

Abstract
Cystathionine β-synthase (CBS) plays a central role in cysteine metabolism, and malfunction of the enzyme leads to homocystinuria, a devastating metabolic disease. CBS contains a pyridoxal 5′-phosphate (PLP) cofactor which catalyzes the synthesis of cystathionine from homocysteine and serine. Mammalian forms of the enzyme also contain a heme group, which is not involved in catalysis. It may, however, play a regulatory role, since the enzyme is inhibited when CO or NO are bound to the heme. We have investigated the mechanism of this inhibition using fluorescence and resonance Raman spectroscopies. CO binding is found to induce a tautomeric shift of the PLP from the ketoenamine to the enolimine form. The ketoenamine is key to PLP reactivity because its imine C=N bond is protonated, facilitating attack by the nucleophilic substrate, serine. The same tautomer shift is also induced by heat inactivation of Fe(II)CBS, or by an Arg266Met replacement in Fe(II)CBS, which likewise inactivates the enzyme; in both cases the endogenous Cys52 ligand to the heme is replaced by another, unidentified ligand. CO binding also displaces Cys52 from the heme. We propose that the tautomer shift results from loss of a stabilizing H-bond from Asn149 to the PLP ring O4 atom, which is negatively charged in the ketoenamine tautomer. This loss would be induced by displacement of the PLP as a result of breaking the salt bridge between Cys52 and Arg266, which resides on a short helix that is also anchored to the PLP via H-bonds to its phosphate group. The salt bridge would be broken when Cys52 is displaced from the heme. Cys52 protonation is inferred to be the rate-limiting step in breaking the salt bridge, since the rate of the tautomer shift, following CO binding, increases with decreasing pH. In addition, elevation of the concentration of phosphate buffer was found to diminish the rate and extent of the tautomer shift, suggesting a ketoenamine-stabilizing phosphate binding site, possibly at the protonated imine bond of the PLP. Implications of these findings for CBS regulation are discussed.
Introduction
Cystathionine β-synthase (CBS) catalyzes the β-replacement reaction between homocysteine and serine to form cystathionine 1-3 (Figure 1). This reaction is part of the metabolic pathway that connects the sulfur containing amino acids methionine and cysteine. Homocysteine is formed from S-adenosylhomocysteine, the product of methyl transfer reactions that utilize S-adenosylmethionine (AdoMet) as the methyl donor. Homocysteine is then either remethylated (by methionine synthase or betainehomocysteine methyltransferase) to regenerate methionine or converted to cystathionine by CBS. Cystathionine is subsequently converted to cysteine by cystathionine γ-lyase 1, 4. CBS also has the capability of producing H2S from cysteine; mounting evidence implicates H2S in significant physiological functions, such as regulation of blood pressure 5-11.
Figure 1.
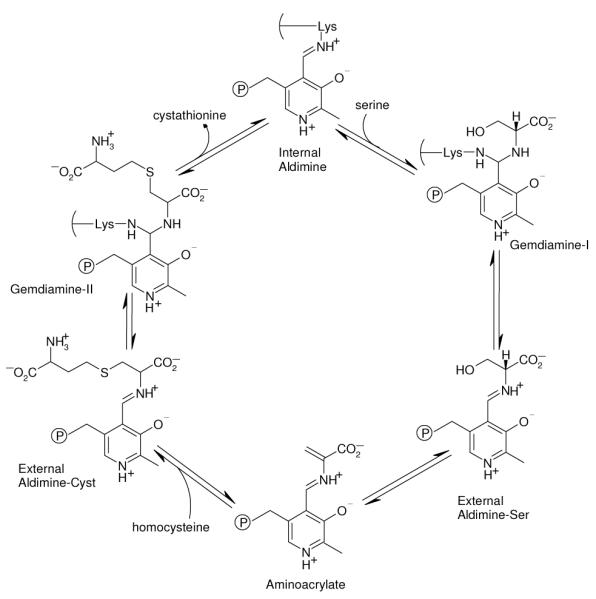
CBS contains a pyridoxal 5′-phosphate (PLP) cofactor which catalyzes the reaction between homocysteine and serine. Mammalian forms of CBS also have a heme cofactor 12-15, and are the only known PLP-dependent enzymes that also contains a heme group. The function of the heme is an intriguing question. The heme is not involved in the catalytic cycle 16, its removal lowers but does not eliminate human CBS (hCBS) activity 4, 17 and it is absent in the yeast 2, 18, 19 and Trypanosoma cruzi 20 CBSs. It was recently reported that exogenous heme stabilizes hCBS in a bacterial expression system 21, indicating a structural role for the heme. In addition, it may play a role in regulating the enzyme’s activity, since binding of NO or CO to the heme inactivates hCBS 22, 23 while redox-dependent ligand switching causes hCBS to lose activity in the ferrous state 14, 15, 24-29. The focus of this investigation is elucidating the mechanism whereby the heme regulates the catalytic activity of the PLP cofactor.
Regulation of enzymatic activity is physiologically important because of the role that hCBS plays in homocystinuria, a metabolic disease in which homocysteine levels are elevated. Mutations in hCBS are the most common cause of homocystinuria 30-32, and over a hundred pathogenic mutations in hCBS have been reported 30, 32, 33. Approximately half of hCBS deficient homocystinuric patients are responsive to treatment with pyridoxine (vitamin B6), a precursor of the PLP cofactor 34. To better understand the pyridoxine responsiveness of some but not other hCBS mutants, a greater knowledge of the interactions between the domains of CBS is needed.
Human CBS is a multimeric protein with 63 kDa subunits each of which contains a PLP cofactor at its active site 3. The C-terminal regulatory domain is thought to be the binding site for AdoMet 35, an allosteric activator that increases hCBS activity approximately two-fold 36. Deletion of the C-terminal domain causes the kcat to increase by a factor of four 37-39 and the responsiveness to AdoMet is lost 37, 38. The C-terminal domain is involved in the aggregation of the full-length protein, which exists as a mixture of tetramer and higher oligomers, while the 45 kDa truncated form lacking the C-terminal domain is a dimer 14, 15, 36, 37. The mammalian forms of CBS have a N-terminal heme binding domain which contains the protoporphyrin IX cofactor 12-15.
In order to study the interactions between the heme and PLP cofactor of hCBS we have used the complementary techniques of fluorescence and resonance Raman spectroscopies. Although PLP fluorescence is reduced by the presence of the heme, it is nevertheless measurable and several studies have been reported. Fluorescence spectroscopy has been used to study the binding of the substrate, serine 23, 38, 40, and of the allosteric activator, AdoMet 38, 41, to characterize the truncated enzyme lacking the C-terminal domain 38, 41-43, and to assess the influence of the I435T, S466L and C431S mutations 41, 43 on the PLP site. Fluorescence spectroscopy reveals distinct spectra from the tautomeric forms of the PLP aldimines and also from some of the other catalytic intermediates 23, 38, 40, 42.
Several studies have used the strong resonance enhancement of the Raman signal from the heme group to identify the heme axial ligands and investigate changes in the heme oxidation and ligation states 25, 26, 44-46. However, to our knowledge, Raman spectroscopy has not been employed to investigate the PLP active site. The Raman spectra in hCBS provide information on the Schiff base linkage between the PLP and the K119 sidechain (Figure 2), the site at which the initial reaction with serine takes place (Figure 1). We have determined the structural changes in the PLP cofactor in hCBS mediated by the heme and found that the changes to the heme that inactivate hCBS do so by triggering tautomerization of the PLP internal aldimine.
Figure 2.

Structure of the PLP and heme cofactors in hCBS. The figure was generated using the PDB coordinates from the PDBID: 1M54 file 15.
Experimental
Materials
Pyridoxal 5′-phosphate (98%), Na2S2O4 (85%), K2HPO4 (98%), KH2PO4 (99%), Na2HPO4 (99%), NaH2PO4 (99%), Tris base (99.9%) and Tris.HCl (99%) were from Sigma-Aldrich. D2O (99.9%) was from Cambridge Isotope Laboratories Inc. All solutions were prepared in 18.2 Ω MilliQ water. Sephadex G25M PD10 desalting columns were from GE Healthcare. All chemicals were used as received.
Sample Preparation
The 45 kDa truncated forms of hCBS and the R266M mutant were expressed and purified as described previously 29, 42. CBS solutions were prepared in 100 mM Tris buffer (pH 8.5-8.6) or phosphate buffer (pH 8.0 or pH 7.0). Unbound PLP was removed from the protein by one of two methods. Method 1: A Sephadex G25M PD10 desalting column (5×1.5 cm) was equilibrated with the desired buffer, the hCBS solution was loaded onto the column and eluted with buffer. The orange-brown hCBS band was collected and if necessary concentrated by using a 30,000 MWCO Centricon centrifuge filter tube (3000 × g, at 4°C 20 min). Method 2 (for D2O solutions of hCBS): The sample was concentrated in a 30,000 MWCO Centricon filter 4°C till the volume was reduced by 80-90%. Fresh buffer (1.8 mL) was added to the sample in the Centricon concentrator and this process was repeated four times.
PLP was removed from ferric hCBS in 50 mM Tris buffer, pH 8.6 by adding 5 mM serine. Under these conditions, the PLP forms an external aldimine with serine, which is no longer covalently linked to CBS and can be removed by extensive washing. The sample was centrifuged using a Centricon concentrator. Fresh buffer containing 5 mM serine (1.7 mL) was added to the sample in the Centricon concentrator and this process was repeated eight times. The resulting solution was used to record the Raman spectrum of the PLP-free hCBS. After recording the Raman spectrum the serine was removed from the hCBS solution by the addition of 50 mM Tris buffer, pH 8.6 and the solution was concentrated at 4°C using a Centricon filter tube (3000 × g, 30 min). Fresh buffer (1.7 mL) was added to the sample in the centrifuge filter tube and this process was repeated three times. The resulting apo-hCBS was reconstituted by addition of PLP (final concentration 150 μM) and incubation of the mixture for 15 h at 4°C. Excess PLP was removed by concentrating in a Centricon and washed 5 times with 1.8 mL of fresh buffer. The resulting hCBS solution was used to record the Raman spectrum of hCBS reconstituted with PLP.
Ferrous hCBS solutions were prepared by adding an excess of sodium dithionite (final concentration ~20 mM) to Ar purged solutions (prepared by blowing a gentle stream of Ar over the sample for ~2h) of ferric hCBS. UV-visible spectroscopy was used to check the redox state of the heme by monitoring the position of the Soret band (λmax = 449 nm in ferrous hCBS) prior to and after the recording of the Raman spectra. Solutions of heat-inactivated ferrous hCBS (the ferrous CBS424 form, λmax = 424 nm) in pH 8.5 Tris buffer were prepared by heating the samples for 20 min at 55°C according to the method of Pazicni et al. 26.
The CO adduct of hCBS was prepared by two different methods. In the first method an excess of sodium dithionite was added to a solution of ferric hCBS saturated with CO. The ferric hCBS was rapidly reduced to the ferrous form, which then binds CO. The second method was to take heat-inactivated ferrous hCBS in the CBS424 state and purge it with CO for 5-10 min. All hCBS-CO samples were maintained under a CO atmosphere during experiments so that any photodissociated products would rebind CO.
Fluorescence spectroscopy
Fluorescence emission spectra were recorded using a Perkin Elmer LS-50B fluorimeter. Solutions of hCBS or the R266M mutant (14-50 μM) in Tris (pH 8.5, 100 mM) or phosphate (pH 8.0, 100 mM; pH 8.0, 5 mM; or pH 7.0 100 mM) buffer were placed in sealed quartz fluorescence cells under Ar (ferrous and heat-inactivated ferrous samples) or CO (CO adduct) atmospheres. Fluorescence spectra were measured using excitation wavelengths of 330 nm and 410 nm. The temperature of the solutions was 22±2 °C. Spectra were recorded of the ferric, ferrous, heat-inactivated ferrous and CO bound forms. The CO adducts formed from the ferrous and heat-inactivated ferrous hCBS were both studied. With 330 nm excitation the emission spectrum was recorded using an excitation slit width=10 nm, emission slit width=10 nm and a scan rate of 25 nm min−1. With 410 nm excitation the emission spectrum was recorded using an excitation slit width=10 nm, emission slit width=20 nm and a scan rate of 100 nm min−1. Changes in fluorescence over time were monitored by recording spectra using 410 nm excitation at 10 min intervals for 3-6 h. Solutions of PLP in Tris buffer (pH 8.5, 100 mM) and phosphate buffer (pH 8.0, 100 mM) were measured as controls.
Raman Spectroscopy
The second harmonic of a Q-switched Nd:YLF laser with a 1 kHz repetition rate (Photonics International GM-30-527) was used to pump a Ti:sapphire laser (Photonics International TU-UV), which gave a narrowed laser frequency output (<0.1 cm−1) tunable between 760 and 920 nm. The excitation wavelengths of 380-420 nm were obtained by frequency doubling the Ti:sapphire output using a non-linear lithium triborate crystal. The laser was focused on to the sample with a pair of cylindrical lenses and the laser power at the sample was 1.0 mW. The scattered light was collected and focused onto a SPEX 14018 spectrograph (2400 grooves/mm) configured as a single stage and equipped with a liquid nitrogen cooled CCD detector (Roper Scientific, model 7375-0001).
Solutions of PLP in H2O ([PLP]=1.5 mM) or D2O ([PLP]=3.8 mM) were prepared in pH (pD) 8.6 Tris buffer (50 mM) in NMR tubes were used to record the Raman spectra. Aliquots of hCBS protein (typical concentration 25-30 μM) in pH (pD) 8.6 Tris buffer (100 mM) were transferred to NMR tubes and used to record the Raman spectra. The NMR tubes were spun at 2000-3000 rpm to mix the solutions and ensure that the volume sampled was replaced between laser pulses. The depolarization ratios were determined for ferric and ferrous hCBS from spectra measured using parallel and perpendicular polarization. This was done to facilitate the assignment of the heme bands, which was primarily based on the work of Hu et al. 47.
N,N-dimethylformamide was used as the calibration standard. Spectral calibration and deconvolution of overlapped bands to determine band positions were carried out with the GRAMS/AI v.7.00 software and band positions are accurate to ±1 cm−1.
UV-visible spectroscopy
The UV-visible spectra of hCBS solutions in a 0.100 cm quartz cuvette or the 0.400 cm quartz fluorescence cuvette were recorded using an Agilent 8453 diode array spectrophotometer. The concentrations of the hCBS solutions were determined using ε428=81 mM−1 cm−1 42 for ferric hCBS. Spectra of solutions used for Raman and fluorescence spectroscopy were also measured on the solutions in the respective sample holders to check the position of the Soret band, these spectra were not used to determine concentrations.
Results
Our experiments utilized the truncated form of hCBS, which is catalytically active, and whose crystal structure has been determined (Figure 2 14, 15). The R266M variant 29 was included in order to assess the role of Arg266, which forms an H-bond with the Cys52 heme ligand. This residue substitution lowers the activity ~5-fold relative to the fully active Fe(III) form of hCBS.
Reduction to Fe(II) inactivates the R266M variant 29, and leads to a gradual loss of activity in WT hCBS 28. On heating or prolonged standing the WT Fe(II)hCBS form converts to an inactive form, CBS424, in which the heme Soret absorption band has shifted from 449 nm to 424 nm, indicating that the Cys52 ligand is no longer bound 26, 28. The identity of the replacing ligand is unknown. For the R266M variant reduction to Fe(II) leads immediately to a 424 nm-absorbing species, indicating loss of the Cys52 ligand. Addition of CO to the Fe(II) form displaces Cys52 in hCBS, or the unidentified sixth ligand in CBS424 and Fe(II)R266M, to produce a 420 nm-absorbing CO adduct.
Fluorescence Emission Spectra
Fluorescence spectra were obtained with excitation at 410 and 330 nm, in order to emphasize responses from the ketoenamine and enolimine tautomers of PLP (Figure 3). The former absorbs at ~420 nm and emits at 480-510 nm, while the latter absorbs at ~330 nm and emits at 450-460 nm 23, 38, 41, 42, 48-53. However, the absorption and emission bands are broad, and overlap. Emission from both tautomers is detectable with either excitation wavelength, but the relative intensities are different.
Figure 3.
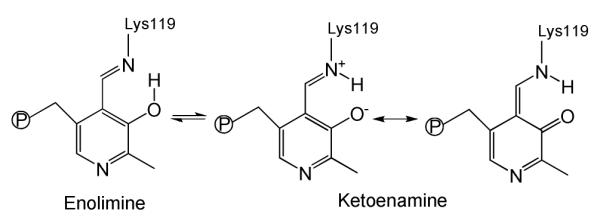
Ketoenamine and enolimine tautomers of the internal aldimine of PLP in hCBS.
Our fluorescence spectra clearly associate hCBS activity with the ketoenamine tautomer. Thus the Fe(III) and Fe(II) forms of hCBS yield weak emission at 483 nm, attributable to ketoenamine, whether excited at 410 or 330 nm (Figure 4). However heat-induced conversion of the Fe(II) form to the inactive CBS424 form shifts the emission band to 462 nm, characteristic of enolimine, with a considerable increase in intensity. The increased intensity is attributed to diminished heme quenching of the emission, though the mechanism is not clear. An emission shoulder is seen at 484 nm for CBS424, indicating a small ketoenamine population. The Fe(II)CO adduct likewise shows elevated emission, at an even shorter wavelength, 456 nm, again with a weak shoulder at 484 nm.
Figure 4.
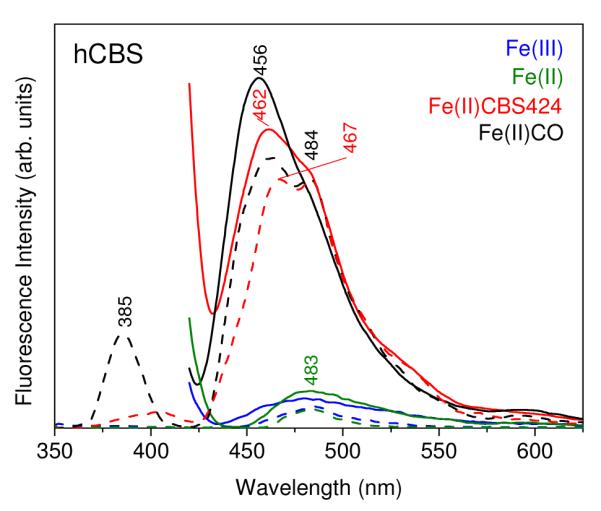
Fluorescence emission spectra of the indicated forms of hCBS (39 μM) in 100 mM phosphate buffer, pH 8.0. Excitation at 410 nm (solid lines) and 330 nm (dotted lines). Spectra were obtained from a single sample that was converted sequentially from Fe(III) to Fe(II) to Fe(II)CBS424 to Fe(II)CO forms.
Excitation at 330 nm gives similar emission envelopes for CBS424 and Fe(II)CO forms, but also reveals an additional emission band at 385 nm for the Fe(II)CO adduct. Assignment of this band is uncertain. A 380 nm emission has been reported 42 upon 330 nm excitation of the aldamine product formed by borohydride reduction of the PLP imine bond in hCBS. A similar band observed for tryptophanase was attributed to formation of a substituted aldamine by reaction with H2O 50 . A similar reaction might explain the present result, although why it would be specific for the Fe(II)CO adduct is unclear.
Emission envelopes are essentially the same for the Fe(II)CO adduct of the R266M variant (Figure 5) as for hCBS. However, for Fe(II)R266M, the emission envelopes are similar to those of CBS424, consistent with the Soret band being at 424 nm and with the lack of activity for Fe(II)R266M. Relative to CBS424, the emission maximum is slightly red-shifted in Fe(II)R266M, 466 vs. 462 nm, and the 483 shoulder is a little more prominent, but the emission envelopes are clearly indicative of a majority enolimine population. In the case of Fe(III)R266M, the emission maximum is at 481 nm, characteristic of ketoenamine, but there is substantial intensity at ~460 nm, and the overall intensity is higher than for Fe(III)hCBS, indicative of partial conversion to enolimine. This behavior is qualitatively consistent with the five-fold lower catalytic activity of Fe(III)R266M than Fe(III)hCBS.
Figure 5.
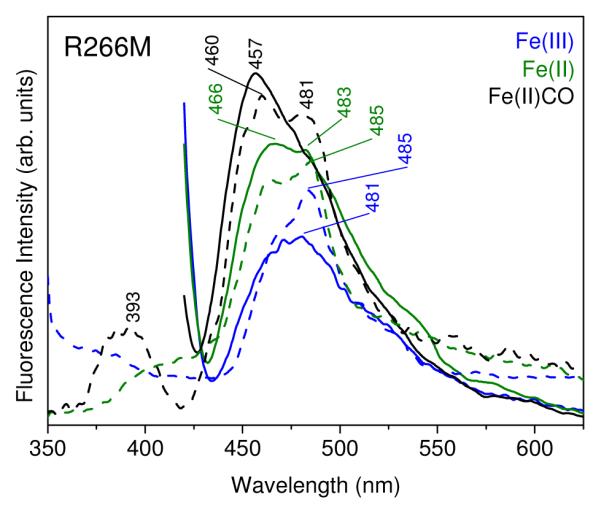
Fluorescence emission spectra of the indicated forms of R266M hCBS (30 μM) in 100 mM phosphate buffer, pH 8.0. Excitation at 410 nm (solid lines) and 330 nm (dotted lines).
Unfortunately, the fluorescence spectra do not permit quantitation of enolimine versus ketoenamine populations, because the intrinsic emission intensities and bandwidths of the tautomers are unknown. However, the data are qualitatively consistent with the hypothesis that ligation changes at the heme shift the tautomer equilibrium, and that hCBS is active only when the PLP is in the ketoenamine tautomer.
We note that PLP in Tris buffer produces a broad emission band at ~500 nm (Figure S1), substantially further to the red than any of the protein emission bands. Thus we are assured that none of the changes observed in the protein spectra are due to release of the PLP into solution.
Raman Spectra
Resonance Raman spectroscopy can monitor the status of the pyridoxine ring of PLP Schiff base, and particularly of the imine νC=N bond 54. In the ketoenamine tautomer the imine N is protonated, and its stretching vibration, at ~1650 cm−1, shifts down 9-12 cm−1 upon H/D exchange in D2O 54.
Unfortunately, the heme chromophore of hCBS is a serious hindrance to Raman monitoring of PLP, because its resonance enhancement is strong with laser excitation near the heme Soret absorption, which overlaps the weaker PLP absorption, ~420 nm in the ketoenamine form. However, by tuning the laser to somewhat shorter wavelengths we were able to detect PLP signals in the presence of the stronger heme RR spectrum (see Supporting Information, Figure S2). With excitation of Fe(III)hCBS at 390 nm (Figure 6), one can see the pyridoxine νCO mode at 1338 cm−1, adjacent to the strongest heme band, ν4 at 1372 cm−1, and a weak imine νC=N band, at 1665 cm−1 (inset). The PLP bands are eliminated when PLP is extracted from the protein with a serine wash (see Methods), and they are restored when the protein is reconstituted with PLP (Supporting Information, Figure S3). The same PLP bands are seen for Fe(II)hCBS (Figure 6), although the heme bands are shifted, as expected for Fe(II) heme 55. The 1665 cm−1 band is identified as arising from a protonated imine bond via its 12 cm−1 downshift in D2O (Figure 6 insets).
Figure 6.

UV-excited Raman spectra of the wild-type ferric hCBS in the indicated forms in 100 mM Tris buffer pH (pD) 8.6. The insets show the H/D shift of the PLP νC=N band. The excitation wavelength was 380 nm for Fe(III)hCBS and 390 nm for Fe(II)hCBS. Accumulation time, 30 min for Fe(III)hCBS and 20 min for Fe(II)hCBS.
When protein-free PLP was examined in Tris buffer, the imine νC=N band was located at 1645 cm−1, downshifting 11 cm−1 in D2O (Supporting Information, Figure S4), consistent with previous reports 54. The 18 cm−1 higher frequency observed in CBS confirms that the signal did not arise from PLP displaced from the protein. It also implicates a substantial protein effect on the strength of the imine C=N bond.
However, the 390 nm-excited RR spectra show no detectable PLP bands for either Fe(II)(CO)hCBS, or for CBS424 (Supporting Information, Figure S5). Their absence is attributable to weak resonance enhancement, since the PLP in these forms of the protein are predominantly in the enolimine tautomer, whose main absorption band, at ~330 nm, is far from the 390 nm excitation wavelength. Likewise there are no detectable PLP bands in the RR spectra of the Fe(II) form of the R266M variant, or of its CO adduct (Figure 7), both of which contain mainly the enolimine tautomer of PLP. The Fe(III) form of R266M does reveal the PLP RR bands, including the characteristic 1663 cm−1 νC=N band (Figure 7), but their intensities, relative to the heme bands, are somewhat lower than in Fe(III)hCBS. The lower intensity is consistent with the partial conversion to enolimine, seen in the fluorescence emission, and with the lower activity of Fe(III)R266M.
Figure 7.

Raman spectra of the indicated forms of R266M hCBS (33 μM) in 100 mM Tris buffer, pH 8.5. Excitation wavelength, 395 nm. The inset shows a weak PLP νC=N band for the Fe(III) form, which was undetectable in the other forms. Accumulation time, 10 min.
The heme RR spectra are in agreement with those reported in earlier RR studies of hCBS 25, 45. They are at positions expected for low-spin Fe(III) and Fe(II) heme, and for the Fe(II)CO adduct. We note a small up-shift of ν4 , from 1357 to 1360 cm−1, upon conversion of the Fe(II) form to CBS424 (Figure 6 and Supporting Information, Figure S5), presumably arising from the replacement of Cys52 with another high field ligand, currently unidentified. The ν4 position in Fe(II)R266M (Figure 7) agrees with that of CBS424, consistent with loss of Cys52 ligation.
Slow Tautomer Conversion after CO binding
In the course of recording fluorescence spectra, we noticed a time lag between addition of CO to Fe(II)hCBS, and the evolution of the characteristic enolimine emission. CO binding to the heme, readily detectable via the color change and the heme absorption spectrum, is complete in <10 min in CO-saturated solutions 46, but the shift in fluorescence from the ketoenamine emission at ~480 nm to the enolimine emission at ~460 nm was much slower. The time course is illustrated in Figure 8 for Fe(II)hCBS in 100 mM Tris buffer, pH 8.5. The intensity loss at 482 nm followed a first-order exponential decay, with a time constant of 73±6 min. RR spectra of an identically prepared solution also showed loss of PLP ketoenamine band intensity (νCO, at 1338 cm−1), and the time courses for the two assays were superimposable (Figure 8 inset).
Figure 8.
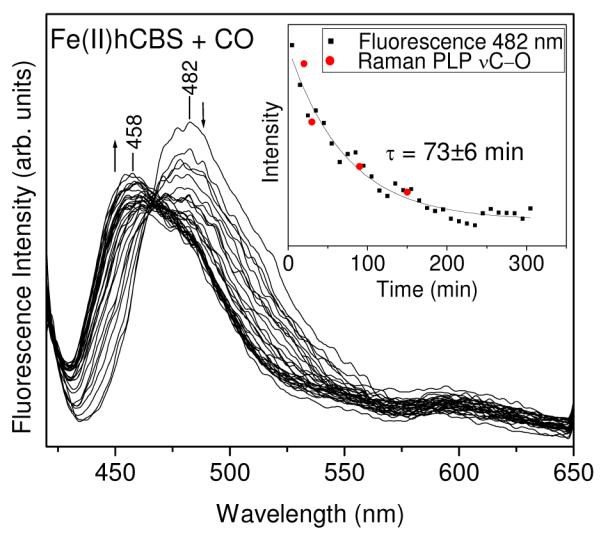
Time-dependent changes in the 410 nm-excited fluorescence spectra of Fe(II)hCBS (48 μM) in 100 mM Tris buffer, pH 8.5 upon CO binding. Spectra were collected every 10 min for 5 h. Inset: Overlay of the fluorescence intensity at 482 nm versus time and intensity of the 1339 cm−1 PLP νC—O Raman band versus time. Fitting the 482 nm fluorescence intensity data with a first order exponential decay function gave a time constant of 73 ± 6 min.
However there was no delay in the fluorescence shift upon addition of CO to the CBS424 form or to Fe(II)R266M (data not shown). Both of these inactive forms have a dominant enolimine tautomer population, prior to CO addition, but the emission intensity increases, and the emission maximum shifts further to the blue when CO is bound (Figures 4, 5) indicating a further shift in the tautomer equilibrium toward enolimine. This emission change occurred within the mixing time (~5 min). Thus the slow CO-induced tautomer shift is specific to the active, Fe(II) WT form, in which the predominant tautomer is the ketoenamine.
The rate and extent of this slow conversion was found to depend on the buffer. A slower conversion (τ = 125±6 min) was observed when the buffer was 100 mM phosphate, pH 8.0 (Figure 9). Moreover, the emission maximum shifted only to 470 nm, remaining there even after standing overnight (Supporting Information, Figure S6), whereas in Tris the maximum reached 456 nm, as it did for the delay-free CBS424 and Fe(II)R266M forms. Thus the extent of the tautomer shift, as well as the rate, were retarded by 100 mM phosphate. But when the concentration of the phosphate was reduced to 5 mM, while retaining the pH at 8.0, the extent of the tautomer shift was restored (λmax = 460 nm — Figure 9), and the rate increased 7-fold (τ = 18±1 min.). Finally, when the phosphate concentration was maintained at 100 mM but the pH was adjusted to 7.0 (Figure 9), the rate was 5-fold faster than at pH 8.0 (τ = 26±1 min.) and the emission maximum shifted even further than it had in Tris at pH 8.5, 450 versus 458 nm.
Figure 9.
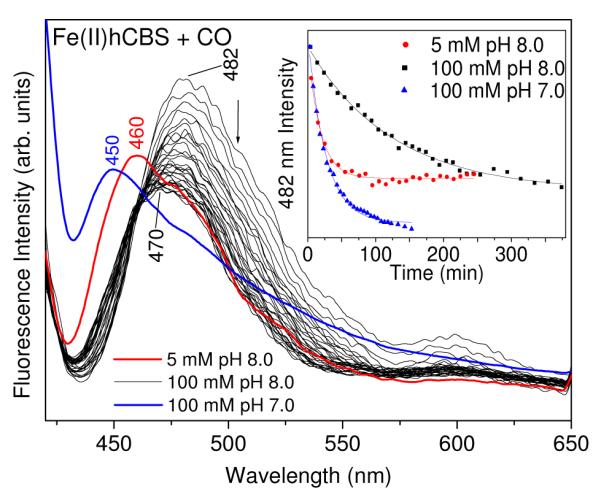
Time-dependent changes in the 410 nm-excited fluorescence spectra of Fe(II)hCBS (36 μM) in 100 mM phosphate buffer pH 8.0 upon CO binding. Spectra were collected every 10 min for 4 h, and then every 20 min for 2 h. Spectra recorded 64 min after initiation of CO binding to Fe(II)hCBS (30 μM) in 5 mM phosphate buffer pH 8.0 (red), and 93 min after initiation of CO binding to Fe(II)hCBS (14 μM) in 100 mM phosphate buffer pH 7.0 (blue) are shown for comparison. Inset: Changes in the normalized fluorescence intensity at 482 nm versus time. Fitting the fluorescence intensity at 482 nm with a first-order exponential decay function gave time constants of 125 ± 6 min for 100 mM phosphate buffer pH 8.0, 18 ± 1 min for 5 mM phosphate buffer pH 8.0 and 26 ± 1 min for 100 mM phosphate buffer pH 7.0.
Thus pH and the composition of the buffer both influence the tautomer shift and its rate. Since phosphate is polyanionic, we infer that it binds to an anion binding site on the protein, inhibiting the tautomer shift. This would account for the greater shift and rate of conversion when the phosphate concentration is reduced. The pH dependence implies that protonation at another site enhances the tautomer shift, and its rate.
Discussion
Inactivation via tautomerization
Our fluorescence spectra establish that a shift from the ketoenamine to the enolimine tautomer of PLP is associated with loss of activity in CBS. This association is consistent with the enzymatic mechanism (Figure 1), whose first step involves attack on the imine C4A carbon atom of PLP by a nucleophile, the amine group of the serine substrate. Protonation of the Schiff base N in the ketoenamine tautomer (Figure 3) makes the imine C4A more reactive toward nucleophilic attack, while conversion to the enolimine tautomer diminishes this reactivity. This principle is well-recognized in the PLP literature. In an early study of pyridoxal imines, Metzler suggested that a key function of PLP-containing enzymes is to facilitate the tautomeric rearrangement of the imine to the reactive form for transamination 56. More recently Karsten et al. postulated that conversion of PLP in serine-glyoxylate aminotransferase to the ketoenamine tautomer is required for the nucleophilic attack on C4A to occur 57. Moreover, DFT calculations on the mechanism of the transimination reaction found enhanced reactivity upon proton transfer from the enolimine O3′ to the imine N to form the ketoenamine tautomer 58.
Additionally, CBS appears to stabilize C=N protonation, as evidenced by the 18 cm−1 higher wavenumber for the imine νC=N band in CBS than in aqueous PLP-Tris Schiff base. The ketoenamine tautomer has two resonance forms (Figure 3), the C=N double bond residing on the zwitterionic form. We note that the protein crystal structure indicates H-bond donation from Asn149 to the pyridoxine ring O atom (Figure 2), which is negatively charged in the zwitterionic resonance form. Stabilization of this form would increase the νC=N frequency, and would also enhance the imine C4A reactivity.
It is, of course, also possible that a protein conformational change induces conversion to the enolimine tautomer, and simultaneously distorts the substrate binding cleft, obstructing access to the PLP C4A atom. However, the lowered enolimine reactivity seems a sufficient explanation.
Structural basis for heme regulation of activity
What is the mechanism for the ketoenamine-enolimine conversion? Since the Asn149/O4 H-bond is a stabilizing interaction for the ketoenamine, we propose that loss of this interaction underlies the tautomer shift. As discussed previously 29, 59 the PLP and heme sites in CBS are connected by a short intervening helix, helix 6 (Figure 2). The PLP is connected to one end of helix 6 by H-bonds to its phosphate group from the side-chains of the helix residues Thr257 and Thr260. In the middle of helix 6, the side-chain of Arg266 forms a salt bridge with the heme ligand Cys52. Disturbing this salt bridge could induce a displacement of helix 6, which in turn could displace the PLP. The Asn149/O4 H-bond would be broken by this displacement, inducing the tautomer shift (Figure 10).
Figure 10.

Proposed mechanism for the slow step in the heme-CO linked inactivation of hCBS.
The present results support this mechanism. The ketoenamine tautomer is predominant in both Fe(III) and Fe(II) forms of hCBS, but shifts to the enolimine when CO displaces Cys52, or when the Cys52 ligand is lost by heat treatment, forming CBS424. This shift is attributed to the loss of the Arg266/Cys52 salt bridge. The influence of this salt bridge is seen in the R266M variant. Replacement of Arg266 with Met results in loss of the Cys52 ligand in the Fe(II) form, in which the tautomer is predominantly enolimine. However, the Cys52 is retained in the Fe(III) form, as is the ketoenamine tautomer, although a significant enolimine population is seen, consistent with the lowered activity of Fe(III)R266M. The negative charge of the Cys52 thiolate balances the charge on the heme in Fe(III)R266M and so the Fe-Cys52 bond is stable even though it can not form a salt bridge with Met266. Reduction to Fe(II) weakens the Fe-Cys52 bond, and leads to Cys52 displacement. This displacement is retarded in hCBS by the Arg266/Cys52 salt bridge.
The slowness of the CO-induced tautomer shift in hCBS, and its buffer dependence, is intriguing. Displacement of Cys52 by CO does not immediately lead to the tautomer-shifting conformation change; some further step is needed. We propose that the slow step is breaking of the Arg266/Cys52 salt bridge. Displacement of Cys52 by CO might actually strengthen this salt bridge, since its sulfur atom would then develop a full negative charge. Maintenance of the salt bridge would inhibit helix 6 displacement, and the attendant tautomer shift. However, the Cys52 would also be subject to protonation, which would break the salt bridge. In aqueous solution the thiolate pKa is ~9 60. Protonation would be linked to reorientation of Cys52, exposing it to solvent (Figure 10). The release of Arg266 would then induce helix 6 displacement and the PLP tautomer shift. Rate-limiting protonation is supported by the observed increase in the rate of the tautomer shift when the pH is lowered from 8.0 to 7.0 in 100 mM phosphate buffer.
In a previous study, we reported that the rate of Cys52 rebinding to the heme following photolysis of the CO adduct is essentially unchanged between pH 7.6 and 10.5 46. This apparent insensitivity to pH was attributed to persistence of the Arg266/Cys52 salt bridge upon CO binding. However, this rate was measured indirectly, as the difference between the recovery rate of photolyzed heme and the CO recombination rate, because the Cys52-heme contribution to the monitoring signal (the RR envelope of the ν4 heme mode) was too small for direct measurement. A subsequent study on the rate of Cys52 rebinding to the heme in the R266M mutant showed that it was also independent of pH over the same range 29. Since R266M has no salt bridge, and the present results imply breaking of the Arg266/Cys52 salt bridge after CO binding to WT hCBS, the pH independence suggests that the measured rebinding rate does not pertain to Cys52 at all. We tentatively reassign it to the auxiliary ligand, currently unidentified, which replaces Cys52 rapidly in Fe(II)R266M, or, slowly, in normal Fe(II)hCBS, to form CBS424. In the altered protein structure of the CO adduct, binding of the auxiliary ligand to the photo-dissociated heme may be fast.
The CO-induced ketoenamine/enolimine tautomer shift is also regulated by buffer binding, as seen in the greater rate, as well as extent of the shift, when the phosphate concentration is reduced from 100 mM to 5 mM, at pH 8.0. The higher phosphate concentration reduces the driving force for conversion to the enolimine tautomer, as evidenced by the reduced extent of the emission wavelength shift, even on prolonged standing. We infer that the conformer favoring ketoenamine has a phosphate binding site, that is missing in the conformer favoring enolimine. Although the location of this binding site is uncertain, the likeliest candidate is the protonated imine C=N bond itself. Since this bond is accessible to the substrate, it is possible that phosphate can reach the positively charged Lys119 N-H, directly stabilizing the ketoenamine.
Heme regulation of CBS
These findings have implications for the role of the heme in physiological regulation of CBS. The CO affinity is in the physiologically relevant micromolar range. CO binding to hCBS is anticooperative, with association constants of 1.5 and 68 μM 46. The reported range of physiological CO concentrations is 3-30 μM, and is under the control of the heme oxygenases, which generate CO in tissues 61, 62. Thus endogenous CO could be an inhibitor of hCBS.
Intriguingly, CBS has recently been implicated as a CO-responsive regulator of bile excretion in rats 63, apparently through its generating H2S using cysteine as a substrate. By shutting down CBS activity CO inhibits H2S production, thereby modulating biliary HCO3- excretion via H2S-sensitive ion channels. Heme-based regulation of CBS activity may also be one of the mechanisms controlling H2S production in other tissues such as the brain, where it is thought to act as a neuromodulator 6, 10, 11.
Our finding that the CO-induced inactivation of hCBS is a delayed response, and that the rate is modulated by pH and buffer concentration adds further dimensions to the regulation issue. The delayed response would protect against transient increases in CO level, while lowering the pH, within the physiologically relevant range, would sensitize hCBS to CO inhibition. On the other hand, elevated phosphate, and possibly other polyanions, would protect the enzyme against CO inhibition, by stabilizing the active conformation.
Finally, we note that CO inhibition requires reduction of CBS to the Fe(II) form. CO does not bind to the Fe(III) form, which remains active. Thus the redox status of the relevant tissue also regulates hCBS activity, and the inhibitory potential of CO. The redox potentials of full-length hCBS (−350±4 mV 29) and truncated hCBS (−287±2 mV 64) differ appreciably, which suggests that the position of the C-terminal domain is an additional factor in the modulation of hCBS activity by CO. Thus the responsiveness of hCBS to CO has multiple points of physiological control.
Supplementary Material
Acknowledgements
The work was supported by grants from the National Institutes of Health (GM33576 to T.G.S. and HL58984 to R.B.).
Footnotes
Supporting Information: Fluorescence spectra of PLP in Tris and phosphate buffers. Additional fluorescence spectra of Fe(III), Fe(II), Fe(II)CBS424 and Fe(II)CO forms of hCBS. Raman spectra of the PLP-Tris Schiff base. Additional Raman spectra of the Fe(III), Fe(II)CBS424 and Fe(II)CO forms of hCBS and detailed band assignments of all the Raman spectra. UV-vis spectra of the Fe(III), Fe(II), Fe(II)CBS424 and Fe(II)CO forms of hCBS and the Fe(III), Fe(II), and Fe(II)CO forms R266M hCBS. Complete Refs. 30 and 63. This material is available free of charge via the Internet at http://pubs.acs.org/.
References
- 1.Stipanuk MH. Annu. Rev. Nutr. 2004;24:539–577. doi: 10.1146/annurev.nutr.24.012003.132418. [DOI] [PubMed] [Google Scholar]
- 2.Jhee K-H, Niks D, McPhie P, Dunn MF, Miles EW. Biochemistry. 2001;40:10873–10880. doi: 10.1021/bi011087j. [DOI] [PubMed] [Google Scholar]
- 3.Banerjee R, Evande R, Kabil Ö, Ojha S, Taoka S. Biochim. Biophys. Acta. 2003;1647:30–35. doi: 10.1016/s1570-9639(03)00044-x. [DOI] [PubMed] [Google Scholar]
- 4.Refsum H, Ueland PM, Nygard O, Vollset SE. Annu. Rev. Medicine. 1998;49:31–62. doi: 10.1146/annurev.med.49.1.31. [DOI] [PubMed] [Google Scholar]
- 5.Yang G, Wu L, Jiang B, Yang W, Qi J, Cao K, Meng Q, Mustafa AK, Mu W, Zhang S, Snyder SH, Wang R. Science. 2008;322:587–590. doi: 10.1126/science.1162667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chen X, Jhee K-H, Kruger WD. J. Biol. Chem. 2004;279:52082–52086. doi: 10.1074/jbc.C400481200. [DOI] [PubMed] [Google Scholar]
- 7.Kimura H. Molec. Neurobiol. 2002;26:13–19. doi: 10.1385/MN:26:1:013. [DOI] [PubMed] [Google Scholar]
- 8.Eto K, Kimura H. J. Biol. Chem. 2002;277:42680–42685. doi: 10.1074/jbc.M205835200. [DOI] [PubMed] [Google Scholar]
- 9.Stipanuk MH, Beck PW. Biochem. J. 1982;206:267–277. doi: 10.1042/bj2060267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Eto K, Ogasawara M, Uemura K, Nagai Y, Kimura H. J. Neurosci. 2002;22:3386–3391. doi: 10.1523/JNEUROSCI.22-09-03386.2002. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 11.Qu K, Lee SW, Bian JS, Low C-M, Wong PT-H. Neurochem. Internat. 2008;52:155–165. doi: 10.1016/j.neuint.2007.05.016. [DOI] [PubMed] [Google Scholar]
- 12.Omura T, Sadano H, Hasegawa T, Yoshida Y, Kominami S. J. Biochem. 1984;96:1491–1500. doi: 10.1093/oxfordjournals.jbchem.a134978. [DOI] [PubMed] [Google Scholar]
- 13.Kery V, Bukovska G, Kraus JP. J. Biol. Chem. 1994;269:25283–25288. [PubMed] [Google Scholar]
- 14.Meier M, Janosik M, Kery V, Kraus JP, Burkhard P. EMBO J. 2001;20:3910–3916. doi: 10.1093/emboj/20.15.3910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Taoka S, Lepore BW, Kabil Ö, Ojha S, Ringe D, Banerjee R. Biochemistry. 2002;41:10454–10461. doi: 10.1021/bi026052d. [DOI] [PubMed] [Google Scholar]
- 16.Bruno S, Schiaretti F, Burkhard P, Kraus JP, Janosik M, Mozzarelli A. J. Biol. Chem. 2001;276:16–19. doi: 10.1074/jbc.C000588200. [DOI] [PubMed] [Google Scholar]
- 17.Oliveriusova J, Kery V, Maclean KN, Kraus JP. J. Biol. Chem. 2002;277:48386–48394. doi: 10.1074/jbc.M207087200. [DOI] [PubMed] [Google Scholar]
- 18.Kabil Ö, Taoka S, LoBrutto R, Shoemaker R, Banerjee R. J. Biol. Chem. 2001;276:19350–19355. doi: 10.1074/jbc.M100029200. [DOI] [PubMed] [Google Scholar]
- 19.Jhee K-H, McPhie P, Miles EW. J. Biol. Chem. 2000;275:11541–11544. doi: 10.1074/jbc.c000056200. [DOI] [PubMed] [Google Scholar]
- 20.Nozaki T, Shigeta Y, Saito-Nakano Y, Imada M, Kruger WD. J. Biol. Chem. 2001;276:6516–6523. doi: 10.1074/jbc.M009774200. [DOI] [PubMed] [Google Scholar]
- 21.Majtan T, Singh LR, Wang L, Kruger WD, Kraus JP. J. Biol. Chem. 2008;283:34588–34595. doi: 10.1074/jbc.M805928200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Taoka S, Banerjee R. J. Inorg. Biochem. 2001;87:245–251. doi: 10.1016/s0162-0134(01)00335-x. [DOI] [PubMed] [Google Scholar]
- 23.Taoka S, West M, Banerjee R. Biochemistry. 1999;38:2738–2744. doi: 10.1021/bi9826052. [DOI] [PubMed] [Google Scholar]
- 24.Taoka S, Ojha S, Shan X, Kruger WD, Banerjee R. J. Biol. Chem. 1998;273:25179–25184. doi: 10.1074/jbc.273.39.25179. [DOI] [PubMed] [Google Scholar]
- 25.Pazicni S, Lukat-Rodgers GS, Oliveriusova J, Rees KA, Parks RB, Clark RW, Rodgers KR, Kraus JP, Burstyn JN. Biochemistry. 2004;43:14684–14695. doi: 10.1021/bi0488496. [DOI] [PubMed] [Google Scholar]
- 26.Pazicni S, Cherney MM, Lukat-Rodgers GS, Oliveriusova J, Rodgers KR, Kraus JP, Burstyn JN. Biochemistry. 2005;44:16785–16795. doi: 10.1021/bi051305z. [DOI] [PubMed] [Google Scholar]
- 27.Banerjee R, Zou C-G. Arch. Biochem. Biophys. 2005;433:144–156. doi: 10.1016/j.abb.2004.08.037. [DOI] [PubMed] [Google Scholar]
- 28.Cherney MM, Pazicni S, Frank N, Marvin KA, Kraus JP, Burstyn JN. Biochemistry. 2007;46:13199–13210. doi: 10.1021/bi701159y. [DOI] [PubMed] [Google Scholar]
- 29.Singh S, Madzelan P, Stasser J, Weeks CL, Becker D, Spiro TG, Penner-Hahn J, Banerjee R. J. Inorg. Biochem. 2009;103:689–697. doi: 10.1016/j.jinorgbio.2009.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kraus JP, et al. Hum. Mutat. 1999;13:362–375. doi: 10.1002/(SICI)1098-1004(1999)13:5<362::AID-HUMU4>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- 31.Mudd SH, Finkelstein JD, Irreverre F, Laster L. Science. 1964;143:1443–1445. doi: 10.1126/science.143.3613.1443. [DOI] [PubMed] [Google Scholar]
- 32.Mudd SH, Levy HL, Kraus JP. Disorders of Transsulfuration. In: Scriver CR, Beaudet AL, Sly WS, Valle D, editors. The Metabolic and Molecular Bases of Inherited Disease. Vol. II. McGraw-Hill; New York: 2001. pp. 2007–2056. [Google Scholar]
- 33.Miles EW, Kraus JP. J. Biol. Chem. 2004;279:29871–29874. doi: 10.1074/jbc.R400005200. [DOI] [PubMed] [Google Scholar]
- 34.Mudd SH, Skovby F, Levy HL, Pettigrew KD, Wilcken B, Pyeritz RE, Andria G, Boers GHJ, Bromberg IL, Cerone R, Fowler B, Groebe H, Schmidt H, Schweitzer L. Am. J. Hum. Genet. 1985;37:1–31. [PMC free article] [PubMed] [Google Scholar]
- 35.Scott JW, Hawley SA, Green KA, Anis M, Stewart G, Scullion GA, Norman DG, Hardie DG. J. Clin. Invest. 2004;113:274–284. doi: 10.1172/JCI19874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bukovska G, Kery V, Kraus JP. Prot. Express. Purif. 1994;5:442–448. doi: 10.1006/prep.1994.1063. [DOI] [PubMed] [Google Scholar]
- 37.Kery V, Poneleit L, Kraus JP. Arch. Biochem. Biophys. 1998;355:222–232. doi: 10.1006/abbi.1998.0723. [DOI] [PubMed] [Google Scholar]
- 38.Taoka S, Widjaja L, Banerjee R. Biochemistry. 1999;38:13155–13161. doi: 10.1021/bi990865t. [DOI] [PubMed] [Google Scholar]
- 39.Jhee K-H, McPhie P, Miles EW. Biochemistry. 2000;39:10548–10556. doi: 10.1021/bi001020g. [DOI] [PubMed] [Google Scholar]
- 40.Jhee K-H, Niks D, McPhie P, Dunn MF, Miles EW. Biochemistry. 2002;41:1828–1835. doi: 10.1021/bi011756t. [DOI] [PubMed] [Google Scholar]
- 41.Janosik M, Kery V, Gaustadnes M, Maclean KN, Kraus JP. Biochemistry. 2001;40:10625–10633. doi: 10.1021/bi010711p. [DOI] [PubMed] [Google Scholar]
- 42.Kery V, Poneleit L, Meyer JD, Manning MC, Kraus JP. Biochemistry. 1999;38:2716–2724. doi: 10.1021/bi981808n. [DOI] [PubMed] [Google Scholar]
- 43.Frank N, Kery V, Maclean KN, Kraus JP. Biochemistry. 2006;45:11021–11029. doi: 10.1021/bi060737m. [DOI] [PubMed] [Google Scholar]
- 44.Taoka S, Green EL, Loehr TM, Banerjee R. J. Inorg. Biochem. 2001;87:253–259. doi: 10.1016/s0162-0134(01)00336-1. [DOI] [PubMed] [Google Scholar]
- 45.Green EL, Taoka S, Banerjee R, Loehr TM. Biochemistry. 2001;40:459–463. doi: 10.1021/bi0010874. [DOI] [PubMed] [Google Scholar]
- 46.Puranik M, Weeks CL, Lahaye D, Kabil Ö, Taoka S, Nielsen SB, Groves JT, Banerjee R, Spiro TG. J. Biol. Chem. 2006;281:13433–13438. doi: 10.1074/jbc.M600246200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hu S, Smith KM, Spiro TG. J. Am. Chem. Soc. 1996;118:12638–12646. [Google Scholar]
- 48.Strambini GB, Cioni P, Peracchi A, Mozzarelli A. Biochemistry. 1992;31:7527–7534. doi: 10.1021/bi00148a013. [DOI] [PubMed] [Google Scholar]
- 49.Strambini GB, Cioni P, Cook PF. Biochemistry. 1996;35:8392–8400. doi: 10.1021/bi952919e. [DOI] [PubMed] [Google Scholar]
- 50.Ikushiro H, Hayashi H, Kawata Y, Kagamiyama H. Biochemistry. 1998;37:3043–3052. doi: 10.1021/bi971995+. [DOI] [PubMed] [Google Scholar]
- 51.Goldberg ME, York S, Stryer L. Biochemistry. 1968;7:3662–3667. doi: 10.1021/bi00850a045. [DOI] [PubMed] [Google Scholar]
- 52.Ahmed SA, McPhie P, Miles EW. J. Biol. Chem. 1996;271:8612–8617. doi: 10.1074/jbc.271.15.8612. [DOI] [PubMed] [Google Scholar]
- 53.McClure GD, Jr., Cook PF. Biochemistry. 1994;33:1674–1683. doi: 10.1021/bi00173a009. [DOI] [PubMed] [Google Scholar]
- 54.Benecky MJ, Copeland RA, Haye TR, Lobenstine EW, Rava RP, Pascal RA, Jr., Spiro TG. J. Biol. Chem. 1985;260:11663–11670. [PubMed] [Google Scholar]
- 55.Spiro TG, Li X-Y. Resonance Raman Spectroscopy of Metalloporphyrins. In: Spiro TG, editor. Biological Applications of Raman Spectroscopy. Vol. 3. John Wiley & Sons; New York: 1988. pp. 1–37. [Google Scholar]
- 56.Metzler DE. J. Am. Chem. Soc. 1957;79:485–490. [Google Scholar]
- 57.Karsten WE, Ohshiro T, Izumi Y, Cook PF. Arch. Biochem. Biophys. 2001;388:267–275. doi: 10.1006/abbi.2001.2294. [DOI] [PubMed] [Google Scholar]
- 58.Salva A, Donoso J, Frau J, Munoz F. J. Phys. Chem. A. 2004;108:11709–11714. [Google Scholar]
- 59.Evande R, Ojha S, Banerjee R. Arch. Biochem. Biophys. 2004;427:188–196. doi: 10.1016/j.abb.2004.04.027. [DOI] [PubMed] [Google Scholar]
- 60.Kortemme T, Creighton TE. J. Mol. Biol. 1995;253:799–812. doi: 10.1006/jmbi.1995.0592. [DOI] [PubMed] [Google Scholar]
- 61.Carraway MS, Ghio AJ, Carter JD, Piantadosi CA. Am. J. Physiol. 2000;278:L806–L812. doi: 10.1152/ajplung.2000.278.4.L806. [DOI] [PubMed] [Google Scholar]
- 62.Ingi T, Chiang G, Ronnett GV. J. Neurosci. 1996;16:5621–5628. doi: 10.1523/JNEUROSCI.16-18-05621.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Shintani T, et al. Hepatology. 2009;49:141–150. doi: 10.1002/hep.22604. [DOI] [PubMed] [Google Scholar]
- 64.Carballal S, Madzelan P, Zinola CF, Grana M, Radi R, Banerjee R, Alvarez B. Biochemistry. 2008;47:3194–3201. doi: 10.1021/bi700912k. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.