 (19). Two additional oligonucleotides were designed to analyze a region of the GAL10 promoter (−290 to −40 bp). Their sequences are as follows: 5′-CACGGAGGAGAGTCTTCCGTCGGAG-3′ and 5′-GGACGCAAAGAAGTTTAATAATCAT-3′.
Growth and Silencing Assay
Growth assays were performed as follows. Yeast strains MSY421 and MBY1587 were transformed with plasmids to express wild type, epitope-tagged, FLAG-Set1 (1–1080), FLAG-Set1 ΔRRM (1–1080 ΔRRM), FLAG-Set1 (780–1080), FLAG-Set1 (829–1080), or blank vector. Cells were grown to mid-log phase (A600 = 1.0), serially diluted 5-fold, and plated on SC-Trp plates. Cells were photographed after 36 h at 30 °C. Telomere silencing assays were performed as described previously (11). Briefly, strains UCC506 (MATa ade2–101 his3-Δ200 leu2-Δ1 lys2–801 trp1-Δ1 ura3–52 URA∷Tel-V-R) and UCC506 set1Δ were transformed with the above plasmids. Individual isolates were grown 3 days to saturation in SC-Trp, normalized for A600, serially diluted (2-fold), and spotted (5 μl/spot) on SC-Trp or SC-Trp plates containing 5-fluoroorotic acid (5-FOA) (100 μg/ml, Bio 101, Inc.). Cell growth was monitored over time at 30 °C. Cells on SC-Trp plates were photographed after 24 h, and cells on SC-Trp + 5-FOA were photographed after 60 h. rDNA, HML, and HMR silencing were performed as described previously (13, 25). For rDNA silencing cells were grown 5 days to saturation, normalized for A600, serially diluted (4-fold), and spotted on SC-Trp or SC-Trp plates containing 5-FOA (100 μg/ml, Bio 101, Inc.). For HML and HMR silencing cells were grown overnight, diluted (5-fold), and spotted as described above. Cells on SC-Trp plates were photographed after 24 h. 5-FOA plates were photographed at 48 h.
RESULTS AND DISCUSSION
To investigate the importance of the N terminus of Set1 in regulating H3 Lys4 methylation, two FLAG-tagged N-terminal set1 deletion constructs (780–1080 and 829–1080) were generated (Fig. 1A) and expressed in a yeast strain in which the endogenous SET1 was deleted (set1Δ). To determine the histone H3 Lys4 methylation status in these cells, yeast set1Δ strains containing blank plasmid (Vector) and the two N-terminal set1 deletion constructs (780–1080 and 829–1080) were grown to an A600 = 1.0 and extracted with 2× SDS-Laemmli sample buffer. Clarified supernatants were run on SDS-polyacrylamide gels, transferred to a PVDF membrane, and probed with methyl-specific antibodies (Upstate Biotechnology) to mono-, di-, and trimethylated forms of histone H3 Lys4. As shown in Fig. 1B, H3 Lys4 trimethylation was nearly abolished in set1Δ strains expressing N-terminal set1 deletion constructs (780–1080 and 829–1080). Trace amounts of H3 Lys4 trimethylation were detected after longer exposure time to film (Fig. 1B). Surprisingly, H3 Lys4 mono- and dimethylation were restored to similar levels to that of a wild-type isogenic strain, indicating that the N terminus of Set1 is specifically required for H3 Lys4 trimethylation. To confirm that the Set1 N-terminal deletion mutants (780–1080 and 829–1080) were expressed, mutant yeast strains were grown to an A600 = 1.0 and lysed using glass beads. Clarified supernatants were immunoprecipitated with an anti-FLAG affinity resin (M2 resin, Sigma). Immunoprecipitates were run on SDS-polyacrylamide gels, transferred to a PVDF membrane, and probed with anti-FLAG monoclonal antibodies (M2, Sigma). Western blots indicated equal expression of the two N-terminal set1 deletion constructs (780–1080 and 829–1080) (Fig. 1B). No FLAG-Set1 deletion mutants were detected in the unbound fraction (data not shown).
Fig. 1. The N terminus of Set1 is required for global histone H3 Lys4 trimethylation.

A, schematic representation of Set1 and Set1 deletion constructs used in this study. Amino acid positions of the RRM domain, n-SET, SET, and post-SET domains are indicated. All constructs are N-terminally tagged with a single FLAG epitope. B and C, Western blots using methyl-specific antibodies shows the methylation status of H3 Lys4 in yeast strains expressing full-length Set1 (1–1080) and Set1 mutants (1–1080 ΔRRM, 780–1080, and 829–1080). Long exposure (Long Exp.) represents the same trimethyl immunoblot (above) but 15× longer exposure to film. Antibodies directed against general histone H3 (Abcam) were used as a loading control. Anti-FLAG immunoblots were performed to examine the protein levels of the Set1 mutants.
Besides the n-SET, SET, and post-SET domains of Set1, which are required for methyltransferase activity, the only other known domain found in Set1 is its putative RRM. The RRM is a domain commonly found in RNA-binding proteins and has been implicated in binding RNA, single-stranded DNA, as well as proteins (28). To test whether the putative RRM domain might be the region in the N terminus of Set1 that regulates H3 Lys4 trimethylation, we constructed a FLAG-tagged Set1 expression construct that lacked this domain (Fig. 1A). This deletion construct was expressed in the set1Δ strain and examined for H3 Lys4 methylation status. As shown in Fig. 1C, the Set1 ΔRRM (1–1080 ΔRRM) mutant restored H3 Lys4 mono- and dimethylation levels similar to wild-type cells and full-length Set1 (1–1080). Similar to the N-terminal Set1 deletion mutants (780–1080 and 829–1080), H3 Lys4 trimethylation was nearly abolished in the Set1 ΔRRM (1–1080 ΔRRM) mutant strain (Fig. 1C). Again, longer exposure time to film indicated the presence of trace amounts of H3 Lys4 trimethylation (Fig. 1C). Together these data suggest the RRM domain is at least one domain within the N terminus of Set1 that is needed for trimethylation. In further support of our data, Schlichter and Cairns (29) recently published a similar result showing that the RRM domain of Set1 is required for H3 Lys4 trimethylation. To determine the levels of protein expression, full-length FLAG-Set1 (1–1080) and FLAG-Set1 ΔRRM (1–1080 ΔRRM) were immunoprecipitated from whole cell extracts and immunoblotted with anti-FLAG antibodies (Fig. 1C). Our results showed expression of both full-length Set1 (1–1080) and Set1 ΔRRM (1–1080 ΔRRM). However, lower levels of Set1 ΔRRM (1–1080 ΔRRM) mutant protein were detected when compared with full-length Set1 (1–1080) (Fig. 1C). Again, no FLAG-Set1 (1–1080) or Set1 ΔRRM (1–1080 ΔRRM) was detected in the unbound fraction (data not shown). Together these data may indicate that the Set1 ΔRRM (1–1080 ΔRRM) mutant protein is less stable. Interestingly, Swd2, a Set1-associated protein, has been implicated in the protein stability of Set1 suggesting the possibility that Swd2 or other Set1-associated proteins (Swd1, Swd3, Spp1, Bre2, Sdc1, or Shg1) interact with the RRM domain to stabilize the protein levels of Set1 (30).
To examine the effects Set1 trimethyl-deficient mutants at the gene level, chromatin immunoprecipitations were preformed using H3 Lys4 di- and trimethyl-specific antibodies. The GAL10 and PYK1 loci, which are known targets of Set1 and H3 Lys4 methylation, were examined (19, 31, 32). A complete loss of H3 Lys4 trimethylation was observed at these loci in our set1Δ strain expressing Set1 829–1080 under conditions in which GAL10 and PYK1 were either induced (Fig. 2) or uninduced (data not shown). Importantly, the level of H3 Lys4 dimethylation was similar to that of wild type cells (Fig. 2). Together these data suggest that our Set1 trimethyl-deficient mutants are still targeted to chromatin and are competent to dimethylate chromatin templates.
Fig. 2. Set1 trimethyl-defective mutants show gene-specific loss of H3 Lys4 trimethylation but not H3 Lys4 dimethylation.
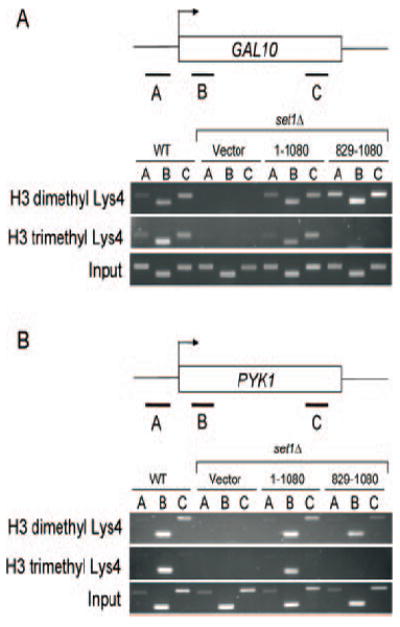
Chromatin immunoprecipitations were performed at GAL10 and PYK1 using antibodies specific to di- or trimethylated Lys4. A and B, chromatin immunoprecipitation eluates were examined by PCR at the indicated regions (A–C) and analyzed by agarose gel electrophoresis. Chromatin immunoprecipitation data shown represent both PYK1 and GAL10 under induced conditions.
With the establishment of Set1 mutants that are defective for H3 Lys4 trimethylation but not mono- or dimethylation, we wanted to determine whether differential methylation of H3 Lys4 played a distinct biological function. Deletion of SET1 in S. cerevisiae leads to the loss of telomere, HML, and rDNA silencing (6, 8, 10-13). In addition, some yeast strains deleted for SET1 also demonstrate a slow growth phenotype (6). To examine the extent of H3 Lys4 methylation (mono-, di-, or trimethylation) associated with these known set1Δ phenotypes, we expressed full-length Set1 (1–1080), Set1 ΔRRM (1–1080 ΔRRM), and both N-terminal Set1 deletions (780–1080 and 829–1080) in set1Δ strains that either have growth or silencing defects (6, 8, 10, 11).
To determine whether slow growth is the result of a loss in H3 Lys4 mono-, di-, or trimethylation, the yeast strain background, MSY421, was used. This strain was previously shown to have a significant growth defect when SET1 was deleted (6). Both full-length Set1 (1–1080), Set1 ΔRRM (1–1080 ΔRRM), and the two N-terminal Set1 deletion mutants (780–1080 and 829–1080) were expressed in the MSY421 set1Δ strain. All cells were grown to A600 = 1.0, and 5-fold serial dilutions were spotted on plates containing synthetic complete media lacking uracil (SC-Ura, Bio 101, Inc.) and incubated at 30 °C. Interestingly, set1Δ strains expressing Set1 trimethylation mutants (1–1080 ΔRRM, 780–1080, and 829–1080) rescued the slow growth phenotype similar to that of full-length Set1 (Fig. 3). Furthermore, these data would suggest that mono- or dimethylation of H3 Lys4 or a combination of both is sufficient for proper cell growth.
Fig. 3. The N terminus of Set1 is not essential for cell growth.

Yeast strains MSY421 and MBY1587 were transformed with the indicated plasmids to determine whether H3 Lys4 trimethylation is essential for growth. Cells were grown to mid-log phase (A600 = 1.0), serially diluted 5-fold, and plated on SC-Trp plates. Cells were photographed after 36 h of incubation at 30 °C.
To determine whether the observed loss of telomere silencing in a set1Δ strain is the result of losing H3 Lys4 mono-, di-, or trimethylation, SET1 was deleted in a strain where a URA3 gene has been integrated at a subtelomere locus (24). Surprisingly, we observed that the set1Δ cells expressing Set1 ΔRRM (1–1080 ΔRRM) or N-terminal Set1 deletions (780–1080 and 829–1080) showed a dramatic sensitivity to 5-FOA (Bio 101, Inc.) as compared with both wild-type cells transformed with blank plasmid (Vector) or set1Δ cells expressing full-length Set1 (1–1080) (Fig. 4). Interestingly, cells expressing Set1 trimethylation mutants (1–1080 ΔRRM, 780–1080, and 829–1080) will eventually grow on 5-FOA plates, albeit in a diminished capacity, upon longer incubation periods at 30 °C (data not shown). This is consistent with our data indicating trace amount of H3 Lys4 trimethylation in cells expressing trimethyl-defective mutants (Fig. 1, B and C). Since these cells contain proper H3 Lys4 mono- and dimethylation, our data suggest that H3 Lys4 trimethylation is needed for proper telomere silencing.
Fig. 4. The N terminus of Set1 is required for silencing.
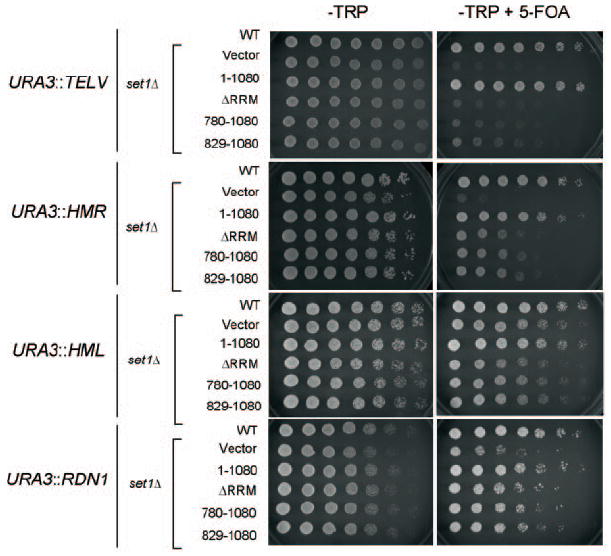
Strains UCC506 and UCC506 set1Δ(TEL-V), UCC7262 and UCC7262 set1Δ(HMR), UCC7266 and UCC7266 set1Δ(HML), and UCC1188 and UCC1188 set1Δ(rDNA) were transformed with the indicated plasmids. For silencing assays, individual isolates were grown to saturation, normalized for A600, and serially diluted and spotted on SC-Trp or SC-Trp/5-FOA plates. Cell growth was monitored at 30 °C. Cells on SC-Trp plates were photographed after 24 h, and cells on SC-Trp + 5-FOA were photographed after 48 h (HMR, HML, rDNA) or after 60 h (TEL).
To determine whether silencing at rDNA, HML, and HMR loci is dependent upon H3 Lys4 trimethylation, similar assays, as described above, were performed in strains lacking Set1 and containing a URA3 gene integrated at the rDNA locus and the HMR and HML silent mating type loci (24, 25). In all instances, cells that expressed Set1 mutants that are deficient for H3 Lys4 trimethylation exhibit sensitivity to 5-FOA as compared with both wild-type cells transformed with blank plasmid (Vector) or set1Δ cells expressing full-length Set1 (1–1080) (Fig. 4). This data again suggests that H3 Lys4 trimethylation is also needed for proper silencing at the rDNA, HML, and HMR loci.
Interestingly, at the different silent loci, silencing seems to be compromised at varying degrees when SET1 is deleted or when expressing the Set1 trimethyl-deficient mutants in a set1Δ. Although the reason for this difference is not known, this has been commonly observed among other protein factors that disrupt silencing. However, various explanations could account for our observed differences. For example, the differences between HML and HMR silencing could be a consequence on how the URA3 gene was previously integrated in these strains. It has been shown previously that the URA3 gene is silenced better at the HML than HMR locus due to the way the gene was inserted (33). Therefore, HMR silencing in this strain has been shown to be more sensitive to silencing defects than HML (33). Another strain difference is that HMR, HML, and rDNA strains contain one gene copy of histones H3 and H4, therefore histone amounts may also contribute to these silencing differences. Although we speculate that some of these differences are due to subtle strain differences, it is also possible these differences are due to the type or amount of protein factors (e.g. Sir proteins) that are required for silencing at each of these distinct loci (34).
Our results demonstrate a strong correlation between H3 Lys4 trimethylation and silencing. Although, the mechanism by which H3 Lys4 trimethylation regulates silencing is still unclear, it has been proposed that loss of histone methylation at H3 Lys4 and/or H3 Lys79 methylation allows promiscuous binding of Sir proteins to euchromatic regions, which results in titrating away Sir proteins from silent loci (35). In support of this model, it has recently been shown that Sir3 localization is disrupted in a yeast strain expressing a catalytically inactive mutant of Set1 and that Sir3 can bind to unmodified histone peptides but not peptides trimethylated at H3 Lys4 (12). It will now be interesting to determine whether similar results are observed using yeast strains lacking only H3 Lys4 trimethylation. However, other possibilities could still exist. For example, protein-protein interactions at the N terminus of Set1 may be needed for proper gene silencing, or Set1-mediated trimethylation may regulate expression of a known or unknown silencing factor(s).
It has been indicated by several groups that H3 Lys4 trimethylation plays a role in transcriptional activation and/or elongation (7, 19-21, 36). To determine the role of Set1 trim-ethyl-deficient mutants in transcription, mRNA levels of known Set1 target genes GAL1, GAL10, and PYK1 (19, 31, 32, 37) were examined under uninduced and induced conditions. Surprisingly, reverse transcription-PCR and quantitative real time PCR analysis revealed no significant changes in GAL1, GAL10, and PYK1 steady state mRNA levels under uninduced or induced conditions in either set1Δ cells or set1Δ cells expressing Set1 trimethyl-deficient mutants (data not shown). These data suggest that specific loss of H3 Lys4 trimethylation is not sufficient to disrupt transcription of these Set1 targeted genes. In addition, set1Δ and Set1 trimethyl-deficient mutant strains do not show a hypersensitive phenotype when plated on media containing 6-azauracil, suggesting they are not defective in transcriptional elongation.2 Further investigation will be needed to assess the precise role of Set1-mediated Lys4 trimethylation in transcriptional activation and elongation.
In summary, this study demonstrates that a region outside the SET domain (i.e. the RRM domain or other domains in the N terminus of Set1) is required for regulation of Set1-mediated trimethylation. This is in contrast to Set7/9 and Dim5 histone methyltransferases in which a conserved tyrosine or phenylalanine residue within the SET domain mediates the degree of histone methylation (38, 39). In addition, Set1 trimethyl-specific mutants have allowed us to discover that differential H3 Lys4 methylation is required for distinct biological functions such as proper cell growth and telomere, rDNA, HML, and HMR silencing. Surprisingly, we were unable to determine a role for H3 Lys4 trimethylation in transcription. Further investigation will be needed to determine precise mechanism of how Set1-mediated Lys4 mono-, di-, and trimethylation can regulate these biological processes. Since Set1 exists in a high molecular weight complex, it is likely that Set1-associated factors will play a significant role in mediating the distinct forms of H3 Lys4 methylation (8, 9, 14). These and other Set1 methyl-specific mutants will be useful in dissecting out the biological mechanism of how the different methylation states in the eukaryotic genome effects chromatin structure and function.
Acknowledgments
We thank Ann Kirchmaier, Joe Ogas, and Harry Charbonneau for helpful discussions; Dan Gottschling for the UCC506, UCC7266, UCC7262, and UCC1188 yeast silencing strains; Mary Bryk for the MBY1198, 1217 and 1587 yeast strains; and Peter Cheung and Doug Mersman for critical review of the manuscript.
Footnotes
This work was supported by the Walther Cancer Institute, Purdue Cancer Center, American Cancer Society Institutional Grant, Indiana Elks Charities, Inc., and the Purdue Department of Biochemistry. This is journal paper number 17682 from the Purdue University Agricultural Experiment Station.
The abbreviations used are: PVDF, polyvinylidene fluoride; 5-FOA, 5-fluoroorotic acid; RRM, RNA recognition motif.
I. M. Fingerman and S. D. Briggs, unpublished observation.
References
- 1.Murray K. Biochemistry. 1964;127:10–15. doi: 10.1021/bi00889a003. [DOI] [PubMed] [Google Scholar]
- 2.Lachner M, O’Sullivan RJ, Jenuwein T. J Cell Sci. 2003;116:2117–2124. doi: 10.1242/jcs.00493. [DOI] [PubMed] [Google Scholar]
- 3.Peterson CL, Laniel MA. Curr Biol. 2004;14:R546–R551. doi: 10.1016/j.cub.2004.07.007. [DOI] [PubMed] [Google Scholar]
- 4.Rea S, Eisenhaber F, O’Carroll D, Strahl BD, Sun ZW, Schmid M, Opravil S, Mechtler K, Ponting CP, Allis CD, Jenuwein T. Nature. 2000;406:593–599. doi: 10.1038/35020506. [DOI] [PubMed] [Google Scholar]
- 5.Jenuwein T, Laible G, Dorn R, Reuter G. Cell Mol Life Sci. 1998;54:80–93. doi: 10.1007/s000180050127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Briggs SD, Bryk M, Strahl BD, Cheung WL, Davie JK, Dent SYR, Winston F, Allis CD. Genes Dev. 2001;15:3286–3295. doi: 10.1101/gad.940201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Santos-Rosa H, Schneider R, Bannister AJ, Sherriff J, Bernstein BE, Emre NCT, Schreiber SL, Mellor J, Kouzarides T. Nature. 2002;419:407–411. doi: 10.1038/nature01080. [DOI] [PubMed] [Google Scholar]
- 8.Nagy PL, Griesenbeck J, Kornberg RD, Cleary ML. Proc Natl Acad Sci U S A. 2002;99:90–94. doi: 10.1073/pnas.221596698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Roguev A, Schaft D, Shevchenko A, Pijnappel WWMP, Wilm M, Aasland R, Stewart AF. EMBO J. 2001;20:7137–7148. doi: 10.1093/emboj/20.24.7137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Krogan NJ, Dover J, Khorrami S, Greenblatt JF, Schneider J, Johnston M, Shilatifard A. J Biol Chem. 2002;277:10753–10755. doi: 10.1074/jbc.C200023200. [DOI] [PubMed] [Google Scholar]
- 11.Nislow C, Ray E, Pillus L. Mol Biol Cell. 1997;8:2421–2436. doi: 10.1091/mbc.8.12.2421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Santos-Rosa H, Bannister AJ, Dehe PM, Geli V, Kouzarides T. J Biol Chem. 2004;279:47506–47512. doi: 10.1074/jbc.M407949200. [DOI] [PubMed] [Google Scholar]
- 13.Bryk M, Briggs SD, Strahl BD, Curcio MJ, Allis CD, Winston F. Curr Biol. 2002;12:165–170. doi: 10.1016/s0960-9822(01)00652-2. [DOI] [PubMed] [Google Scholar]
- 14.Miller T, Krogan NJ, Dover J, Erdjument-Bromage H, Tempst P, Johnston M, Greenblatt JF, Shilatifard A. Proc Natl Acad Sci U S A. 2001;98:12902–12907. doi: 10.1073/pnas.231473398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Milne TA, Briggs SD, Brock HW, Martin ME, Gibbs D, Allis CD, Hess JL. Mol Cell. 2002;10:1107–1117. doi: 10.1016/s1097-2765(02)00741-4. [DOI] [PubMed] [Google Scholar]
- 16.Yokoyama A, Wang Z, Wysocka J, Sanyal M, Aufiero DJ, Kitabayashi I, Herr W, Cleary ML. Mol Cell Biol. 2004;24:5639–5649. doi: 10.1128/MCB.24.13.5639-5649.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wysocka J, Myers MP, Laherty CD, Eisenman RN, Herr W. Genes Dev. 2003;17:896–911. doi: 10.1101/gad.252103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hughes CM, Rozenblatt-Rosen O, Milne TA, Copeland TD, Levine SS, Lee JC, Hayes DN, Shanmugam KS, Bhattacharjee A, Biondi CA, Kay GF, Hayward NK, Hess JL, Meyerson M. Mol Cell. 2004;13:587–597. doi: 10.1016/s1097-2765(04)00081-4. [DOI] [PubMed] [Google Scholar]
- 19.Ng HH, Robert F, Young RA, Struhl K. Mol Cell. 2003;11:709–719. doi: 10.1016/s1097-2765(03)00092-3. [DOI] [PubMed] [Google Scholar]
- 20.Bernstein BE, Humphrey EL, Erlich RL, Schneider R, Bouman P, Liu JS, Kouzarides T, Schreiber SL. Proc Natl Acad Sci U S A. 2002;99:8695–8700. doi: 10.1073/pnas.082249499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Krogan NJ, Dover J, Wood A, Schneider J, Heidt J, Boateng MA, Dean K, Ryan OW, Golshani A, Johnston M, Greenblatt JF, Shilatifard A. Mol Cell. 2003;11:721–729. doi: 10.1016/s1097-2765(03)00091-1. [DOI] [PubMed] [Google Scholar]
- 22.Xiao T, Kao CF, Krogan NJ, Sun ZW, Greenblatt JF, Osley MA, Strahl BD. Mol Cell Biol. 2005;25:637–651. doi: 10.1128/MCB.25.2.637-651.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Morgan BA, Mittman BA, Smith MM. Mol Cell Biol. 1991;11:4111–4120. doi: 10.1128/mcb.11.8.4111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gottschling DE, Aparicio OM, Billington BL, Zakian VA. Cell. 1990;63:751–762. doi: 10.1016/0092-8674(90)90141-z. [DOI] [PubMed] [Google Scholar]
- 25.van Leeuwen F, Gafken PR, Gottschling DE. Cell. 2002;109:745–756. doi: 10.1016/s0092-8674(02)00759-6. [DOI] [PubMed] [Google Scholar]
- 26.Carvin CD, Kladde MP. J Biol Chem. 2004;279:33057–33062. doi: 10.1074/jbc.M405033200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kuo MH, Allis CD. Methods. 1999;19:425–433. doi: 10.1006/meth.1999.0879. [DOI] [PubMed] [Google Scholar]
- 28.Stefl R, Skrisovska L, Allain FH. EMBO Rep. 2005;6:33–38. doi: 10.1038/sj.embor.7400325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Schlichter A, Cairns BR. EMBO J. 2005;24:1222–1231. doi: 10.1038/sj.emboj.7600607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dichtl B, Aasland R, Keller W. RNA (N Y) 2004;10:965–977. doi: 10.1261/rna.7090104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ezhkova E, Tansey WP. Mol Cell. 2004;13:435–442. doi: 10.1016/s1097-2765(04)00026-7. [DOI] [PubMed] [Google Scholar]
- 32.Daniel JA, Torok MS, Sun ZW, Schieltz D, Allis CD, Yates JR, III, Grant PA. J Biol Chem. 2004;279:1867–1871. doi: 10.1074/jbc.C300494200. [DOI] [PubMed] [Google Scholar]
- 33.Singer MS, Kahana A, Wolf AJ, Meisinger LL, Peterson SE, Goggin C, Mahowald M, Gottschling DE. Genetics. 1998;150:613–632. doi: 10.1093/genetics/150.2.613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rusche LN, Kirchmaier AL, Rine J. Annu Rev Biochem. 2003;72:481–516. doi: 10.1146/annurev.biochem.72.121801.161547. [DOI] [PubMed] [Google Scholar]
- 35.van Leeuwen F, Gottschling DE. Curr Opin Cell Biol. 2002;14:756–762. doi: 10.1016/s0955-0674(02)00393-9. [DOI] [PubMed] [Google Scholar]
- 36.Hampsey M, Reinberg D. Cell. 2003;113:429–432. doi: 10.1016/s0092-8674(03)00360-x. [DOI] [PubMed] [Google Scholar]
- 37.Henry KW, Wyce A, Lo WS, Duggan LJ, Emre NC, Kao CF, Pillus L, Shilatifard A, Osley MA, Berger SL. Genes Dev. 2003;17:2648–2663. doi: 10.1101/gad.1144003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhang X, Yang Z, Khan SI, Horton JR, Tamaru H, Selker EU, Cheng X. Mol Cell. 2003;12:177–185. doi: 10.1016/s1097-2765(03)00224-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Collins RE, Tachibana M, Tamaru H, Smith KM, Jia D, Zhang X, Selker EU, Shinkai Y, Cheng X. J Biol Chem. 2005;280:5563–5570. doi: 10.1074/jbc.M410483200. [DOI] [PMC free article] [PubMed] [Google Scholar]