

Abstract
The present report identifies the enzymatic substrates of a member of the mammalian nitrilase-like (Nit) family. Nit2, which is widely distributed in nature, has been suggested to be a tumor suppressor protein. The protein was assumed to be an amidase based on sequence homology to other amidases and on the presence of a putative amidase-like active site. This assumption was recently confirmed by the publication of the crystal structure of mouse Nit2. However, the in vivo substrates were not previously identified. Here we report that rat liver Nit2 is ω-amidodicarboxylate amidohydrolase (E.C. 3.5.1.3; abbreviated ω-amidase), a ubiquitously expressed enzyme that catalyzes a variety of amidase, transamidase, esterase and transesterification reactions. The in vivo amidase substrates are α-ketoglutaramate and α-ketosuccinamate, generated by transamination of glutamine and asparagine, respectively. Glutamine transaminases serve to salvage a number of α-keto acids generated through non-specific transamination reactions (particularly those of the essential amino acids). Asparagine transamination appears to be useful in mitochondrial metabolism and in photorespiration. Glutamine transaminases play a particularly important role in transaminating α-keto-γ-methiolbutyrate, a key component of the methionine salvage pathway. Some evidence suggests that excess α-ketoglutaramate may be neurotoxic. Moreover, α-ketosuccinamate is unstable and is readily converted to a number of hetero aromatic compounds that may be toxic. Thus, an important role of ω-amidase is to remove potentially toxic intermediates by converting α-ketoglutaramate and α-ketosuccinamate to biologically useful α-ketoglutarate and oxaloacetate, respectively. Despite its importance in nitrogen and sulfur metabolism, the biochemical significance of ω-amidase has been largely overlooked. Our report may provide clues regarding the nature of the biological amidase substrate(s) of Nit1 (another member of the Nit family), which is a well-established tumor suppressor protein), and emphasizes a) the crucial role of Nit2 in nitrogen and sulfur metabolism, and b) the possible link of Nit2 to cancer biology.
Keywords: ω-Amidase, Nit2, α-ketoglutaramate, glutamine transaminases, methionine salvage pathway
1. Introduction
The nitrilase superfamily contains enzymes that are widely distributed in nature [1–3]. The family is divided into 13 branches, only one of which (Branch 1) actually possesses nitrilase catalytic activity [nitrile + 2H2O → carboxylate + 2NH4+]. The remaining branches contain amidases [amide + H2O → carboxylate + NH4+] or enzymes with closely related activities [1–3]. Branch 10 contains two proteins that are found in mammalian tissues, designated Nit1 (nitrilase-like protein 1) and Nit2 (nitrilase-like protein 2). The two proteins share ~40% sequence identity [4] and ~55% homology [5], and a somewhat lower homology to β-ureidopropionase (β-alanine synthase) [4]. Although the substrate specificities of Nit1 and Nit2 were unknown they were assumed to be amidases based on sequence homology to known amidases and on the presence of a canonical Cys-Glu-Lys catalytic triad in the putative active site. Nit1 knockout in mouse tissues/cells results in increased cell proliferation, enhanced survival of cells exposed to DNA-damaging agents and an increased incidence of N-nitrosomethylbenzylamine-induced tumors [6]. Conversely, Nit1 overexpression leads to decreased cancer cell viability and increased caspase-3-dependent apoptosis [6]. These findings suggest that Nit1 has tumor suppressor properties that enhance the apoptotic responsiveness in cancer cells. In an analogous fashion, Nit2 may be a candidate protein as a tumor suppressor, although in this case, the protein appears to arrest cells in their G2 phase without inducing apoptosis [5]. The authors showed that overexpression of Nit2 in HeLa cells up-regulates the expression of the 14-3-3σ gene and down-regulates that of the 14-3-3β gene. This finding suggests that Nit2 affects growth suppression by this double mechanism [5]. The suggestion was based on the previously reported findings that upregulation of the 14-3-3σ gene triggers cell cycle arrest in G2 [7] and inhibits Akt-activated cell growth [8], and that overexpression of 14-3-3β protein induces tumorigenesis [9], whereas its reduction results in tumor suppression [10]. Elucidating Nit2 function in terms of its enzymology, expression, and protein interactions may help delineate the nature of G2M cell cycle arrest and uncover important mechanisms of tumor progression and control. Interestingly, genotype analysis in four types of human primary tumors showed 12.5–38.5% allelic imbalance surrounding the Nit2 genomic locus [5], and Nit1 protein expression is reduced or lost in 50% of adenocarcinomas of the esophagus [11].
Several studies of Nit2 are of biomedical interest. For example, in a recent study, Duthie et al. showed that the level of catechol O-methyltransferase and Nit2, proteins linked to malignant transformations, were greatly reduced in cells grown in folate-deficient medium as assessed by 2-D gel electrophoresis [12]. Low folate intake is associated with cell cycle entrapment and with increased risk of cancer development [reviewed in ref. 12]. Wang et al. noted that human MCF7 breast cancer cells when transfected with ErbB2 (erythroblastic leukemia viral oncogene homologue 2; also known as human epidermal growth factor receptor 2 (HER2)) exhibited a 70% reduction in Nit2 protein compared to the level in untransfected cells as inferred from 2-D gel analysis [13]. [ErbB2 is involved in signal transduction and growth differentiation and is frequently overexpressed in breast cancers. Overexpression of ErbB2 leads to aggressive cell growth and insufficient signaling can promote tumor development and progression or create resistance to chemotherapy.] Interestingly, the transfected cells (but not the untransfected cells) were reported to contain a phosphorylated form of the Nit2 protein [13]. Finally, a 2-D gel proteomic analysis of human cortical brain tissue revealed significant differences in nine proteins between control fetuses and Down syndrome fetuses at early second trimester of development [14]. One of the proteins identified as being reduced by at least two fold was Nit2 [14]. Whether decreased expression of Nit2 is related to asynchronous expression of ErbB2 observed in cells from Down syndrome fetuses is not known [15].
Here we identify Nit2 as ω-amidase (an enzyme first discovered more than 55 years ago [16,17]) that catalyzes the deamidation of α-ketoglutaramate (αKGM) and α-ketosuccinamate (αKSM) to ketoglutarate and oxaloacetate, respectively. We also show that Nit1 is unlikely to be an ω-amidase. αKGM and αKSM are generated in vivo by transamination of glutamine and asparagine, respectively. Nit2/ω-amidase is important in nitrogen and sulfur metabolism (see the Discussion). By contrast to Nit2, the natural substrate(s) of Nit1 remain(s) unknown.
After our work was finished and a manuscript completed, it came to our attention that Jaisson et al. also arrived at the same conclusions that ω-amidase is identical to Nit2 and that Nit1 does not have ω-amidase activity. The authors used a different approach from that used here, thus corroborating the two conclusions. The work by Jaisson et al. [18] is presented in an accompanying article in this issue of Biochimie.
Some of the work described herein was presented at the 40th Annual Meeting of the American Society for Neurochemistry, Charleston, South Carolina, USA, March 7–11, 2009 [19].
2. Materials and Methods
2.1. Reagents
Dithiothreitol (DTT), 1-chloro-2,4-dinitrobenzene (CDNB), beef blood hemoglobin (type 1) and bovine heart cytochrome c were obtained from Sigma Aldrich Chemical Company (St. Louis, MO). 2,4-Dinitrophenylhydazine was obtained from Eastman Kodak (Rochester, NY). Succinamic acid was from Aldrich Chemical Company (Milwaukee, WI).
2.2. Enzyme assay
In most cases, ω-amidase activity was determined by the 2,4-dinitrophenylhydrazone procedure [20] as adapted to a 96-well plate spectrophotometer [21]. The procedure relies on the fact that the substrate, α-ketoglutaramate, exists overwhelmingly in a cyclized form that does not possess a reactive carbonyl group [22], whereas, the product, α-ketoglutarate, possesses a reactive carbonyl (α-keto) group that reacts with 2,4-dinitrophenylhydrazine. In some cases, ω-amidase activity was assayed by determining the formation of succinyl hydroxamate in a reaction mixture containing hydroxylamine and succinamate (hydroxaminolysis reaction) [21]. This reaction was shown to be exclusively catalyzed by ω-amidase in rat liver cytosol [21]. Protein was measured by the Bradford procedure using bovine serum albumin as a standard. Specific activities are expressed as μmol·mg−1·min−1 of protein at 37°C.
2.3. Purification of ω-amidase from rat liver cytosol
The enzyme was purified by the method of Hersh [22] with some modifications. In the final step of the purification a hydroxylapatite column was used instead of a batch procedure. Fractions that possessed the highest specific activity of ω-amidase were combined. SDS-PAGE analysis of the combined fractions revealed a major band with a Mr of ~26,000. Mass spectral analysis showed this band to contain glutathione S-transferases (GSTs). The presence of GST activity was confirmed by using a standard GST substrate (i.e. CDNB) [23]. We therefore used glutathione-immobilized onto cross-linked beaded agarose (Sigma, St. Louis, MO) packed into a small column to remove active GSTs. Analysis of the eluate showed no GST activity, as assessed by the CDNB method, and no loss of ω-amidase activity. The specific activity of ω-amidase in the eluate (αKGM as substrate) was 15.1 μmol·mg−1·min−1. SDS-PAGE analysis of this preparation showed a low intensity band with Mr of ~32,000 and a higher intensity band with a Mr of ~26,000. Mass spectral analysis revealed that the lower intensity (higher Mr) band is Nit2, and that the higher intensity (lower Mr) band is composed of GSTs (sections 3.2, 3.3). Possibly, these GSTs were inactivated during the purification or were tightly bound to endogenous ligands at the active sites (as a result of their ability to bind to endogenous compounds, GSTs were originally named ligandins) and were therefore incapable of binding to the glutathione column.
2.4. Polyacrylamide gel electrophoresis
For SDS-PAGE, samples were mixed with an equal volume of Laemmli buffer, heated at 100°C for 5 min, cooled and individually loaded into wells of a 4–20 % gradient Tris-HCl precast gel. The reservoir buffer was 25 mM Tris, 192 mM glycine and 0.1% (w/v) SDS, pH 8.3 (Sigma, St. Louis, MO). Molecular markers were loaded into a separate well. Electrophoresis was carried out at 160 V for 1.5 h at 4°C using a mini-gel unit. For native-PAGE, samples were mixed with an equal volume of native sample buffer and individually loaded into wells of a 4–20 % gradient Tris-HCl precast gel. The reservoir buffer was 25 mM Tris, 192 mM glycine, pH 8.3. Electrophoresis was carried out at 40 V for 8 h at 4°C using a mini-gel unit. The gels were removed and washed twice with distilled water. Protein bands were revealed by silver staining. The gel unit, precast gels, Laemmli and native sample buffers, and silver staining kit were from Bio-Rad (Hercules, CA).
2.5. Mass spectrometry
Excised gel bands were washed 3 times with deionized water, diced, de-stained with aliquots of 300 μl of 200 mM ammonium bicarbonate buffer (pH 8.9) (ABC)/50% acetonitrile (v/v), and then washed with 300 μl of 100% acetonitrile. The gel pieces were vacuum-dried, and then swelled with 300 μl of 10 mM DTT/0.1 M ABC (v/v) at 56°C for 45 minutes. After cooling to room temperature and removal of supernatant, the gel pieces were incubated with 300 μl of freshly prepared 55 mM iodoacetamide/0.1 M ABC (v/v) for 30 minutes in the dark at room temperature, washed three times with 300 μl of 200 mM ABC/50% acetonitrile (v/v), followed by a wash with 300 μl of 100% acetonitrile. After vacuum drying, trypsin (20 μl, 25 ng/μl) in 50 mM ABC was added and the gel pieces were kept on ice for 45 minutes. The unabsorbed trypsin solution was removed, 50 μl of 50 mM ABC was added to the gel pieces, which were digested overnight at 37°C in a shaker. An aliquot (1.0 μl) of 10% aqueous trifluoroacetic acid was added, and the tryptic peptides were purified and concentrated using a C18 Ziptip (Millipore, Billerica, MA) following the manufacturer’s instructions. The bound peptides were eluted with 1 μl of α-cyano-4-hydroxycinnamic acid matrix (saturated solution in 50% acetonitrile, 0.1% TFA) onto a MALDI-TOF target plate and allowed to crystallize at room temperature. For MS/MS, an AB 4800 TOF/TOF mass spectrometer (Applied Biosystems, Foster City, CA) was used. A total of 1000 shots were summed to obtain the spectrum of the 10 most abundant peptide ions in each gel band. Protein identification was accomplished through database searching (NCBI) using Mascot (in-house). The search parameters were as follows: mammalian; monoisotopic mass tolerance of 50 ppm for peptides and 0.8 Da for product ions; carbamidomethyl cysteines; one maximum missed cleavage; variable modifications included oxidized methionine, deamidated Asn or Gln, Gln → pyro-Glu (N-term Q) and Glu → pyro-Glu (N-term E). The Mascot automatic decoy database search was utilized for all searches to obtain a false discovery rate along with a decoy score. Individual ion scores >44 indicate identity or extensive homology (p<0.05). Significant protein hits are listed in Table 1 which includes for each gel band: the protein hit number; protein name, accession number, mass and percent coverage; Mascot score; number of peptides having individual ion scores of 44 or greater (the most likely peptide assignment); and expectation value(s) or a range of expectation values (the number of matches with equal or better scores that are expected to occur by chance alone; the lower the expectation value the more significant the score). The Mascot false discovery rate obtained for all gel bands was 0.00%.
Table 1.
Mass spectral analysis of purified cytosolic rat liver ω-amidase.
| Gel Band # | Protein Hit # | Accession # | Protein Name | Mascot Score | Protein Mass | % Coverage | # of Significant Peptides | Expectation Values Range |
|---|---|---|---|---|---|---|---|---|
| 1a | 1 | 77628000 | Nitrilase family, member 2 | 584 | 31024 | 26 | 7 | 5.3 × 10−10 to 0.003 |
| 2a | 1 | 204499 | Glutathione S-transferase Y-b subunit (EC 2.5.1.18) | 104 | 22042 | 17 | 2 | 1.2 × 10−6 to 0.011 |
| 2 | 33356830 | Crystal structures of class Mu chimeric GST Isoenzymes M1-2 And M2-1 | 84 | 25858 | 14 | 2 | 0.00017 to 0.011 | |
| 3 | 77628000 | Nitrilase family, member 2 | 57 | 31024 | 3 | 1 | 0.0021 | |
| 1b | 1 | 77628000 | Nitrilase family, member 2 | 323 | 31024 | 25 | 6 | 3.7 × 10−9 to 0.0002 |
| 2b | 1 | 33356830 | Crystal structures of class Mu chimeric GST Isoenzymes M1-2 And M2-1 | 246 | 25858 | 26 | 4 | 1.5 × 10−12 to 0.024 |
| 2 | 204499 | Glutathione S-transferase Y-b subunit (EC 2.5.1.18) | 210 | 22042 | 36 | 5 | 4.7 × 10−6 to 0.024 | |
| 3 | 204501 | Glutathione S-transferase (EC 2.5.1.18) | 162 | 25840 | 11 | 2 | 1.5 × 10−12 to 0.0043 | |
| 4 | 77628000 | Nitrilase family, member 2 | 67 | 31024 | 3 | 1 | 0.00027 | |
| 3 | 1 | 77628000 | Nitrilase family, member 2 | 161 | 31024 | 15 | 3 | 2 × 10−7 to 0.0096 |
| 2 | 136429 | Trypsin precursor | 61 | 25078 | 8 | 1 | 0.0093 | |
| 3 | 204499 | Glutathione S-transferase Y-b subunit (EC 2.5.1.18) | 58 | 22042 | 7 | 1 | 0.0029 | |
| 4 | 220684 | Cytosolic aspartate aminotransferase | 46 | 46627 | 2 | 1 | 0.028 |
Blank piece of SDS gel – no significant hits
Blank piece of Native gel – no significant hits
2.6. Estimation of the native Mr of ω-amidase in rat liver cytosol
Rat liver cytosol, prepared by the method of Krasnikov et al. [24], was applied to a Sephadex G-200 column (0.7 × 46 cm, superfine grade; Fluka Chemie, Buchs, Switzerland) equilibrated with 5 mM potassium phosphate buffer (pH 7.4) containing 1 mM DTT and eluted with the same buffer at 4°C. Fractions (~0.5 ml) were collected and assayed for ω-amidase activity. Glutamine transaminase K (GTK, Mr ~97,000), cystathionine γ-lyase (Mr ~160,000) and GST (Mr ~52,000) in the eluted fractions were in situ Mr markers. These enzymes were assayed as described [23,25]. In a separate experiment, hemoglobin (Mr ~68,000; 1 mg) and cytochrome c (Mr ~12,500; 1 mg) were used as additional Mr markers. Elution volumes of these two proteins were determined by measuring absorbance at 450 nm in the eluate.
2.7. Purification of human ω-amidase/Nit2
Nit2-amplified DNA was obtained by PCR amplification of Nit2 cDNA cloned from human liver cDNA as described [5]. The amplified Nit2 cDNA was subcloned into pFlag-CMV2 plasmids (at the EcoRI/BamHI site). HeLa cells were then transfected with pFlag-CMV2-Nit2 [5]. The sequence of the hNit2 clone was found to be exactly the same as that shown in GenBank [BC107890.1. Homo sapiens nitrilase family, member 2, mRNA (cDNA clone MGC:111199 IMAGE:6648910), complete coding sequence]. A proofreading DNA polymerase [Invitrogen, Carlsbad, CA, USA] was used for cloning.
HeLa cells were plated in 60 × 15 mm culture dishes at a density of 106 cells per dish and then transfected as described [5]. The transfected HeLa cells expressing human Nit2 were cultured in 100 dishes for 96 h at 37°C, the cells were combined and then lysed with a phosphate buffer containing Nonidet-P40, 1 mM EDTA and a complete set of proteinase inhibitors. The background level of ω-amidase in the non-transfected cells was very low. For the non transfected cells, the specific activity of ω-amidase was 0.006 μmol·mg−1·min−1. For the transfected cells the specific activity was about 0.012 μmol·mg−1·min−1. Based on the assumption that Nit2 is identical to ω-amidase, the recombinant human Nit2 was purified from the lysed HeLa cell homogenate by a previous method used by one of the present authors (AJLC) to purify ω-amidase from rat liver cytosol [20].
2.8. Mouse models
Fragile histidine triad (Fhit)−/−/Nit1−/− double knockout (DKO) mice were generated by mating Fhit−/− mice with Nit1−/− mice [26]. The F1 generation was self-crossed and the progeny genotyped by PCR. Fhit−/− mice and Fhit−/−/Nit1−/− mice were sacrificed at the age of 34 weeks. Liver samples were snap frozen at −80°C and shipped on dry ice from the Ohio State University to New York Medical College where analysis for ω-amidase activity was performed. The liver was gently thawed and separated into subcellular fractions as described [24]. The cytosolic fraction was analyzed for ω-amidase activity. Kidney cells from the DKO mice were established in culture as described [27]. Briefly, the kidneys were dissected and cultured with minimal essential medium (Sigma, St. Louis, MO) supplied with 20% fetal bovine serum containing 100 μg/ml of gentamicin (Sigma, St. Louis, MO) for the first three generations. Thereafter, cells were subcultured in the same medium but with 10% FBS. The experiments were approved by the Ohio State University Institutional Animal Care and Use Committee.
3. Results
3.1. Kinetic properties of purified rat liver cytosolic ω-amidase
Previously no information was available concerning the sequence of this enzyme, except that the active site contains a cysteine residue [22]. We reasoned that identification of the ω-amidase gene within mammalian genomes would provide valuable insights into the relationship of the expressed enzyme to other amidases. ω-Amidase was previously purified about 50-fold from rat liver by Meister [28] and later about 150-fold from rat liver cytosol by Hersh [22].
We used the Hersh procedure with some modifications to purify ω-amidase from rat liver cytosol (see the Materials and Methods section). Hersh reported that the purified ω-amidase exhibited a specific activity of 11.7 μmol·mg−1·min−1 in a 1-ml reaction mixture containing 5 mM αKGM, 1 mM DTT, 10 mM ammonium chloride, 0.128 mM NADH and 50 μg of glutamate dehydrogenase in 50 mM Tris-HCl buffer (pH 8.5) incubated at 30°C [22]. The high concentration of glutamate dehydrogenase (indicator enzyme) in the reaction mixture ensures that α-ketoglutarate generated from αKGM is rapidly converted to glutamate by reductive amination. The rate of disappearance of NADH was followed spectrophotometrically (ε340nm = 6220 M−1·cm−1).
The purified ω-amidase preparation was stated to be pure as judged by the occurrence of a single band on native gel electrophoresis. However, three bands were noted on sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) with Mr values of ~58,000, ~30,000 and ~27,000. Hersh also stated that the purified enzyme has a Mr of ~58,000 and is composed of two subunits [22]. However, it was unknown at that time whether the enzyme exists as a homodimer (two identical subunits with Mr values of ~28,000 – 30,000) or a heterodimer composed of one subunit with a Mr of ~28,000 and another subunit with a Mr of ~30,000. The enzyme was reported to exhibit an apparent Km of 3.3 mM and a Vmax of 16.0 μmol·mg−1·min−1 [22]. In solution, αKGM is in equilibrium with a cyclic form (2-hydroxy-5-oxoproline) [22,28]. Hersh reported that at neutral pH, the equilibrium position greatly favors the cyclic (lactam) form (99.7%) over the open-chain form (0.3%) [22]. Conversion between the two forms is specific-base (OH−) catalyzed. At levels of ω-amidase commonly used in assay procedures, the rate of ring opening below pH 8.0 limits the enzymatic reaction. However, at pH 8.0 or above, the rate of ring opening is no longer limiting for the ω-amidase reaction [22]. Thus, at pH 8.5 the “true” Km exhibited by rat liver cytosolic ω-amidase for the open-chain form of αKGM according to Hersh’s data is about 10 μM [22].
In the present study we used a reaction mixture that contained 5 mM αKGM, 5 mM DTT and 100 mM Tris-HCl buffer (pH 8.5) in a final volume of 50 μl. After incubation at 37°C, α-ketoglutarate was measured in an end-point assay as its 2,4-dinitrophenylhydrazone. Under these conditions, the purified enzyme exhibited a specific activity of 15.1 μmol·mg−1·min−1 (Methods). A Lineweaver-Burk plot yielded an apparent Km of 1.8 mM, a corrected Km of 5.4 μM and a Vmax of 20.6 μmol·mg−1·min−1. These values compare favorably with those obtained by Hersh [22]. However, the true Vmax value is likely to be much higher because our preparation (see the next section) and that of Hersh, were not homogeneous. Indeed, we have recently, succeeded in obtaining a preparation of ω-amidase purified from rat liver cytosol with a specific activity (αKGM as substrate) of ~126 μmol·mg−1·min−1 at 37°C [21].
3.2. Identification of ω-amidase as Nit2
Our preparation of ω-amidase purified from rat liver cytosol exhibited one broad protein-stained band on native gel electrophoresis (Fig. 1A, band marked 3). However, SDS-PAGE analysis of our preparation showed the occurrence of two major bands (Fig. 1B, lane marked ω-A). Comparison of the relative mobilities of the two bands with those of Mr markers (lane M) suggested that the apparent Mr values of the two bands are ~30,000 (band marked 1) and ~26,000 (band marked 2). These are presumably equivalent to two of the three bands noted on SDS-PAGE for purified rat liver cytosolic ω-amidase in the Hersh preparation [22]. The protein band noted on the native gel was excised, subjected to limited proteolysis and mass spectral analysis (Table 1; gel band #3). Mass spectral analysis showed a highly significant probability that the band in the non-denaturing gel contains Nit2 (Table 1). Also present were peptides derived from trypsin precursor (presumably originating from the trypsin used to digest the band), a member of the glutathione S-transferase (GST) family, and cytosolic aspartate aminotransferase. Since Nit2 is listed in the human and rodent genome data banks as an amidase (albeit of unknown specificity), the findings were very convincing that ω-amidase is Nit2. Further confirmation for this conclusion was obtained from analysis of the two bands obtained on SDS-PAGE (Fig. 1B, lane marked ω-A). When the tryptic peptides of band 1 were analyzed by tandem mass spectrometry of peptide fragments (Table 1; duplicates labeled 1a and 1b) the data indicated a highly significant probability that the band is Nit2. When band 2 was similarly analyzed (Table 1; duplicates labeled 2a and 2b), the data indicated a highly significant probability that the band contained GST isozymes and some Nit2. Thus, two of the bands in the purified preparation of ω-amidase observed by Hersh [22] and by us (present work) are Nit2/ω-amidase [Mr ~31,000 (calculated from the sequence)] and GST isozymes [Mr ~26,000 (calculated from the sequence)]. Aspartate aminotransferase is highly expressed in rat liver [29] and traces of this enzyme presumably co-purified with the ω-amidase and co-migrated with ω-amidase in the native gel.
Fig. 1. Analysis of rat liver ω-amidases by gel electrophoresis.
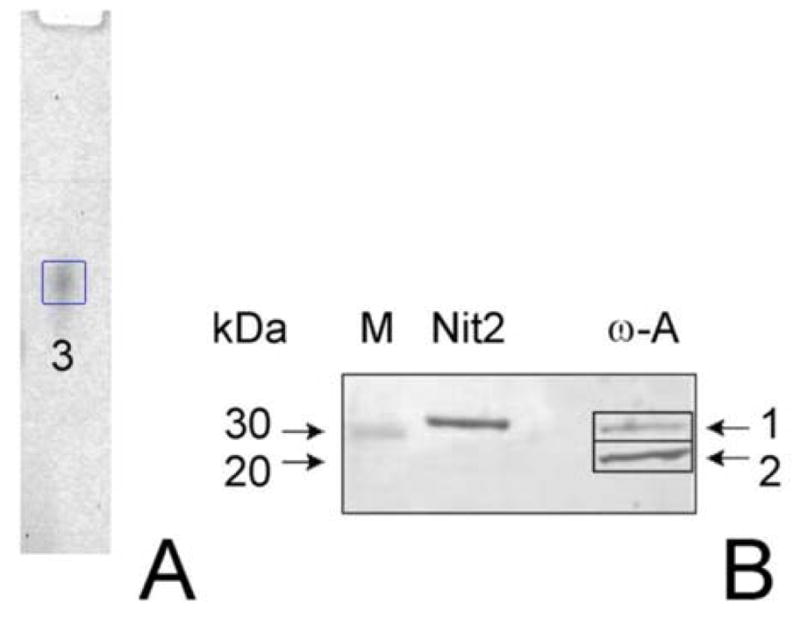
A) Electrophoresis of purified rat cytosolic rat liver ω-amidase on a native gel. B) SDS-PAGE. Lanes are labeled as follows: M, Mr markers; Nit2, purified human ω-amidase/Nit2; ω-A, purified cytosolic rat liver ω-amidase. Equal amounts of protein (1.0 μg) were applied to the native gel and to lanes labeled Nit2 and ω-A in the SDS-PAGE gel. Blank lanes contained an equal volume of the sample buffer lacking enzyme. Protein bands were visualized by silver staining.
Additional evidence confirming that Nit2 is ω-amidase was obtained by purifying Nit2 from HeLa cells overexpressing human Nit2. This enzyme preparation contained considerable ω-amidase activity (sections 3.3, 3.4) and exhibited only a single band on SDS-PAGE (Fig. 2, lane marked Nit2).
Fig. 2. Determination of the Mr of ω-amidase in rat liver cytosol.

An aliquot (0.2 ml; 4.6 mg protein) of the cytosol was subjected to Sephadex G-200 chromatography and the elution volume of ω-amidase activity was compared to the Mr value of standards. The elution volume indicated a Mr of ~60,000. Standards (gray filled circles) were: cystathionine γ-lyase (Mr ~140,000), GTK (Mr ~97,000), hemoglobin (Mr ~68,000), GSTs (Mr ~52,000), and cytochrome c (Mr ~12,500). The arrow indicates a Mr of ~60,000.
3.3. Subunit composition of rat and human ω-amidases
When rat liver cytosol was chromatographed on Sephadex G-200, ω-amidase activity was eluted with an apparent Mr of ~60,000 (Fig. 2). This finding indicates that in rat liver cytosol active ω-amidase is a dimer. The finding, however, cannot distinguish between a homodimer (Mr ~62,000) and a heterodimer composed of a catalytic subunit (Nit2; Mr ~31,000) and a GST isoform (e.g. GST class Mu; Mr ~26,000) as a structural component. Such a heterodimer would have a Mr ~57,000. However, highly purified human Nit2 exhibits robust ω-amidase activity but does not contain any GSTs (see section 3.4). Most likely the GSTs in our preparation (and presumably also in the preparation obtained by Hersh [22]) are fortuitous contaminants and not a component necessary for ω-amidase activity. In this regard, 3–10% of all the soluble protein in mammalian liver, depending on the species, is accounted for by GSTs [30], so that even a trace of GSTs can noticeably contaminate a protein preparation obtained from mammalian liver.
To determine the subunit composition of human Nit2/ω-amidase we subjected 0.2 ml of human Nit2/ω-amidase (0.1 mg/ml) to size-exclusion chromatography on the same column as that used to determine the Mr of ω-amidase in rat liver cytosol. In the presence of 5 mM potassium phosphate buffer (pH 7.4) and 1 mM DTT (4°C), the enzyme was found to elute with an apparent Mr of ~62,000. Thus, the purified human enzyme is naturally a homodimer.
Barglow et al. found that mouse Nit2 crystallizes as a dimer with two crystallographically independent, almost identical monomers in the asymmetric unit [4]. Interestingly, however, based on the results of size-exclusion chromatography the authors stated that their preparation of mouse Nit2 appears to be mostly a monomer in solution, but with occasional observations of a small amount of dimer [4]. In the studies of Barglow et al. the enzyme was expressed with a his6 tag and a gene 10 leader sequence, which were not removed. These additions increase the Mr from ~31,000 to ~34,900. Moreover, rat liver cytosolic ω-amidase is sensitive to air oxidation and must be stored in the presence of a reducing agent [20,22]. All these factors may have contributed to a weakened attraction between two mouse Nit2 monomers. However, we cannot rule out the possibility that the native mouse Nit2/ω-amidase is different from the rat and human homologues regarding subunit composition.
3.4. Kinetic characteristics of human Nit2 as ω-amidase
This preparation of purified human Nit2 (0.1 mg/ml) exhibited considerable ω-amidase activity and was judged to be >98% pure by the criterion of SDS-PAGE (Figure 1B, lane marked Nit2). A single, prominent band with a Mr of ~30,000 was observed. Importantly, no bands corresponding to GST isozymes were visible. Therefore, the presence of GST is not associated with enzymatic properties of human ω-amidase. A Lineweaver-Burk analysis (Fig. 3) indicated an apparent Km of ~3 mM, Km (open-chain form) of ~9 μM and a Vmax of ~5.9 μmol·mg−1·min−1 (confirmed in both the AJLC and CHC laboratories). The affinity of the human enzyme toward αKGM is similar to that reported for the cytosolic rat liver enzyme (section 3.1 and reference [22]). However, the Vmax is lower. The lower specific activity of the purified recombinant human enzyme compared to that of the rat enzyme may simply be due to species differences, i.e., the human enzyme is inherently less active than the rat enzyme. On the other hand, it should be noted that the recombinant human enzyme was expressed with a hydrophilic pFLAG extension (DYKDDDDK) at the C terminus, and we cannot rule out the possibility that the presence of the pFLAG affected the specific activity.
Fig. 3. Determination of kinetic characteristics of human ω-amidase/Nit2.

Enzyme (0.82 μg) was incubated with varying concentrations of αKGM in 100 mM Tris-HCl buffer (pH 8.5) at 37°C. α-Ketoglutarate was measured as its 2,4-dinitrophenylhydrazone. Each point is the average ± s.d., N=3.
3.5. ω-Amidase specific activity is not altered in liver cytosol of Fhit−/− or Fhit−/−/Nit1−/− double knockout mice
In order to determine whether the expression of Nit2 protein is affected by Fhit knock-out we determined the specific activity of ω-amidase in liver cytosol prepared from Fhit−/− knock-out (FKO), Fhit−/−/Nit1−/− double knock-out (DKO) and control mouse tissues (Table 2). No difference in specific activity of ω-amidase (αKGM as substrate) in liver cytosol was observed among the three tissues. Similar results were also obtained when the mouse liver cytosolic fractions were analyzed using the hydroxaminolysis reaction (Table 2). As noted in the Methods section, this reaction in rat cytosol is catalyzed exclusively by ω-amidase. Moreover, when we analyzed for ω-amidase activity (αKGM as substrate) in FKO and DKO mouse kidney-derived cells there was no difference in ω-amidase specific activity versus control kidney-derived cells. All three exhibited a specific activity of ~18 nmol·mg−1·min−1. Taken together, these data are consistent with the hypothesis that Nit1 does not possess ω-amidase activity.
Table 2.
Specific Activity of ω-amidase in mouse liver cytosol
| Substrate | Fhit−/− (FKO) | Fhit−/−/Nit−/− (DKO) | Control | |
|---|---|---|---|---|
| Experiment 1 | αKGM | 15.9±0.7 | 14.7±0.7 | 17.5±0.5 |
| Experiment 2 | αKGM | 18.5±1.3 | 18.7±1.5 | 18.5±1.1 |
| Experiment 3 | Succinamate | 33.5±0.5 | 30.9±0.9 | 30.9±1.4 |
Specific activity is expressed as nmol/min/mg of protein; 37°C. Each value is the average ± s.d.; N = 3. Conversion of αKGM to α-ketoglutarate was measured in experiments 1 and 2. Hydroxaminolysis of succinamate was measured in experiment 3. Experiment 1. The reaction mixture contained liver extract (25 μg protein) incubated for 45 min. Experiment 2. The reaction mixture contained liver extract (100 μg of protein) incubated for 10 min. Experiment 3. The reaction mixture contained liver extract (100 μg of protein) incubated for 2 h.
4. Discussion
The present report identifies Nit 2 as ω-amidase, an enzyme that catalyzes the hydrolytic deamidation of αKGM and αKSM, the α-keto acids derived from transamination of glutamine and asparagine, respectively. One of the functions of the glutamine transaminases is to salvage the α-keto acids of essential amino acids [including α-keto-γ-methiolbutyrate (KMB), the α-keto acid analogue of methionine], arising via non-specific transamination reactions, at the expense of readily available glutamine [31,32]. In this regard, it should be noted that KMB is an exceptionally active α-keto acid substrate of both of the two known mammalian glutamine transaminases [31]. This has important consequences for the methionine salvage pathway, which is widely distributed in nature. During polyamine biosynthesis, the C1-4 carbons of methionine are lost and the methyl and sulfur are incorporated into 5′-methylthioadenosine (MTA). If MTA were not recycled, a scarcity of methyl groups and biologically useful sulfur may ensue. Nature has gone to extraordinary lengths to salvage this methyl and sulfur [e.g. 33,34]. The salvage pathway converts MTA to KMB, through a pathway that involves some complex and unique biochemical transformations. The last step in the pathway is a transamination of KMB to methionine. Various Bacillus species can utilize a number of amino acids, including glutamine, as transamination partners with KMB [35]. (See also the accompanying article by Jaisson et al. [18].) In rats the preferred amino donor is glutamine and to a lesser extent asparagine [36]. The net result of the salvage pathway in rats is retention of the sulfur and methyl group of methionine, formation of C1-4 anew from readily available ribose moieties, and provision of amine nitrogen by transamination of KMB with expendable glutamine.
The role of glutamine transaminases plus ω-amidase in closing the methionine salvage pathway in mammals is shown in Fig. 4. The α-ketoglutarate generated from the transamination of glutamine plus ω-amidase can be converted back to glutamine by the action of aminotransferases (or glutamate dehydrogenase) plus glutamine synthetase. Thus, the cycling of five-carbon units (i.e. α-ketoglutarate, αKGM, glutamate, glutamine) can be linked to the cycling of methionine in a process that we have termed the “methionine-glutamine bicycle” (Fig. 4). The advantage of using glutamine in transamination reactions is that, unlike most aminotransferase reactions, which are freely reversible, transamination reactions involving glutamine will be drawn in the direction of α-keto acid conversion to the corresponding amino acid as a result of cyclization of αKGM followed by removal of αKGM by ω-amidase [31,32] (Fig. 4).
Fig. 4. Proposed key role of ω-amidase/Nit2 in the “methionine-glutamine bicycle”.

During polyamine biosynthesis the methyl and sulfur of methionine are incorporated into 5′-methylthioadenosine (MTA). The methionine salvage pathway converts MTA to α-keto-γ-methiolbutyrate. In mammals, the pathway is completed by transamination with glutamine (cycle 1). The product of glutamine transamination (i.e. αKGM, α-ketoglutaramate) is hydrolyzed to α-ketoglutarate plus ammonium ion by ω-amidase/Nit2. The α-ketoglutarate is converted to glutamate by transamination and/or reductive amination, which in turn is converted to glutamine via glutamine synthetase, thus closing cycle 2. Ado Met, S-adenosyl-L-methionine. Note that αKGM reversibly cyclizes to a lactam (2-hydroxy-5-oxoproline) in which the concentration of lactam is greatly favored over the open-chain form.
In addition to roles in salvaging α-keto acids, glutamine transamination is also important in the synthesis of certain antibiotics. Thus, synthesis of aminocyclitol antibiotics, such as gentamicin and neomycin, produced by Micromonospora purpurea and Streptomyces fradia, respectively, requires a glutamine transaminase (L-glutamine:keto-scyllo-inositol aminotransferase). This enzyme catalyzes the transamination of keto-scyllo-inositol with glutamine to yield aminodeoxy-scyllo-inositol and αKGM [37–40]. αKGM was detected as a major metabolite generated during the production of the aminocyclitol antibiotics [40].
Some researchers have suggested that the glutamine transaminase/ω-amidase pathway is part of a glutamine cycle in certain microorganisms, including Neurospora and Rhizobium species. The cycle is proposed to regulate nitrogen flux depending on the nutritional needs of the cell [41,42]. In this regard, it is interesting that Nit2 in Neurospora activates a series of unlinked enzymes in the nitrogen regulatory circuit [43].
In addition to glutamine transamination, asparagine transamination is also widespread in nature. For example, transamination of asparagine with glyoxylate is important in photorespiration in higher plants [44]. Asparagine is preferentially metabolized by asparaginase in rat liver cytosol, but by an asparagine transaminase coupled to ω-amidase in rat liver mitochondria [45].
Despite the obvious biological importance in bacteria, plants and mammals, transamination of glutamine and asparagine, however, may have a downside in mammals. αKGM has been detected in rat tissues and human cerebrospinal fluid (CSF) in μM concentrations [46]. The concentration of αKGM is greatly increased in the CSF of patients with liver disease, in some cases by as much as fifty-fold [47]. The level of αKGM in the CSF correlates remarkably well with the degree of encephalopathy in these patients [47], and there is some evidence that excess αKGM is toxic to rat brain [48]. αKSM is unstable and readily dimerizes [28,49,50] to a product that undergoes further reactions to form a plethora of hetero-aromatic products, some of which may be toxic [50]. Thus, ω-amidase may have evolved to metabolize potentially toxic αKGM and αKSM to biologically useful α-ketoglutarate and oxaloacetate, respectively.
Liver disease is associated with increased ammonia levels in blood and brain, which, in turn, results in increased cerebral glutamine formation. ω-amidase/Nit2 is product-inhibited by ammonia [20,22]. Thus, the increased αKGM in the CSF of patients with liver disease presumably reflects in part increased glutamine transamination coupled with ammonia-induced inhibition of ω-amidase/Nit2. Most researchers in the area of liver disease now regard excess glutamine formation in the astrocyte compartment as a major contributor to the encephalopathy associated with liver disease. However, the exact mechanism is under debate [51]. The possibility that a product of glutamine metabolism (i.e. αKGM) contributes to the encephalopathy, however, has only rarely been considered [52] since the initial report more than 35 years ago that αKGM may be neurotoxic. We suggest that it is time for a re-evaluation of the role of ω-amidase and αKGM in liver disease. Given the potential toxicity of αKGM and αKSM, it is perhaps not surprising that ω-amidase has evolved to convert these products into biochemically useful molecules, and that the activity is widespread in mammalian tissues, plants and bacteria [28].
One other aspect of αKGM metabolism deserves discussion. Nit proteins have been discussed in terms of the “Rosetta Stone” hypothesis. This hypothesis asserts that a fusion protein (i.e. a protein with two distinct biologically active domains) in the genome of one organism, when expressed as two separate proteins in another organism will function in the same cellular or biochemical pathway [53]. Often two separated proteins in a metabolic pathway are coordinately expressed. A possible Rosetta Stone partner of Nit is Fhit. These proteins are fused in Drosophila melanogaster and Caenorhabditis elegans. However, Nit proteins (i.e. Nit1 and Nit2) are separated from Fhit in mammals. Members of the Fhit superfamily bind and cleave diadenosine polyphosphates (ApnA), such as AppppA and ApppA, thereby generating AMP plus ATP or ADP, respectively [54]. The possibility that the Rosetta Stone hypothesis holds for the Fhit/Nit1 pair and Fhit/Nit2 pair in mammals has been discussed previously [5,6]. There is, however, only moderate correlation of message expression between Fhit and Nit1 or between Fhit and Nit2 in mouse tissues [5,6]. In the present work, we determined that Fhit knockout has no effect on the expression of Nit2 as assessed by no change in ω-amidase activity. Taken together, the finding suggests that the Rosetta Stone theory may not hold for the Nit2/Fhit pair. Moreover, it is difficult to ascertain any metabolic relationship between the reactions catalyzed by ω-amidase/Nit2 and those catalyzed by Fhit.
Although our work shows that Nit2 isolated from rat liver cytosol is identical to ω-amidase, and Jaisson et al. have shown that the Nit2 gene in the mouse genome codes for ω-amidase [18], these findings do not rule out a priori the possibility that other genes encode enzymes that can also catalyze hydrolysis of αKGM. In this regard, liver glutaminase was previously shown to have no activity with αKGM [17], and ω-amidase has no activity with glutamine [17,18]. We also considered the possibility that Nit1 might have some ω-amidase activity. However, our work and that of Jaisson et al. [18] shows that Nit1 does not possess ω-amidase activity. Thus, the identity of the Nit1 amidase is still unknown. As noted in the Introduction, both Nit1 and Nit2 show some homology to the metalloprotein β-ureidopropionase [4]. In addition to αKGM and αKSM, ω-amidase exhibits amidase activity with glutaramate and succinamate [21,22,28]. The structure of glutaramate [H2NC(O)CH2CH2CH2CO2−] is remarkably similar to that of ureidopropionate (N-carbamyl-β-alanine) [H2NC(O)NHCH2CH2CO2−]. Although a mammalian Nit1 has not yet been crystallized, the C. elegans NitFhit protein has been crystallized and the putative amidase active site has been mapped [54]. Moreover, Drosophila and yeast β-ureidopropionase have been crystallized [55]. Of interest is the recent determination of the crystal structure of mouse Nit2 by Barglow et al. [4]. Remarkably, although these authors constructed a detailed topology of the amidase active site and pinpointed the position of the canonical Cys-Glu-Lys catalytic triad (but see our comment in 3.3), they were unable to determine the actual substrate(s). Our work and that of Jaisson et al. [18] now establishes the identity of the Nit2 amidase. We suggest that a detailed comparative molecular modeling of the active sites of β-ureidopropionase and ω-amidase/Nit2, together with the known crystal structure of C. elegans NitFhit will provide clues as to the natural substrate(s) of mammalian Nit1.
Finally, Cooper et al. previously noted the presence of ω-amidase activity in rat liver mitochondria [20]. Whether the gene coding for mitochondrial ω-amidase is identical to Nit2 or is another gene is not yet known.
Possibly as a result of its widespread expression nature appears to have chosen the ω-amidase protein for another function, namely as a tumor suppressor protein. It remains to be determined whether the tumor suppressor properties of Nit2 are related to the amidase activity or reside in another domain of the protein that is not involved in amidase catalysis. Questions also remain about the possible regulation of the properties ω-amidase/Nit2 protein by phosphorylation. In other words, does phosphorylation alter ω-amidase activity, anti-tumor activity or both? For Nit1, at least, there is a hint that its tumor suppressor function is unaffected by mutation of the active site cysteine [6], suggesting that the enzymatic function of Nit1 is not required for suppressor activity, much as Fhit enzymatic function is not required for suppressor activity [56]. These findings further support the idea that nature has evolved multiple functions for proteins encoded by the surprisingly few mammalian genes identified in mammalian genomes.
In conclusion, given the burgeoning biomedical interest in mammalian nitrilase-like (Nit) proteins it was important to identify the in vivo natural substrates and establish their biological and biomedical roles. The present findings, together with those of Jaisson et al. [18], and the recent determination of the crystal structure of mouse Nit2 to 1.4Å resolution [4], should provide an impetus for further studies into the roles of ω-amidase/Nit2 in 1) nitrogen and sulfur metabolism, 2) tumor suppression, and 3) brain diseases. A comparative analysis of the topologies of the active sites of Nit1 and Nit2, together with the known amidase substrates of Nit2, may provide clues as to the nature of the Nit1 biological substrate(s).
Acknowledgments
This work was supported by NIH grants RO1ES008421 (to AJLC) and CA111842 (to JTP).
Abbreviations
- ABC
ammonium bicarbonate buffer
- ω-amidase
ω-amidodicarboxylate amidohydrolase
- αKGM
α-ketoglutaramate
- αKSM
α-ketosuccinamate
- ApnA
diadenosine polyphosphates
- CDNB
1-chloro-2,4-dinitrobenzene
- CSF
cerebrospinal fluid
- DKO
Fhit−/−/Nit1−/− double knockout
- DTT
dithiothreitol
- ErbB2
erythroblastic leukemia viral oncogene homologue 2
- GST
glutathione S-transferase
- Fhit
fragile histidine triad
- FKO
Fhit−/− knock-out
- KMB
α-keto-γ-methiolbutyrate
- MTA
5′-methylthioadenosine
- Nit1
nitrilase-like protein 1
- Nit2
nitrilase-like protein 2
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Bork P, Koonin EV. A new family of carbon-nitrogen hydrolases. Protein Sci. 1994;3:1344–1346. doi: 10.1002/pro.5560030821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pace HC, Brenner C. The nitrilase superfamily: classification, structure and function. Genome Biol. 2001;2:1–9. doi: 10.1186/gb-2001-2-1-reviews0001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Brenner C. Catalysis in the nitrilase superfamily. Curr Opin Struct Biol. 2002;12:775–782. doi: 10.1016/s0959-440x(02)00387-1. [DOI] [PubMed] [Google Scholar]
- 4.Barglow KT, Saikatendu KS, Bracey MH, Huey R, Morris GM, Olson AJ, Stevens RC, Cravatt BF. Functional proteomic and structural insights into molecular recognition in the nitrilase family enzymes. Biochemistry. 2008;47:13514–13523. doi: 10.1021/bi801786y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lin CH, Chung MY, Chen WB, Chien CH. Growth inhibitory effect of the human NIT2 gene and its allelic imbalance in cancer. FEBS J. 2007;274:2946–2956. doi: 10.1111/j.1742-4658.2007.05828.x. [DOI] [PubMed] [Google Scholar]
- 6.Semba A, Han SY, Qin HR, McCorkell KA, Iliopoulos D, Pekarsky Y, Druck T, Trapasso F, Corce CC, Huebner K. Biological functions of Mammalian Nit1, the counterpart of the invertebrate NitFhit Rosetta Stone protein, a possible tumor suppressor. J Biol Chem. 2007;281:28244–28253. doi: 10.1074/jbc.M603590200. [DOI] [PubMed] [Google Scholar]
- 7.Laronga C, Yang HY, Neal C, Lee MH. Association of the cyclin-dependent kinases and 14-3-3 sigma negatively regulates cell cycle progression. J Biol Chem. 2000;275:23106–23112. doi: 10.1074/jbc.M905616199. [DOI] [PubMed] [Google Scholar]
- 8.Yang H, Wen YY, Zhao R, Lin YL, Fournier K, Yang HY, Qiu Y, Diaz J, Laronga C, Lee MH. DNA damage-induced protein 14-3-3 sigma inhibits protein kinase B/Akt activation and suppresses Akt-activated cancer. Cancer Res. 2006;66:3096–3105. doi: 10.1158/0008-5472.CAN-05-3620. [DOI] [PubMed] [Google Scholar]
- 9.Takihara Y, Matsuda Y, Hara J. Role of the β isoform of 14-3-3 proteins in cellular proliferation and oncogenic transformation. Carcinogenesis. 2000;21:2073–2077. doi: 10.1093/carcin/21.11.2073. [DOI] [PubMed] [Google Scholar]
- 10.Sugiyama A, Miyagi Y, Komiya Y, Kurabe N, Kitanaka C, Kato N, Nagashima Y, Kuchino Y, Tashiro F. Forced expression of antisense 14-3-3β RNA suppresses tumor cell growth in vitro and in vivo. Carcinogenesis. 2003;24:1549–1559. doi: 10.1093/carcin/bgg113. [DOI] [PubMed] [Google Scholar]
- 11.Sun J, Okumura H, Yearsley M, Frankel W, Fong LY, Druck T, Huebner K. Nit1 and Fhit tumor suppressor activities are additive. J Cell Biochem. 2009 doi: 10.1002/jcb.22207. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Duthie SJ, Mavrommatis Y, Rucklidge G, Reid M, Duncan G, Moyer MP, Pirie LP, Bestwick CS. The response of human colonocytes to folate deficiency in vitro: Functional and proteomic analyses. J Proteome Res. 2008;7:3254–3266. doi: 10.1021/pr700751y. [DOI] [PubMed] [Google Scholar]
- 13.Wang D, Jensen RH, Williams KE, Pallavicini MG. Differential protein expression in MCF7 breast cancer cells transfected with ErbB2, neomycin resistance and luciferase plus yellow fluorescent protein. Proteomics. 2004;4:2175–2183. doi: 10.1002/pmic.200300728. [DOI] [PubMed] [Google Scholar]
- 14.Myung JK, Gulesserian T, Fountoulakis M, Lubec G. Deranged hypothetical proteins Rik protein, Nit protein 2 and mitochondrial inner membrane protein, mitofilin, in fetal Down syndrome brain. Cell Mol Biol. 2003;49:739–746. [PubMed] [Google Scholar]
- 15.Amiel A, Avivi L, Gaber E, Fejgin MD. Asynchronous replication of allelic loci in Down syndrome. Eur J Hum Genet. 1998;6:359–364. doi: 10.1038/sj.ejhg.5200199. [DOI] [PubMed] [Google Scholar]
- 16.Meister A, Sober HA, Tice SV, Fraser PE. Transamination and associated deamidation of asparagine and glutamine. J Biol Chem. 1952;197:319–330. [PubMed] [Google Scholar]
- 17.Meister A, Levintow L, Greenfield RE, Abendschein PA. Hydrolysis and transfer reactions catalyzed by ω-amidase. J Biol Chem. 1955;215:441–460. [PubMed] [Google Scholar]
- 18.Jaisson S, Veiga-da-Cunha M, Van Schaftingen E. Molecular identification of ω-amidase, the enzyme that is functionally coupled with glutamine transaminases. Biochimie. 2009 doi: 10.1016/j.biochi.2009.07.002. this issue. [DOI] [PubMed] [Google Scholar]
- 19.Cooper AJ, Krasnikov BF, Nostramo R, Nieves E, Callaway M, Chien CH, Pinto JT. Ubiquitously expressed ω-amidase is a potential neuroprotectant identical to tumor suppressor Nit2. J Neuochem. 2009;108(Suppl 1):156. Abstract. [Google Scholar]
- 20.Cooper AJL, Duffy TE, Meister A. α-Keto acid ω-amidase from rat liver. Methods Enzymol. 1985;113:350–358. doi: 10.1016/s0076-6879(85)13048-x. [DOI] [PubMed] [Google Scholar]
- 21.Krasnikov BF, Nostramo R, Pinto JT, Cooper AJL. ω-amidase/Nit2, a ubiquitously expressed. Assay and purification of putative tumor suppressor that catalyzes the deamidation of the α-keto acid analogues of glutamine and asparagine. Anal Biochem. 2009;391:144–150. doi: 10.1016/j.ab.2009.05.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hersh LB. Rat liver ω-amidase: Purification and properties. Biochemistry. 1971;10:2884–2891. doi: 10.1021/bi00791a014. [DOI] [PubMed] [Google Scholar]
- 23.Jensson H, Ålin P, Mannervik B. Glutathione transferase isoenzymes. Methods Enzymol. 1985;113:504–507. doi: 10.1016/s0076-6879(85)13066-1. [DOI] [PubMed] [Google Scholar]
- 24.Krasnikov BF, Zorov DB, Antonenko YN, Zaspa AA, Kulikov IV, Kristal BS, Cooper AJL, Brown AM. Comparative kinetic analysis reveals that inducer-specific ion release precedes the mitochondrial transition. Biochim Biophys Acta (Bioenergetics) 2005;1708:375–392. doi: 10.1016/j.bbabio.2005.05.009. [DOI] [PubMed] [Google Scholar]
- 25.Cooper AJL, Pinto JT. Aminotransferase, L-amino acid oxidase and beta-lyase reactions involving L-cysteine S-conjugates found in allium extracts. Relevance to biological activity? Biochem Pharmacol. 2005;69:209–220. doi: 10.1016/j.bcp.2004.08.034. [DOI] [PubMed] [Google Scholar]
- 26.Fong LYY, Fidanza V, Zanesi N, Lock LF, Siracusa LD, Mancini R, Siprashvili Z, Ottey M, Martin SE, Druck T, McCue PA, Croce CM, Huebner K. Muir-Torre-like syndrome in fhit-deficient mice. P Natl Acad Sci USA. 2000;97:4742–4747. doi: 10.1073/pnas.080063497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ottey M, Han SY, Druck T, Barnoski BL, McCorkell KA, Croce CM, Raventos-Suarez C, Fairchild CR, Wang Y, Huebner K. Fhit-deficient normal and cancer cells are mitomycin C and UVC resistant. Br J Cancer. 2004;91:1669–1677. doi: 10.1038/sj.bjc.6602058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Meister A. Preparation and enzymatic reactions of the keto analogues of glutamine and asparagine. J Biol Chem. 1953;200:571–589. [PubMed] [Google Scholar]
- 29.Parli JA, Godfrey DA, Ross CD. Separate microassays for aspartate aminotransferase isoenzymes. Biochim Biophys Acta. 1987;925:175–184. doi: 10.1016/0304-4165(87)90107-3. [DOI] [PubMed] [Google Scholar]
- 30.Ketterer B, Christodoulides LG. Enzymology of cytosolic glutathione S-transferases. Adv Pharmacol. 1994;27:37–69. doi: 10.1016/s1054-3589(08)61029-7. [DOI] [PubMed] [Google Scholar]
- 31.Cooper AJL, Meister A. Comparative studies of glutamine transaminases from rat tissues. Comp Biochem Physiol. 1981;69B:137–145. [Google Scholar]
- 32.Cooper AJL. The role of -keto acid metabolism in the_glutamine transaminase K (GTK) in sulfur and brain, and in the possible bioactivation of neurotoxicants. Neurochem Int. 2004;44:557–577. doi: 10.1016/j.neuint.2003.12.002. [DOI] [PubMed] [Google Scholar]
- 33.Dai Y, Wensink PC, Abeles RH. One protein, two enzymes. J Biol Chem. 1999;274:1193–1195. doi: 10.1074/jbc.274.3.1193. [DOI] [PubMed] [Google Scholar]
- 34.Dai Y, Pochapsky TC, Abeles RH. Mechanistic studies of two dioxygenases in the methionine salvage pathway of Klebsiella pneumoniae. Biochemistry. 2001;40:6379–6387. doi: 10.1021/bi010110y. [DOI] [PubMed] [Google Scholar]
- 35.Berger BJ, English S, Chan G, Knodel MH. Methionine regeneration and aminotransferases in Bacillus subtilis, Bacillus cereus, and Bacilis anthracis. J Bacteriol. 2003;185:2418–2431. doi: 10.1128/JB.185.8.2418-2431.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Backlund PS, Jr, Chang CP, Smith RA. Identification of 2-keto-4-methylthiobutyrate as an intermediate compound in methionine synthesis from 5′-methylthioadenosine. J Biol Chem. 1982;257:4196–4202. [PubMed] [Google Scholar]
- 37.Walker JB. Pathways of biosynthesis of guanidated inositol moieties of streptomycin and bluensomycin. Methods Enzymol. 1975;43:429–470. doi: 10.1016/0076-6879(75)43097-x. [DOI] [PubMed] [Google Scholar]
- 38.Walker JB, Walker MS. Streptomycin biosynthesis. Transamination reactions involving inosamines and inosadiamines. Biochemistry. 1969;8:763–770. doi: 10.1021/bi00831a003. [DOI] [PubMed] [Google Scholar]
- 39.Chen Y, Walker JB. Transamination involving keto- and amino-inositols and glutamine in actinomycetes which produce gentamycin and neomycin. Biochem Biophys Res Commun. 1977;77:688–692. doi: 10.1016/s0006-291x(77)80033-8. [DOI] [PubMed] [Google Scholar]
- 40.Lucher LA, Chen YM, Walker JB. Reactions catalyzed by purified L-glutamine keto-scyllo-Inositol aminotransferase, an enzyme required for biosynthesis of aminocyclitol antibiotics. Antimicrob Agents Chemother. 1989;33:452–459. doi: 10.1128/aac.33.4.452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Calderón J, Morett E, Mora J. ω-Amidase pathway in the degradation of glutamine in Neurospora crassa. J Bacteriol. 1985;161:807–809. doi: 10.1128/jb.161.2.807-809.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mora J. Glutamine metabolism and cycling in Neurospora crassa. Microbiol Rev. 1990;54:293–304. doi: 10.1128/mr.54.3.293-304.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chiang TY, Marzluf GA. DNA recognition by the NIT2 nitrogen regulatory protein: Importance of the number, spacing, and orientation of GATA core elements and their flanking sequences upon Nit2 binding. Biochemistry. 1994;33:576–582. doi: 10.1021/bi00168a024. [DOI] [PubMed] [Google Scholar]
- 44.Ta TC, Joy KW, Ireland RJ. Role of asparagine in the photorespiratory nitrogen metabolism of pea leaves. Plant Physiol. 1985;78:334–337. doi: 10.1104/pp.78.2.334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Moraga-Amador DA, MacPhee-Quiggley KM, Keefer JF, Schuster SM. Asparagine catabolism in rat liver mitochondria. Arch Biochem Biophys. 1989;258:314–326. doi: 10.1016/0003-9861(89)90593-6. [DOI] [PubMed] [Google Scholar]
- 46.Duffy TE, Cooper AJL, Meister A. Identification of α-ketoglutaramate in rat liver, kidney and brain. Relationship to glutamine transaminase and ω-amidase activities. J Biol Chem. 1974;249:7603–7606. [PubMed] [Google Scholar]
- 47.Vergara F, Plum F, Duffy TE. α-Ketoglutaramate: increased concentrations in the cerebrospinal fluid of patients in hepatic coma. Science. 1974;183:81–83. doi: 10.1126/science.183.4120.81. [DOI] [PubMed] [Google Scholar]
- 48.Vergara F, Duffy TE, Plum F. α-Ketoglutaramate, a neurotoxic agent in hepatic coma. Trans Assoc Am Phys. 1973;86:255–263. [PubMed] [Google Scholar]
- 49.Stephani RA, Meister A. Structure of the dimeric α-keto acid analogue of asparagine. J Biol Chem. 1971;246:7115–7118. [PubMed] [Google Scholar]
- 50.Cooper AJL, Raps SP, Meister A. Fluorometric determination of α-ketosuccinamic acid in rat tissues. Anal Biochem. 1987;167:312–320. doi: 10.1016/0003-2697(87)90170-9. [DOI] [PubMed] [Google Scholar]
- 51.Zwingmann C, Butterworth R. An update on brain glutamine synthesis and its relation to cell-specific energy metabolism in the hyperammonemic brain: Further studies using NMR spectroscopy. Neurochem Int. 2005;47:19–30. doi: 10.1016/j.neuint.2005.04.003. [DOI] [PubMed] [Google Scholar]
- 52.Hawkins RA, Jessy J, Mans A, DeJoseph MR. Effect of reducing brain glutamine synthesis on metabolic symptoms of hepatic encephalopathy. J Neurochem. 1993;60:1000–1006. doi: 10.1111/j.1471-4159.1993.tb03247.x. [DOI] [PubMed] [Google Scholar]
- 53.Marcotte EM, Pellegrini M, Ng HL, Rice DW, Yeates TO, Eisenberg D. Detecting protein function and protein-protein interactions from genome sequences. Science. 1999;285:751–753. doi: 10.1126/science.285.5428.751. [DOI] [PubMed] [Google Scholar]
- 54.Pace HC, Hodawadekar SC, Draganescu A, Huang J, Bieganowski P, Pekarsky Y, Croce CM, Brenner C. Crystal structure of the worm NitFhit Rosetta Stone protein reveals a Nit tetramer binding two Fhit dimers. Current Biol. 2000;10:907–917. doi: 10.1016/s0960-9822(00)00621-7. [DOI] [PubMed] [Google Scholar]
- 55.Lundgren S, Lohkamp B, Andersen B, Piskur J, Dobritzsc D. The crystal structure of β-alanine synthase from Drosophila melanogaster reveals a homooctameric helical turn-like assembly. J Mol Biol. 2008;377:1544–1559. doi: 10.1016/j.jmb.2008.02.011. [DOI] [PubMed] [Google Scholar]
- 56.Trapasso F, Krakowiak A, Cesari R, Arkles J, Yendamuri S, Ishii H, Vecchione A, Kuroki T, Bieganowski P, Pace HC, Huebner K, Croce CM, Brenner C. Designed FHIT alleles established that Fhit-induced apoptosis in cancer cells is limited by substrate-binding. Proc Natl Acad Sci USA. 2003;100:1592–1597. doi: 10.1073/pnas.0437915100. [DOI] [PMC free article] [PubMed] [Google Scholar]