

Abstract
RET/PTC (RET/papillary thyroid carcinoma) oncoproteins result from the in-frame fusion of the RET receptor tyrosine kinase domain with protein dimerization motifs encoded by heterologous genes. Here we show that RET/PTC stimulates the β-catenin pathway. By stimulating PI3K/AKT and Ras/ERK, RET/PTC promotes GSK3β phosphorylation, thereby reducing GSK3β-mediated N-terminal β-catenin (Ser33/Ser37/Thr41) phosphorylation. In addition, RET/PTC physically interacts with β-catenin, and increases its phosphotyrosine content. The increased free pool of S/T(nonphospho)/Y(phospho)β-catenin is stabilized as a result of the reduced binding affinity for the Axin/GSK3β complex and activates the T-cell factor/lymphoid enhancer factor (TCF/LEF) transcription factor. Moreover, through the ERK pathway, RET/PTC stimulates cAMP-responsive element binding protein (CREB) phosphorylation and promotes the formation of a β-catenin-CREB-CBP/p300 transcriptional complex. Transcriptional complexes containing β-catenin are recruited to the cyclin D1 promoter and a cyclin D1 gene promoter reporter is active in RET/PTC expressing cells. Silencing of β-catenin by siRNA inhibits proliferation of RET/PTC transformed PC thyrocytes, whereas a constitutively active form of β-catenin stimulates autonomous proliferation of thyroid cells. Thus, multiple signaling events downstream from RET/PTC converge on β-catenin to stimulate cell proliferation.
Keywords: thyroid cancer, tyrosine kinase, oncogene
Introduction
Papillary thyroid carcinoma (PTC) features chromosomal aberrations that result in the fusion of the tyrosine kinase domain of the RET receptor with the N-terminus of heterologous proteins, thereby generating the RET/papillary thyroid carcinoma (PTC) oncoproteins (1). Although there is large variation according to the geographic area and the detection method, the fraction of PTC samples positive for RET/PTC oncogenes is estimated to be 20-40%; this fraction increases up to 80% in PTC developed in radiation-exposed individuals (1). RET/PTC1 (H4-RET) and RET/PTC3 (NCOA4-RET) are the most prevalent variants (1). Fusion with protein partners that have protein-protein interaction motifs provides RET/PTC kinases with dimerizing interfaces, thereby resulting in ligand-independent dimerization and constitutive kinase activation. Autophosphorylation of RET Y1062 plays a particularly important role in cell transformation (2). Accordingly, when phosphorylated, Y1062 acts as the binding site for several protein tyrosine binding (PTB) proteins namely, Shc, IRS1/2, FRS2, and DOK1/4/5 (2). This mediates recruitment of Grb2/SOS complexes (2-5) and Gab family adaptors (2, 6-8) leading to Ras/RAF/ERK and PI3K/AKT signaling (9-11). Moreover, RET/PTC depends on the phosphorylation of Y1062 for recruitment at the inner surface of the cell membrane (2-5).
RET/PTC-mediated transformation requires the Ras/RAF/ERK cascade (11-13). In line with this observation, oncogenic conversion of a member of the RAF family, BRAF, is frequently detected in PTC (1, 14). Also deregulation of the PI3K signaling, through activation of PI3K and AKT serine/threonine kinase or loss of PTEN phosphatase, is prevalent in thyroid cancer (15, 16). PI3K signaling is mitogenic for thyrocytes (15, 16).
β-catenin is a multifunctional protein that plays an important role in signal transduction. In normal resting cells, β-catenin is mainly localized to the adherens junctions, whereas free cytosolic β-catenin is recruited to a “destruction” complex that includes the scaffolding protein Axin, the tumor suppressor adenomatous polyposis coli (APC), and glycogen synthase kinase 3 (GSK3β). This complex facilitates the N-terminal serine/threonine-phosphorylation of β-catenin by GSK3β, thereby targeting β-catenin for degradation by the ubiquitin-proteasome (17). When the pathway is activated, as it occurs in the presence of Wnt ligands, β-catenin is stabilized. Stabilization of β-catenin results in its nuclear accumulation and interaction with T-cell factor/lymphoid enhancer factor (TCF/LEF) or other transcription factors, and activation of genes required for cell proliferation (e.g., c-Myc and cyclin D1) (18). PI3K/AKT induces Ser9/21 GSK3β phosphorylation and inhibition of GSK3β-mediated phosphorylation of β-catenin (19). p90RSK, a Ras/ERK downstream kinase, also phosphorylates and inhibits GSK3β thereby leading to β-catenin upregulation (17, 20, 21). In this pathway, ERK associates with a docking motif, FKFP (residues 291-294), of GSK3β and phosphorylates it at Thr43 to prime it for subsequent phosphorylation at Ser9/21 by p90RSK (22).
Upregulation of β-catenin occurs in a variety of cancers, namely colorectal, breast and ovarian cancers (17, 18). Mutations that activate β-catenin occur late in thyroid tumor progression being detected in undifferentiated (anaplastic) thyroid carcinomas (23). Increased free β-catenin pools have been observed in thyroid carcinomas secondary to reduced E-cadherin expression (24). It has been recently reported that oncogenic point mutants of RET (2A-RET and 2B-RET), which are associated with medullary thyroid cancer (MTC), phosphorylate β-catenin on Y654 thereby promoting β-catenin escape from APC/Axin/GSK3β-mediated destruction (25). This prompted us to investigate the functional connection between RET/PTC and the β-catenin signaling cascades.
Materials and Methods
Cell lines
HEK293T cells were grown in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal calf serum (Invitrogen Groningen, The Netherlands). The human thyroid cancer cell lines, derived from papillary (TPC-1, BCPAP) or anaplastic (OCUT-1, 8505C) thyroid carcinoma, were grown in DMEM containing 10% fetal bovine serum. 8505C were from DSMZ (Deutsche Sammlung von Mikroorganismen und Zellkulturen GmbH, Braunschweig, Germany). OCUT-1 were a gift of K. Hirakawa and N. Onoda. TPC-1 harbour the RET/PTC1 rearrangement; BCPAP, OCUT-1 and 8505C harbour the BRAF V600E mutation (26). Normal human thyrocytes (NT) were isolated from normal thyroid tissue obtained from a patient who underwent thyroid surgery and cultivated in RPMI supplemented with 20% fetal bovine serum. PC Cl 3 (hereafter referred to as “PC”) is a differentiated thyroid follicular cell line derived from 18-month-old Fischer rats. PC cells were cultured in Coon's modified Ham F12 medium supplemented with 5% calf serum and a mixture of 6 hormones (6H), including thyrotropin (TSH, 10 mU/ml), hydrocortisone (10 nM), insulin (10 μg/ml), apo-transferrin (5 μg/ml), somatostatin (10 ng/ml) and glycyl-histidyl-lysine (10 ng/ml) (Sigma Chemical Co., St. Louis, MO). RET/PTC-expressing PC cells have been described previously (11). In order to obtain PC cells stably expressing the RET/PTC(4F) or (3F) mutants, transfections were performed by the calcium phosphate co-precipitation technique as described previously (11) and mass populations of several ten cell clones were isolated for each transfection by G418 selection. Equal levels of expression of RET/PTC mutants was confirmed by immunoblotting. Transient transfections were carried out with the lipofectamine reagent according to the manufacturer's instructions (GIBCO, Paisley, PA).
Cell growth and staining
For growth curves, 0.5×105 cells were seeded in triplicate and counted at the indicated time points. DNA synthesis rate was measured by the 5′-bromo-3′-deoxyuridine (BrdU) Labeling and Detection Kit from Boehringer Mannheim (Germany). Briefly, cells were seeded on glass coverslips, pulsed for 1 h with BrdU (final concentration of 10 μM), fixed and permeabilized. Coverslips were incubated with anti-BrdU mouse monoclonal and rhodamine-conjugated secondary antibodies (Jackson ImmunoResearch Laboratories, Philadelphia, Pennsylvania) and mounted in Moviol on glass slides. Cell nuclei were identified by Hoechst 33258 (final concentration 1 μg/ml; Sigma Chemicals Co) staining. The fluorescent signal was visualized with an epifluorescent microscope (Axiovert 2, Zeiss) (equipped with a 100X lens) interfaced with the image analyzer software KS300 (Zeiss). At least 100 cells were counted in five different microscopic fields; results were average fractions of BrdU-positive cells ± SD.
Tissue samples
Archival frozen thyroid tissue samples from 12 patients affected by PTC (T1-T12) and 2 normal thyroids (N1-2) were retrieved from the files of the Pathology Department of the University of Pisa (Italy). The study was approved by the Institutional Ethics Committee. Sections (4-μM thick) of paraffin-embedded samples were stained with hematoxylin and eosin for histological examination to ensure that the samples fulfilled the diagnostic criteria required for the identification of PTC (enlarged nuclei with fine dusty chromatin, nuclear grooves, single or multiple micro/macro nucleoli and intranuclear inclusions) (27). According to a previous characterization, 3 of these PTC samples had a RET/PTC1 rearrangement (T2, T7, T12) and 6 had a BRAF V600E mutation (T1, T4, T5, T6, T8, T10) (11). Snap-frozen tissue samples were kept in liquid nitrogen for storage at −80°C until protein extraction was performed.
Plasmids
Unless otherwise specified, RET/PTC3 is the RET/PTC form used in this study. The RET/PTC3 constructs were cloned in pBABE and pCDNA3.1 (Invitrogen). They encode the short (RET-9) RET/PTC3 spliced form and are described elsewhere (11). For simplicity, we numbered the residues of RET/PTC proteins according to the corresponding residues in unrearranged RET. Briefly, RET/PTC(K-) is a kinase-dead mutant carrying the substitution of the catalytic lysine (residue 758 in full-length RET) with a methionine. RET/PTC(4F) is a mutant in which the 4 autophosphorylation sites (Y826, Y1015, Y1029, Y1062) of the carboxyl-terminal tail are mutated to phenylalanines; in RET/PTC(3F), the Y1062 has been added back. Plasmids encoding the dominant-negative Ras(N17), MEK(DN) and AKT(DN), the constitutively active Ras(V12), BRAF(V600) and β-catenin(S374A), and the TCF/LEF reporter system are described elsewhere (28).
Antibodies and compounds
Anti-RET is an affinity-purified polyclonal antibody raised against the tyrosine kinase protein fragment of human RET. Anti-β-catenin (610153) and anti E-cadherin (610181) were purchased from Becton Dickinson (BD Transduction Laboratories, Heidelberg, Germany). Anti-phospho p44/42 MAPK (9102), recognizing MAPK (ERK1/2) when phosphorylated either individually or dually on Thr202 and Tyr204, anti-p44/42 MAPK (9101), anti-phospho AKT (9271), specific for AKT phosphorylated at Ser 473, anti-AKT (9272), anti-phospho Ser 21/9 GSK (9331), anti-GSK (9338), anti phospho-β-catenin (Ser33/37/Thr41) (9561S) were purchased from Cell Signaling (Beverly, MA). Anti-tubulin (T9036) was from Sigma Chemical Company (St. Louis, MO). Monoclonal anti-phospho (Y654)-β-catenin antibody was from Abcam (Cambridge, UK). Anti-phospho-CREB (S133) (06-519), anti-CREB (06-863) and anti-Tcf4 (05-511) were from Upstate Biotechnology Inc. (Lake Placid, NY). Anti CBP (sc-1211), anti-p300 (sc-584), anti-Sp1 (sc-14027) and anti-c-Myc antibody (sc-40) were from Santa Cruz Biotechnology (Santa Cruz, CA). Secondary antibodies coupled to horseradish peroxidase were from Amersham Pharmacia Biotech (Little Chalfort, UK). Cycloheximide was purchased from Sigma Chemical Company, and used at 10 μg/ml final concentration. LY294002 was from Calbiochem (Merck Chemicals Ltd. Nottingham UK) and used at 10 μM final concentration. MEK1/2 inhibitor UO126 was from Cell Signaling and used at 10 μM final concentration.
Protein studies
Immunoblotting experiments were performed according to standard procedures. Protein concentration was estimated with a modified Bradford assay (Bio-Rad, Munich, Germany). Immune complexes were detected with the enhanced chemiluminescence kit (ECL, Amersham Pharmacia Biotech. Signal intensity was analyzed with the Phosphorimager (Typhoon 8600, Amersham Pharmacia Biotech) interfaced with the ImageQuant software. For immunoprecipitations, total lysates were incubated with 2 μg antibody for 2 h at 4°C. Antibody-antigen complexes were collected with 30 μl of protein-G or protein-A sepharose beads overnight at 4°C with gentle rotation. The samples were centrifuged, washed, eluted in sample buffer and run on SDS- polyacrylamide gel. Nuclear extraction was performed as described elsewhere (29). Briefly, cells were harvested in lysis buffer (10 mM Tris-HCl pH 7.9, 10 mM KCl, 1.5 mM MgCl2, 1 mM dithiothreitol (DTT), 1 mM phenylmethyl-sulphonylfluoride (PMSF), supplemented with 60 mM NaF, 60mM β-glycerophosphate and protease inhibitors (aprotinin, leupeptin and pepstatin; 40 mg/ml) and lysed by shearing with 15 passages through a 26-gauge needle mounted in a 1 ml syringe. Nuclei were recovered by centrifugation at 3,000g for 10 min. Nuclear proteins were extracted in 50 mM Tris-HCl, pH 7.5, containing 0.3M sucrose, 0.42 M KCl, 5 mM MgCl2, 0.1 mM EDTA, 20% glycerol, 2 mM DTT, 0.1 mM PMSF, 60 mM NaF, 60 mM β-glycerophosphate, leupeptin and aprotinin. Cytosolic fractions were recovered after membrane fraction removal by 100,000 g ultracentrifugation.
Pull down assay
The GST-RET/PTC3 and GST-RET/TK plasmids were generated by PCR amplification of full length RET/PTC3 or the isolated RET component of the RET/PTC3 protein (RET residues 718-1072) and fusion to the GST coding sequence into the pEBG vector kindly provided by S. Meakin (30). GST-RET/PTC3 and GST-RET/TK fusion proteins were purified from transiently-transfeccted cell lysates using glutathione Sepharose according to standard procedures. HEK293T cells were serum starved for 18 h and lysed in an ice-cold buffer. Protein lysates (2 mg) were incubated over-night with 30 μg of GST-RET/PTC3 and GST-RET/TK fusion proteins. Pellet beads were collected by centrifugation (14,000 × g) and washed with lysis buffer. The beads were resuspended in 2X Laemmli buffer and subjected to western blotting.
Reporter assay
To evaluate the TCF/LEF transcriptional activity, we used a pair of luciferase reporter constructs, TOP-FLASH and the negative control FOP-FLASH (Upstate Biotechnology). TOP-FLASH contains three copies of the TCF/LEF binding site (AAGATCAAAGGGGGT) upstream of the thymidine kinase minimal promoter; FOP-FLASH contains a mutated TCF/LEF binding site (AAGGCCAAAGGGGGT). Cells were transiently transfected by one of these reporters together with pRL-TK (encoding the Renilla luciferase) in triplicate, as instructed by the manufacturer (Promega Corporation, Madison, WI). Luciferase activity was measured 48 h after transfection with the Dual-luciferase reporter assay system (Promega Corporation). TCF/LEF activity was determined by the TOP-FLASH:FOP-FLASH ratio, after normalization for Renilla luciferase activity. Light emission was quantified with a Berthold Technologies luminometer (Centro LB 960) (Bad Wildbad, Germany). Data were represented as average fold change ± S.D. with respect to the negative control.
RNA silencing
Pre-designed duplex siRNA against Rat β-catenin (190086, 190087, 190088) were provided by Ambion (Austin, TX). The scrambled oligonucleotide was synthesized by Proligo (Boulder, CO) and the sequence was 5′-AGGAUAGCGUGGAUUUCGGUTT-3′. The day before transfection, cells were plated in six-well dishes at 30–40% confluency. Transfection was performed using 5 μg of duplex RNA and 6 μl of oligofectamine reagent (Invitrogen). Cells were harvested at 48 h post-transfection or counted at different time points for evaluating cell growth.
Chromatin immunoprecipitation (ChIP)
Chromatin was extracted from RET/PTC or empty vector transfected HEK293T cells. ChIP assay was performed by using the chromatin immunoprecipitation assay kit (Upstate Biotechnology), following manufacturer's instructions. Chromatin was fixed by directly adding formaldehyde (1% v/v) to the cell culture medium. Nuclear extracts were isolated and then fragmented through sonication. Transcription factor-bound chromatin was immunoprecipitated with β-catenin, CREB or TCF antibodies, cross-linking was reversed, and the isolated genomic DNA amplified by quantitative PCR using primers spanning either the CRE site or the TCF/LEF binding site (not shown) of the human cyclin D1 promoter:
CRE-forward (5′-AACGTCACACGGACTACAGG-3′);
CRE-reverse (5′-TGTTCCATGGCTGGGGCTCTT-3′);
TCF-forward (5′-GAGCGCATGCTAAGCTGAAA-3′);
TCF-reverse (5′-GGACAGACGGCCAAAGAATC-3′).
Fluorescent threshold values (Ct) were measured in triplicate for immunoprecipitated samples as well as for an aliquot of the input DNA. Results were calculated by the formula: 2- [ΔCt/Ctinput DNA], where ΔCt is the difference between the Ct of the specific antibody-immunoprecipitated DNA and the Ct of mock-immunoprecipitated DNA. Results are average ± S.D. of triplicate samples.
Statistical analysis
The Student's t test was used for statistical analysis. All p values were two sided and differences were significant when p < 0.05.
Results
RET/PTC promotes the nuclear accumulation of β-catenin
Thyroid follicular PC cells transiently transfected with RET/PTC accumulated nuclear β-catenin, a hallmark of β-catenin activation (Fig. 1A). By biochemical fractionation, although total β-catenin levels were increased upon RET/PTC expression, nuclear beta-catenin levels were proportionally increased to a greater extent, thereby accounting for the increase in the nuclear β-catenin fraction in HEK293T or PC cells (7-fold, p< 0.001, or 3.6-fold, p< 0.01, respectively), upon transient or stable RET/PTC expression (Fig. 1B). β-catenin nuclear accumulation depended on RET/PTC kinase activity, because it was reduced by a kinase-dead mutant (K-). The residual, albeit not significant, activity of PTC (K-) might depend on RET kinase rescue operated by other tyrosine kinases, as EGFR, as recently demonstrated (31). Moreover, β-catenin accumulation depended on RET/PTC autophosphorylation, because it was not exerted by a RET/PTC mutant (PTC-4F) whose major autophosphorylation sites (Y826, Y1015, Y1029, Y1062) were mutated to phenylalanine (Fig. 1B). Y1062 was essential, because nuclear accumulation of β-catenin was restored when tyrosine Y1062 was added-back to the 4F mutant (PTC-3F) (p< 0.01) (Fig. 1B). There was no detectable difference in the mRNA levels of β-catenin between RET/PTC-positive and -negative cells, which suggests that the increase in β-catenin occurred at a post-transcriptional level (data not shown). Accordingly, the half-life of β-catenin increased in RET/PTC expressing cells (> 24 h) versus control HEK293T (about 12 h) (Fig. 1C).
Figure 1. RET/PTC stabilizes β-catenin and promotes its nuclear accumulation.
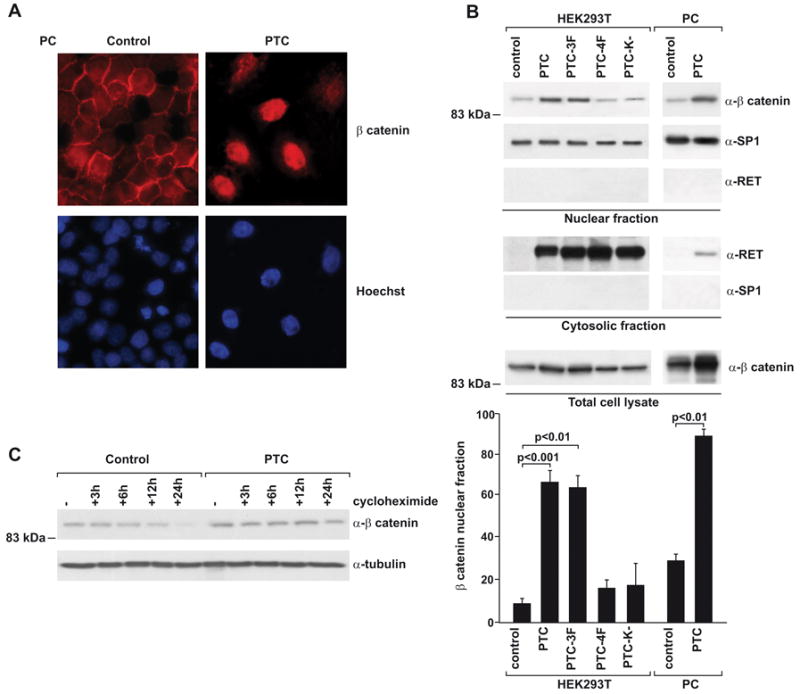
A) Immunofluorescence for β-catenin in PC cells transiently transfected with RET/PTC. Note the increase of nuclear staining in transfected cells. Hoechst stain was used to visualize cell nuclei; parallel transfection with GFP showed that about 20% of the cells expressed exogenous DNA (not shown). B) Nuclear translocation of β-catenin upon RET/PTC expression. HEK293T were transfected with RET/PTC or the indicated mutants (mock-transfected: control). PC-RET/PTC cells or parental PC were also used. Subcellular fractions were obtained and nuclear extracts (50 μg) probed with anti-β-catenin. The purity of the fractions was assessed by verifying the absence of SP1 in the cytosolic fraction and of RET/PTC in the nuclear fraction. Total β-catenin levels were also measured in unfractionated extracts. The percentage of nuclear β-catenin with respect to total levels are reported in the bar-graph (average results ± SD of 5 independent determinations). Reported p values were calculated by the Student's T test. C) HEK293T cells transiently expressing RET/PTC or a control vector were treated with cycloheximide for different time points. Lysates (30 μg) were run on SDS-PAGE, and β-catenin levels were determined by immunoblot.
RET/PTC targets the Axin-GSK3β-β-catenin complex
The pathway leading to β-catenin activation involves a series of events that result in the dissociation of β-catenin from Axin, a scaffold protein that forms a large molecular complex with APC, Dsh, and GSK3β (17, 18). Transient RET/PTC expression in HEK293T cells decreased the amount of β-catenin co-precipitating with myc-tagged Axin; this effect depended on Y1062, as demonstrated when we used the PTC-4F mutant (Fig. 2A). Secondary to the assembly of the Axin-GSK3β-β-catenin complex, phosphorylation of β-catenin by GSK3β in N-terminal Ser33, Ser37 and Thr41 promotes its ubiquitin-dependent proteolytic degradation (19-22). RET/PTC expression in HEK293T reduced amounts of S/T phsophorylated β-catenin co-precipitating with Axin (Fig. 2A). Moreover, transient RET/PTC expression in HEK293T (Fig. 2A and 2B) and stable expression in PC (Fig. 2C) reduced overall S/T β-catenin phosphorylation by immunoblot, an effect that depended on RET/PTC kinase and Y1062 (Fig. 2B). β-catenin phosphorylation is affected by GSK3β. In turn, the activity of GSK3β can be blocked by both AKT (19) and ERK pathway-mediated phosphorylation at Ser9 (22). RET/PTC stimulates both the PI3K/AKT and the ERK pathways by recruiting several adaptors to phosphorylated Y1062 (2). RET/PTC-triggered phosphorylation of AKT and ERK paralleled the phosphorylation of GSK3β-Ser9 and the consequent reduction of phospho-S/T β-catenin (Fig. 2B and 2C). PTC-4F had a significantly reduced activity with respect to wild type RET/PTC (Fig. 2B). Also PTC-3F (although expressed at lower levels) had a reduced effect on GSK3β phosphorylation, suggesting that C-terminal tyrosines other than Y1062 may partecipate to this pathway. LY294002, a PI3K inhibitor, partially impaired GSK3β phosphorylation and increased β-catenin S/T phosphorylation; these effects were associated with a reduction in AKT phosphorylation (Fig. 2B). Also, treatment with U0126, a MEK inhibitor, reduced phosphorylation of GSK-3β and partially rescued phospho-S/T β-catenin (Fig. 2B). Finally, treatment with LY294002 or U0126 reduced nuclear accumulation of β-catenin (Fig. 2D).
Figure 2. RET/PTC induces the dissociation of the β-catenin degradation complex.
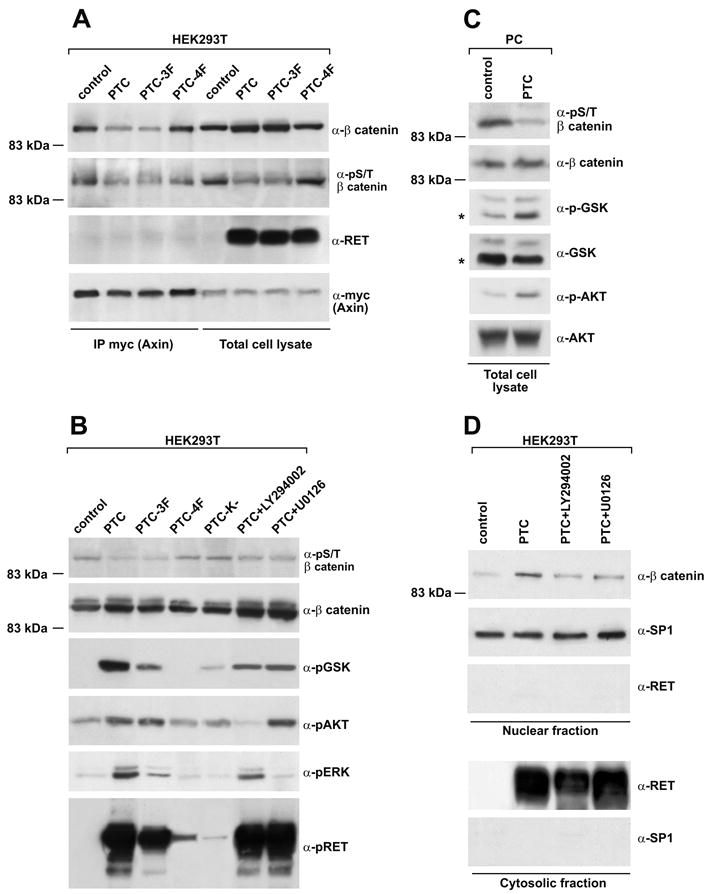
A) HEK293T cells were transfected with a myc-tagged Axin together with RET/PTC or the indicated mutants (mock-transfected: control). Protein complexes containing Axin were recovered by immunoprecipitation (1 mg) with anti-myc tag and blotted against β-catenin or S/T-phosphorylated β-catenin (phospho-Ser33/37/Thr41). Myc-Axin and RET/PTC protein levels in total cell lysates are shown for normalization. B-C) Phosphorylation levels of GSK3β, β-catenin (phospho-Ser33/37/Thr41), AKT, ERK and RET (Y905) were measured by immunoblot of total cell lysates (50 μg) harvested from HEK293T (B) or PC (C) cells expressing the indicated RET/PTC variants or treated with the indicated compounds. GSK3β antibodies may recognize both GSK-3α (51 kDa) and GSK3β (46 kDa) proteins; in (C) an asterisk indicates GSK3β migration. D) Nuclear accumulation of β-catenin (measured as in Fig. 1B) in HEK293T cells transfected with RET/PTC and treated or not with chemical PI3K or MEK inhibitors. The data are representative of at least three independent experiments.
RET/PTC increases β-catenin phosphotyrosine content
β-catenin interacts directly with the oncogenic tyrosine kinases c-src (32), c-MET (33), RON (34), c-erbB-2 (35) and BCR-ABL (36), thereby resulting in β-catenin phosphorylation on tyrosine residues. More importantly, Gujral and co-workers recently reported that MEN2-associated RET point mutants bind to β-catenin and phosphorylate it on tyrosine 654 (25). We tested whether this mechanism was used also by RET/PTC. In HEK293T cells, RET/PTC co-immunoprecipitated with β-catenin (Fig. 3A) and upon RET/PTC expression tyrosine-phosphorylated β-catenin (pY654) increased; overall increased β-catenin levels may partially account for these effects (Fig. 3A). The binding of β-catenin to RET/PTC paralleled its dissociation from E-cadherin (Fig. 3A). Moreover, recombinant GST-RET/PTC3 and GST-RET/TK proteins were able to pull-down β-catenin from HEK293T cell lysates, demonstrating an interaction, either direct or mediated by intermediate protein(s), between the RET component of RET/PTC and β-catenin (Fig. 3B).
Figure 3. RET/PTC interaction with β-catenin.
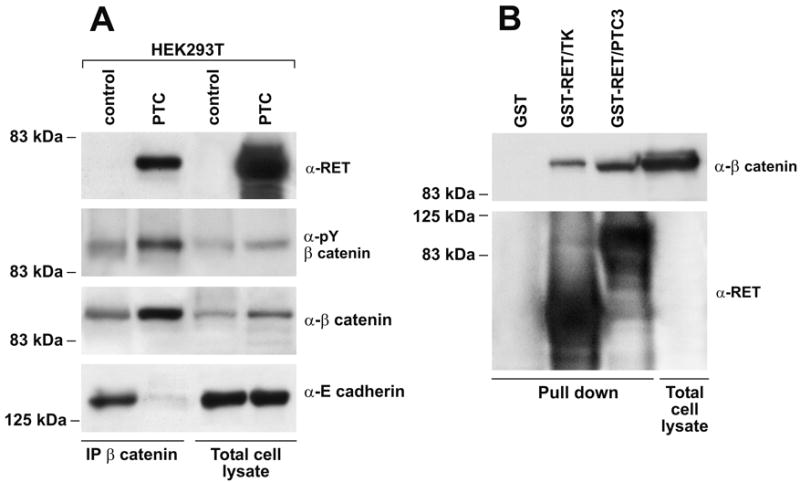
A) Protein lysates (2 mg) from mock-transfected or RET/PTC-transfected HEK293T cells were immunoprecipitated with anti-β-catenin and probed with tyrosine-phosphorylated β-catenin (pY654), RET, or E-cadherin antibodies. Expression levels in total cell lysates are shown for normalization. B) Full-length RET/PTC3 as well as the isolated RET component of the RET/PTC3 (deprived of the NCOA4 region) protein were produced as GST-fusion proteins and used to pull-down beta-catenin from HEK293T protein lysates. Immunoblot was stained with anti-β-catenin; recombinant RET proteins input is also reported.
RET/PTC stimulates TCF/LEF- and CREB-mediated cyclin D1 transcription
Nuclear β-catenin forms complexes with members of the T cell factor (TCF) and lymphoid enhancer factor-1 (LEF) family of DNA-binding proteins, and this process results in activation of target gene promoters. RET/PTC stimulated the activity of a luciferase TCF/LEF-dependent reporter gene system (TOPflash) in both HEK293T and PC cells (p<0.01) (Fig. 4A). The mutant FOPflash reporter, bearing a mutated TCF/LEF site, was used to subtract background. This activity depended on the integrity of the RET/PTC kinase and of the Y1062 multidocking site (p<0.05) (Fig. 4A, left). Again, other tyrosines besides Y1062 likely played a role because the PTC-3F mutant was impaired with respect to wild type RET/PTC (Fig. 4A, left). TOPflash expression was partially reduced by treatment with LY294002 and U0126 and by the co-expression of dominant negative mutants for Ras (N17), MEK (MEKDN) and AKT (AKTDN) (p<0.05). Constitutively active BRAF (V600E) and Ras (V12) stimulated the TCF/LEF reporter in HEK293T cells, although to a lesser extent than RET/PTC. A transcriptionally active form of β-catenin (S374A) served as a positive control (Fig. 4A).
Figure 4. RET/PTC-mediated activation of TCF/LEF and CREB transcription factors.
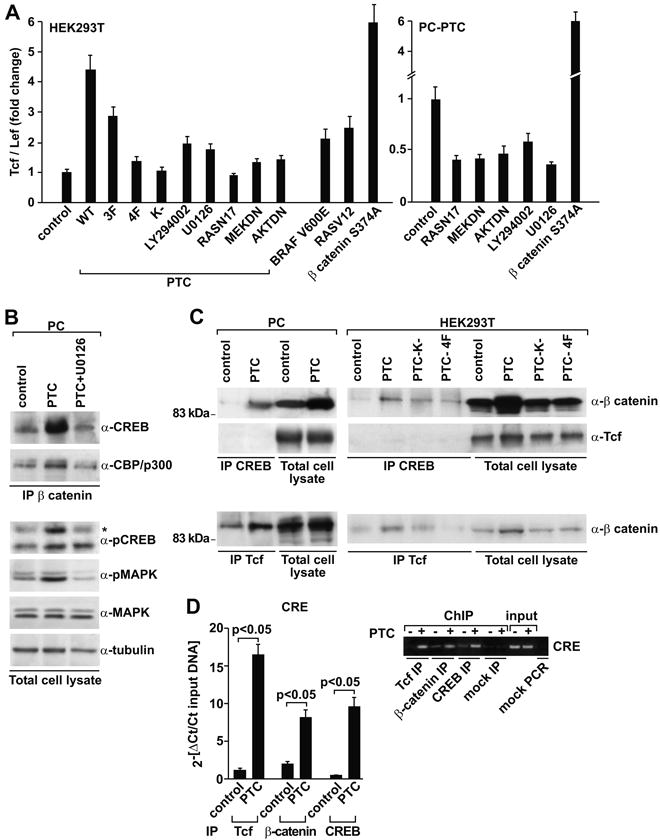
A) HEK293T (left) and PC-PTC cells (right) were transfected with the indicated plasmids together with the TOPflash reporter and when indicated treated with the chemical inhibitors. Luciferase activity was expressed as fold increase with respect to mock-transfected cells. Average results of three independent assays ± SD are indicated. Reported p values were calculated by the Student's T test. B) Cellular lysates (1 mg) from PC and PC-PTC cells, treated or not with U0126, were immunoprecipitated with anti-β-catenin and probed with CREB and CBP/p300 antibodies. The CREB antibody may recognize, besides CREB (43 kDa), also CREM (30 kDa) and ATF-1 (38 kDa); an asterisk indicates CREB migration. Phosphorylation of CREB (S133) and ERK (MAPK) was measured in total cell lysates by immunoblot. Tubulin was used for normalization. C) Lysates from PC or PC-PTC cells (left) and HEK293T cells transiently expressing the indicated RET/PTC constructs (right) were immunoprecipitated with CREB and TCF antibodies and probed for β-catenin or TCF antibodies, as reported. D) Chromatin immunoprecipitation was performed in HEK293T cells transfected with RET/PTC or the empty vector (control). Processed DNA was immunoprecipitated with TCF, β-catenin or CREB antibodies and subjected to real-time PCR (bar graphs) with amplimers spanning the CRE site of the cyclin D1 promoter. Results are average ± S.D. of triplicate samples. Fluorescent threshold values (Ct) were measured for immunoprecipitated samples as well as for an aliquot of the input DNA. Reported p values were calculated by the Student's T test. Semi-quantitative PCR (ethidium bromide stain) was also performed with the same primers by using 1 μl of the DNA and 25 cycles of amplification. Amplification of the expected fragments in the input samples indicated equal input; mock PCR was perfomed without template.
Besides TCF/LEF, β-catenin recruits the activated form (serine 133 phosphorylated) of cAMP-responsive element binding protein (CREB), thereby resulting in a transcriptionally active complex, which in turn binds CREB-binding protein (CBP/p300) (37-40). TCF/LEF and CREB collaborate in regulating transcription of TCRα (41) and WISP-1 (42). Finally, RET signaling through Y1062 and Ras/ERK increases S133 phospho-CREB levels (2). To determine whether RET/PTC affected the binding of CREB to β-catenin, we immunoprecipitated β-catenin from PC cells in the presence or absence of RET/PTC and probed the immunocomplexes with anti-CREB and anti-CBP/p300 antibodies. Expression of RET/PTC significantly increased the amounts of β-catenin bound to CREB and to CBP/p300 (Fig. 4B). Chemical blockade of MEK by U0126 reduced this association, and this effect was paralleled by reduction in CREB phosphorylation on Ser 133 and ERK phosphorylation (Fig. 4B). In a mirror experiment, RET/PTC expression not only increased TCF/LEF-β-catenin (Fig. 4C, bottom) but also CREB-β-catenin protein complexes (Fig. 4C, upper). In HEK293T cells, formation of both complexes was reduced when kinase-dead and 4F mutants were used (Fig. 4C, right). Instead, by immunoprecipitating CREB and staining with TCF/LEF we did not detect TCF/LEF-CREB interaction, suggesting that β-catenin-containing TCF/LEF and CREB complexes are distinct (Fig. 4C, middle).
Cyclin D1 promoter contains adjacent TCF/LEF (at -81 bp) and CREB (at -58 bp) binding sites (40). We used chromatin-immunoprecipitation (ChIP) to measure β-catenin, TCF/LEF and CREB binding to the region of the cyclin D1 promoter that contains CREB and TCF/LEF binding sites. For PCR, we used two primer pairs spanning the CRE (Fig. 4D) and the TCF (not shown) site, respectively. With both primer sets, binding of β-catenin, TCF/LEF and CREB to the cyclin D1 promoter was greatly increased upon RET/PTC expression (p<0.05) (Fig. 4D). Since the binding sites for CREB and TCF/LEF are just few nucleotides apart on the cyclin D1 promoter, this experiment left open possibilities that the two β-catenin-containing (CREB- and TCF/LEF) complexes can be either identical or distinct; however, the lack of co-IP in Figure 4C favours the possibility that the two complexes are distinct.
RET/PTC-mediated mitogenic signaling depends on β-catenin
In PC cells, RET/PTC-mediated increase in β-catenin activity was paralleled by accumulation of the TCF/LEF and CREB target protein cyclin D1 and, more weakly, c-Myc (Fig. 5A). We knocked-down β-catenin by RNAi in parental and RET/PTC expressing PC cells. As shown in Figure 5A, β-catenin specific siRNA, but not the scrambled control, reduced the expression of β-catenin by >50% in both PC and PC-PTC cells. Moreover, β-catenin siRNA reduced cyclin D1 and, more weakly, c-Myc levels in RET/PTC expressing cells. β-catenin RNAi reduced hormone-independent proliferation of PC-PTC cells (Fig. 5B, lower panel), whereas a constitutively active form of β-catenin (S374A) stimulated hormone-independent growth of PC cells (Fig. 5B, upper panel). We generated mass populations of PTC-4F and PTC-3F expressing PC cells. Upon hormone-deprivation, we counted S-phase cells upon a 1 h BrdU pulse after transient transfection with scrambled or β-catenin specific siRNA. Similarly to wt PTC, PTC-3F, but not PTC-4F, expressing cells, incorporated BrdU in the absence of hormones in a β-catenin dependent manner (p< 0.01). Taken together, these findings demonstrate that β-catenin is a mediator of the RET/PTC mitogenic signaling in thyrocytes.
Figure 5. β-catenin contributes to autonomous growth of RET/PTC-expressing thyroid cells.
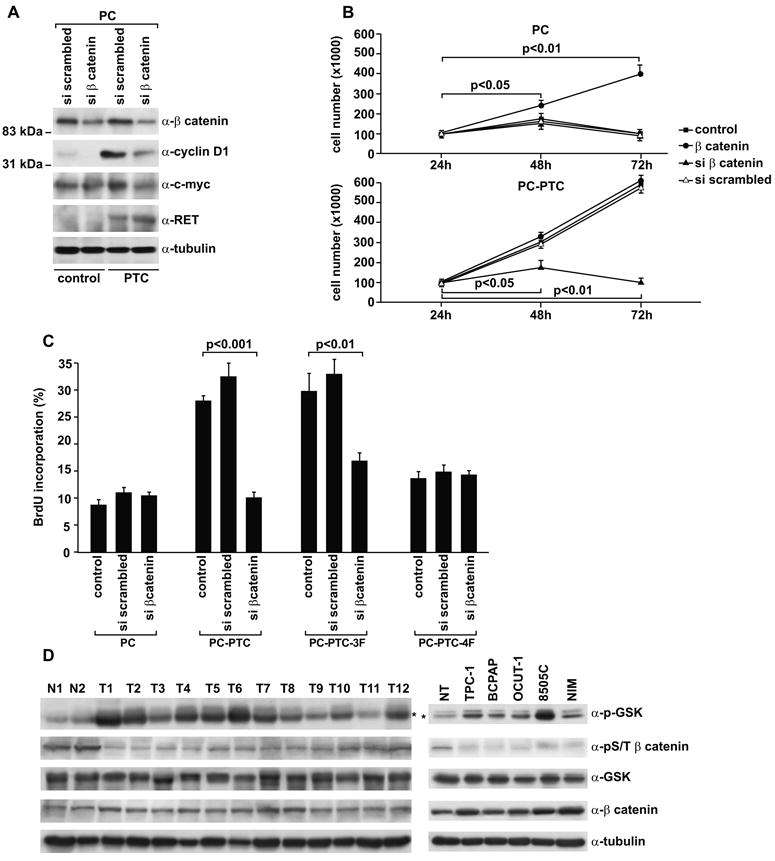
A) β-catenin was knocked-down by transient transfection with specific siRNA in parental or RET/PTC-expressing PC cells. As a control, cells were treated with scrambled siRNA. Protein levels were measured by immunoblot with the indicated antibodies. B) Proliferation in the absence of hormones of parental, constitutively active β-catenin (S374A) or β-catenin siRNA transiently transfected PC or PC-PTC cells was measured by cell counts (average of triplicate samples ± SD). Reported p values were calculated by the Student's T test. C) Mass populations of PTC-4F and PTC-3F expressing PC cells were generated by stable transfection and marker selection. Upon hormone-deprivation and transfection with scrambled or β-catenin specific siRNA (as in A), cells were pulsed (1h) with BrdU and BrdU-positive cells analysed by immunofluorescence. At least 100 cells were counted in five different microscopic fields; results in the bar graph are the average fractions of BrdU-positive cells ± SD. Reported p values were calculated by the Student's T test. D) Phosphorylation levels of GSK3β and β-catenin (phospho-Ser33/37/Thr41) were measured by immunoblot of total cell lysates (50 μg) harvested from PTC (T1-12) and normal thyroid (N1-2) tissue samples (left) or thyroid carcinoma and normal human primary cells (NT) (right). GSK3β antibodies may recognize both GSK-3α (51 kDa) and GSK3β (46 kDa) proteins; an asterisk indicates GSK3β migration.
Finally, to study the β-catenin pathway in human PTC samples, we measured levels of GSK3β-Ser9 and S/T β-catenin phosphorylation in comparison to normal thyroid samples (n. 2) in a small set of PTC (n. 12) samples (3 RET/PTC1-positive, 6 BRAF V600E-positive and the remainder RET/PTC and BRAF negative). Reduced levels of beta catenin S/T phosphorylation was visible in virtually all the tumor samples; increased levels of GSK3β-Ser9 phosphorylation and total β-catenin were detectable in about 75% and 50% of them, respectively (Fig. 5D, left). Similarly, thyroid carcinoma cell lines either positive for RET/PTC1 (TPC-1) or for BRAF V600E (BCPAP, 8505C, OCUT-1) showed reduced levels of S/T β-catenin and increased GSK3β-Ser9 phosphorylation when compared with a primary culture of normal thyrocytes (Fig. 5D, right).
Discussion
Here we describe the functional interaction between the RET/PTC and the β-catenin signaling pathways. β-catenin activation by RET/PTC occurred through several coordinated mechanisms (Fig. 6). As previously reported by Gujral and colleagues for point mutant RET (25), RET/PTC induced the tyrosine phosphorylation of β-catenin, thereby mobilizing the fraction of β-catenin associated to E-cadherin and increasing its free cytosolic pool. RET/PTC-mediated activation of PI3K/AKT and Ras/ERK contributed to promote β-catenin stabilization through inactivation of GSK3β. However, neither pathway was probably sufficient because chemical blockade of either one reduced but did not abrogate β-catenin accumulation. Finally, RET/PTC signaling favoured the formation of transcriptional complexes containing β-catenin by triggering ERK-mediated CREB phosphorylation. Ser133-phosphorylated CREB formed a protein complex with CBP and β-catenin, which participated in the transcription of cyclin D1. TCF/LEF and CREB binding sites co-exist in the promoters of some β-catenin target genes such as cyclin D1 (40), TCRalpha (41) and WISP-1 (42), and collaborate in their firing. Accordingly, our findings suggest that downstream of RET/PTC, β-catenin participates in two distinct transcriptional complexes, one with TCF/LEF and another one with CREB, and that both are recruited to cyclin D1 promoter. The particularly important role played by Y1062 in signaling to β-catenin in the case of RET/PTC with respect to full-length RET mutants (25) may be explained by differential signaling mechanisms between the distinct RET oncogenic forms as well as the cytosolic localization of RET/PTC that relies on Y1062 to be recruited to the cell membrane (2-4). Finally, β-catenin expression is required for RET/PTC-mediated autonomous proliferation of PC thyrocytes.
Figure 6. A summary of the pathways leading to β-catenin induction by RET/PTC.
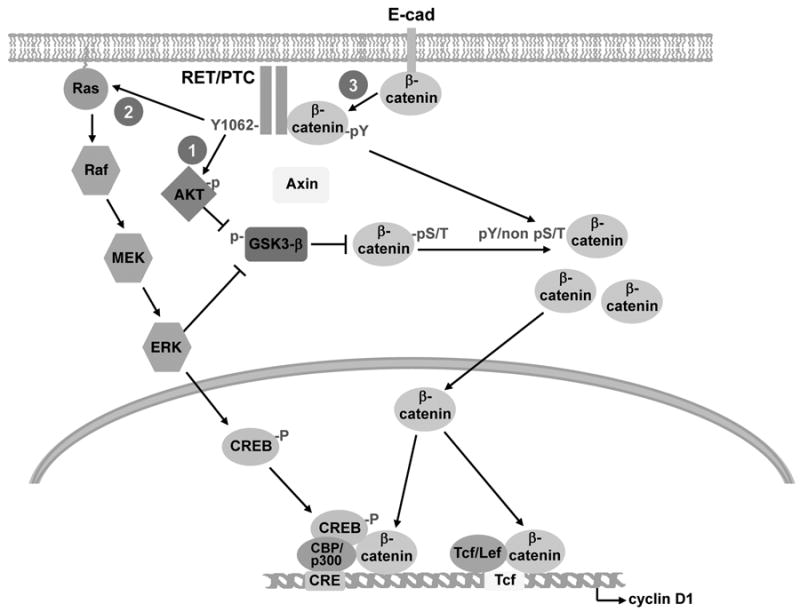
1, 2) RET/PTC activates PI3K/AKT and Ras/MEK/ERK. This leads to GSK3β phosphorylation thereby relieving its negative control on β-catenin, and to an increase of the free β-catenin protein pool. 2) Through ERK activation, RET/PTC induces S133 phosphorylation of CREB, which associates in a complex with β-catenin and CBP/p300 to stimulate transcription of cyclin-D1. 3) RET/PTC directly binds β-catenin and increases its phosphotyrosine content, thereby decreasing the E-cadherin-bound pool and increasing the free β-catenin pool. Free β-catenin enters the nucleus and participates in at least two different transcription complexes with CREB/p300 and with TCF/LEF, both ultimately involved in the regulation of cyclin D1.
It is possible that β-catenin activity cooperates with other signaling cascades activated by RET/PTC to mediate mitogenic activity. Cytosolic β-catenin is recruited to a “destruction” complex that includes APC and GSK3β. Loss of APC (adenomatous polyposis coli) tumor suppressor reduces the activity of the β-catenin destruction complex (17, 21). Of direct relevance to thyroid tumorigenesis, patients affected by familial adenomatous polyposis coli (FAP) and harboring APC mutations are predisposed to thyroid carcinoma (43). Furthermore, the RET/PTC oncogene is activated in some FAP-associated thyroid carcinomas (44). Therefore, our data support a model whereby two different lesions concomitantly present in FAP-associated thyroid carcinoma, i.e. RET/PTC activation and APC loss-of-function, may converge to enhance the activity of the β-catenin signaling cascade. On the other hand, direct β-catenin mutations are restricted to aggressive and undifferentiated types of thyroid carcinomas (23).
β-catenin targeting approaches for cancer therapy are currently being explored. For example, a conditionally replicative adenovirus, which kills only cells with a hyperactive β-catenin pathway, significantly inhibited the growth of undifferentiated (anaplastic) thyroid cancers (45). ICG-001, an inhibitor of the β-catenin/CBP transcriptional complex, efficiently induced apoptosis of colon cancer cells (46). Prostaglandin E2 (PGE2) also promotes β-catenin signaling (28) and prostaglandin E2 synthesis inhibitors, NSAIDs, in an adjuvant or preventive setting are being tested already in cancer. Our data, together with those reported by Gujral et al. (25), suggest that β-catenin targeting approaches could have therapeutic potential in thyroid cancer.
Acknowledgments
We gratefully acknowledge F. Carlomagno, R.M. Melillo, G. Salvatore, P. Salerno and G. Vecchio for their support. We are grateful to K. Hirakawa and N Onoda for OCUT-1 cells. We thank Jean Ann Gilder for text editing. This study was supported by the Associazione Italiana per la Ricerca sul Cancro (AIRC), the NOGEC (Naples OncoGEnomic Center), the Istituto Superiore di Oncologia (ISO), the Italian Ministero dell'istruzione, Universita' and Ricerca (MIUR), Ministero della Salute, the Project Applicazioni Biotecnologiche dalle molecole all'uomo (MoMa) and the E.C. Contract 03695 (GenRisk-T). M.D. Castellone was recipient of a fellowship from the Accademia Nazionale dei Lincei, Roma, Italy. D.M. Rao and M. Muthu were recipients of fellowships from MIUR in the frame of an India-Italy cooperation program.
References
- 1.Ciampi R, Nikiforov YE. RET/PTC rearrangements and BRAF mutations in thyroid tumorigenesis. Endocrinology. 2007;148:936–41. doi: 10.1210/en.2006-0921. [DOI] [PubMed] [Google Scholar]
- 2.Hayashi H, Ichihara M, Iwashita T, et al. Characterization of intracellular signals via tyrosine 1062 in RET activated by glial cell line-derived neurotrophic factor. Oncogene. 2000;19:4469–75. doi: 10.1038/sj.onc.1203799. [DOI] [PubMed] [Google Scholar]
- 3.Asai N, Murakami H, Iwashita T, Takahashi M. A mutation at tyrosine 1062 in MEN2A-Ret and MEN2B-Ret impairs their transforming activity and association with shc adaptor proteins. J Biol Chem. 1996;271:17644–9. doi: 10.1074/jbc.271.30.17644. [DOI] [PubMed] [Google Scholar]
- 4.Alberti L, Borrello MG, Ghizzoni S, Torriti F, Rizzetti MG, Pierotti MA. Grb2 binding to the different isoforms of Ret tyrosine kinase. Oncogene. 1998;17:1079–87. doi: 10.1038/sj.onc.1202046. [DOI] [PubMed] [Google Scholar]
- 5.Melillo RM, Santoro M, Ong SH, et al. Docking protein FRS2 links the protein tyrosine kinase RET and its oncogenic forms with the mitogen-activated protein kinase signaling cascade. Mol Cell Biol. 2001;21:4177–87. doi: 10.1128/MCB.21.13.4177-4187.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Besset V, Scott RP, Ibanez CF. Signaling complexes and protein-protein interactions involved in the activation of the Ras and phosphatidylinositol 3-kinase pathways by the c-Ret receptor tyrosine kinase. J Biol Chem. 2000;275:39159–66. doi: 10.1074/jbc.M006908200. [DOI] [PubMed] [Google Scholar]
- 7.Hadari YR, Gotoh N, Kouhara H, Lax I, Schlessinger J. Critical role for the docking-protein FRS2 alpha in FGF receptor-mediated signal transduction pathways. Proc Natl Acad Sci U S A. 2001;98:8578–83. doi: 10.1073/pnas.161259898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.De Falco V, Guarino V, Malorni L, et al. RAI(ShcC/N-Shc)-dependent recruitment of GAB 1 to RET oncoproteins potentiates PI 3-K signalling in thyroid tumors. Oncogene. 2005;24:6303–13. doi: 10.1038/sj.onc.1208776. [DOI] [PubMed] [Google Scholar]
- 9.Miyagi E, Braga-Basaria M, Hardy E, et al. Chronic expression of RET/PTC 3 enhances basal and insulin-stimulated PI3 kinase/AKT signaling and increases IRS-2 expression in FRTL-5 thyroid cells. Mol Carcinog. 2004;41:98–107. doi: 10.1002/mc.20042. [DOI] [PubMed] [Google Scholar]
- 10.Segouffin-Cariou C, Billaud M. Transforming ability of MEN2A-RET requires activation of the phosphatidylinositol 3-kinase/AKT signaling pathway. J Biol Chem. 2000;275:3568–76. doi: 10.1074/jbc.275.5.3568. [DOI] [PubMed] [Google Scholar]
- 11.Melillo RM, Castellone MD, Guarino V, et al. The RET/PTC-RAS-BRAF linear signaling cascade mediates the motile and mitogenic phenotype of thyroid cancer cells. J Clin Invest. 2005;115:1068–81. doi: 10.1172/JCI22758. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 12.Knauf JA, Kuroda H, Basu S, Fagin JA. RET/PTC-induced dedifferentiation of thyroid cells is mediated through Y1062 signaling through SHC-RAS-MAP kinase. Oncogene. 2003;22:4406–12. doi: 10.1038/sj.onc.1206602. [DOI] [PubMed] [Google Scholar]
- 13.Mitsutake N, Miyagishi M, Mitsutake S, et al. BRAF mediates RET/PTC-induced mitogen-activated protein kinase activation in thyroid cells: functional support for requirement of the RET/PTC-RAS-BRAF pathway in papillary thyroid carcinogenesis. Endocrinology. 2006;147:1014–9. doi: 10.1210/en.2005-0280. [DOI] [PubMed] [Google Scholar]
- 14.Xing M. BRAF mutation in papillary thyroid cancer: pathogenic role, molecular bases, and clinical implications. Endocr Rev. 2007;28:742–62. doi: 10.1210/er.2007-0007. [DOI] [PubMed] [Google Scholar]
- 15.Shinohara M, Chung YJ, Saji M, Ringel MD. AKT in thyroid tumorigenesis and progression. Endocrinology. 2007;148:942–7. doi: 10.1210/en.2006-0937. [DOI] [PubMed] [Google Scholar]
- 16.Coulonval K, Vandeput F, Stein RC, Kozma SC, Lamy F, Dumont JE. Phosphatidylinositol 3-kinase, protein kinase B and ribosomal S6 kinases in the stimulation of thyroid epithelial cell proliferation by cAMP and growth factors in the presence of insulin. Biochem J. 2000;348(Pt 2):351–8. [PMC free article] [PubMed] [Google Scholar]
- 17.Reya T, Clevers H. Wnt signalling in stem cells and cancer. Nature. 2005;434:843–50. doi: 10.1038/nature03319. [DOI] [PubMed] [Google Scholar]
- 18.Peifer M, Polakis P. Wnt signaling in oncogenesis and embryogenesis--a look outside the nucleus. Science. 2000;287:1606–9. doi: 10.1126/science.287.5458.1606. [DOI] [PubMed] [Google Scholar]
- 19.Rubinfeld B, Albert I, Porfiri E, Fiol C, Munemitsu S, Polakis P. Binding of GSK3beta to the APC-beta-catenin complex and regulation of complex assembly. Science. 1996;272:1023–6. doi: 10.1126/science.272.5264.1023. [DOI] [PubMed] [Google Scholar]
- 20.Desbois-Mouthon C, Cadoret A, Blivet-Van Eggelpoel MJ, et al. Insulin and IGF-1 stimulate the beta-catenin pathway through two signalling cascades involving GSK-3beta inhibition and Ras activation. Oncogene. 2001;20:252–9. doi: 10.1038/sj.onc.1204064. [DOI] [PubMed] [Google Scholar]
- 21.Brembeck FH, Rosario M, Birchmeier W. Balancing cell adhesion and Wnt signaling, the key role of beta-catenin. Curr Opin Genet Dev. 2006;16:51–9. doi: 10.1016/j.gde.2005.12.007. [DOI] [PubMed] [Google Scholar]
- 22.Ding Q, Xia W, Liu JC, et al. Erk associates with and primes GSK-3beta for its inactivation resulting in upregulation of beta-catenin. Mol Cell. 2005;19:159–70. doi: 10.1016/j.molcel.2005.06.009. [DOI] [PubMed] [Google Scholar]
- 23.Garcia-Rostan G, Tallini G, Herrero A, D'Aquila TG, Carcangiu ML, Rimm DL. Frequent mutation and nuclear localization of beta-catenin in anaplastic thyroid carcinoma. Cancer Res. 1999;59:1811–5. [PubMed] [Google Scholar]
- 24.Motti ML, Califano D, Baldassarre G, et al. Reduced E-cadherin expression contributes to the loss of p27kip1-mediated mechanism of contact inhibition in thyroid anaplastic carcinomas. Carcinogenesis. 2005;26:1021–34. doi: 10.1093/carcin/bgi050. [DOI] [PubMed] [Google Scholar]
- 25.Gujral TS, van Veelen W, Richardson DS, et al. A novel RET kinase-beta-catenin signaling pathway contributes to tumorigenesis in thyroid carcinoma. Cancer Res. 2008;68:1338–46. doi: 10.1158/0008-5472.CAN-07-6052. [DOI] [PubMed] [Google Scholar]
- 26.Schweppe RE, Klopper JP, Korch C, et al. DNA profiling analysis of 40 human thyroid cancer cell lines reveals cross-contamination resulting in cell line redundancy and misidentification. J Clin Endocrinol Metab. 2008 Aug 19; doi: 10.1210/jc.2008-1102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hedinger C, Williams ED, Sobin LH. The WHO histological classification of thyroid tumors: a commentary on the second edition. Cancer. 1989;63:908–11. doi: 10.1002/1097-0142(19890301)63:5<908::aid-cncr2820630520>3.0.co;2-i. [DOI] [PubMed] [Google Scholar]
- 28.Castellone MD, Teramoto H, Williams BO, Druey KM, Gutkind JS. Prostaglandin E2 promotes colon cancer cell growth through a Gs-axin-beta-catenin signaling axis. Science. 2005;310:1504–10. doi: 10.1126/science.1116221. [DOI] [PubMed] [Google Scholar]
- 29.Feliciello A, Li Y, Avvedimento EV, Gottesman ME, Rubin CS. A-kinase anchor protein 75 increases the rate and magnitude of cAMP signaling to the nucleus. Curr Biol. 1997;7:1011–4. doi: 10.1016/s0960-9822(06)00424-6. [DOI] [PubMed] [Google Scholar]
- 30.Carlomagno F, Vitagliano D, Guida T, et al. ZD6474, an orally available inhibitor of KDR tyrosine kinase activity, efficiently blocks oncogenic RET kinases. Cancer Res. 2002;62:7284–90. [PubMed] [Google Scholar]
- 31.Croyle M, Akeno N, Knauf JA, et al. RET/PTC-induced cell growth is mediated in part by epidermal growth factor receptor (EGFR) activation: evidence for molecular and functional interactions between RET and EGFR. Cancer Res. 2008;68:4183–91. doi: 10.1158/0008-5472.CAN-08-0413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Piedra J, Martinez D, Castano J, Miravet S, Dunach M, de Herreros AG. Regulation of beta-catenin structure and activity by tyrosine phosphorylation. J Biol Chem. 2001;276:20436–43. doi: 10.1074/jbc.M100194200. [DOI] [PubMed] [Google Scholar]
- 33.Rasola A, Fassetta M, De Bacco F, et al. A positive feedback loop between hepatocyte growth factor receptor and beta-catenin sustains colorectal cancer cell invasive growth. Oncogene. 2007;26:1078–87. doi: 10.1038/sj.onc.1209859. [DOI] [PubMed] [Google Scholar]
- 34.Danilkovitch-Miagkova A, Miagkov A, Skeel A, Nakaigawa N, Zbar B, Leonard EJ. Oncogenic mutants of RON and MET receptor tyrosine kinases cause activation of the beta-catenin pathway. Mol Cell Biol. 2001;21:5857–68. doi: 10.1128/MCB.21.17.5857-5868.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kanai Y, Ochiai A, Shibata T, et al. c-erbB-2 gene product directly associates with beta-catenin and plakoglobin. Biochem Biophys Res Commun. 1995;208:1067–72. doi: 10.1006/bbrc.1995.1443. [DOI] [PubMed] [Google Scholar]
- 36.Coluccia AM, Vacca A, Dunach M, et al. Bcr-Abl stabilizes beta-catenin in chronic myeloid leukemia through its tyrosine phosphorylation. Embo J. 2007;26(5):1456–66. doi: 10.1038/sj.emboj.7601485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.D'Amico M, Hulit J, Amanatullah DF, et al. The integrin-linked kinase regulates the cyclin D1 gene through glycogen synthase kinase 3beta and cAMP-responsive element-binding protein-dependent pathways. J Biol Chem. 2000;275:32649–57. doi: 10.1074/jbc.M000643200. [DOI] [PubMed] [Google Scholar]
- 38.Takemaru KI, Moon RT. The transcriptional coactivator CBP interacts with beta-catenin to activate gene expression. J Cell Biol. 2000;149:249–54. doi: 10.1083/jcb.149.2.249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mayr BM, Canettieri G, Montminy MR. Distinct effects of cAMP and mitogenic signals on CREB-binding protein recruitment impart specificity to target gene activation via CREB. Proc Natl Acad Sci U S A. 2001;98:10936–41. doi: 10.1073/pnas.191152098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pradeep A, Sharma C, Sathyanarayana P, et al. Gastrin-mediated activation of cyclin D1 transcription involves beta-catenin and CREB pathways in gastric cancer cells. Oncogene. 2004;23:3689–99. doi: 10.1038/sj.onc.1207454. [DOI] [PubMed] [Google Scholar]
- 41.Giese K, Kingsley C, Kirshner JR, Grosschedl R. Assembly and function of a TCR alpha enhancer complex is dependent on LEF-1-induced DNA bending and multiple protein-protein interactions. Genes Dev. 1995;9:995–1008. doi: 10.1101/gad.9.8.995. [DOI] [PubMed] [Google Scholar]
- 42.Xu L, Corcoran RB, Welsh JW, Pennica D, Levine AJ. WISP-1 is a Wnt-1- and beta-catenin-responsive oncogene. Genes Dev. 2000;14:585–95. [PMC free article] [PubMed] [Google Scholar]
- 43.Eng C. Familial papillary thyroid cancer--many syndromes, too many genes? J Clin Endocrinol Metab. 2000;85:1755–7. doi: 10.1210/jcem.85.5.6632. [DOI] [PubMed] [Google Scholar]
- 44.Cetta F, Chiappetta G, Melillo RM, et al. The ret/ptc1 oncogene is activated in familial adenomatous polyposis-associated thyroid papillary carcinomas. J Clin Endocrinol Metab. 1998;83:1003–6. doi: 10.1210/jcem.83.3.4614. [DOI] [PubMed] [Google Scholar]
- 45.Abbosh PH, Li X, Li L, Gardner TA, Kao C, Nephew KP. A conditionally replicative, Wnt/beta-catenin pathway-based adenovirus therapy for anaplastic thyroid cancer. Cancer Gene Ther. 2007;14:399–408. doi: 10.1038/sj.cgt.7701024. [DOI] [PubMed] [Google Scholar]
- 46.Emami KH, Nguyen C, Ma H, et al. A small molecule inhibitor of beta-catenin/CREB-binding protein transcription. Proc Natl Acad Sci U S A. 2004;101:12682–7. doi: 10.1073/pnas.0404875101. [DOI] [PMC free article] [PubMed] [Google Scholar]