

Abstract
With incomplete lineage sorting (ILS), the genealogy of closely related species differs along their genomes. The amount of ILS depends on population parameters such as the ancestral effective population sizes and the recombination rate, but also on the number of generations between speciation events. We use a hidden Markov model parameterized according to coalescent theory to infer the genealogy along a four-species genome alignment of closely related species and estimate population parameters. We analyze a basic, panmictic demographic model and study its properties using an extensive set of coalescent simulations. We assess the effect of the model assumptions and demonstrate that the Markov property provides a good approximation to the ancestral recombination graph. Using a too restricted set of possible genealogies, necessary to reduce the computational load, can bias parameter estimates. We propose a simple correction for this bias and suggest directions for future extensions of the model. We show that the patterns of ILS along a sequence alignment can be recovered efficiently together with the ancestral recombination rate. Finally, we introduce an extension of the basic model that allows for mutation rate heterogeneity and reanalyze human–chimpanzee–gorilla–orangutan alignments, using the new models. We expect that this framework will prove useful for population genomics and provide exciting insights into genome evolution.
BIOLOGICAL sequence data, and particularly the variation therein, contain information about the evolutionary processes that shaped the present-day organisms. Coalescent theory provides tools for comparative sequence analysis to investigate the history of populations, by studying the genealogy of the sampled sequences (Hein et al. 2005). More recently, with the rapid accumulation of molecular data, multiple-loci studies have become possible, allowing for the estimation of population genetics parameters such as speciation times and ancestral population sizes (Rannala and Yang 2003; Burgess and Yang 2008). The availability of complete genome sequences for closely related species opens a new area of research, by providing virtually as many loci as possible, yet for a single sequence from a limited number of species. While such data cannot be used to study contemporary populations, they contain information about ancestral population processes, particularly when speciation events are sufficiently close in time that incomplete lineage sorting (ILS) occurs.
Consider a site in an alignment of human, chimpanzee, and gorilla. The most likely evolutionary scenario is that going backward in time, the human and chimpanzee sequences coalesce first within the human–chimpanzee (HC) ancestral population and then meet the gorilla sequence within the human–chimpanzee–gorilla (HCG) ancestral population (Figure 1, case HC1). Because of genetic drift, we expect the sequences to have an older common ancestor in some regions, falling back in the HCG ancestral population. The two lineages would have been passed to and survived within the ancestral population (ancestral polymorphism), potentially having a genealogy different from the phylogeny. There are three equiprobable scenarios: the human and chimpanzee sequences coalesce first (HC2), the human and gorilla (HG) sequences coalesce first, or the chimpanzee and gorilla (CG) sequences coalesce first. In addition to these four scenarios, the timing of the coalescence events also varies along the genome. This phenomenon is illustrated in Figure 2, showing a partial alignment simulated using a coalescent with recombination process, with parameters close to the currently accepted values for the ape populations. The theory of coalescence allows us to predict quantities like the proportions of sites in each type of genealogy according to ancestral effective population sizes, speciation times, and recombination rates. Reciprocally, the pattern of variation along the alignment carries information on the underlying genealogies, and this variation enables the estimation of population parameters.
Figure 1.—

The four different types that the genealogy of four sequences can take. The four species are H, human; C, chimpanzee; G, gorilla; and O, orangutan (outgroup). Following Hobolth et al. (2007), these genealogies are labeled HC1, HC2, HG, and CG.
Figure 2.—

The genealogy of sequences varies along the genome. This graph shows the changes in topologies by colors (see Figure 1) together with the divergence between human and chimpanzee along 20,000 positions simulated using a coalescent with recombination (see data and methods).
Our goal is to extract population genetics information in ancestral species by explicitly modeling the changes in genealogy, using the coalescent with recombination process. Several approaches have been conducted to extract this information (see Chen and Li 2001; Nichols 2001; Patterson et al. 2006; Burgess and Yang 2008). Hobolth et al. (2007) introduced the use of hidden Markov models in combination with results from coalescent theory to estimate population genetics parameters from genomic alignments, an approach they named the coalescent hidden Markov model (CoalHMM). Contrary to previous approaches, this model-based, maximum-likelihood methodology makes use of all the data, not only certain loci as in Chen and Li (2001) or parsimony informative sites as in Patterson et al. (2006).
The purpose of this article is to describe two extensions of the Hobolth et al. (2007) model and to investigate model assumptions and properties in detail. The most novel extension is a reparameterization of the hidden states and transition probabilities according to parameters from the demographic model and recombination rate (see Figure 3). This reparameterization offers a more natural way of estimating population parameters and allows for the estimation of the ancestral recombination rate. The second extension accounts for mutation rate heterogeneity along the alignment. We investigate the ability of the model to infer population parameters, using simulations under the coalescent with recombination, and apply the new method to the data sets used by Hobolth et al. (2007).
Figure 3.—

The CoalHMM framework. The basic demographic model for human (H), chimpanzee (C), and gorilla (G) with the orangutan (O) as outgroup is used to design a set of candidate genealogies (topology plus coalescence times) and a corresponding transition matrix. These are computed according to the parameters of the model, namely speciation times (τX), ancestral population sizes (θX), and recombination rate ρ. The genealogies and transition probabilities are then used within a standard hidden Markov model to compute the likelihood and perform the posterior decoding.
THE COALHMM FRAMEWORK
The information on ancestral population history lies in the succession of the distinct genealogies of the sequences along the genome. To retrieve this information, one needs to infer the genealogy for each site of the genome alignment. Therefore, it is tempting to use standard phylogenetic reconstruction methods to infer the site-specific lineage relationships, with the limit that there are few sequences (four in this study). Patterson et al. (2006) uses the maximum-parsimony method, which restricts their analysis to informative sites only. Conversely, the CoalHMM approach uses maximum-likelihood (ML) inference, following Felsenstein's (1981) work. Although one site carries only little information on the local genealogy, positions in close proximity of the genome are likely to share the same genealogy. Accounting for the across-site correlation of genealogies is hence an important source of information. The methodology is exemplified using the human, chimpanzee, and gorilla species, with the orangutan as an outgroup.
Hidden Markov model:
Reconstructing a sequence of correlated features along a sequence is the overall goal of hidden Markov models. Such models have been successfully used to model correlation of mutation rates along the genome (Yang 1995), to infer isochores (Melodelima et al. 2006), gene content (Stanke and Waack 2003), or secondary structure prediction (Goldman et al. 1996), for instance, and are now a standard tool in biological sequence analysis. The HMM methodology consists of a Markov model along the sequence, with states as features to reconstruct. These features are not directly observable and are hence named “hidden states,” but can be predicted from the “observed states” in the data. In the CoalHMM approach, the observed states are the distinct columns in the alignment, and the hidden states are the genealogies.
We denote by 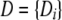 the set of alignment columns and
the set of alignment columns and 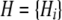 the sequence of hidden states. The alphabet of different states taken by Hi is denoted
the sequence of hidden states. The alphabet of different states taken by Hi is denoted 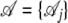 ; in our case
; in our case 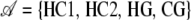 . The use of four archetypal genealogies is an approximation of the real ancestral recombination graph, as there is an infinite set of putative genealogies due to variation in coalescent times. We further denote by Θ the set of parameters in the model. In the following, and for the sake of clarity, we note PrΘ(X) = Pr(X | Θ).
. The use of four archetypal genealogies is an approximation of the real ancestral recombination graph, as there is an infinite set of putative genealogies due to variation in coalescent times. We further denote by Θ the set of parameters in the model. In the following, and for the sake of clarity, we note PrΘ(X) = Pr(X | Θ).
The joint probability of a particular sequence H of hidden states and the data D is given by
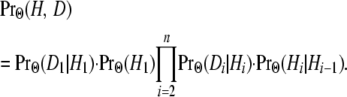 |
(1) |
Here it is assumed that the process of state changes along the sequence is Markovian and that the observed states are independent given the hidden states; i.e.,
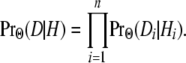 |
(2) |
The probability of the data thus depends on two major components, namely 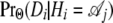 and PrΘ(Hi | Hi−1). The first probability is called the emission probability and the second the transition probability, and they are the core ingredients in a hidden Markov model (Figure 3).
and PrΘ(Hi | Hi−1). The first probability is called the emission probability and the second the transition probability, and they are the core ingredients in a hidden Markov model (Figure 3).
Emission probabilities:
We denote by 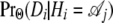 the probability of the alignment column Di conditional on the genealogy at the site being
the probability of the alignment column Di conditional on the genealogy at the site being 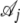 . These are computed as the probability of a column in the alignment conditioned on a given genealogy. They depend on the branch lengths of the genealogies (a, b, c,
. These are computed as the probability of a column in the alignment conditioned on a given genealogy. They depend on the branch lengths of the genealogies (a, b, c, 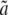 ,
, 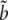 ,
,  ; see Figure 4) and a substitution model. These probabilities are then computed using standard approaches developed in phylogenetics, following Felsenstein's (1981) work.
; see Figure 4) and a substitution model. These probabilities are then computed using standard approaches developed in phylogenetics, following Felsenstein's (1981) work.
Figure 4.—

Population and genealogy parameter notations.
Transition probabilities:
The probabilities of change between genealogies as we move along the alignment depend on the demographic and coalescent parameters θHC, θHCG, τHC, and τHCG and on the recombination rate ρ. These probabilities are functions of the lineage-specific recombination rates, ρH, ρC, and ρG for the human, chimpanzee, and gorilla lineages, respectively. In some models, however, we assume that these recombination rates are identical. We denote the transition probability 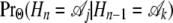 by pk,j. While there are potentially 12 transition probabilities, symmetry considerations reduce the number of parameters. Most importantly, the probability distribution of the state of a single site is independent of position,
by pk,j. While there are potentially 12 transition probabilities, symmetry considerations reduce the number of parameters. Most importantly, the probability distribution of the state of a single site is independent of position,
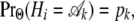 |
(3) |
for all i, and the relative order of the sites is immaterial:
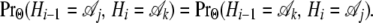 |
As a consequence, a relationship exists between transition probabilities across the diagonal,
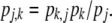 |
(4) |
In addition, within the HCG ancestor, the human and chimpanzee lineages have exchangeable histories, implying
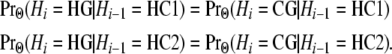 |
These considerations imply that determination of the full transition matrix
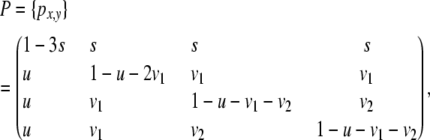 |
(5) |
in which u is determined from s using (4), requires specification of only three free transition probabilities (s, v1, and v2), together with the probabilities for single sites (3).
Hobolth et al. (2007) provided the single-site probabilities
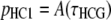 |
(6a) |
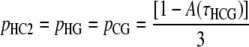 |
(6b) |
in which
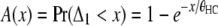 |
(7) |
represents the cumulative distribution function (cdf) of the time to the coalescence of a pair of lineages in the human–chimpanzee ancestor (compare Hudson 1983; Rannala and Yang 2003).
We note that while H and C are exchangeable from the perspective of G, they are distinguishable from their own perspective:
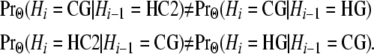 |
This aspect of our model differs from that of Hobolth et al.(2007), who set v1 = v2. The details of the equations can be found in the appendix.
Likelihood recursion:
Since the “true” sequence of hidden states is unknown, the likelihood of the data is the sum over all possible sequences:
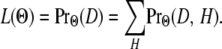 |
(8) |
L(Θ) can be efficiently computed by recursion using the general equation for a HMM (e.g., Durbin et al. 1998),
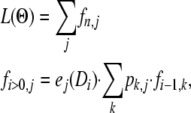 |
(9) |
with 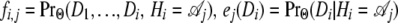 , and
, and 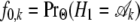 , the initial probability that is set to the equilibrium frequency of the chain.
, the initial probability that is set to the equilibrium frequency of the chain.
Reconstructing local genealogies:
Of particular interest are also the posterior probabilities of each hidden state at each position in the alignment (Durbin et al. 1998),
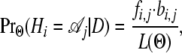 |
(10) |
where the 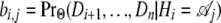 are computed using a recursion similar to the fi,j, using the backward algorithm. The hidden state with the maximum posterior value at each position provides a reconstruction of the sequence of hidden states (an approach referred to as “posterior decoding”).
are computed using a recursion similar to the fi,j, using the backward algorithm. The hidden state with the maximum posterior value at each position provides a reconstruction of the sequence of hidden states (an approach referred to as “posterior decoding”).
COALHMM PARAMETERIZATION
There are several choices for emission and transition probability parameterization, and they are distinct targets for model improvement. Below, we first describe the approach developed in Hobolth et al. (2007). Second, we show how these calculations can be extended to provide direct estimates of the population parameters, via a reparameterization of the model. Third, we describe the mutation rate variation extension.
The basic model:
Following Hobolth et al. (2007), we consider the case of three ingroup species plus one outgroup. The use of the outgroup sequence allows us to reconstruct the ancestral state of the HCG ancestor and extract information from the informative sites (Hobolth et al. 2007). There are four distinct types of possible genealogies, as shown in Figure 1 with example species of human, chimpanzee, gorilla, and orangutan as an outgroup. The distribution of the coalescent times in each case can be expressed as a function of the speciation times (τHC for the human–chimpanzee speciation, τHCG for the HC–gorilla speciation) and ancestral effective population sizes (θHC for the HC ancestral population and θHCG for the HCG ancestral population size); see Figure 4 for notations. A fifth parameter noted τHCGO is needed. It corresponds to the divergence with the outgroup, which is approximated to be constant and confounded with the speciation time. All parameters are scaled by 2Ne · u, Ne being the effective population size of one of the extant species and u the mutation rate. In practice this scaling factor is estimated using a calibration point in the phylogeny. In this article, we use Greek letters to refer to the scaled parameters and roman letters for the unscaled parameters (see Table 1).
TABLE 1.
Parameter notations with their corresponding scaled name
| Unscaled | Scaled | Description |
|---|---|---|
| u | Per-nucleotide, per-generation mutation rate | |
| g | Generation time in years | |
| Ne | Effective population size of the human population | |
| THC | τHC = THCu/2/Ne/g | Human–chimpanzee speciation time |
| THCG | τHCG = THCGu/2/Ne/g | HC–gorilla speciation time |
| NHC | θHC = NHCu/2/Ne | HC effective ancestral population size |
| NHCG | θHCG = NHCGu/2/Ne | HCG effective ancestral population size |
| r | ρ = r/u | Per nucleotide, per generation, recombination rate (per mutation for the scaled version) |
HC and HCG: human–chimpanzee and human–chimpanzee–gorilla ancestral populations.
Within a population of effective size θ, the coalescence time of two lineages follows an exponential distribution (Exp) with mean θ. We note Δ1 as the coalescence time for the human and chimpanzee lineages in the HC ancestor, that is, assuming we are in the HC1 genealogy. Δ1 hence follows a right-truncated exponential distribution (TExp):
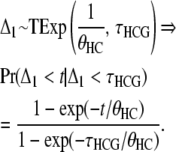 |
(11) |
Let Δ2 be the second coalescence event, involving the HC ancestor lineage and the gorilla, following an exponential distribution:
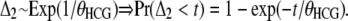 |
(12) |
In the alternative genealogies, all coalescence events occur within the HCG ancestor. 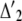 denotes the time distribution of the first two lineages to coalesce,
denotes the time distribution of the first two lineages to coalesce,
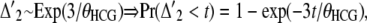 |
(13) |
and 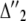 denotes the distribution of the last two ones,
denotes the distribution of the last two ones,
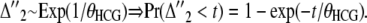 |
(14) |
In the work by Hobolth et al. (2007), the branch lengths for each genealogy were computed by taking the mean of each distribution, leading to
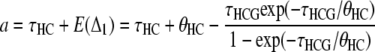 |
(15) |
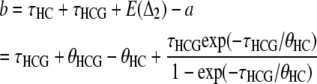 |
(16) |
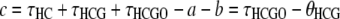 |
(17) |
for the standard genealogy and
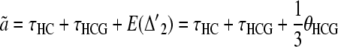 |
(18) |
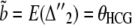 |
(19) |
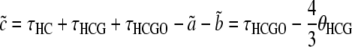 |
(20) |
for the alternative genealogies. These parameters are, however, not independent and can be reduced to four parameters, for instance a, b, c, and 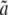 . Hobolth et al. (2007) estimated these parameters directly from the data, together with the transition probabilities s, u, v1, and v2, further assuming that v1 = v2 = v. To translate these estimates into population parameters, they used Equations 15, further noting that
. Hobolth et al. (2007) estimated these parameters directly from the data, together with the transition probabilities s, u, v1, and v2, further assuming that v1 = v2 = v. To translate these estimates into population parameters, they used Equations 15, further noting that
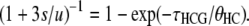 |
(21) |
Together with Equations 15–20, this can be used to provide estimates for τHC, τHCG, θHC, and θHCG from the estimated values.
Reparameterization of the basic model:
The approach previously introduced has two major drawbacks. First, this model is potentially overparameterized since s, u, and v are functions of τHC, τHCG, θHC, θHCG, and the recombination rate ρ, resulting in several constraints on the parameters not accounted for in the model. An additional drawback is that one must rely on the delta method to obtain confidence intervals for the parameters. To overcome these limitations, we expressed the likelihood function directly from the population parameters. Branch lengths parameters a, b, c, 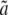 ,
, 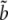 , and
, and  were deducted from τHC, τHCG, τHCGO, θHC, and θHCG to compute the emission probabilities, using the above equations. To avoid the constraint on τHCGO (it must be
were deducted from τHC, τHCG, τHCGO, θHC, and θHCG to compute the emission probabilities, using the above equations. To avoid the constraint on τHCGO (it must be 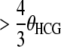 ), we parameterized the likelihood according to
), we parameterized the likelihood according to  instead, whose only constraint is to be positive. The transition parameters s, u, v1, and v2 are no longer independent parameters, but expressed as functions of τHC, τHCG, θHC, θHCG, and the recombination rate ρ. The full details of these equations are given in the appendix. The new parameterization removes one parameter and contains only directly interpretable population parameters in addition to the nucleotide substitution parameters that remain unchanged. The reparameterization allows the estimation of an average ancestral recombination rate, which was not possible in the previous model. We refer to the Hobolth et al. (2007) implementation as the “07” model, as opposed to the “09” model for the new parameterization.
instead, whose only constraint is to be positive. The transition parameters s, u, v1, and v2 are no longer independent parameters, but expressed as functions of τHC, τHCG, θHC, θHCG, and the recombination rate ρ. The full details of these equations are given in the appendix. The new parameterization removes one parameter and contains only directly interpretable population parameters in addition to the nucleotide substitution parameters that remain unchanged. The reparameterization allows the estimation of an average ancestral recombination rate, which was not possible in the previous model. We refer to the Hobolth et al. (2007) implementation as the “07” model, as opposed to the “09” model for the new parameterization.
Accounting for across-site mutation rate variation:
Variation of mutation rate along genomes is a common phenomenon, and several models have been developed for phylogenetic inference (see pioneer work by Yang 1993). We introduce here an extension to the model that uses Yang's (1994) method for correcting emission probabilities. A prior distribution of mutation rate is assumed, in most cases a discretized Gamma distribution whose shape is estimated from the data, together with the substitution parameters. This rate across site (RAS) model increases only the complexity of the calculation of emission probabilities, multiplying it by the number of rate categories considered in the discretization, and adds only one parameter.
DATA AND METHODS
Optimization and confidence intervals:
A modified Newton–Raphson algorithm was used to find the maximum of the likelihood function. The first- and second-order derivatives with respect to population parameters τHC, τHCG,  , θHC, θHCG, and ρ, together with the substitution parameters, were computed numerically using the three-points method. We used the Fisher method to compute confidence intervals for the estimated parameters (Efron and Tibshirani 1998). The variance of the τHC + τHCG sum was derived from the estimated variances and covariance of τHC and τHCG, and we used the delta method for the intervals of the 07 method, as in Hobolth et al. (2007).
, θHC, θHCG, and ρ, together with the substitution parameters, were computed numerically using the three-points method. We used the Fisher method to compute confidence intervals for the estimated parameters (Efron and Tibshirani 1998). The variance of the τHC + τHCG sum was derived from the estimated variances and covariance of τHC and τHCG, and we used the delta method for the intervals of the 07 method, as in Hobolth et al. (2007).
Simulations:
Simulating data for this study involves two steps: (i) simulating an ancestral recombination graph, i.e., a set of trees corresponding to different regions of the data set, and (ii) simulating alignments by applying a substitution process on the graph. The latter step uses standard phylogenetic tools. Alignments with 500,000 sites were simulated using a general time reversible model with parameters a = 1.49, b = 0.67, c = 0.38, d = 0.35, e = 0.6, πA = 0.27, πC = 0.25, πG = 0.27, and πT = 0.21, using the bppSeqGen program (Dutheil and Boussau 2008). A mutation rate of 0.1% change per million years per nucleotide was used.
The former step can be achieved in two ways. One can use a coalescent with recombination (Hudson 2002; Mailund et al. 2005) model to generate an ancestral recombination graph according to speciation times, contemporary and ancestral population sizes, and recombination rates, as in Hobolth et al. (2007). Another option consists of simulating the ancestral recombination graph from the hidden Markov model, by drawing states sequentially along the genome, using the matrix of transition probabilities. Both approaches were used for comparison.
We used two simulation setups. In the first one, we simulated 100 data sets with 500,000 positions each. Parameters were chosen to be, respectively, 4 and 5.5 MY for the first and second speciation times, with a generation time of 25 years. Effective sizes were set to 40,000 individuals for the ancestral populations and 30,000 for the extant ones. The divergence with the outgroup was assumed to be 18 MY, and the recombination rate r = 1.5 cM/Mb. In the second setup, we tested various combinations of parameter values to assess their interaction in the estimation process. We used the parameter values THC = {2 MY, 4 MY, 6 MY}, THCG = {2 MY, 4 MY, 6 MY}, NHC = {20,000, 40,000, 60,000}, NHCG = {20,000, 40,000, 60,000}, r = {0.5 cM/Mb, 1.5 cM/Mb, 2.5 cM/Mb}, and five replicates in each case, resulting in 35 · 5 = 1215 simulated alignments.
Model fitting for the bias correction:
We conducted a large set of simulations, with five replicates for all parameters combinations with THC = {3 MY, 5 MY, 7 MY}, THCG = {1 MY, 3 MY, 5 MY}, NHC = {20,000, 40,000, 60,000, 80,000}, NHCG = {20,000, 40,000, 60,000, 80,000}, r = 1.5 cM/Mb, hence resulting in 32 · 42 · 5 = 720 simulated data sets. The bias was computed for each parameter X as
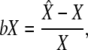 |
(22) |
where X and 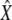 are the true and estimated values of parameter X, respectively. The resulting data sets containing bias values for the various parameter combinations were used to compute corrected estimators and their confidence intervals, as follows: Five linear models were fitted for parameters τHC, τHCG, θHC, θHCG, and ρ, with the corresponding bias as the response variable, and the estimated values of parameters τHC, τHCG and θHC as the explanatory variable, using the R software (R Development Core Team 2008). Fifteen simulated data sets for which the optimization failed were removed, and a stepwise model selection was performed. All points with a Cook distance >0.01 were ignored and considered as outliers (20 points in the worst case). These models were then used to predict the bias for the estimated values in real data:
are the true and estimated values of parameter X, respectively. The resulting data sets containing bias values for the various parameter combinations were used to compute corrected estimators and their confidence intervals, as follows: Five linear models were fitted for parameters τHC, τHCG, θHC, θHCG, and ρ, with the corresponding bias as the response variable, and the estimated values of parameters τHC, τHCG and θHC as the explanatory variable, using the R software (R Development Core Team 2008). Fifteen simulated data sets for which the optimization failed were removed, and a stepwise model selection was performed. All points with a Cook distance >0.01 were ignored and considered as outliers (20 points in the worst case). These models were then used to predict the bias for the estimated values in real data:
Let
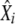 and
and 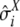 , where X stands for τHC, τHCG, θHC, θHCG, and ρ, be the mean and standard deviation of estimated parameters, computed as described in Optimization and confidence intervals. Draw a random number
, where X stands for τHC, τHCG, θHC, θHCG, and ρ, be the mean and standard deviation of estimated parameters, computed as described in Optimization and confidence intervals. Draw a random number 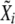 from a normal distribution
from a normal distribution 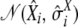 , for each parameter.
, for each parameter.Predict the expected bias
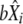 from the obtained values, together with their standard error
from the obtained values, together with their standard error 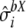 , from the values of
, from the values of 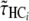 ,
,  , and
, and 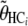 and using the previously adjusted linear model.
and using the previously adjusted linear model.Draw a random number
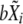 from normal distribution
from normal distribution 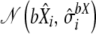 .
.Compute
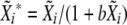 . Repeat the procedure 1000 times, and get the 95% confidence interval from the distribution of the
. Repeat the procedure 1000 times, and get the 95% confidence interval from the distribution of the 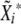 .
.
Data and program availability:
The data sets from Hobolth et al. (2007) were reanalyzed for comparison with previous models, using a coalescent with recombination process. The data contain five targets, including one from the X chromosome, and comprise 2.1 Mbp. The CoalHMM program was developed in C++ using the Bio++ libraries (Dutheil et al. 2006) and is available upon request.
RESULTS AND DISCUSSION
A large set of simulations was performed to assess the properties of the different models. This simulation procedure allows the test of model assumptions, since the simulated data sets do not rely on the Markov assumption of the HMM, but result from the true coalescent process. The simulations were used to assess the estimation of the population parameters, including the recombination rate. The parameter values used in the simulation procedure are close to the values of the human, chimpanzee, orangutan, and macaque data set analyzed in this work.
Population parameters estimation:
The substitution parameters from the GTR model are recovered with very good precision (supporting information, Figure S1). This accuracy is explained by the estimation being performed under the same model as the simulations, resulting in no model misspecification. The high precision results from the large amount of data used for the estimation: 500,000 sites simulated in each replicate.
Conversely, the inference of population parameters appears to be biased. The most recent speciation time (THC) is found to be underestimated by ∼0.5 MY (12.5%) and the ancestral population size of the corresponding ancestral population (NHC) to be overestimated by 20,000 individuals (50%, Figure 5, a and b). The second split (THC + THCG) and the most ancient population size (NHCG) are recovered with good precision. It is also noteworthy that the variance of NHC is larger than the one of NHCG. Results from the 07 implementation and the 09 implementation, which contains one parameter less due to the reparameterization, display the same amount of bias.
Figure 5.—

Population parameters recovery: (a) 07 implementation; (b–d) 09 implementation. (a and b) Ancestral recombination graph (ARG) simulated using a coalescent with recombination; (c) ARG simulated from the HMM; (d) ARG with averaged coalescent times. The dashed lines show the true values.
To further investigate the origin of these biases, we simulated data with various combinations of parameters. We used three values (2, 4, and 6 MY) for THC and THCG, three values (20,000, 40,000, and 60,000) for NHC and NHCG, and three distinct recombination rates (0.5 cM/Mb, 1.5 cM/Mb, 2.5 cM/Mb). Five replicates were performed for each combination, resulting in 1215 simulated data sets. The results show that parameters THCG, THC, and NHC have an effect on the biases (Figure S2). The relative bias on the first speciation time is larger for small values of THC and larger for large values of THCG. It is also larger for large values of NHC. The bias on the population size NHC is also larger for large values of THC. The bias on the recombination rate is proportional to the value of THC.
We conducted a second set of simulations directly from the Markov chain, to assess whether the bias was due to an optimization problem or to one or several assumptions of the model. This procedure consists of first simulating a sequence of genealogies under the Markov assumption by sampling from the chain and then simulating sites from these genealogies as before. Results are shown in Figure 5c and display no bias, in agreement with the maximum-likelihood principle. It is noteworthy that the variance of the most recent ancestral effective population size, NHC, is still twice as large as the most ancient one, NHCG, as observed by Hobolth et al. (2007) on real data.
There are two differences between the two simulation procedures, which are two possible causes for the observed bias: (i) the Markov dependency between genealogies along the alignment and (ii) the number of candidate genealogies, coerced to four in the CoalHMM setup while the actual coalescent times take values from a continuous range in the real ancestral recombination graph (ARG). We assessed the effect of the number of candidate genealogies by simulating ARGs from the coalescent with recombination, as in Figure 5, a and b. The resulting trees have distinct branch lengths corresponding to distinct coalescent times, taken from the expected distribution (see Equations 11–14). We then “forced” the genealogies to four categories, by pulling the coalescent times to the average of their distributions. The resulting ARGs have only four “average” states, as in the fitted model, yet without the Markovian property. Results are shown in Figure 5d and show no bias in population parameters, demonstrating that the major component of the biases is the restricted set of genealogies (that is, hidden states) of the model. These results suggest that using more realistic—yet more complex—models will certainly improve our estimation of population parameters, although at a high cost in terms of computer resources: Multiplying the number of genealogies by a factor λ multiplies the memory usage and computation time by a factor λ2.
Recombination rate estimation:
The new parameterization of the 09 model has recombination rates as explicit parameters, therefore allowing for the estimation of ancestral, potentially lineage-specific, recombination rates. Figure 6a shows that the recombination rate is recovered up to a scaling factor. The bias is removed when simulating the ARG from the HMM, but persists when coercing the coalescent times to their mean (as in Figure 5d), suggesting that this bias has a different origin than the one on population parameters. Additional simulations show that the underestimation depends on the human–chimpanzee speciation time: The more ancient the speciation is, the more recombination events we miss (results not shown). A possible explanation comes from an assumption made in the calculation of the transition probabilities. For mathematical tractability, we assume that once a recombination event occurs between two nucleotides, the actual genealogy at both nucleotides is independent. If a recombination event, however, happens early in one of the human or chimpanzee lineages, it is likely that the two new lineages recoalesce, leading to a nonindependent choice of topology for the two positions, potentially likely to bias the transition probabilities. More work is needed to assess the relative importance of this effect. It is, however, likely to depend on the most recent speciation time. This type of bias, however, does not prevent comparisons of estimates along genome alignments, since the speciation time is constant.
Figure 6.—

(a) Estimation of recombination rates. Ten data sets were simulated for each value of the recombination rate. The recombination rate was fixed to the same value in all lineages, and the 09 implementation with ρH = ρC = ρG was used for the estimation. Other parameters are set to the same values as in Figure 5. The dashed line shows the 1:1 ratio, and the solid line is fitted to the data. The slope is estimated to be 0.29. (b) Global vs. lineage-specific recombination rates. One hundred simulations were performed in each case, with parameter values identical to those in Figure 5. Dashed lines represent the true value of the recombination rate. Overlapping notches show nonsignificant differences in the medians.
The transition probabilities calculations allow for lineage-specific recombination rates. Simulations show that there is only little power to distinguish between these parameters, which are recovered with a larger variance than when assumed to be equal (Figure 6b). In nearly 20% of cases, the human- or chimpanzee-specific recombination rates could not be estimated and were found to be zero.
Bias correction:
To assess the effect of each parameter on the bias, we conducted a large set of simulations, with different parameter values (including mutation rate) encompassing the real ones (see data and methods). This procedure showed that the biases depend on the values of the τHC, τHCG, and θHC parameters and are independent of θHCG and ρ. Furthermore, the relation between the relative bias and the parameters appears to be linear on the ranges of parameters tested, allowing us to predict its amount using a linear model. We propose here a simple empirical correction to improve the estimators from the current model. We fitted one linear model for each biased parameter, with the corresponding relative bias as a response variable and 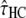 ,
, 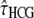 , and
, and 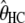 , the estimated parameter values, as explanatory variables. These models were then used to predict the bias components and correct the estimators. The resulting confidence intervals, taking into account the variance in the prediction of the bias, are computed using a parametric bootstrap approach (see data and methods). We applied this method to the 100 previously simulated data sets, to assess its efficiency. Results are shown in Figure 7 and show that this method is successful in correcting the bias.
, the estimated parameter values, as explanatory variables. These models were then used to predict the bias components and correct the estimators. The resulting confidence intervals, taking into account the variance in the prediction of the bias, are computed using a parametric bootstrap approach (see data and methods). We applied this method to the 100 previously simulated data sets, to assess its efficiency. Results are shown in Figure 7 and show that this method is successful in correcting the bias.
Figure 7.—

Population parameters recovery when the bias correction is used. The parameter values used in the simulation are the same as in Figure 5.
Hidden state recovery:
In addition to the parameter estimation, an important feature of the CoalHMM approach is the inference of patterns of incomplete lineage sorting along the genomes. It is achieved using the posterior decoding of the HMM, which aims at calculating the probabilities of each hidden state—that is, candidate genealogy—at each position, and by taking the state with the maximum probability (see data and methods).
We first investigated the distribution of segment length, according to the underlying genealogy. Under the Markov assumption, this distribution is expected to be geometric, with the parameter equal to the corresponding diagonal element in the transition matrix (Equation 5). Figure 8 compares the distribution of segment lengths over 100 simulations, computed from the posterior decoding and the full ARG, with the theoretical expectation under a Markov assumption. Surprisingly, the fit between the distribution from the ARG and the expected geometric distribution is very good, suggesting that the Markov assumption is a reasonable approximation of the real process. The discrepancy with the posterior distribution is due to a deficiency in small fragments. Such small fragments have very little information and are hence most often missed.
Figure 8.—

Distribution of posterior fragment lengths. Shaded bars show the posterior distribution, hatched bars show the distribution from the ancestral recombination graph, and the solid lines show the geometric distribution (expected under the Markov assumption).
We then investigated the ability of the CoalHMM to recover the correct genealogy class. We used the same simulation setup as in Figure 5a and derived the sequence of true genealogies from the ARG. This sequence was then compared to posterior decoding. Figure 9 displays the average results for 100 simulations. It shows that the posterior decoding is quite efficient in recovering the correct genealogy: 82% of HC1 topologies are found to have the maximum posterior probability (recall measure, Figure 9b). This proportion is 17% for HC2 and 58% for HG and CG, leading to an average of 64% of the genealogies being correctly inferred. These numbers are in all cases significantly higher than the random expectation, computed by random permutations of the states in the true and inferred sequences of genealogies. The efficiency of the method appears quite high, knowing that under the simulation parameter values, the probability of a parsimony informative site is only 0.28% under HC1 and 0.2% under any alternative genealogy. The proportion of correctly inferred states (precision measure, Figure 9b) is also very high for state HC1: 73%. HG and CG are quite efficiently recovered (average precision of 56%), compared to HC2 (precision of 34%), meaning that many HC2 genealogies are assigned to another category, in most case HC1 (Figure 9a). This results in a global underestimation of the proportion of incomplete lineage sorting, defined as the proportion of sites with alternative genealogies (Figure 9c).
Figure 9.—

Comparison of the posterior decoding with the known ancestral recombination graph in 100 simulated data sets. (a) Joint distribution of reconstructed states (by column) and real states (by row). The numbers indicate the average percentage of each configuration. The proportions in all combinations sum to 100%, and the frequencies on the top-left/bottom-right diagonal correspond to the correctly inferred cases. The sums over each row give the true frequencies of each state, and the sums over columns provide the frequencies inferred by the CoalHMM method. (b) Precision and recall measures. The precision measure corresponds, for each column of panel 9a, to the diagonal proportion divided by the sum of the column (number of true positives over number of positives). The recall measure corresponds, for each row of panel 9a, to the diagonal proportion divided by the sum of the row (number of true positives over actual number of sites in a given genealogy). (c) Comparison of the reconstructed and real frequencies of hidden states. The proportion of incomplete lineage sorting is taken as the frequency of sites in alternative genealogies.
Reanalysis of ape alignments:
We reanalyzed the data sets of Hobolth et al. (2007) and compared the different models and implementations. We conducted several model comparisons on target 1, the largest alignment. We compared the 07 and 09 implementations with lineage-specific recombination rates (three-rates model: ρH, ρC, ρG), human and chimpanzee recombination rates (two-rates model, ρH = ρC, ρG), and the one-rate model (ρH = ρC = ρG), with and without mutation rate heterogeneity (constant or RAS model). We used the Bayesian information criterion (BIC) and the likelihood-ratio test (LRT) when relevant for comparing models. The two criteria gave identical conclusions in all cases (Figure 10). Results show that (i) the RAS model is always preferred over the constant rate model and (ii) there is very little difference between the 07 and the 09 implementations when mutation rate heterogeneity is taken into account (Figure 11, a–c). The 09 implementation with one recombination rate, the one with the smaller number of parameters, is preferred by the two criteria. When comparing models with a constant rate, the 07 implementation is better than the 09 with one recombination rate and closer to the two-recombination rates model. Figure 11 shows that the transition probabilities s, u, v1, and v2 are different in the 07 and the 09-1ρ model, the transition between alternative genealogies (v1 and v2 parameters) being 15-fold higher than the transitions from and to the “standard” genealogy. In the 09 implementation, these transition probabilities are not free but functions of the lineage-specific recombination rates, assuming a single recombination parameter does not allow us to catch this characteristic of the data, whereas adding a second one can do so. However, this high transition probability disappears when mutation rate heterogeneity is taken into account, together with the support for the need of different recombination parameters, suggesting that it is an artifact resulting from the variation of mutation rates along the genome. As a result, accounting for mutation rate heterogeneity leads to different estimates of transition parameters, the 07+RAS model being very similar to the 09 implementation in that respect. The 09+RAS model differs from the 09 model mostly in the estimation of the recombination rate, which is found to be lower (Figure 11 for target 1, Figure S3 for other targets). More generally, distinguishing between variation due to recombination and variation due to mutation rate heterogeneity is quite difficult, particularly for small data sets (Husmeier 2005). Target 121 is an example: Convergence is very slow with the RAS model and results in a likelihood similar to the constant rate model, the latter being favored by the BIC. The estimates obtained by the RAS model are also particularly unrealistic on this target.
Figure 10.—

Model comparison on target 1 of the human–chimpanzee–gorilla–orangutan alignment. LRT: likelihood-ratio test. ΔBIC: difference in Bayesian information criterion. The arrows show the direction of the model comparison, pointing toward the alternative model. BIC is the difference in the BIC indexes between the two models. A negative value hence favors the alternative model, whereas a positive value favors the null model. NS: nonsignificant. ***P-value <1e − 16. Δ ln L: logarithm of the likelihood compared to the value of the 09 model. Γ: Gamma-distributed mutation rates.
Figure 11.—

Parameter estimates obtained with different models applied to target 1. The parameter estimate for the gorilla recombination rate with the 09 model is 24 cM/Mb.
For all ape alignments, we compared the 09 and 09+RAS models using the Bayesian information criterion, which takes into account the size of the data sets. The model accounting for mutation rate heterogeneity is favored for the three largest alignments (Table 2). We then applied the previously introduced bias correction. We ran a set of simulations with parameter ranges likely to encompass the estimates of the data: τHC ∈ [3 MY, 7 MY], τHCG ∈ [1 MY, 5MY], θHC ∈ [4e4, 8e4], θHCG ∈ [2e4, 8e4] (see data and methods). The corrected estimates are noted 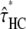 ,
, 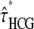 ,
, 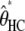 , and
, and 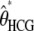 and are shown in Figure 11 for target 1 and summarized in Table 2 for all targets. As expected, the corrected (Table 2, footnote a) estimates lead to a speciation time more ancient for the speciation of human and chimpanzee, which is found to be on average 4.38 MY (95% confidence interval: [3.86; 5.05]), whereas the speciation with the gorilla is found to be 6.69 MY ([6.22; 7.39]). The ancestral population sizes of HC and HCG are found to be ∼57,000 ([45,000; 69,000]) and 35,000 ([30,000; 41,000]), respectively. We find a more ancient date for the speciation of human and chimpanzee and smaller ancestral population sizes for the HC and HCG ancestral species than previously reported. All the estimated values, however, are up to a scaling factor. Speciation times and ancestral population sizes depend on the calibration point used, here the divergence from the Orangutan, taken to be 18 MY. The ancestral population sizes and the recombination rate further depend on the generation time, here assumed to be 25 years. We chose these values to be the same as in Hobolth et al. (2007) for the sake of comparison, but different values can be plugged in. The use of the full genome of gorilla, when available, will also provide better estimates for these quantities.
and are shown in Figure 11 for target 1 and summarized in Table 2 for all targets. As expected, the corrected (Table 2, footnote a) estimates lead to a speciation time more ancient for the speciation of human and chimpanzee, which is found to be on average 4.38 MY (95% confidence interval: [3.86; 5.05]), whereas the speciation with the gorilla is found to be 6.69 MY ([6.22; 7.39]). The ancestral population sizes of HC and HCG are found to be ∼57,000 ([45,000; 69,000]) and 35,000 ([30,000; 41,000]), respectively. We find a more ancient date for the speciation of human and chimpanzee and smaller ancestral population sizes for the HC and HCG ancestral species than previously reported. All the estimated values, however, are up to a scaling factor. Speciation times and ancestral population sizes depend on the calibration point used, here the divergence from the Orangutan, taken to be 18 MY. The ancestral population sizes and the recombination rate further depend on the generation time, here assumed to be 25 years. We chose these values to be the same as in Hobolth et al. (2007) for the sake of comparison, but different values can be plugged in. The use of the full genome of gorilla, when available, will also provide better estimates for these quantities.
TABLE 2.
Estimated values for the speciation times, ancestral population sizes, and recombination rate with various methods
| Model | THC | THC + THCG | NHC | NHCG | r | BIC |
|---|---|---|---|---|---|---|
| Target 1 (1,255,492 sites, rp = 0.9, u = 0.090) | ||||||
| 09 | 3.4 | 6.69 | 108,000 | 34,000 | 0.528 | 4,098,983 |
| 09+RAS | 3.67 | 6.97 | 98,000 | 26,000 | 0.17 | 4,098,680 |
| 09+RASa | 4.45 | 6.87 | 54,000 | 28,000 | 0.559 | |
| Target 106 (257,420 sites, rp = 0.2, u = 0.094) | ||||||
| 09 | 2.93 | 6.81 | 120,000 | 43,000 | 0.28 | 850,185 |
| 09+RAS | 2.76 | 7.11 | 131,000 | 34,000 | 0.143 | 850,071 |
| 09+RASa | 3.8 | 7.02 | 70,000 | 37,000 | 0.414 | |
| Target 121 (230,666 sites, rp = 0.6, u = 0.100) | ||||||
| 09 | 1.97 | 5.95 | 401,000 | 46,000 | 1.064 | 774,799 |
| 09+RAS | 0.65 | 5.99 | 627,000 | 44,000 | 2.394 | 774,802 |
| 09a | 4.56 | 6.51 | 131,000 | 36,000 | 2.942 | |
| Target 122 (92,240 sites, rp = 1.9, u = 0.097) | ||||||
| 09 | 4.08 | 5.91 | 61,000 | 48,000 | 2.336 | 308,087 |
| 09+RAS | 3.76 | 6.15 | 79,000 | 42,000 | 2.366 | 308,090 |
| 09a | 4.46 | 5.92 | 42,000 | 50,000 | 8.008 | |
| Target X (263,100 sites, u = 0.108) | ||||||
| 09 | 3.49 | 6.16 | 40,000 | 47,000 | 2.282 | 877,589 |
| 09+RAS | 4.26 | 5.91 | 20,000 | 51,000 | 1.273 | 877,565 |
| 09+RASa | 4.41 | 5.99 | 21,000 | 51,000 | 4.282 | |
| Average (2,098,918 sites) | ||||||
| 4.38 | 6.69 | 59,000 | 34,000 | |||
BIC: Bayesian information criterion. The value of the best model according to the BIC is in boldface type for each target. Average estimates are taken as the mean of estimates of the best model over all targets, weighted by their respective numbers of sites. For each target, the number of sites used is reported, together with the broad recombination rates in centimorgans per megabase (rp), as estimated from pedigree data, and the mutation rate (u) estimated from this analysis (in percentage of change per million years).
The parameters after correction for the bias.
Perspectives:
The present study introduces a new parameterization of the CoalHMM approach by Hobolth et al. (2007) and adds the recombination rate to quantities that can be directly estimated in ancestral species. It should therefore be possible in genomewide surveys to investigate how far back in time properties of the genetic map are conserved. The broad-scale recombination rate (Kong et al. 2002) is expected to be more conserved than the fine-scale recombination map (Myers et al. 2005). It should be possible to directly correlate recombination rate estimates in the CoalHMM with fine-scale estimates to investigate how far back the impact of present recombination hot spots can be observed. It is trivial to extend the HMM to include spatially variable recombination rate but extensive simulation studies, as presented here, would be necessary to investigate how powerful estimation would be.
The assumption of a single coalescent time within each HMM state is restrictive and the basis for the bias in the estimation of parameters. It would be desirable to allow for a continuous distribution of coalescence times or at least to allow several coalescence times within each state. This would require coalescent calculations of transitions between these substates as a function of the recombination rate and this is presently under investigation. It would allow for inference on changes in population size over time by appropriate posterior decoding. For instance, more than expected coalescence in a given time interval could suggest a restricted population size. It would also allow a more detailed genomewide scanning for anomalous regions of either very recent coalescent times (within the HC1 state) or a rapid change among alternative states. Such regions could represent selective sweeps in the HC ancestor and balancing selection, respectively, particularly if deviating strongly from the estimated recombination rate of the region. Regions of recent introgression might also be identified in this way given proper modeling. Recent work suggests natural selection in the ancestor of human and chimpanzee to be prevalent (McVicker et al. 2009), and a scan of posterior decoding in a genomewide human–chimpanzee–gorilla–orangutan alignment would be an alternative test of this suggestion. This awaits the full sequencing of the gorilla genome.
The genomes of more species will soon be sequenced and the phenomenon of incomplete lineage sorting will occur on many internal branches. Analysis by models in the spirit of the one presented here might answer general questions on the speciation process and its differences in different groups of organisms, e.g., animals vs. plants, that may not be addressable by other means, opening the way for ancestral population genomics.
Acknowledgments
We gratefully acknowledge Asbjørn T. Brask for helping with the data preprocessing and Mathieu Emily and Carsten Wiuf for helpful discussions. This work is funded by European Research Area in Plant Genomics (ERA-PG) ARelatives, Forskningsrådet for Teknologi og Produktion (FTP) research council.
APPENDIX: DETAILED EQUATIONS FOR COMPUTING THE TRANSITION MATRIX OF THE COALHMM
We derive expressions for transition probabilities among hidden states,
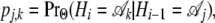 |
as explicit functions of the population parameters Θ. Transition probabilities take the form
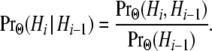 |
(A1) |
Differences between Hi−1 and Hi entail separation of the genealogical histories of the sites by crossing over. We make the large-population assumption that lineages presently residing on the same haplotype have independent histories ancestral to the most recent crossover event between them.
Transition to HC1:
From (6a), site Hi−1 has a genealogy other than HC1 with probability
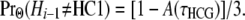 |
We consider separately the segment of a genealogy from the present to the most recent common ancestor (MRCA) of the three species and the remainder,
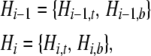 |
in which t denotes the top (more ancient) segment and b the bottom (more recent) segment. In
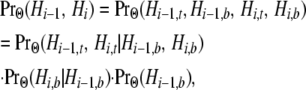 |
(A2) |
Hi−1,b denotes the persistence of all three lineages into the MRCA of the three species and Hi,b the coalescence of the human and chimpanzee lineages in their common ancestral gene pool. Because only two lineages at site i exist in the MRCA of the three species, only one topology (coalescence of the pair) can exist. This observation, together with (6b), implies
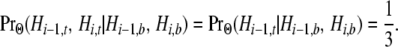 |
From (7), we obtain
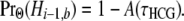 |
Consequently, we need only determine the probability of the transition of the bottom segment of the genealogy:
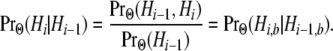 |
That the human and chimpanzee lineages coalesce in the human–chimpanzee ancestor at site i but not at site i − 1 entails a crossover in either the human or the chimpanzee lineage more recently than the common ancestor of all three species and then coalescence between the human and chimpanzee lineages at site i. The cdf of the time to the most recent crossover event in the human lineage is
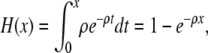 |
with an identical expression for the corresponding quantity for the chimpanzee lineage. The cdf of the waiting time to the most recent crossover event in either lineage is
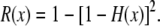 |
For r(t), the probability density function of the time of the most recent crossover event in either the human or the chimpanzee lineage, we obtain an expression for u in the transition probability matrix (5),
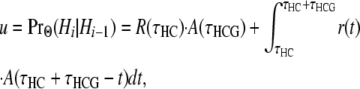 |
(A3) |
in which the first major term on the right denotes a crossover more recently than the human–chimpanzee ancestor and the second major term a crossover in the human–chimpanzee ancestor.
As we have not had to specify the particular nonstandard genealogy at site i − 1, the three transition rates to the HC1 topology occur at this same rate u. This identity implies identity among the three transition rates from the HC1 topology to the nonstandard topologies (s), which we obtain from u using (4).
Transitions among nonstandard topologies:
As the final two transition rates in (5), v1 and v2, represent transitions among nonstandard topologies, they entail the absence of a coalescence event between the human and chimpanzee lineages at both site i − 1 and site i. From (A2), we have
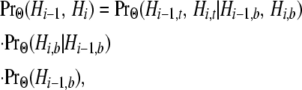 |
in which
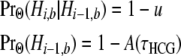 |
[see (7) and (A3)]. To obtain v1 and v2, we require
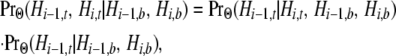 |
in which
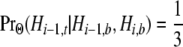 |
for any particular nonstandard topology.
We obtain expressions for v1 and v2 using straightforward arguments similar to those given for (A3). Because the derivations require the consideration of a large number of cases, we describe an algorithmic approach for the generation of all possible joint configurations of the genealogies of site i − 1 and i.
Let type 3 denote a haplotype that carries an ancestral lineage at both site i − 1 and site i, type 2 denote a haplotype that carries an ancestral lineage only at i − 1 and not at i, and type 1 denote a haplotype that carries an ancestral lineage only at i and not at i − 1 (compare Kaplan and Hudson 1985; Griffiths 1991). The initial sample comprises three type 3 haplotypes (one sampled from each of human, chimpanzee, and gorilla), and changes in the relative numbers of the types of haplotypes reflect evolutionary events. For example, coalescence of two type 3 haplotypes simultaneously reduces the number of lineages at both sites, and recombination in a type 3 haplotype reduces the number of type 3 haplotypes by one and generates one type 1 and one type 2 haplotype.
Coalescence or crossing over occurs independently, with an exponentially distributed waiting time. Cumulative distribution functions similar to (7) provide expressions for the probability that these events occur more recently than the common ancestor of all three species. At least one of the events necessarily occurs in that common ancestor.
For example, consider the situation in which four haplotypes occur at the point of divergence between the human–chimpanzee ancestor and the common ancestor of all three species: two type 2 haplotypes (chimpanzee and gorilla) together with one type 1 and one type 2 haplotype representing the human lineage. Evolutionary events that may occur include recombination in the chimpanzee or the gorilla haplotype (each at rate ρ) and coalescence at rate 1/θHCG between any of five pairs of haplotypes (our assumption of independent histories ancestral to any recombination event excludes the possibility of reformation of a type 3 haplotype from the type 1 and type 2 haplotypes). The probability that the most recent event is coalescence between the two type 3 haplotypes is
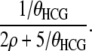 |
(A4) |
Given the configuration of haplotypes at the point of divergence of the human–chimpanzee ancestor from the human–chimpanzee–gorilla ancestor, expressions similar to (A4) give the probabilities of all of the possible next most recent states. We represent the states as nodes in a decision tree, with the probabilities of transitions between states as weights on the branches. All possible routes to the coalescence of all site i − 1 lineages and all site i lineages can easily be enumerated, with the total probability of each terminal state given by the product of the branches connecting it to the state at the speciation point. Expressions for all elements of the transition probability matrix (5), including v1 and v2, are given in the R file provided as File S1.
Supporting information is available online at http://www.genetics.org/cgi/content/full/genetics.109.103010/DC1.
References
- Burgess, R., and Z. Yang, 2008. Estimation of hominoid ancestral population sizes under Bayesian coalescent models incorporating mutation rate variation and sequencing errors. Mol. Biol. Evol. 25: 1979–1994. [DOI] [PubMed] [Google Scholar]
- Chen, F. C., and W. H. Li, 2001. Genomic divergences between humans and other hominoids and the effective population size of the common ancestor of humans and chimpanzees. Am. J. Hum. Genet. 68: 444–456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durbin, R., S. Eddy, A. Krogh and G. Mitchison, 1998. Biological Sequence Analysis: Probabilistic Models of Proteins and Nucleic Acids. Cambridge University Press, Cambridge/London/New York.
- Dutheil, J., and B. Boussau, 2008. Non-homogeneous models of sequence evolution in the Bio++ suite of libraries and programs. BMC Evol. Biol. 8: 255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dutheil, J., S. Gaillard, E. Bazin, S. Glémin, V. Ranwez et al., 2006. Bio++: a set of C++ libraries for sequence analysis, phylogenetics, molecular evolution and population genetics. BMC Bioinformatics 7: 188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Efron, B., and R. J. Tibshirani, 1998. An Introduction to the Bootstrap. Chapman & Hall/CRC, Boca Raton, FL/London/New York/Washington, DC.
- Felsenstein, J., 1981. Evolutionary trees from DNA sequences: a maximum likelihood approach. J. Mol. Evol. 17: 368–376. [DOI] [PubMed] [Google Scholar]
- Goldman, N., J. L. Thorne and D. T. Jones, 1996. Using evolutionary trees in protein secondary structure prediction and other comparative sequence analyses. J. Mol. Biol. 263: 196–208. [DOI] [PubMed] [Google Scholar]
- Griffiths, R. C., 1991. The two-locus ancestral graph, pp. 100–117 in Selected Proceedings of the Sheffield Symposium on Applied Probability, Vol. 18, edited by I. V. Basawa and R. L. Taylor. Institute of Mathematical Statistics, Hayward, CA.
- Hein, J., M. Schierup and C. Wiuf, 2005. Gene Genealogies, Variation and Evolution: A Primer in Coalescent Theory. Oxford University Press, London/New York/Oxford.
- Hobolth, A., O. F. Christensen, T. Mailund and M. H. Schierup, 2007. Genomic relationships and speciation times of human, chimpanzee, and gorilla inferred from a coalescent hidden Markov model. PLoS Genet. 3: e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hudson, R. R., 1983. Testing the constant-rate neutral allele model with protein sequence data. Evolution 37: 203–217. [DOI] [PubMed] [Google Scholar]
- Hudson, R. R., 2002. Generating samples under a Wright-Fisher neutral model of genetic variation. Bioinformatics 18: 337–338. [DOI] [PubMed] [Google Scholar]
- Husmeier, D., 2005. Discriminating between rate heterogeneity and interspecific recombination in DNA sequence alignments with phylogenetic factorial hidden Markov models. Bioinformatics 21(Suppl. 2): ii166–ii172. [DOI] [PubMed] [Google Scholar]
- Kaplan, N. L., and R. R. Hudson, 1985. The use of sample genealogies for studying a selectively neutral m-loci model with recombination. Theor. Popul. Biol. 28: 382–396. [DOI] [PubMed] [Google Scholar]
- Kong, A., D. F. Gudbjartsson, J. Sainz, G. M. Jonsdottir, S. A. Gudjonsson et al., 2002. A high-resolution recombination map of the human genome. Nat. Genet. 31: 241–247. [DOI] [PubMed] [Google Scholar]
- Mailund, T., M. H. Schierup, C. N. S. Pedersen, P. J. M. Mechlenborg, J. N. Madsen et al., 2005. CoaSim: a flexible environment for simulating genetic data under coalescent models. BMC Bioinformatics 6: 252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McVicker, G., D. Gordon, C. Davis and P. Green, 2009. Widespread genomic signatures of natural selection in hominid evolution. PLoS Genet. 5: e1000471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melodelima, C., L. Guéguen, D. Piau and C. Gautier, 2006. A computational prediction of isochores based on hidden Markov models. Gene 385: 41–49. [DOI] [PubMed] [Google Scholar]
- Myers, S., L. Bottolo, C. Freeman, G. Mcvean and P. Donnelly, 2005. A fine-scale map of recombination rates and hotspots across the human genome. Science 310: 321–324. [DOI] [PubMed] [Google Scholar]
- Nichols, R., 2001. Gene trees and species trees are not the same. Trends Ecol. Evol. 16: 358–364. [DOI] [PubMed] [Google Scholar]
- Patterson, N., D. J. Richter, S. Gnerre, E. S. Lander and D. Reich, 2006. Genetic evidence for complex speciation of humans and chimpanzees. Nature 441: 1103–1108. [DOI] [PubMed] [Google Scholar]
- R Development Core Team, 2008. R: A Language and Environment for Statistical Computing. R Foundation for Statistical Computing, Vienna.
- Rannala, B., and Z. Yang, 2003. Bayes estimation of species divergence times and ancestral population sizes using DNA sequences from multiple loci. Genetics 164: 1645–1656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanke, M., and S. Waack, 2003. Gene prediction with a hidden Markov model and a new intron submodel. Bioinformatics 19(Suppl. 2): ii215–ii225. [DOI] [PubMed] [Google Scholar]
- Yang, Z., 1993. Maximum-likelihood estimation of phylogeny from DNA sequences when substitution rates differ over sites. Mol. Biol. Evol. 10: 1396–1401. [DOI] [PubMed] [Google Scholar]
- Yang, Z., 1994. Maximum likelihood phylogenetic estimation from DNA sequences with variable rates over sites: approximate methods. J. Mol. Evol. 39: 306–314. [DOI] [PubMed] [Google Scholar]
- Yang, Z., 1995. A space-time process model for the evolution of DNA sequences. Genetics 139: 993–1005. [DOI] [PMC free article] [PubMed] [Google Scholar]