

Abstract
PTEN is a dual function phosphatase with tumor suppressor function compromised in a wide spectrum of cancers. Because tissue polarity and architecture are crucial modulators of normal and malignant behavior, we postulated that PTEN may play a role in maintenance of tissue integrity. We used two non-malignant human mammary epithelial cell lines (HMECs) that form polarized, growth-arrested structures (acini) when cultured in 3-dimensional laminin-rich extracellular matrix gels (3D lrECM). As acini begin to form, PTEN accumulates in both the cytoplasm, and at cell-cell contacts where it co-localizes with E-cadherin/β-catenin complex. Reduction of PTEN levels by shRNA in lrECM prevents formation of organized breast acini and disrupts growth arrest. Importantly, disruption of acinar polarity and cell-cell contact by E-cadherin function-blocking antibodies reduces endogenous PTEN protein levels and inhibits its accumulation at cell-cell contacts. Conversely, in SKBR3 breast cancer cells lacking endogenous E-cadherin expression, exogenous introduction of E-cadherin gene causes induction of PTEN expression and its accumulation at sites of cell interactions. These studies provide evidence that E-cadherin regulates both the PTEN protein levels and its recruitment to cell-cell junctions in 3D lrECM indicating a dynamic reciprocity between architectural integrity and the levels and localization of PTEN. This interaction thus appears to be a critical integrator of proliferative and morphogenetic signaling in breast epithelial cells.
Keywords: 3D culture, microenvironment, PTEN, E-cadherin, Breast cancer
INTRODUCTION
The dual function phosphatase, PTEN (MMAC/TEP-1), is one of the most common targets of mutations in human cancers (1–4). Yet, in the context of breast cancer, PTEN mutations are not common, affecting less than 5% of patients. Nevertheless 30–50% of breast cancer patients have reduced PTEN expression, which in turn, is associated with poor clinical outcome (5, 6). Approximately 50% of patients with breast cancer have a mutation in or loss of at least one copy of the PTEN gene, which results in the activation of the phosphoinositide 3-kinase (PI3K) signaling (7, 8). The severity of PTEN mutations strongly correlates with the tumor stage and grade. For example, complete loss of PTEN is more frequent in metastatic cancer than in primary tumors. The loss of one copy of PTEN increases the risk that a tumor will develop; and the level of expression of PTEN dramatically affects the initiation and progression of tumors in mice models (9). Low expression of PTEN correlates also with unresponsiveness to breast cancer therapies such as trastuzumab (Herceptin) (10), Tamoxifen (11) and Gefitinib (12). Thus an understanding of how PTEN levels and its subcellular localization are regulated is important for understanding the mechanism by which PTEN may be involved in protecting breast cells from proceeding to malignancy.
PTEN acts as a tumor suppressor, in part, by regulating or attenuating the activity of the oncogene phosphoinositide 3-kinase (PI3K). There is some evidence that it does so through catalyzing the degradation of a critical second messenger, phosphotidylinositol-(3,4,5)-triphosphate (PIP3), generated by PI3K (7). PTEN activity negatively influences multiple PI3K downstream targets, most notably PKB/Akt (13–15) and exogenous addition of PTEN suppresses the growth of tumor cells (16–18) by upregulation of p27kip1 and downregulation of cyclin D1 in an Akt-dependent manner (19, 20). PTEN functions also in regulating dynamic cell surface interactions that involve integrins, focal adhesion kinase (FAK), cell migration, and the cytoskeleton (21–24). Moreover, it has been demonstrated that PTEN interacts with cell adhesion molecules such as β-catenin and E-cadherin through MAGI-2 protein to inhibit migration and proliferation (25–28). In all these studies, however, PTEN’s involvement has been studied on two dimensional (2D) tissue cultures. We hypothesized that the multiple consequences of loss of PTEN function observed in breast cancer could be related to a crucial but epigenetic role of PTEN in tissue-specific integration of form and function.
Recently, several reports have indicated that PTEN shuttles between the nuclear and cytoplasmic compartments. Nuclear PTEN is primarily found in normal cells and directly correlates with cell differentiation (29). A short sequence in the N-terminal region of PTEN has been identified to localize PTEN in the cytoplasm (30). Neither the cause(s) nor the consequences of these differences in PTEN sub-cellular localization have been studied in a physiologically relevant model.
The phosphorylation state of PTEN contributes to the regulation of PTEN sub-cellular localization and function. PTEN phosphorylation at serine 380 (ser380) and threonines 382/383 (thr382/383) within its C-terminal tail strongly influences PTEN protein stability and its localization to the cell membrane. Several kinases, including casein kinase-2, LKB1, RhoA-associated kinase, the microtubule associated kinase MAT205 and GSK 3β have been reported to phosphorylate PTEN (29).
One of the earliest manifestations of breast cancer is the loss of cellular organization within a tissue. This loss of function can be mimicked in 3D culture systems where its causes can be readily studied. In 3D lrECM, non-malignant HMEC form polarized, self-organizing structures that closely resemble structures observed in the mammary gland in vivo, whereas malignant cells form structures that remain disorganized (31). We have shown that signaling pathways respond to microenvironmental cues analogous to those that occur in mammary acini in vivo (32). Recently we demonstrated additional utility of 3D lrECM cultures of non-malignant HMEC for identification of molecular signatures predictive of clinical outcome in breast cancer (33). The expression patterns of genes significantly down modulated during acinar morphogenesis in 3D cultures could be used to distinguish groups of patients with poor-versus good-prognosis. Thus, 3D lrECM cultures of HMEC offer an opportunity to study the regulation of endogenous PTEN in a manipulatable and physiologically relevant context.
Here, we used the 3D lrECM cultures of non-malignant HMECs to investigate possible new roles for PTEN and how its level is regulated by microenvironment cues. We found that PTEN is indeed involved in acini formation and growth arrest. We also show that its level and localization is modulated by E-cadherin.
MATERIALS AND METHODS
Cell Culture
Immortalized non-malignant HMT-3522 S1 (S1) HMEC (34) were cultured in H14 medium (DMEM/F12 containing 250 ng/ml insulin, 10 μg/ml transferrin, 2.6 ng/ml sodium selenite, 10-10 M estradiol, 1.4 × 10-6 M hydrocortisone, 10 ng/ml EGF and 5 μg/ml prolactin). Finite life span 184 HMEC were grown in serum-free MCDB 170 medium (MEGM; Clonetics Division of BioWhittaker, Walkersville, MD), as described previously (35). Skbr-3 cells were obtained from the American Type Culture Collection and cultured in DMEM/F12 and supplemented with insulin (5 μg/ml; Sigma Chemical) and 5% (v/v) FCS. Both S1 and 184 cells (1 × 106 cells/ml) were cultured in 3D lrECM (Matrigel, BD Biosciences, Franklin Lakes, NJ), as previously described (36). Mouse anti-E-cadherin (clone SHE78-7; Zymed Laboratories Inc, San Francisco, CA) was added to lrECM to a final concentration of 5 μg/ml prior to cell seeding for function-blocking experiments. Colonies were isolated from the lrECM in ice-cold phosphate buffered saline (PBS)/5 mM EDTA for 60 min at 4°C after 3, 5, 7, and 15 days. For immunoblot analysis, the colonies were lysed in buffer containing 150 mM NaCl, 1 % NP-40, 50 mM Tris (pH 8.0). Nuclear and cytoplasmic cell extracts were prepared using NE-PER nuclear and cytoplasmic extract reagents kit (Pierce, Rockford, IL) according to the manufacturer’s instructions.
mRNA profiling
The full microarray results were published in a previous study (33) and data can be retrieved at the public database links GEOSeries GSE8096 and ArrayExpress E-MEXP-1006. In short, cell samples were harvested in duplicate at 3, 5, and 7 days post-seeding in lrECM. Purified total cellular RNA was biotin-labeled and hybridized to human oligonucleotide microarrays (Affymetrix HG-U133A), as previously described (33). Experiments with Affymetrix-present P-call rates of >30% were included in the analysis. Signal values from each of the 22,283 probe sets were calculated by means of robust multi-array analysis (RMA) (37) and genes were normalized to the mean of the 3-day time point for each cell type. Transcripts exhibiting greater than 2-fold differences in 2 independent microarray experiments were analyzed by 1-way ANOVA as a function of time in 3D cultures. Genes that were significantly differential (p<0.05) were selected. From these, we identified genes that were modulated early in the time-course (by our definition, on day 5) in both cell types. Three probe sets on Affymetrix HG-U133A GeneChips uniquely match PTEN: including 204054_at, 204053_x_at, and 211711_s_at (GeneCards/GeneTide data). Results represent the mean +/− the standard error of the three probes.
Indirect immunofluorescence and image acquisition
Acinar structures were fixed on 4 well chamber glass slides in methanol-acetone (1:1) at −20°C for 10 minutes and air-dried. A primary block was performed in IF Buffer (130 mM NaCl, 7 mM Na2HPO4, 3.5 mM NaH2PO4, 7.7 mM NaN3, 0.1% bovine serum albumin, 0.2% Triton X-100, 0.05% Tween-20) + 10% goat serum (Sigma, St. Louis, MO) for 1 hour at room temperature. A secondary block was performed in IF Buffer + 10% goat serum + 20μg/ml goat anti-mouse F(ab′)2 fragment (Jackson Immune Research, West Grove, PA) for 30 minutes. The primary antibodies, rat anti-alpha-6 integrin (clone NKI-GoH3; Chemicon International, Billerica, MA), rabbit anti-Ki67 (Sigma, St. Louis, MO), mouse anti-PTEN (26H9), rabbit anti-phosphorylated-PTEN at serine 380 (Cell Signaling, Danvers, MA), rabbit anti-MAGI-2 (H-60), (Santa Cruz, Santa Cruz, CA) and mouse anti-β-Catenin, mouse anti-E-cadherin (BD Transduction Laboratories, Franklin Lakes, NJ), were diluted 1:50 in the latter blocking buffer and incubated overnight (15–18 hours) at 4°C. Secondary antibodies conjugated with fluorescein or Texas Red (Sigma, St. Louis, MO) were diluted 1:100 in blocking buffer and incubated 1 hour at room temperature. Nuclei were counter-stained with DAPI. Immunofluorescent images were acquired using a Nikon (DIAPHOT 300) inverted microscope equipped with a digital camera and SPOT software. Confocal analysis was performed using a Zeiss 410 confocal microscopy system and software. The images presented are representative of two or more independent experiments.
Flow cytometry
Acini were dispersed into single cell suspensions by treatment with 0.25% trypsin at 37°C for exactly 10 minutes. The dispersed cells were washed three times in PBS and fixed in 40% ethanol at 4°C overnight. They were then incubated with 500 μg/ml RNase A in PBS at 37°C for 30 minutes and stained at room temperature for 30 minutes with a 69 μM propidium iodide solution prepared in PBS. The DNA content was determined by flow cytometry using a FACScan (Becton Dickinson, Franklin Lakes, NJ), and the data were analyzed with Cell Quest software (Becton Dickinson, Franklin Lakes, NJ).
Immunoblot and immunoprecipitation
Cell lysates (20–100 μg protein) were separated by SDS-PAGE gel electrophoresis and transferred to PVDF membranes (Millipore, Billerica, MA). After blocking in 10% nonfat dry milk for 90 minutes at room temperature, the membranes were incubated with primary antibodies at 1:500 to 1:1000 dilution in 2% nonfat dry milk for 2 hours at room temperature or overnight at 4°C. The blots were then incubated with HRP-conjugated sheep anti-mouse IgG or anti-rabbit IgG at 1:2000 in 2% nonfat dry milk for 90 minutes at room temperature. The blots were developed using Super Signal West Pico Chemilumiscent Substrate (Pierce, Rockford, IL). Immunoprecipitation studies were carried out as previously reported (36), with minor modifications, using rabbit anti-β-catenin (Santa Cruz Biotechnology, Santa Cruz, CA) or rabbit IgG at a dilution of 1:100–300. Signal was captured with the FluorChem 8900 analysis system (Alpha Innotech, San Leandro, CA).
Retroviral vectors and virus production
The retroviral vectors containing GFP and PTEN shRNA were obtained from Open Biosystems (Huntsville, AL), while the LXSN retroviral vector encoding full length E-cadherin cDNA was obtained from Dr. John Muschler (California Pacific Medical Center). Retroviruses were prepared by transient co-transfection of 293 cells with retroviral constructs and packaging plasmids. Retrovirus-containing supernatants were collected at 24 and 48 hrs following transfection and stored at −80°C. HMEC were plated at 5 × 105 cells per 60 mm dish. After 24 hours, parallel cultures were transduced with experimental or control viruses. Stable populations were obtained by selection in 1 μg/ml puromycin for 7 – 10 days. Skbr-3 cells were infected with LXSN-E-cadherin or LXSN viruses and selected with 500 μg/ml G418 (Genetecin; Invitrogen Corp. Carlsbad, CA). Experiments involved pooled populations of selected cells.
RESULTS
1. PTEN becomes bound to β-catenin/E-cadherin complexes during HMEC acinar morphogenesis
Profiling of global mRNA expression as a function of time in 3D lrECM indicated that PTEN expression increased significantly (p<0.05) in two non-malignant HMEC undergoing acinar morphogenesis in 3D lrECM cultures (Figure 1A) (33). To confirm the microarray results and analyze the distribution of endogenous PTEN protein in non-malignant HMEC undergoing acinar morphogenesis, we isolated cytoplasmic and nuclear extracts from S1 cells cultured for 3, 5 and 7 days in 3D lrECM. Consistent with microarray results we observed that both total PTEN and PTEN (phospho-ser380) protein levels were elevated in S1 3D cultures as a function of time. The bulk of endogenous PTEN and its serine 380-phosphorylated form (PTEN-ser380) were localized to the cytoplasm (Figure 1B) but PTEN-ser380 levels in the nucleus increased with time (Figure 1B). We chose to focus further analysis on PTEN-ser380 because it displayed the greatest relative increase in cytoplasm and nuclear extracts. We detected increases in the levels of FOXO3a, a protein downstream of PTEN that translocates to the nucleus and up-regulates a series of target genes, including p27, that promote cell cycle arrest (38). Not surprisingly, p27 protein increased over time during acini formation in 3D, consistent with results from the microarray data and with its function as a promoter of cell cycle arrest (33). Parallel samples were collected and the percentage of cells in S phase of the cell cycle was determined by propidium iodide staining and flow cytometry. Cell-cycle analysis showed a 64 percent decrease in the number of cells in S-phase between days 3 and 7 consistent with cultures undergoing cell cycle arrest (Figure 1B).
Figure 1. Induction of PTEN during HMEC acinar morphogenesis.

(A) PTEN RNA expression levels were investigated by microarray analysis during acinar formation in S1 and 184 HMECs cultured in 3D lrECM. Three probe sets on Affymetrix HG-U133A GeneChips uniquely match PTEN: including 204054_at, 204053_x_at, and 211711_s_at (GeneCards/GeneTide data). Results represent the mean +/− the standard error of the three probes. The p-values (t-test comparing day 3 versus days 5 and 7) were 0.0025 and 0.015 for S1 and 184 cells, respectively. (B) S1, an immortal non-malignant HMEC line was seeded in 3D lrECM and cytoplasmic (c) and nuclear (n) cell extracts were obtained on days (d) 3, 5 and 7 post-seeding. Proteins were separated by SDS-PAGE and detected by immunoblotting. The data shown are representative of two independent experiments. Results of cell cycle analysis at day (d) 3, 5 and 7 are shown below.
We showed previously that both PI3K and PIP3 were polarized to the basal surface of mammary acini (39). To determine whether PTEN-ser380 is co-localized to this surface in growth-arrested acini as well, we performed indirect immunofluorescence and confocal microscopy. We observed that PTEN-ser380 indeed was enriched at cell-cell contacts where E-cadherin, β-catenin and MAGI-2 was localized (Figure 2A). This suggested that PTEN may be part of the E-cadherin/β-catenin complex at cell-cell junctions. To explore this possibility, we performed co-immunoprecipitation studies using a polyclonal antibody against β-catenin. We observed increased ratio of PTEN protein complexed with β-catenin/E-cadherin in cells incubated in lrECM compared to conventional adherent (2D) conditions (PTEN/β-catenin ratio: 0.4 in 3D vs 0.1 in 2D) (Figure 2B).
Figure 2. Co-localization of β-catenin/E-cadherin with PTEN and MAGI-2.

(A) S1 cells were cultured in 3D lrECM for 7 days and sub-cellular localization of endogenous PTEN-ser380 or MAGI-2 (red) and β-catenin or E-cadherin (green) was recorded by confocal microscopy. The images shown are representative of two independent experiments. Scale bars in the left panels equal 25 μm and right panels 10 μm. (B) Cell lysates from S1 cells obtained at indicated intervals post-seeding were immunoprecipitated with a polyclonal rabbit β-catenin antibody, followed by immunobloting with monoclonal mouse anti-total PTEN, anti-E-cadherin and anti-β-catenin antibodies. Representative results from one of two independent experiments are shown.
2. Reduced PTEN levels during mammary acini formation abrogate lrECM-induced growth arrest
To critically test the contribution of increased PTEN expression to the lrECM-mediated growth arrest and self-organization of non-malignant HMEC in 3D cultures, we expressed PTEN shRNA stably in 184 HMEC using retroviruses. Immunoblotting of total cell extracts confirmed that PTEN protein levels were inhibited by almost 50% in cells expressing PTEN shRNA compared to cells expressing a control scrambled shRNA (Figure 3A). We found that shRNA-mediated reduction of PTEN expression led to the formation of large disorganized structures composed of abundant cells expressing the proliferation marker Ki67, compared to the well organized, largely Ki67(-) structures present in parallel control cultures (Figure 3B). The numbers of DAPI-stained nuclei per acini cross section were calculated and acinar structures were classified by size as of 1–4; 5–8; 9–12 or greater than 12 cells per acini cross section. Nearly twice as many of the structures contained more than 12 cells in the PTEN shRNA colonies in cross section compared to control cultures (Figure 3C). These results indicate that PTEN expression is required for both growth arrest and formation of organized structure in 3D lrECM cultures of breast epithelial cells. In concordance to our shRNA studies, conditional loss of PTEN in the mouse mammary gland demonstrated hyper proliferative phenotype (40)
Figure 3. Reduced PTEN levels abrogate lrECM-induced growth arrest.

(A) 184 cells stably expressing PTEN shRNA or a scrambled control sequence were incubated in 3D lrECM for 10 days. The cells were then collected and processed for immunoblotting with PTEN antibodies. Relative PTEN protein levels were calculated after normalization to a prominent Ponceau-stained band. (B) The expression of Ki67 (red) was detected by indirect immunofluorescence in control and shPTEN treated cultures. Nuclei were stained with DAPI (blue). Scale bar = 50 μm. (C) The numbers of DAPI-stained nuclei per acini cross section were calculated and acini classified by size.
3. E-cadherin function regulates acinus polarity, cellular proliferation, and PTEN levels
Given the increased association among PTEN, β-catenin, and E-cadherin during HMEC acinar morphogenesis, we hypothesized that E-cadherin may directly modulate the levels and subcellular localization of PTEN. E-cadherin plays a key role in the structural organization of cell-cell adhesion and serves as a widely recognized suppressor of invasion and proliferation of epithelial cancers. In addition, its functional elimination represents a key step in the acquisition of the invasive phenotype for many tumors, including breast cancers. Consistent with previous reports (36, 41, 42), we observed that blocking E-cadherin-dependent cell-cell contact using neutralizing antibodies, disrupted the cell cycle arrest and polarity of HMEC cultured in 3D lrECM. Cultures treated with E-cadherin blocking antibodies lost basal-restricted staining for α-6 integrin (Figure 4A) and had strong nuclear staining for Ki67 (Figure 4B). Moreover, the number of cells/acinus cross-section was increased in cultures treated with the E-cadherin blocking antibody compared to those treated with a control IgG antibody (Figure 4C). Immunoblot analysis showed that the levels of cytoplasmic PTEN-ser380 in E-cadherin blocking antibody-treated cultures were reduced by approximately 30% compared to those in control IgG-treated cultures (Figure 5A). Total PTEN protein levels were also reduced as a result of neutralizing E-cadherin function (Figure 5B). Conversely, we observed robust induction of cyclin E in cytoplasmic and nuclear extracts of HMEC in the same cultures. In both S1 (p53 negative) and 184 (p53 wild type) HMEC cultures treated with E-cadherin blocking antibody, we observed a reduction of PTEN levels at cell-cell interactions (Figure 5C). These results indicate that blocking E-cadherin function in HMEC in 3D lrECM has profound consequences for PTEN activation, acinar polarity and loss of growth control.
Figure 4. E-cadherin function regulates acinar polarity and cellular proliferation.

S1 cells were cultured in 3D lrECM for 10 days in the presence of 5 μg/ml function-blocking antibody against E-cadherin or IgG control. Treated cells were fixed in methanol:acetone (1:1) and immunostained for (A) Alpha-6 integrin (green) or (B) ki67 (red). Nuclei were stained with DAPI (blue). Scale bars equal 50 μm (C) The numbers of DAPI-stained nuclei per acini cross sections were determined, and acini were classified by size as indicated.
Figure 5. E-cadherin function regulates PTEN protein levels.

S1 cells were cultured in 3D lrECM for 10 days in the presence of 5 μg/ml function-blocking antibody against E-cadherin or IgG control. Parallel cultures were grown to obtain (A) cytoplasmic (c) and nuclear (n) cell extracts and (B) total cell extract. Levels of indicated proteins were determined by immunoblotting. Ponceau was used for loading control. (C) S1 and 184 cells were grown in 3D lrECM in the presence of isotype IgG or E-cadherin function-blocking antibody for 10 days. The treated cells were fixed as described in Methods and immunostained for PTEN-ser380 (red). Images are representative of three independent experiments. Scale bars equal 25 μm.
4. Restoration of E-cadherin function in the malignant breast cancer cell line SKBR3 restores PTEN expression
Having shown that E-cadherin function is important for maintaining PTEN expression levels, we examined whether re-expression of E-cadherin could restore PTEN-ser380 protein expression patterns in a breast cancer cell line, Skbr-3, that lacks E-cadherin. Expression of vector control or E-cadherin gene in Skbr-3 cells was confirmed by immunofluorescent analysis (Figure 6 and Suppl Figure 1). Figure 6 shows that in Skbr-3 cells expressing E-cadherin, PTEN-ser380 immunofluorescence was prominent at sites of cell-cell contact where it co-localized with E-cadherin (arrowhead). In contrast, cells negative for E-cadherin expression (small arrow) displayed reduced levels of only diffuse PTEN-ser380 immunofluorescence.
Figure 6. Restoration of E-cadherin function in the malignant breast cancer cell line, Skbr-3, restores PTEN expression.
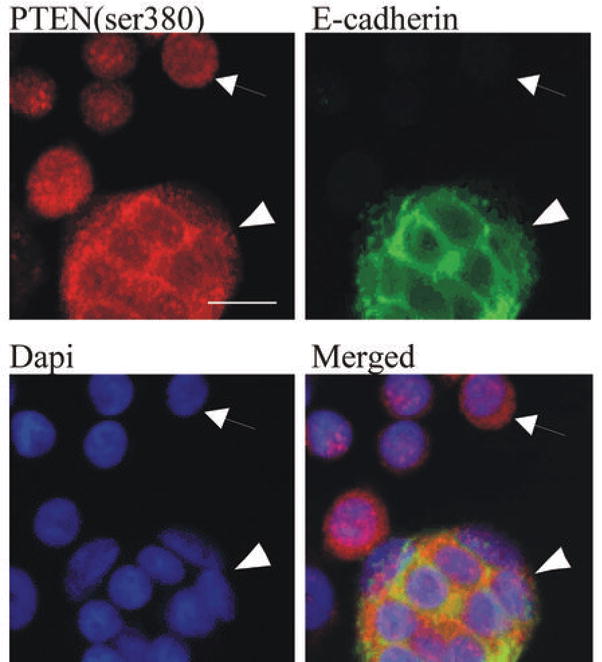
Skbr-3 cells stably expressing vector control or E-cadherin were grown in 2D-cultures. Cells were plated in slide chambers, fixed and immunostained for PTEN-ser380 (red) and E-cadherin (green). Nuclei were stained with DAPI (blue). Co-localization of endogenous PTEN-ser380 with E-cadherin was observed under confocal microscopy. Arrowheads indicate cells displaying E-cadherin expression. Small arrows indicate cells negative for E-cadherin expression. Images are representative of three independent experiments.
DISCUSSION
Our results indicate that growth arrest in a physiologically relevant model of acinar morphogenesis is dependent on E-cadherin-mediated up-regulation and accumulation of PTEN. Although PTEN has been shown previously to interact with cell adhesion complexes and to stabilize intercellular junctions, allowing a reduction of invasiveness in a range of cancer cells (43), regulation of endogenous PTEN in non-malignant cells and its ability to contribute to the formation or blockage of intercellular junctions has not been previously demonstrated. Our work ties together two tumor suppressor pathways, and suggests that PTEN may constitute a key internal node at which extra-cellular signals for growth arrest are integrated. Aberrations in E-cadherin expression or function may be the proximal cause of loss of PTEN expression in cancers, such as those that frequently occur in breast, where PTEN expression is lost without identifiable mutations in the PTEN gene itself. Our work has shown that even partial abrogation of PTEN expression is sufficient to cause continued proliferation and to impede the formation of organized acinar structures in the 3D lrECM model of HMEC morphogenesis. The surprising sensitivity of mammary cells to the PTEN regulatory node may explain why it is so low in a variety of solid tumors where tissue structure is compromised.
Previous work performed in non-malignant HMEC cultured in 3D lrECM has indicated that signaling pathways influenced by PTEN play key roles during mammary acini formation. For example, constitutive Akt activation has been shown to disrupt the organization and maintain proliferation of MCF-10A cells in 3D lrECM (44).
Conversely, inhibition of PI3K signaling was shown to cause cell cycle arrest and restoration of baso-apical polarity in breast cancer cell lines in 3D lrECM (39). Interestingly, PI3K and its lipid product, PIP3, are re-localized to the basal surfaces, away from regions of cell-cell contact and PTEN-ser380 accumulation, in organized structures formed when malignant HMEC are reverted in lrECM (39). This suggests that the recruitment of PTEN-ser380 to regions of cell-cell contact is part of a larger reorganization of cytoplasmic components orchestrated by the formation of adherens junctions between adjacent epithelial cells, and that a primary consequence of localized PTEN may be to allow compartmentalization of PI3K activity as well as other signaling molecules.
PTEN-dependent changes in PI3K signaling may operate in conjunction with other E-cadherin-dependent processes to cause cessation of cell growth. In the canonical model of E-cadherin-mediated growth suppression, E-cadherin sequesters β-catenin in cell adhesion complexes where the latter plays an essential role in the structural organization and function of cadherins by linking them to the actin cytoskeleton. In the presence of stimulatory signals, dissociation of cadherin-catenin complexes is mediated by activation of receptor tyrosine kinases and cytoplasmic tyrosine kinases. The free β-catenin is then available to bind to TCF/LEF transcription factors and to initiate events such as cyclin D expression and cell proliferation (45). PTEN can inhibit this process indirectly through stimulation of GSK-3 mediated phosphorylation and subsequent degradation of β-catenin (46).
Although other studies have indicated that phosphorylated PTEN has decreased affinity for cell membranes (47), our study indicates that in 3D cultures of HMECs PTEN-ser380 is recruited to regions of cell–cell contacts where it may be stabilized by interactions with E-cadherin/β-catenin complexes. PTEN phosphorylation at serine residues has been associated with an increase in PTEN protein stability (48, 49). Additional studies will be required to determine the mechanism(s) by which PTEN mRNA and protein are up regulated by E-cadherin, as well as the role of PTEN phosphorylation in the model of lrECM-mediated morphogenesis.
Supplementary Material
Acknowledgments
We are grateful to Virginia Spencer and Alain Beliveau for critical reading of the manuscript and for fruitful discussions. We thank Martha Stampfer for the gift of 184 cells, David Knowles for providing Zeiss 410 confocal microscopy system, Tarlochan Nijjar for retrovirus preparation, John Muschler for pLXSN-Ecadherin, and Jessie Lee for technical assistance. The work from MJB’s laboratory is supported by grants from the U.S. Department of Energy, OBER Office of Biological and Environmental Research, DE-AC02-05CH11231, a Distinguished Fellow Award and Low Dose Radiation Program and the Office of Health and Environmental Research, Health Effects Division, 03-76SF00098; by National Cancer Institute awards R01CA064786, R01CA057621, U54CA126552 and U54CA112970, and by grants from the U.S. Department of Defense W81XWH0810736 and W81XWH0510338.
References
- 1.Li DM, Sun H. TEP1, encoded by a candidate tumor suppressor locus, is a novel protein tyrosine phosphatase regulated by transforming growth factor beta. Cancer Res. 1997;57:2124–9. [PubMed] [Google Scholar]
- 2.Steck PA, Pershouse MA, Jasser SA, et al. Identification of a candidate tumour suppressor gene, MMAC1, at chromosome 10q23.3 that is mutated in multiple advanced cancers. Nat Genet. 1997;15:356–62. doi: 10.1038/ng0497-356. [DOI] [PubMed] [Google Scholar]
- 3.Li J, Yen C, Liaw D, et al. PTEN, a putative protein tyrosine phosphatase gene mutated in human brain, breast, and prostate cancer. Science. 1997;275:1943–7. doi: 10.1126/science.275.5308.1943. [DOI] [PubMed] [Google Scholar]
- 4.Myers MP, Stolarov JP, Eng C, et al. P-TEN, the tumor suppressor from human chromosome 10q23, is a dual-specificity phosphatase. Proc Natl Acad Sci U S A. 1997;94:9052–7. doi: 10.1073/pnas.94.17.9052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Depowski PL, Rosenthal SI, Ross JS. Loss of expression of the PTEN gene protein product is associated with poor outcome in breast cancer. Mod Pathol. 2001;14:672–6. doi: 10.1038/modpathol.3880371. [DOI] [PubMed] [Google Scholar]
- 6.Tsutsui S, Inoue H, Yasuda K, et al. Reduced expression of PTEN protein and its prognostic implications in invasive ductal carcinoma of the breast. Oncology. 2005;68:398–404. doi: 10.1159/000086981. [DOI] [PubMed] [Google Scholar]
- 7.Myers MP, Pass I, Batty IH, et al. The lipid phosphatase activity of PTEN is critical for its tumor supressor function. Proc Natl Acad Sci U S A. 1998;95:13513–8. doi: 10.1073/pnas.95.23.13513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Parsons R, Simpson L. PTEN and cancer. Methods Mol Biol. 2003;222:147–66. doi: 10.1385/1-59259-328-3:147. [DOI] [PubMed] [Google Scholar]
- 9.Trotman LC, Niki M, Dotan ZA, et al. Pten dose dictates cancer progression in the prostate. PLoS Biol. 2003;1:E59. doi: 10.1371/journal.pbio.0000059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nagata Y, Lan KH, Zhou X, et al. PTEN activation contributes to tumor inhibition by trastuzumab, and loss of PTEN predicts trastuzumab resistance in patients. Cancer Cell. 2004;6:117–27. doi: 10.1016/j.ccr.2004.06.022. [DOI] [PubMed] [Google Scholar]
- 11.Shoman N, Klassen S, McFadden A, Bickis MG, Torlakovic E, Chibbar R. Reduced PTEN expression predicts relapse in patients with breast carcinoma treated by tamoxifen. Mod Pathol. 2005;18:250–9. doi: 10.1038/modpathol.3800296. [DOI] [PubMed] [Google Scholar]
- 12.She QB, Solit D, Basso A, Moasser MM. Resistance to gefitinib in PTEN-null HER-overexpressing tumor cells can be overcome through restoration of PTEN function or pharmacologic modulation of constitutive phosphatidylinositol 3′-kinase/Akt pathway signaling. Clin Cancer Res. 2003;9:4340–6. [PubMed] [Google Scholar]
- 13.Dahia PL. PTEN, a unique tumor suppressor gene. Endocr Relat Cancer. 2000;7:115–29. doi: 10.1677/erc.0.0070115. [DOI] [PubMed] [Google Scholar]
- 14.Hlobilkova A, Knillova J, Bartek J, Lukas J, Kolar Z. The mechanism of action of the tumour suppressor gene PTEN. Biomed Pap Med Fac Univ Palacky Olomouc Czech Repub. 2003;147:19–25. [PubMed] [Google Scholar]
- 15.Sulis ML, Parsons R. PTEN: from pathology to biology. Trends Cell Biol. 2003;13:478–83. doi: 10.1016/s0962-8924(03)00175-2. [DOI] [PubMed] [Google Scholar]
- 16.Furnari FB, Huang HJ, Cavenee WK. The phosphoinositol phosphatase activity of PTEN mediates a serum-sensitive G1 growth arrest in glioma cells. Cancer Res. 1998;58:5002–8. [PubMed] [Google Scholar]
- 17.Weng L, Brown J, Eng C. PTEN induces apoptosis and cell cycle arrest through phosphoinositol-3-kinase/Akt-dependent and -independent pathways. Hum Mol Genet. 2001;10:237–42. doi: 10.1093/hmg/10.3.237. [DOI] [PubMed] [Google Scholar]
- 18.Weng LP, Smith WM, Dahia PL, et al. PTEN suppresses breast cancer cell growth by phosphatase activity-dependent G1 arrest followed by cell death. Cancer Res. 1999;59:5808–14. [PubMed] [Google Scholar]
- 19.Weng LP, Brown JL, Eng C. PTEN coordinates G(1) arrest by down-regulating cyclin D1 via its protein phosphatase activity and up-regulating p27 via its lipid phosphatase activity in a breast cancer model. Hum Mol Genet. 2001;10:599–604. doi: 10.1093/hmg/10.6.599. [DOI] [PubMed] [Google Scholar]
- 20.Radu A, Neubauer V, Akagi T, Hanafusa H, Georgescu MM. PTEN induces cell cycle arrest by decreasing the level and nuclear localization of cyclin D1. Mol Cell Biol. 2003;23:6139–49. doi: 10.1128/MCB.23.17.6139-6149.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tamura M, Gu J, Danen EH, Takino T, Miyamoto S, Yamada KM. PTEN interactions with focal adhesion kinase and suppression of the extracellular matrix-dependent phosphatidylinositol 3-kinase/Akt cell survival pathway. J Biol Chem. 1999;274:20693–703. doi: 10.1074/jbc.274.29.20693. [DOI] [PubMed] [Google Scholar]
- 22.Tamura M, Gu J, Matsumoto K, Aota S, Parsons R, Yamada KM. Inhibition of cell migration, spreading, and focal adhesions by tumor suppressor PTEN. Science. 1998;280:1614–7. doi: 10.1126/science.280.5369.1614. [DOI] [PubMed] [Google Scholar]
- 23.Tamura M, Gu J, Takino T, Yamada KM. Tumor suppressor PTEN inhibition of cell invasion, migration, and growth: differential involvement of focal adhesion kinase and p130Cas. Cancer Res. 1999;59:442–9. [PubMed] [Google Scholar]
- 24.Tamura M, Gu J, Tran H, Yamada KM. PTEN gene and integrin signaling in cancer. J Natl Cancer Inst. 1999;91:1820–8. doi: 10.1093/jnci/91.21.1820. [DOI] [PubMed] [Google Scholar]
- 25.Wu X, Hepner K, Castelino-Prabhu S, et al. Evidence for regulation of the PTEN tumor suppressor by a membrane-localized multi-PDZ domain containing scaffold protein MAGI-2. Proc Natl Acad Sci U S A. 2000;97:4233–8. doi: 10.1073/pnas.97.8.4233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Subauste MC, Nalbant P, Adamson ED, Hahn KM. Vinculin controls PTEN protein level by maintaining the interaction of the adherens junction protein beta-catenin with the scaffolding protein MAGI-2. J Biol Chem. 2005;280:5676–81. doi: 10.1074/jbc.M405561200. [DOI] [PubMed] [Google Scholar]
- 27.Tolkacheva T, Boddapati M, Sanfiz A, Tsuchida K, Kimmelman AC, Chan AM. Regulation of PTEN binding to MAGI-2 by two putative phosphorylation sites at threonine 382 and 383. Cancer Res. 2001;61:4985–9. [PubMed] [Google Scholar]
- 28.Hu Y, Li Z, Guo L, et al. MAGI-2 Inhibits cell migration and proliferation via PTEN in human hepatocarcinoma cells. Arch Biochem Biophys. 2007;467:1–9. doi: 10.1016/j.abb.2007.07.027. [DOI] [PubMed] [Google Scholar]
- 29.Gericke A, Munson M, Ross AH. Regulation of the PTEN phosphatase. Gene. 2006;374:1–9. doi: 10.1016/j.gene.2006.02.024. [DOI] [PubMed] [Google Scholar]
- 30.Denning G, Jean-Joseph B, Prince C, Durden DL, Vogt PK. A short N-terminal sequence of PTEN controls cytoplasmic localization and is required for suppression of cell growth. Oncogene. 2007;26:3930–40. doi: 10.1038/sj.onc.1210175. [DOI] [PubMed] [Google Scholar]
- 31.Petersen OW, Ronnov-Jessen L, Howlett AR, Bissell MJ. Interaction with basement membrane serves to rapidly distinguish growth and differentiation pattern of normal and malignant human breast epithelial cells. Proc Natl Acad Sci U S A. 1992;89:9064–8. doi: 10.1073/pnas.89.19.9064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bissell MJ, Kenny PA, Radisky DC. Microenvironmental regulators of tissue structure and function also regulate tumor induction and progression: the role of extracellular matrix and its degrading enzymes. Cold Spring Harb Symp Quant Biol. 2005;70:343–56. doi: 10.1101/sqb.2005.70.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Fournier MV, Martin KJ, Kenny PA, et al. Gene expression signature in organized and growth-arrested mammary acini predicts good outcome in breast cancer. Cancer Res. 2006;66:7095–102. doi: 10.1158/0008-5472.CAN-06-0515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Briand P, Petersen OW, Van Deurs B. A new diploid nontumorigenic human breast epithelial cell line isolated and propagated in chemically defined medium. In Vitro Cell Dev Biol. 1987;23:181–8. doi: 10.1007/BF02623578. [DOI] [PubMed] [Google Scholar]
- 35.Hammond SL, Ham RG, Stampfer MR. Serum-free growth of human mammary epithelial cells: rapid clonal growth in defined medium and extended serial passage with pituitary extract. Proc Natl Acad Sci U S A. 1984;81:5435–9. doi: 10.1073/pnas.81.17.5435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Weaver VM, Petersen OW, Wang F, et al. Reversion of the malignant phenotype of human breast cells in three-dimensional culture and in vivo by integrin blocking antibodies. J Cell Biol. 1997;137:231–45. doi: 10.1083/jcb.137.1.231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Irizarry RA, Hobbs B, Collin F, et al. Exploration, normalization, and summaries of high density oligonucleotide array probe level data. Biostatistics. 2003;4:249–64. doi: 10.1093/biostatistics/4.2.249. [DOI] [PubMed] [Google Scholar]
- 38.Arden KC. FoxO: linking new signaling pathways. Mol Cell. 2004;14:416–8. doi: 10.1016/s1097-2765(04)00213-8. [DOI] [PubMed] [Google Scholar]
- 39.Liu H, Radisky DC, Wang F, Bissell MJ. Polarity and proliferation are controlled by distinct signaling pathways downstream of PI3-kinase in breast epithelial tumor cells. J Cell Biol. 2004;164:603–12. doi: 10.1083/jcb.200306090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Li G, Robinson GW, Lesche R, et al. Conditional loss of PTEN leads to precocious development and neoplasia in the mammary gland. Development. 2002;129:4159–70. doi: 10.1242/dev.129.17.4159. [DOI] [PubMed] [Google Scholar]
- 41.St Croix B, Sheehan C, Rak JW, Florenes VA, Slingerland JM, Kerbel RS. E-Cadherin-dependent growth suppression is mediated by the cyclin-dependent kinase inhibitor p27(KIP1) J Cell Biol. 1998;142:557–71. doi: 10.1083/jcb.142.2.557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Weaver VM, Lelievre S, Lakins JN, et al. beta4 integrin-dependent formation of polarized three-dimensional architecture confers resistance to apoptosis in normal and malignant mammary epithelium. Cancer Cell. 2002;2:205–16. doi: 10.1016/s1535-6108(02)00125-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kotelevets L, van Hengel J, Bruyneel E, Mareel M, van Roy F, Chastre E. The lipid phosphatase activity of PTEN is critical for stabilizing intercellular junctions and reverting invasiveness. J Cell Biol. 2001;155:1129–35. doi: 10.1083/jcb.200105109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Debnath J, Walker SJ, Brugge JS. Akt activation disrupts mammary acinar architecture and enhances proliferation in an mTOR-dependent manner. J Cell Biol. 2003;163:315–26. doi: 10.1083/jcb.200304159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Nelson WJ, Nusse R. Convergence of Wnt, beta-catenin, and cadherin pathways. Science. 2004;303:1483–7. doi: 10.1126/science.1094291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Persad S, Troussard AA, McPhee TR, Mulholland DJ, Dedhar S. Tumor suppressor PTEN inhibits nuclear accumulation of beta-catenin and T cell/lymphoid enhancer factor 1-mediated transcriptional activation. J Cell Biol. 2001;153:1161–74. doi: 10.1083/jcb.153.6.1161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Das S, Dixon JE, Cho W. Membrane-binding and activation mechanism of PTEN. Proc Natl Acad Sci U S A. 2003;100:7491–6. doi: 10.1073/pnas.0932835100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Vazquez F, Matsuoka S, Sellers WR, Yanagida T, Ueda M, Devreotes PN. Tumor suppressor PTEN acts through dynamic interaction with the plasma membrane. Proc Natl Acad Sci U S A. 2006;103:3633–8. doi: 10.1073/pnas.0510570103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Birle D, Bottini N, Williams S, et al. Negative feedback regulation of the tumor suppressor PTEN by phosphoinositide-induced serine phosphorylation. J Immunol. 2002;169:286–91. doi: 10.4049/jimmunol.169.1.286. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.