

Summary
The major histocompatibility complex class I molecules display peptides (pMHC I) on the cell surface for immune surveillance by CD8+ T cells. These peptides are generated by proteolysis of intracellular polypeptides by the proteasome in the cytoplasm and then in the endoplasmic reticulum (ER) by the ER aminopeptidase associated with antigen processing (ERAAP). To define the unknown mechanism of ERAAP function in vivo, we analyzed naturally processed peptides in cells with or without appropriate MHC I and ERAAP. In the absence of MHC I, ERAAP degraded the antigenic precursors in the ER. However, MHC I molecules could bind proteolytic intermediates and were essential for generation of the final peptide by ERAAP. Thus, ERAAP synergizes with MHC I to generate the final pMHC I repertoire.
Introduction
The display of peptides representing endogenous proteins by the major histocompatibility complex class I molecules (MHC I) is essential for immune surveillance (Shastri et al., 2002; Yewdell et al., 2003; Trombetta and Mellman, 2005). Normally, the peptides are derived from self-proteins, but if the cells are infected with viruses or bacteria or harbor mutated genes, novel nonself peptides from these sources are also generated. The presence of these foreign peptides in the mix displayed by the MHC class I molecules allows CD8+ T cells to detect intracellular abnormalities and to cause the elimination of these cells. Effective immune surveillance by the CD8+ T cell repertoire therefore depends upon the antigen-processing pathway to efficiently generate the peptide-MHC I (pMHC I) repertoire.
MHC I molecules are highly polymorphic in every species examined (Klein and Figueroa, 1986). Since the early 1990s it has been known that each MHC I molecule displays a set of peptides that number in the thousands but share a distinct consensus motif defined by length and two or three conserved amino acids. For example, the Kb MHC I displays 8mer peptides with the consensus motif XXXX[Y,F]XX[L,M,V,I] (where X = any amino acid), and the Ld MHC I displays a different set of 9mer peptides with the consensus motif X[P,S]XXXXXX[F,L,M] (Falk et al., 1991; Corr et al., 1992; Rammensee et al., 1997). Yet all the known components of the MHC processing pathway are essentially conserved within and across species (Paulsson, 2004). This raises the intriguing question of how the same antigen-processing machinery can efficiently generate the diverse sets of precisely cut peptides that conform to the distinct consensus motifs of all the different MHC I molecules.
The MHC I antigen-processing pathway begins in the cytoplasm (Townsend et al., 1986). Many antigenic precursors enter the pathway when they are synthesized as defective or cryptic ribosomal products or when they are retrieved from other subcellular compartments into the cytoplasm (Shastri et al., 2002; Yewdell et al., 2003; Trombetta and Mellman, 2005). The notion that the final antigenic peptides are generated in the cytoplasm itself prior to their transport into the ER appears to have lost its initial appeal (Saveanu et al., 2002; Shastri et al., 2002; Rock et al., 2004; Trombetta and Mellman, 2005). For most antigenic precursors, it is now believed that proteolysis in the cytoplasm generates the C terminus of the final antigenic peptide, but its N terminus is generated only after its transport by the transporter associated with antigen processing (TAP) in the ER compartment (Cascio et al., 2001; Kunisawa and Shastri, 2003). The ER enzyme that trims the N-terminally extended peptide precursors to their final length has also been identified as the ER aminopeptidase associated with antigen processing (ERAAP) in mice (Serwold et al., 2002) and as ER aminopeptidase 1 (ERAP1) in humans (Saric et al., 2002). Indeed, inhibition of ERAAP expression with RNA interference or by homologous recombination disrupts the pMHC I repertoire (Serwold et al., 2002; York et al., 2002; Hammer et al., 2006; Yan et al., 2006). In the absence of ERAAP, some peptides remained unchanged while others were either dramatically up- or downregulated. There are thus compelling reasons to believe that trimming N termini from antigenic precursors in the ER is a key step in generating the normal pMHC I repertoire on the cell surface.
The molecular mechanism for ERAAP function, however, remains obscure. Based upon ERAAP’s ability to trim peptides in vitro, it has been argued that ERAAP alone or in concert with another related aminopeptidase, leukocyte-derived arginine aminopeptidase (L-RAP), is sufficient to generate the final peptide (York et al., 2002; Saveanu et al., 2005). In another study that used a panel of synthetic peptides and recombinant ERAP1, it was found that ERAP1 exhibits substrate preferences defined by peptide length and the C-terminal residue (Chang et al., 2005). Thus, it has been proposed that ERAP1 acts as a unique “molecular ruler” to generate the correct 8–10 amino acid peptides that can then bind the available MHC I molecules. On the other hand, analysis of ERAAP-deficient mice also revealed substrate preferences for ERAAP (Hammer et al., 2006; Yan et al., 2006) but did not support the molecular ruler mechanism. In vivo, ERAAP generated octa- and nona-peptides presented by different MHC I, yet inexplicably degraded other similarly sized peptides. These discrepancies make it difficult to extrapolate the results of in vitro studies to the normal function of ERAAP in the ER of living cells. Furthermore, because the in vitro analysis did not include MHC I molecules, it is not known whether ERAAP activity is influenced by MHC I. Thus, the intriguing question of whether MHC I serve an active or a passive role in the generation of the final pMHC I repertoire has remained an enduring mystery for more than 15 years (Elliott et al., 1990; Falk et al., 1990).
Here we use cells derived from ERAAP-deficient mice to analyze the N-terminal trimming of peptides by ERAAP in the presence or absence of relevant MHC I molecules. We find that MHC molecules play a key role in determining whether ERAAP’s enzymatic activity eliminates antigenic precursors or generates the final pMHC I complex. Thus, ERAAP and MHC I synergize to generate the customized peptide repertoires appropriate for each of the polymorphic MHC I molecules.
Results
ERAAP Generates pMHC I from N-Terminally Extended Precursors in the ER
First we established the model system for studying ERAAP function in the MHC class I processing pathway. The antigenic precursors were targeted directly into the ER by appending their coding sequence downstream of an ER translocation signal (ES). The DNA constructs were then transfected into a fibroblast cell line derived from C57BL/6 (H-2b) mice lacking either the transporter associated with antigen processing alone (TAP-deficient) or both ERAAP and TAP (ERAAP-TAP double-deficient) described earlier (Hammer et al., 2006). This strategy allows antigen processing to be confined to the ER itself, because the products generated by normal cytoplasmic proteolysis cannot enter the ER in the absence of TAP. Expression of the pMHC I on the cell surface was measured by coculturing the transfected cells with appropriate lacZ-inducible T cell hybridomas. The SHL8-Kb complex (peptide SHL8 = SIINFEHL in single amino acid code) recognized by the B3Z T cell hybridoma was efficiently generated in TAP-deficient cells when they were transfected with the ER-targeting constructs. The ES-X5[SHL8] precursor, containing five N-terminal flanking residues (AIVMK), or the ES-[SHL8] precursor without any N-terminal flanking residues yielded comparable responses (Figure 1A, left). The ES-[SHL8] precursor yields the final SHL8 peptide after the ES sequence is cleaved off by the signal peptidase in the ER and thus does not require any further trimming. In contrast, the ES-X5[SHL8] construct yields the X5[SHL8] peptide after the ES sequence is removed and thus requires that the five N-terminal flanking residues be trimmed to generate the final SHL8 peptide. The comparable activity of the trimming-dependent ES-X5[SHL8] and the trimming-independent ES-[SHL8] precursors shows that trimming occurred efficiently in the presence of ERAAP in the ER of TAP-deficient cells.
Figure 1. ERAAP Is Essential for Generating pMHC I on the Cell Surface from ER-Targeted N-Terminally Extended Precursors.
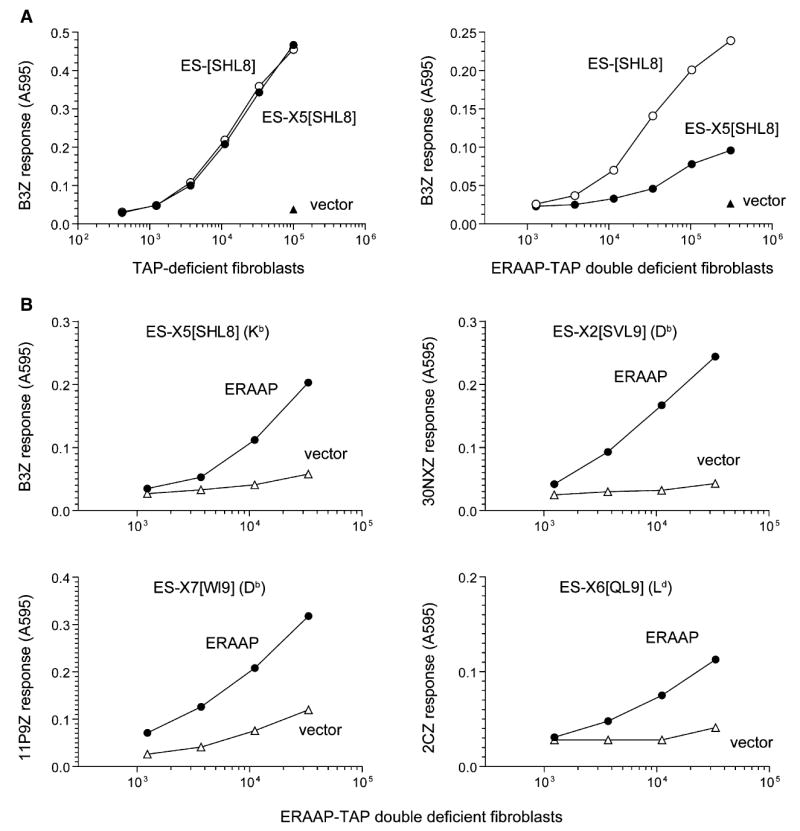
(A) The DNA constructs encoding the vector alone or the SHL8 (SIINFEHL) antigenic peptide downstream of an ER-targeting signal sequence (ES) were transiently transfected into fibroblasts derived from either TAP-deficient or ERAAP-TAP double-deficient mice. The constructs included either no (ES-[SHL8]) or five (ES-X5[SHL8], X5 = AIVMK) additional amino acids between the signal peptidase cleavage site of the ES-signal sequence and the SHL8 peptide. 2 days later, indicated number of transfected cells were incubated overnight with the lacZ-inducible, SHL8-Kb-specific B3Z T cell hybridoma. The lacZ activity was measured by the conversion of the substrate chlorophenolred-β-D-galacto-pyrannoside into chlorophenol red, which absorbs light at 595 nm.
(B) ER-targeted, N-terminally extended versions of the antigenic peptides SHL8, SVL9 (SSVVGVWYL), WI9 (WMHHNMDLI), or QL9 (QLSPFPFDL) were cotransfected with either vector alone or mouse ERAAP into ERAAP-TAP double-deficient fibroblasts. The cDNA encoding Ld was also included with the ES-X6[QL9] construct. 2 days later, varying number of transfected cells were used as APCs for the indicated T cells, and their lacZ response was measured as in (A). The MHC molecule presenting the peptides is shown in parentheses. Data are representative of at least three independent experiments.
When fibroblasts lacking both ERAAP and TAP were transfected with the same constructs, cells expressing the trimming-dependent ES-X5[SHL8] precursor were deficient in their ability to generate the B3Z stimulating SHL8-Kb complex (Figure 1A, right). However, the same cells expressing the trimming-independent ES-[SHL8] precursor did generate the SHL8-Kb complex, demonstrating that the absence of ERAAP did not affect other aspects of MHC loading and transport to the cell surface.
To directly establish that ERAAP expression was required for generating the final peptide from its N-terminally extended precursors, we cotransfected antigenic constructs into ERAAP-TAP double-deficient cells together with either vector alone or the cDNA encoding mouse ERAAP. Reconstitution of ERAAP expression in the ERAAP-TAP double-deficient cells dramatically enhanced the presentation of the B3Z-stimulating SHL8-Kb complex from the ES-X5[SHL8] construct. Likewise, the presentation of the final pMHC I (SVL9-Db, WI9-Db, and QL9-Ld) from three other N-terminally extended precursors was also markedly improved in the presence of ERAAP (Figure 1B). Thus, ERAAP was required for trimming the N-terminal flanking residues from the final peptides presented by Kb, Db, or Ld MHC I molecules. The source of the low T cell-stimulating activity of transfected cells in the absence of ERAAP is presently unclear. It is possible that a small amount of the final peptide might have been generated because of occasional inaccuracy with which the signal peptidase cleaved the ES sequence. Alternatively, an ERAAP-independent pathway might have generated the final peptides. Regardless of the reason for the background activity, the presence of ERAAP reproducibly and markedly enhanced the presentation of all the ER-targeted precursors tested here.
Generation of the Final pMHC I Requires ERAAP Localization in the ER
To determine whether the trimming of antigenic precursors required ERAAP activity in the ER compartment itself, we tested mutant ERAAP molecules that could not enter the ER or were functionally inactive (Figure 2A). We first deleted the predicted N-terminal ER translocation signal of ERAAP by generating a cDNA construct lacking nucleotides encoding residues 2–20 (ERAAPΔES). Unlike expression of wild-type ERAAP (ERAAP WT), which clearly allowed the efficient presentation of the N-terminally extended ES-X9[SHL8] precursor in ERAAP-TAP double-deficient cells, the presentation activity of cells expressing the ERAAPΔES was comparable to those expressing vector alone (Figure 2B). To confirm that the deletion of the ERAAP signal sequence did result in its exclusion from the ER, we used immunofluorescent labeling to detect ERAAP. The ERAAP polyclonal antibodies readily reacted with ERAAP in transfected cells but did not crossreact with the endogenous monkey ERAAP presumably present in vector-transfected cells (Figure 2C). Furthermore, the punctate staining pattern in ERAAP WT transfected cells suggested that it was localized in the ER. The ER location of ERAAP was further established when ERAAP was found to colocalize with other ER-resident proteins containing the KDEL motif detected with the KDEL antibody (Figure 2D). In contrast, cells expressing the ERAAPΔES mutant showed a distinct staining pattern consistent with its localization to the cytoplasm and it clearly did not colocalize with the ER proteins containing the KDEL motif (Figure 2D). To further biochemically confirm these differences in intracellular locations, we tested the sensitivity of ERAAP WT and ERAAPΔES to endoglycosidase H (Endo H). Immunoblot analysis of Endo H-treated lysates showed a decrease in the molecular weight of ERAAP WT protein, reflecting glycosylation consistent with ER localization (Figure 2E). In contrast, the lysate of ERAAPΔES-transfected cells contained ERAAP of a lower molecular weight than ERAAP WT and was not decreased by treatment with Endo H, indicating cytoplasmic localization where N-linked glycosylation does not occur (Figure 2A). Although these results do not address what role, if any, ERAAP could play in the cytoplasm, they provide direct evidence that the putative ES signal is functional. It allows ERAAP’s entry into the ER wherein ERAAP is glycosylated and generates the final SHL8 peptide from its N-terminally extended precursor.
Figure 2. The Generation of pMHC I from N-Terminally Extended Precursors Requires ERAAP Expression in the ER Compartment.
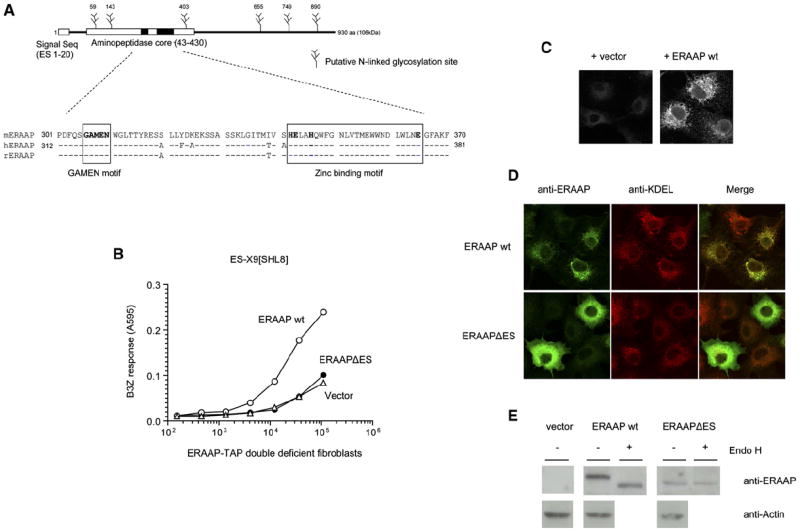
(A) Schematic of mouse ERAAP containing the putative ER translocation signal (ES), the aminopeptidase core, and potential N-linked glycosylation sites. The sequence alignment of the aminopeptidase core with the human and rat orthologs is shown with the conserved GAMEN and zinc binding motifs (boxed).
(B) Deletion of the ES sequence eliminates ERAAP function. The wild-type ERAAP (ERAAP WT), its ES deletion mutant (ERAAPΔES), or vector alone were cotransfected with the ER-targeted antigenic precursor ES-X9[SHL8]. Surface expression of SHL8-Kb complex was measured with B3Z T cells.
(C) Polyclonal rabbit anti-mouse ERAAP antibodies specifically stain ERAAP-transfected COS cells. The COS cells were transfected with either vector or ERAAP cDNA and stained with the primary and secondary antibodies 2 days later as described in the Experimental Procedures.
(D) Deletion of ES signal alters the intracellular location of ERAAP from the ER to the cytoplasm. COS cells transfected with either ERAAP WT or ERAAPΔES were stained with the ERAAP (green) or the ER marker KDEL (red) antibody. The merged image shows an overlay of green and red stains as yellow.
(E) ERAAP without the ES signal remains unglycosylated. The extracts of COS cells transfected with ERAAP WT or ERAAPΔES were either untreated or treated with Endo H. The samples were separated by SDS-PAGE, transferred to the nitrocellulose membranes, and immunoblotted with the ERAAP antibody. Data are representative of at least two independent experiments.
Generation of the Final pMHCI Requires ERAAP Activity
Next we assessed whether the enzymatic activity of ERAAP was required to generate the final peptide. Previous studies had identified two key sequence motifs in aminopeptidases similar to ERAAP (Figure 2A): the GAMEN (aa 306–310 in mouse and aa 317–321 in human) and the HExxHx18E motifs that are involved in binding the substrate and the metal atoms that serve as cofactors for enzymatic activity. We substituted the nucleotides GAA for GCC in human ERAAP, thus changing the glutamic acid (E) at residue 320 to an alanine (A). To test its function, the E320A ERAAP mutant was cotransfected with the ES-X5[SHL8] precursor into ERAAP-TAP double-deficient cells (Figure 3A). Like its mouse counterpart, the wild-type human ERAAP (hERAAP WT) was fully capable of restoring the impaired antigen presentation function of transfected cells, but the activity of the E320A mutant was similar to that of vector alone. Both the hERAAP WT and the E320A mutants were expressed at similar amounts as judged by immunoblot analysis of total cell lysates (Figure 3B). The results show that ERAAP function was dependent upon the presence of an intact GAMEN motif and thus on its enzymatic activity.
Figure 3. The Generation of pMHC I from N-Terminally Extended Precursors in the ER Requires Enzymatically Active ERAAP.
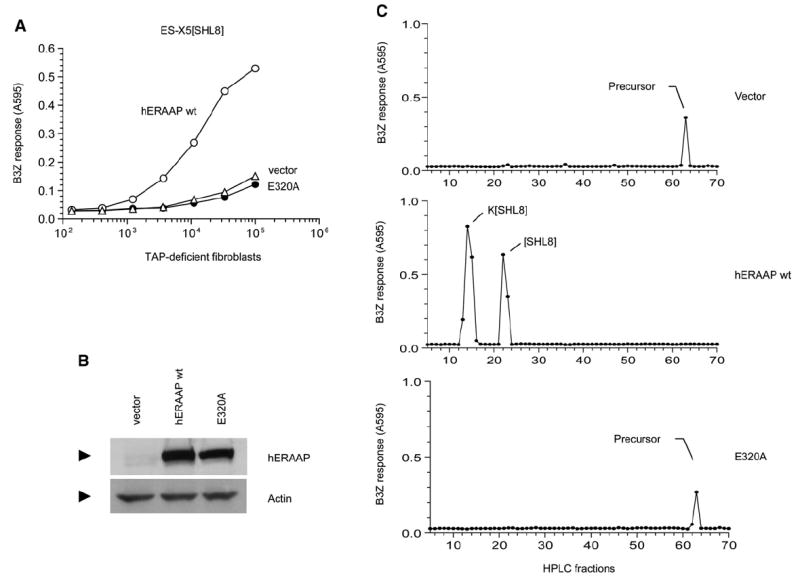
(A) The cDNAs encoding either vector alone, human ERAAP WT, or its single amino acid mutant in the GAMEN motif (E320A) were cotransfected into ERAAP-TAP double-deficient fibroblasts together with the ES-X5[SHL8] construct. After 2 days, expression of SHL8-Kb complex was measured with B3Z T cells.
(B) The hERAAP WT and its E320A mutant are expressed at comparable amounts. The amounts of hERAAP WT, its E320A mutant, or vector alone as a negative control were determined by immunoblotting transfected COS cell lysates with the hERAAP antibody.
(C) The conversion of the N-terminally extended precursor to the final SHL8 or K[SHL8] peptides presented by Kb or Db MHC occurs only in the presence of hERAAP WT. The peptide extracts from ERAAP-TAP double-deficient fibroblasts transfected with ES-X5[SHL8] and either vector alone, hERAAP WT, or its E320A mutant were fractionated by RP-HPLC. The SHL8-containing, N-terminally extended intermediates were detected after trypsin treatment of each fraction and incubating them with B3Z T cells in the presence of the appropriate APC. The peaks were identified, where known, by comparison with synthetic peptides run under identical conditions. Data are representative of at least two independent experiments.
To directly assess the fate of the antigenic precursors in the presence or absence of ERAAP in the transfected cells, we analyzed cell extracts by HPLC fractionation (Figure 3C). To allow the detection of N-terminally extended precursors, each fraction was treated with trypsin, which cleaves the carboxyl terminus of the lysine (K) residue flanking the N terminus of the SHL8 peptide. This allows the release of the optimally active SHL8 octapeptide from its precursors and allows their detection despite their poor antigenicity in the in vitro peptide assay (Paz et al., 1999). In ERAAP-TAP double-deficient cells transfected with the ES-X5[SHL8] precursor and vector alone, a single peak of activity was detected in fraction 63 representing the ER precursor of SHL8 peptide. By contrast, in cells cotransfected with the hERAAP WT, this late eluting activity was not detected and instead two new peptide peaks were found that coeluted with the Kb and Db binding SHL8 octapeptide and K[SHL8] nonapeptide, respectively (Malarkannan et al., 1995). Notably, the HPLC profile of extracts from cells cotransfected with the E320A mutant was identical to that of cells transfected with vector alone. This analysis directly demonstrates (1) that it is possible to detect precursor peptides in the ER, (2) that human ERAAP is functionally equivalent to mouse ERAAP, and (3) that ERAAP’s enzymatic activity is necessary for generating the final MHC-bound peptides from the N-terminally extended precursor.
ERAAP Degrades ER Peptides but Generates the Final Peptide in the Presence of Appropriate MHC I
To define the potential relationship between the MHC molecules and ERAAP function, we used the QL9-Ld-specific T cell hybridoma 2CZ because the ERAAP-TAP double-deficient cells, derived from the B6 (H2b) background, do not express the Ld MHC molecule. This makes it possible to analyze the fate of antigenic precursors in the presence or absence of ERAAP and Ld MHC. The ERAAP-TAP double-deficient cells generated the QL9-Ld complex from the ES-X6[QL9] precursor only when they were cotransfected with ERAAP and the Ld MHC (Figure 4A). In the absence of ERAAP or in its presence with the irrelevant Kd MHC molecule, the cells were unable to serve as APCs for the 2CZ T cells. Thus, both ERAAP and Ld MHC were essential for generating the QL9-Ld complex from its ER-targeted ES-X6[QL9] precursor.
Figure 4. The Fate of N-Terminally Extended X6[QL9] and the Final QL9 Peptide Is Determined by ERAAP and the Ld MHC Molecule.
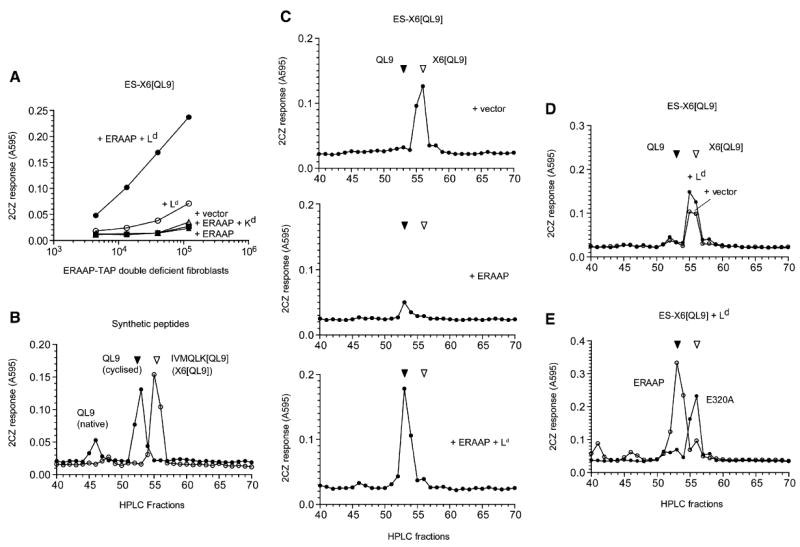
(A) Generation of the QL9-Ld complex in ERAAP-TAP double-deficient fibroblasts from the ER-targeted ES-X6[QL9] precursor requires ERAAP and the Ld MHC. The ERAAP-TAP double-deficient fibroblasts were cotransfected with the ES-X6[QL9] precursor and other cDNAs shown. Expression of the QL9-Ld complex was detected with lacZ-inducible 2CZ T cell hybridoma as in Figure 1.
(B) Synthetic IVMQLK[QL9] and the QL9 peptides can be resolved by HPLC and detected by 2CZ T cells. 10 fmoles of indicated peptides were injected into the HPLC. Each fraction was treated with trypsin and assayed with 2CZ T cells and Ld-L cells as APCs.
(C) ERAAP generates the final QL9 peptide from the X6[QL9] precursor in the presence of Ld but degrades the precursor peptide in the absence of Ld. The ERAAP-TAP double-deficient fibroblasts were cotransfected with the ES-X6[QL9] precursor construct and either vector alone, ERAAP alone, or ERAAP plus Ld. The peptides were extracted from the transfected cells and fractionated by HPLC. Each fraction was treated with trypsin and used to stimulate 2CZ T cells in the presence of Ld-L cells as APCs.
(D) The absence of ERAAP cannot be compensated by other proteases. The ERAAP-TAP double-deficient fibroblasts were transfected with the ES-X6[QL9] construct with either vector alone or Ld, and cell extracts were analyzed for 2CZ activation after HPLC fractionation and trypsin treatment of each fraction.
(E) Generation of the final QL9 peptide from its X6[QL9] precursor in the presence of Ld requires enzymatically active ERAAP. The ERAAP-TAP double-deficient fibroblasts were cotransfected with the ES-X6[QL9] precursor, Ld, and either hERAAP WT or its enzymatically inactive mutant E320A. The cell extracts were analyzed for 2CZ activation after HPLC fractionation and trypsin treatment. Data are representative of at least three (C) or two independent experiments.
To determine whether ERAAP trimmed peptides in the ER in the presence or absence of the cognate Ld MHC, we analyzed naturally processed peptides extracted from cells expressing the ES-IVMQLK[QL9] (ES-X6[QL9]) precursor. This precursor, similar to the ES-X5[SHL8] precursor (Figure 3), also contains a N-terminal flanking lysine residue that allows the release of optimally active QL9 peptide after trypsin treatment of the HPLC fractions. In acid pH, the N-terminal glutamine (Q) residue cyclizes to a pyroglutaminyl residue and thus migrates slower than the native QL9 peptide (Figure 4B; Kageyama et al., 2001). The cyclized QL9 is nevertheless a potent stimulator of 2CZ T cells and can be clearly distinguished from its 15mer X6[QL9] precursor after HPLC fractionation.
In trypsin-treated HPLC fractions of the extract from vector and ES-X6[QL9]-transfected ERAAP-TAP double-deficient cells, we found a single peak of antigenic activity that coeluted with the X6[QL9] peptide (Figure 4C, top). Clearly, the cells generated the X6[QL9] antigenic precursor and it was readily detected in the cell extracts. Remarkably, when the same cells were cotransfected with the ES-X6[QL9] construct and ERAAP, the amount of X6[QL9] precursor was dramatically reduced (Figure 4C, middle). Notably, the loss of X6[QL9] precursor was not accompanied by comparable amount of its QL9 product (Figure 4C, middle versus top). To determine whether the trimming activity of ERAAP could be influenced by MHC I, we analyzed extracts of cells cotransfected with the antigenic construct ERAAP and the Ld MHC. In contrast to cells expressing ERAAP without Ld, approximately 6-fold higher amount of QL9 peptide was now found in cells expressing ERAAP and Ld (Figure 4C, bottom). Thus, ERAAP was capable of trimming the X6[QL9] precursor, but instead of efficiently converting it to the final QL9 peptide, the precursor was degraded to a length that was no longer recognized by the 2CZ T cells. However, in the presence of the ERAAP and the appropriate Ld, the QL9 peptide was efficiently generated from its X6[QL9] precursor. We conclude that the appropriate MHC I protected the antigenic precursor from degradation by ERAAP and facilitated the generation of the final peptide.
To determine whether ERAAP was uniquely required to generate QL9 from its X6[QL9] precursor, we also analyzed extracts of cells transfected with the ES-X6[QL9] construct with or without Ld (Figure 4D). In either case, only the X6[QL9] precursor was detected in the cells, indicating that Ld by itself was insufficient to generate the QL9 peptide and that other proteases did not compensate for the absence of ERAAP. Finally, we cotransfected cells with the E320A active site mutant of ERAAP with the antigenic construct and Ld (Figure 4E). Again, the final QL9 peptide was predominant in extracts of cells expressing Ld and wild-type ERAAP, but only the X6[QL9] precursor was predominant in cells expressing Ld and the inactive E320A mutant. We conclude that the generation of the final QL9 peptide requires both the relevant Ld MHC molecule as well as enzymatically active ERAAP.
Recombinant ERAAP Degrades Synthetic Peptides In Vitro
To establish that ERAAP could directly trim antigenic precursor and the final peptides, we used recombinant ERAAP (rERAAP) purified from baculovirus-infected insect cells. Like the material purified from mouse microsomes in a previous study (Serwold et al., 2002), rERAAP also efficiently cleaved its synthetic Leucine p nitroanilide (LpNA) substrate, and this activity was completely inhibited by the aminopeptidase inhibitor leucinethiol (Figure 5A). We incubated synthetic QL9 and X6[QL9] peptides with rERAAP and analyzed the peptide products for their ability to stimulate the 2CZ T cells. After 90 min incubation at 37°C, ERAAP trimmed the QL9 peptides into shorter products that were no longer recognized by the 2CZ T hybridoma (Figure 5B). Likewise, the X6[QL9] precursor peptide was also trimmed into peptides shorter than QL9 because longer peptides, if present, are detectable after trypsin treatment. Although it is not possible to rule out undetectable contaminants, in our hands both the precursor and final peptides were ERAAP substrates and ERAAP trimmed them beyond the nine QL9 residues.
Figure 5. Recombinant ERAAP Trims the Final QL9 as well as Its X6[QL9] Precursor In Vitro, and Ld MHC Can Protect QL9 Peptide.
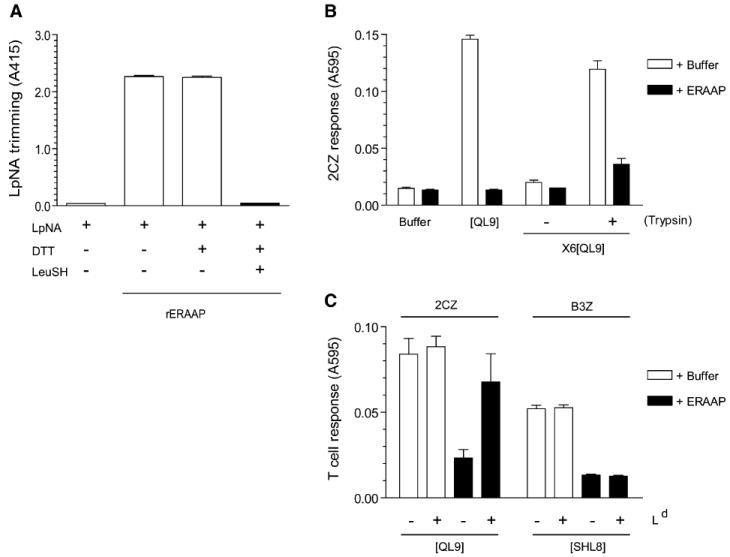
(A) Purified recombinant ERAAP (rERAAP) is active in trimming the model LpNA substrate, and leucinethiol inhibits this activity. The rERAAP purified from baculovirus-infected insect cells was incubated with Leucine pnitroanilide (LpNA) with or without leucinethiol (LeuSH) and DTT, which is required to keep LeuSH in its reduced and active form. The LpNA hydrolysis was measured as light absorbance at 415 nm.
(B) The indicated synthetic peptides were incubated with either buffer alone or with rERAAP. The peptides were assayed by their ability to stimulate 2CZ T cells in the presence of Ld-L cells as APCs. To detect the generation of QL9 peptide from its N-terminally extended X6[QL9] precursor, the peptides were either assayed as such or after trypsin treatment.
(C) The Ld MHC protects QL9 but not SHL8 peptide from degradation by rERAAP. Synthetic QL9 or SHL8 peptides were incubated with buffer or rERAAP in the presence or absence of recombinant Ld-Ig fusion protein. The QL9 and SHL8 peptides were assayed with 2CZ or B3Z T cells and Ld or Kb-L cells as APCs, respectively. The graph shows the mean values with SD from three independent experiments (A–C).
Next we tested the influence of Ld MHC in this in vitro peptide-trimming reaction. Inclusion of recombinant Ld in the reaction mixture increased the recovery of antigenic QL9 peptide (Figure 5C). The ability of Ld to protect the QL9 peptide was specific because inclusion of Ld did not prevent the degradation of another final SHL8 peptide by ERAAP. Likewise, recombinant Kb protected only the SHL8 peptide (see Figure S1A in the Supplemental Data available online). In other time course experiments, the inclusion of Ld did not, however, enhance the rate at which QL9 was generated from its precursor, suggesting that at least in vitro Ld does not bind and protect N-terminally extended peptides (Figure S1B). We conclude that ERAAP can degrade the precursor as well as the final peptide and that the presence of the appropriate MHC I molecules slows the degradation of the final peptide.
Model for Detecting MHC I-Bound Antigenic Precursors
The profound influence of the MHC I on the recovery of the final peptide suggested that the N-terminally extended peptide precursors might be trimmed while they were bound to the MHC I molecule. Indeed, in an in vitro peptide-trimming model with purified microsomes, we had shown earlier that N-terminally extended peptides could bind MHC I as potential intermediates for the generation of the final pMHC I (Brouwenstijn et al., 2001). However, other studies that have examined peptide trimming in microsomes have either supported a role for MHC I (Komlosh et al., 2001) or have argued against it (Fruci et al., 2001). Nevertheless, whether MHC I molecules interact with antigenic precursors in living cells remains unknown. This is the case possibly because such interactions, even if they occur, are likely to be transient and would make the complexes too rare to be detected in conventional assays.
To detect the putative complexes containing N-terminally extended intermediates bound to MHC molecules in living cells, we made several modifications to our model system. First, we altered the antigenic precursor to make its products resistant to ERAAP. We took advantage of the fact that peptides with the structure “X-P-Xn” (P = proline and X = any amino acid) are inefficiently trimmed by ERAAP in the ER (Serwold et al., 2001, 2002). A proline residue was inserted within the N-terminal residues flanking QL9 in its ER-targeted precursor ES-X3-EPK[QL9]. We reasoned that the presence of the proline residue within the N-terminal flanking residues would prevent ERAAP from trimming beyond the penultimate glutamic acid (E) residue, resulting in the 12mer peptide EPK[QL9].
We transfected the QL9 precursor constructs in ERAAP-TAP double-deficient cells. As before, cells expressing the ES-X6[QL9] construct as well as the Ld MHC and ERAAP generated the 2CZ-stimulating QL9-Ld complex on their surface (Figure 6A). In contrast, the same cells failed to generate the QL9-Ld complex from the ES-X3-EPK[QL9] precursor despite the presence of both Ld and ERAAP. Thus, the presence of the proline residue effectively inhibited the cells’ ability to generate the final QL9 peptide.
Figure 6. Model for Detecting MHC I-Bound Antigentic Precursors.
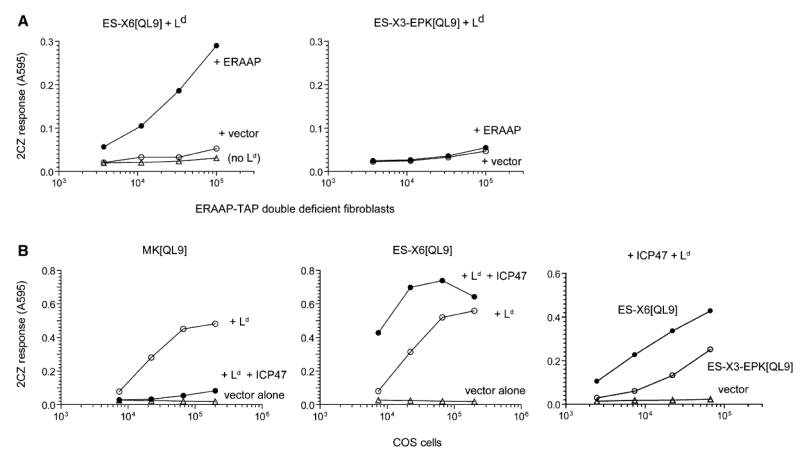
(A) The presence of a proline residue among the N-terminal flanking residues inhibits ERAAP’s ability to generate the final QL9-Ld complex. The ERAAP-TAP double-deficient fibroblasts or COS cells were cotransfected with ER-targeted, N-terminally extended QL9 precursors without (ES-X6[QL9]) or with (ES-X3-EPK[QL9]), a proline residue (P), the Ld MHC, and either ERAAP or vector alone. The presence of the QL9-Ld complex on the cell surface was measured with 2CZ T cells.
(B) The TAP inhibitor ICP47 blocks presentation of cytoplasmic but not ER-targeted QL9 precursors in COS cells. Simian COS cells were cotransfected with constructs encoding cytoplasmic (MK[QL9]) or ER-targeted (ES-X6[QL9], ES-X3-EPK[QL9]) precursors, and the indicated cDNAs encoding Ld, ICP47, or vector alone. The presence of the QL9-Ld complex on the cell surface was measured with 2CZ T cells. Data are representative of at least two independent experiments. The similar results were confirmed by using either ES-X3-LPK[QL9] or -QPK[QL9] instead of -EPK[QL9] (data not shown).
Second, to ensure high expression of the antigenic precursor and the Ld MHC, we used monkey COS cells as APCs. The COS cells express high amounts of endogenous MHC and can be efficiently transfected with mouse MHC and antigenic precursors to serve as APCs (Karttunen et al., 1992). Furthermore, as in human cells, the monkey TAP in COS cells can be inhibited with the TAP inhibitor ICP47 (Hill et al., 1995; Serwold et al., 2001). Indeed, when cells were cotransfected with Ld and the construct expressing MK[QL9], a cytoplasmic QL9 precursor, the presentation of QL9-Ld complex was strongly inhibited in the presence of ICP47 (Figure 6B). In contrast, when COS cells were cotransfected with the ER-targeted ES-X6[QL9] construct and ICP47, the presentation of QL9-Ld complex was enhanced. The enhanced presentation is likely due to reduced competition from the pool of Ld binding precursors that are normally transported by TAP. As noted above, the presentation activity of the precursor with a proline residue in the N-terminal flanking position was lower than one without a proline residue. Thus, as in TAP-deficient fibroblasts, expression of ICP47 in COS cells allows processing of ER-targeted antigenic precursors.
N-Terminally Extended Proteolytic Intermediate Is Bound by Ld MHC I
We cotransfected COS cells with the proline precursor ES-X3-EPK[QL9] together with Ld and ICP47. Like the other N-terminally extended peptides used above, the ES-X3-EPK[QL9] peptide also contains a lysine (K) residue flanking the final QL9 peptide. As seen by the dramatic increase in antigenicity of the synthetic EPK[QL9] peptide after trypsin treatment (Figure 7A), all putative N-terminally extended QL9 intermediates should be detectable after trypsin treatment of the HPLC fractions. Indeed, after the cell extract was HPLC fractionated and each fraction treated with trypsin and assayed for activating 2CZ T cells, we detected three major peaks containing the QL9 peptide (Figure 7B). The first and highest peak coeluted with synthetic EPK[QL9] peptide and the third activity peak with the final QL9 peptide. The identity of the middle peak is presently unknown. The presence of the final QL9 peptide is consistant with the low presentation activity of this precursor observed in direct presentation assay (Figure 6B). Most importantly, the predicted ERAAP product EPK[QL9] was abundant in the cells.
Figure 7. The N-Terminally Extended EPK[QL9] Proteolytic Intermediate Is Bound by the Ld MHC Molecules.
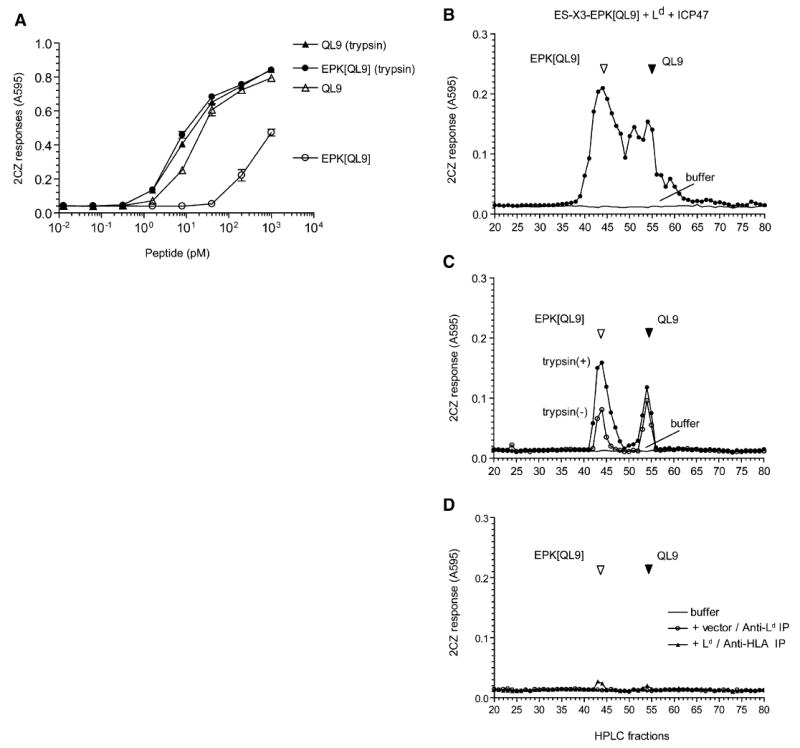
(A) Trypsin treatment of N-terminally extended QL9 precursor EPK[QL9] enhances its antigenicity. Varying concentrations of synthetic QL9 and its N-terminally extended analog EPK[QL9] peptide were tested for their ability to stimulate 2CZ T cells with Ld-L cells as APC. The peptides were tested as such or after preincubation with trypsin.
(B) The final antigenic QL9 peptide, its N-terminally extended precursors and intermediates present in the total peptide extract of COS cells transfected with ES-X3-EPK[QL9], Ld, and ICP47. The extract was fractionated by HPLC and antigenic peptides detected in the fractions by their ability to stimulate 2CZ response after trypsin treatment. HPLC fractions collected after injection of buffer alone were tested in parallel to rule out carryover between samples.
(C) The Ld MHC was immunoprecipitated from detergent lysate of COS cells transfected with the ES-X3-EPK[QL9] construct, Ld, and ICP47 cDNAs. The peptides contained in the immunoprecipitated material were fractionated by HPLC and assayed with 2CZ T cells with (closed circle) or without (open circle) trypsin treatment.
(D) As controls, the same cell lysate was immunoprecipitated with the pan HLA (W6/32) antibody (triangle), or the lysate from cells without Ld transfection was immunoprecipitated with the Ld antibody (circle). The peptide content of the immunoprecipitated material was analyzed as above. Data are representative of at least three independent experiments.
To determine whether any of these peptides were associated with the Ld MHC, we immunoprecipitated the endogenous monkey MHC or Ld MHC and analyzed their peptide content after HPLC fractionation. The material immunoprecipitated by the Ld monoclonal antibody (28.14.8S), specific for its α3 domain, from cells expressing the antigenic precursor, Ld, and ICP47 contained two distinct antigenic peaks that coeluted with the EPK[QL9] and QL9 peptides, respectively (Figure 7C). Trypsin treatment of the HPLC fractions enhanced 2CZ response to the EPK[QL9] peptide, further confirming that this material in the Ld immunoprecipitate contained additional N-terminal residues. In contrast, antigenic activity was barely detected in the material immunoprecipitated from the same cells with the HLA monoclonal antibody W6/32, which binds to the monkey MHC I molecules (Figure 7D). Likewise, the material immunoprecipitated with the same Ld antibody 28.14.8S from cells expressing the antigenic precursor and ICP47 but no Ld MHC did not contain detectable antigenic activity. Similar results were obtained when we analyzed cells in which peptides were first stripped from the cell surface before extraction, indicating that the antigenic material was largely intracellular (Figure S2). We conclude that the EPK[QL9] is the major product generated after proteolysis of the ES-X3-EPK[QL9] precursor in the ER and that it is specifically bound to the Ld MHC molecule.
Discussion
The mechanism of ERAAP function and whether MHC I play any role in generating the final antigenic peptides are controversial issues. Rammensee and colleagues first showed that the presence of appropriate MHC molecules was essential for detecting the final antigenic peptides in cell extracts (Falk et al., 1990). They suggested that MHC I molecules influenced then-unknown proteases or that MHC I could even serve as templates for directing protease activity. However, an alternative hypothesis suggested that the requirement for the MHC to detect antigenic peptides was instead due to their protective role toward independently generated peptides (Elliott et al., 1990). It was argued that antigenic peptides were extremely labile, and therefore impossible to detect unless the MHC I molecules bound and protected them from certain degradation. Resolving these opposing “template” versus “protection” models has remained a challenge.
The analysis of proteolytic events in the MHC I antigen-processing pathway is fraught with technical difficulties. First and foremost is the problem that antigenic peptides are generated in vanishing amounts in the APCs. This is of course not an issue when the final pMHC I is detected on the cell surface by CD8+ T cells because they have evolved to recognize rare pMHC I (Purbhoo et al., 2004). Second, the proteolytic intermediates, before they are converted to their final form, are also rare and even harder to detect by conventional assays (Paz et al., 1999; Kunisawa and Shastri, 2003). Third, the pMHC I are generated at a very rapid rate in the APCs or microsomes in vitro, making it difficult to capture intermediates (Brouwenstijn et al., 2001; Fruci et al., 2001). Finally, until recently, the molecular mechanism of antigen processing in the ER, the site where MHC I are loaded with peptides, was unknown and therefore difficult to manipulate.
The discovery of the ERAAP aminopeptidase as the catalyst for converting antigenic precursors into their final peptide products presented by the MHC I established the importance of ER proteolysis in the MHC I antigen-processing pathway (Serwold et al., 2002; York et al., 2002). When ERAAP-deficient mice also showed disruptions in their pMHC I repertoire, they provided further confirmation of this view as well as new tools for studying antigen processing in the ER (Hammer et al., 2006; Yan et al., 2006). To our knowledge, no mammalian cell line is known to lack ERAAP, and therefore the availability of cells from mice deficient in ERAAP and/or TAP are key to the analysis of proteolysis that occurs exclusively in the ER.
All four ER-targeted, N-terminally extended antigenic precursors required ERAAP expression for generation of pMHC I on the cell surface. Furthermore, generation of pMHC I required the presence of an enzymatically active form of ERAAP in the ER compartment itself and thus provided the model system in which normal ERAAP function could be studied in intact cells. To track the proteolytic intermediates, we used ER-targeted antigenic precursors with a lysine residue flanking the N terminus of the final peptides presented by the appropriate MHC molecule (Paz et al., 1999). The presence of this lysine residue allowed us to directly detect all possible N-terminally extended precursors or intermediates in HPLC-fractionated cell extracts because the optimally active final peptides are released by trypsin.
The fate of the X6[QL9] precursor and the generation of the final QL9 peptide was profoundly influenced by the presence of ERAAP and Ld in the cells. The X6[QL9] precursor was readily detected in transfected cells lacking ERAAP with or without the Ld molecule. In contrast, when the cells expressed both ERAAP and Ld, the X6[QL9] precursor was undetectable and only the final QL9 peptide was detected. This shows the remarkable efficiency with which ERAAP trimmed the X6[QL9] precursor to its final QL9 product. Moreover, when ERAAP was expressed in the absence of Ld, the amount of X6[QL9] precursor or any of its N-terminally trimmed products was dramatically reduced. An in vitro analysis of peptide trimming with recombinant ERAAP also showed that ERAAP was capable of trimming the X6[QL9], QL9, or SHL8 peptides beyond the minimal length required for pMHC I expression. Note that in contrast to other in vitro studies that used micromolar amounts, we used picomolar amounts of peptide substrates, and most importantly we analyzed antigen processing in living cells that included the appropriate MHC molecules (Saric et al., 2002; York et al., 2002; Chang et al., 2005; Saveanu et al., 2005). Thus, both in vitro and in living cells, ERAAP did not stop trimming the 15 amino acid X6[QL9] precursor when it reached a length of 9 amino acids, but trimmed it to a length no longer detected by the T cells. Our analysis therefore does not support the “molecular ruler” model for ERAAP function (Chang et al., 2005).
That antigenic precursors for the SHL8 or the QL9 peptides existed in the ER also demonstrates that these precursors are not impossible to detect, as proposed by the “protection” model (Elliott et al., 1990). Rather, the difficulty in detecting these peptides can now be attributed to their poor antigenicity in conventional assays, as well as their rapid degradation by ERAAP in the absence of the relevant MHC I. Paradoxically, this view suggests that MHC I molecules do serve an important “protection” function, but instead it is for the precursors of antigenic peptides by saving them from total degradation by ERAAP. As a corollary, ERAAP can also be seen as serving a “janitorial” function of keeping the ER free of worthless peptides that arrive from the cytoplasm but have no future in the absence of appropriate MHC I molecules.
Our results confirmed the key prediction of the template model that MHC molecules bind N-terminally extended intermediates (Falk et al., 1990; Brouwenstijn et al., 2001; Komlosh et al., 2001). We took advantage of the inability of ERAAP to trim the penultimate amino-terminal residue preceding a proline residue. As expected, the presence of a proline prevented ERAAP from trimming the precursor, and the presentation of the QL9-Ld complex was inhibited on the cell surface. Yet, when Ld MHC was immunoprecipitated with an antibody specific for its α3 domain, it was found in specific association with the predicted 12mer EPK[QL9] peptide. Given that most reagents (such as antibodies and T cells) that detect pMHC I are strongly biased toward recognizing the final pMHC I, it is perhaps not surprising that such complexes have hitherto gone undetected. From this role of the MHC I molecule in binding longer antigenic precursors, we predict that ERAAP will eventually be found to interact, if only transiently, with the peptide-loading complex in the ER.
Taken together, our findings strongly support the original template model in which MHC I molecules themselves define the peptides (Falk et al., 1990; Brouwenstijn et al., 2001). Such a role for the MHC I molecules is attractive because it can also resolve the efficiency conundrum inherent in the antigen-processing pathway: namely, how can the conserved antigen-processing machinery independently generate the vast collections of exact peptides to satisfy the binding preferences of highly polymorphic MHC I molecules? This is no longer a challenge because the antigen-processing machinery is not responsible for producing an enormously wasteful collection of all conceivable peptides that can be presented by different MHC I (Rammensee et al., 1997). In an elegant solution, the MHC I and ERAAP can together produce only those peptides that are suitable for the MHC I molecules that exist in that APC.
In conclusion, we show here that ERAAP can trim and eliminate antigenic precursors in the ER. When appropriate MHC I are present, they can bind the N-terminally extended antigenic intermediates and protect them from elimination. Concomitantly, this precursor-bound MHC I complex could serve as a template for ERAAP to allow removal of only the extra N-terminal flanking residues. Thus, rather than being mutually exclusive, the “protection” and “template” models come together to generate the final pMHC I repertoire.
Experimental Procedures
Cell Lines and T Cell Activation Assay
β-galactosidase (lacZ)-inducible T cell hybridomas B3Z (SHL8-Kb), 11p9Z (WI9-Db), 30NXZ (SVL9-Db), 2CZ (QL9-Ld), and L-cells expressing Kb, Db, or Ld have been described (Sanderson and Shastri, 1994; Shastri et al., 1998; Serwold et al., 2001). The generation of tail or embryonic fibroblast cell lines from mice (wild-type, ERAAP-deficient, and ERAAP-TAP double-deficient) has been described (Hammer et al., 2006). The T cells were incubated with the APCs overnight and produced lacZ in response to the cognate pMHC I expressed by the transfected cells or by APCs incubated with exogenous synthetic or extracted peptides. LacZ activity was measured by the conversion of the substrate chlorophenolred-β-D-galacto-pyrannoside as absorbance at 595 nm (Sanderson and Shastri, 1994).
DNA Constructs and Transfection
The ES-[SHL8], ES-X5[SHL8], ES-X9[SHL8], ES-X7[WI9], ES-X2[SVL9], ES-X6[QL9], and ES-X3-EPK[QL9] DNA constructs all encode the ER targeting signal sequence followed by N-terminally extended (X) antigenic peptides and have been described or were constructed similarly (Serwold et al., 2001; Hammer et al., 2006). The human ERAAP cDNA was isolated by PCR from HeLa cell mRNA and was subcloned into pcDNA I vector. The ERAAPΔES is a mouse ERAAP mutant lacking amino acids 2–20, wherein the first Met residue is followed by the 21 Gln residue. ERAAPΔES insert was amplified by PCR. The E320A is a single amino acid substitute of human ERAAP made by site-directed mutagenesis (Glu320 to Ala320). For transient transfection, the DNA constructs were introduced into host cells by FuGENE6 (Roche) according to the manufacturer’s instruction and assayed after 2 days.
Immunohistochemistry and Immunoblots
Expression of ERAAP and its mutants in transfected COS cells was analyzed as described earlier (Serwold et al., 2002)
Peptides, Peptide Extracts, and HPLC Analysis
All synthetic peptides were prepared by D. King (University of California at Berkeley) by solid phase synthesis and their purity confirmed by mass spectrometry. The preparation of peptide extracts and their analysis after HPLC fractionation has been described (Paz et al., 1999). For immunoprecipitation, the 1% NP40 lysates of COS cells transfected with indicated DNA constructs were coincubated with the Ld monoclonal antibody (28.14.8S) or the pan HLA monoclonal antibody (W6/32) followed by binding to protein A Sepharose 4B (Zymed). After washing, the peptides were eluted by 10% formic acid, fractionated by HPLC, and assayed as described above with or without trypsin treatment.
In Vitro Peptide Trimming by Recombinant ERAAP
Recombinant mouse ERAAP (rERAAP) was prepared by Bac-to-Bac Baculovirus Expression System (Invitrogen) according to the manufacturer’s instruction. Recombinant ERAAP was purified by first ammonium sulfate precipitation followed by ion-exchange HPLC and was tested for hydrolysis of the chromogenic substrate Leucine p-nitroanilide (LPNA) in the presence or absence of the inhibitor leucinethiol (Sigma) as described (Serwold et al., 2002). The purity of the rERAAP was further confirmed by silver staining and immunoblotting with the ERAAP polyclonal antibody (data not shown). For in vitro peptide trimming assay, synthetic peptides (30 pM) were incubated with 1.7 μg of rERAAP in 50 μl of 50 mM Tris (pH 7.6) for 90 min or 60 min at 37°C in Figures 5B and 5C, respectively. Reaction was stopped by adding acetic acid and samples were dried by vacuum centrifugation. To detect the N-terminally extended peptides, the samples were treated with 50 μg/ml trypsin, and lac Z response of appropriate T cells was measured as above. In some experiments, soluble recombinant Ld-Ig fusion protein (3 nM, BD Pharmingen) was incubated with peptides for 3 hr at 37°C before the trimming assay.
Supplementary Material
Acknowledgments
We thank D. King for peptide synthesis and T. Serwold and D. Cado for generating the 2CZ hybridoma and embryonic fibroblasts, respectively. Supported by grants from the NIH to N.S. and The International Human Frontier Science Program Organization to N.B.
Footnotes
The authors declare that they have no conflicts of interest.
Supplemental Data Two Supplemental Figures can be found with this article online at http://www.immunity.com/cgi/content/full/25/5/795/DC1/.
References
- Brouwenstijn N, Serwold T, Shastri N. MHC class I molecules can direct proteolytic cleavage of antigenic precursors in the endoplasmic reticulum. Immunity. 2001;15:95–104. doi: 10.1016/s1074-7613(01)00174-1. [DOI] [PubMed] [Google Scholar]
- Cascio P, Hilton C, Kisselev AF, Rock KL, Goldberg AL. 26S proteasomes and immunoproteasomes produce mainly N-extended versions of an antigenic peptide. EMBO J. 2001;20:2357–2366. doi: 10.1093/emboj/20.10.2357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang SC, Momburg F, Bhutani N, Goldberg AL. The ER aminopeptidase, ERAP1, trims precursors to lengths of MHC class I peptides by a “molecular ruler” mechanism. Proc Natl Acad Sci USA. 2005;102:17107–17112. doi: 10.1073/pnas.0500721102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corr M, Boyd LF, Frankel SR, Kozlowski S, Padlan EA, Margulies DH. Endogenous peptides of a soluble major histocompatibility complex class I molecule, H-2Lds: sequence motif, quantitative binding, and molecular modeling of the complex. J Exp Med. 1992;176:1681–1692. doi: 10.1084/jem.176.6.1681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elliott T, Townsend A, Cerundolo V. Antigen presentation: naturally processed peptides. Nature. 1990;348:195–197. doi: 10.1038/348195a0. [DOI] [PubMed] [Google Scholar]
- Falk K, Rötzschke O, Rammensee HG. Cellular peptide composition governed by major histocompatibility complex class I molecules. Nature. 1990;348:248–251. doi: 10.1038/348248a0. [DOI] [PubMed] [Google Scholar]
- Falk K, Rotzschke O, Stevanovic S, Jung G, Rammensee H-G. Allele-specific motifs revealed by sequencing of self-peptides eluted from MHC molecules. Nature. 1991;351:290–296. doi: 10.1038/351290a0. [DOI] [PubMed] [Google Scholar]
- Fruci D, Niedermann G, Butler RH, van Endert PM. Efficient MHC class I-independent amino-terminal trimming of epitope precursor peptides in the endoplasmic reticulum. Immunity. 2001;15:467–476. doi: 10.1016/s1074-7613(01)00203-5. [DOI] [PubMed] [Google Scholar]
- Hammer GE, Gonzalez F, Champsaur M, Cado D, Shastri N. The aminopeptidase ERAAP shapes the peptide repertoire displayed by major histocompatibility complex class I molecules. Nat Immunol. 2006;7:103–112. doi: 10.1038/ni1286. [DOI] [PubMed] [Google Scholar]
- Hill A, Jugovic P, York I, Russ G, Bennink J, Yewdell J, Ploegh H, Johnson D. Herpes simplex virus turns off the TAP to evade host immunity. Nature. 1995;375:411–415. doi: 10.1038/375411a0. [DOI] [PubMed] [Google Scholar]
- Kageyama S, Tsomides TJ, Fukusen N, Papayannopoulos IA, Eisen HN, Sykulev Y. Potent cytolytic response by a CD8+ CTL clone to multiple peptides from the same protein in association with an allogeneic class I MHC molecule. J Immunol. 2001;166:3028–3034. doi: 10.4049/jimmunol.166.5.3028. [DOI] [PubMed] [Google Scholar]
- Karttunen J, Sanderson S, Shastri N. Detection of rare antigen presenting cells by the lacZ T-cell activation assay suggests an expression cloning strategy for T-cell antigens. Proc Natl Acad Sci USA. 1992;89:6020–6024. doi: 10.1073/pnas.89.13.6020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein J, Figueroa F. The evolution of class I MHC genes. Immunol Today. 1986;7:41–44. doi: 10.1016/0167-5699(86)90123-4. [DOI] [PubMed] [Google Scholar]
- Komlosh A, Momburg F, Weinschenk T, Emmerich N, Schild H, Nadav E, Shaked I, Reiss Y. A role for a novel luminal endoplasmic reticulum aminopeptidase in final trimming of 26 S proteasome-generated major histocompatability complex class I antigenic peptides. J Biol Chem. 2001;276:30050–30056. doi: 10.1074/jbc.M103177200. [DOI] [PubMed] [Google Scholar]
- Kunisawa J, Shastri N. The group II chaperonin TRiC protects proteolytic intermediates from degradation in the MHC class I antigen processing pathway. Mol Cell. 2003;12:565–576. doi: 10.1016/j.molcel.2003.08.009. [DOI] [PubMed] [Google Scholar]
- Malarkannan S, Goth S, Buchholz DR, Shastri N. The role of MHC class I molecules in the generation of endogenous peptide/MHC complexes. J Immunol. 1995;154:585–598. [PubMed] [Google Scholar]
- Paulsson KM. Evolutionary and functional perspectives of the major histocompatibility complex class I antigen-processing machinery. Cell Mol Life Sci. 2004;61:2446–2460. doi: 10.1007/s00018-004-4113-0. [DOI] [PubMed] [Google Scholar]
- Paz P, Brouwenstijn N, Perry R, Shastri N. Discrete proteolytic intermediates in the MHC class I antigen processing pathway and MHC I-dependent peptide trimming in the ER. Immunity. 1999;11:241–251. doi: 10.1016/s1074-7613(00)80099-0. [DOI] [PubMed] [Google Scholar]
- Purbhoo MA, Irvine DJ, Huppa JB, Davis MM. T cell killing does not require the formation of a stable mature immunological synapse. Nat Immunol. 2004;5:524–530. doi: 10.1038/ni1058. [DOI] [PubMed] [Google Scholar]
- Rammensee HG, Bachmann J, Stevanovic S. MHC Ligands and Peptide Motifs. Austin, TX: Landes Bioscience; 1997. [Google Scholar]
- Rock KL, York IA, Goldberg AL. Post-proteasomal antigen processing for major histocompatibility complex class I presentation. Nat Immunol. 2004;5:670–677. doi: 10.1038/ni1089. [DOI] [PubMed] [Google Scholar]
- Sanderson S, Shastri N. LacZ inducible peptide/MHC specific T-hybrids. Int Immunol. 1994;6:369–376. doi: 10.1093/intimm/6.3.369. [DOI] [PubMed] [Google Scholar]
- Saric T, Chang SC, Hattori A, York IA, Markant S, Rock KL, Tsujimoto M, Goldberg AL. An IFN-g-induced aminopeptidase in the ER, ERAP1, trims precursors to MHC class I-presented peptides. Nat Immunol. 2002;3:1169–1176. doi: 10.1038/ni859. [DOI] [PubMed] [Google Scholar]
- Saveanu L, Fruci D, van Endert P. Beyond the proteasome: trimming, degradation and generation of MHC class I ligands by auxiliary proteases. Mol Immunol. 2002;39:203–215. doi: 10.1016/s0161-5890(02)00102-5. [DOI] [PubMed] [Google Scholar]
- Saveanu L, Carroll O, Lindo V, Del Val M, Lopez D, Lepelletier Y, Greer F, Schomburg L, Fruci D, Niedermann G, van Endert PM. Concerted peptide trimming by human ERAP1 and ERAP2 aminopeptidase complexes in the endoplasmic reticulum. Nat Immunol. 2005;6:689–697. doi: 10.1038/ni1208. [DOI] [PubMed] [Google Scholar]
- Serwold T, Gaw S, Shastri N. ER aminopeptidases generate a unique pool of peptides for MHC class I molecules. Nat Immunol. 2001;2:644–651. doi: 10.1038/89800. [DOI] [PubMed] [Google Scholar]
- Serwold T, Gonzalez F, Kim J, Jacob R, Shastri N. ERAAP customizes peptides for MHC class I molecules in the endoplasmic reticulum. Nature. 2002;419:480–483. doi: 10.1038/nature01074. [DOI] [PubMed] [Google Scholar]
- Shastri N, Serwold T, Paz P. Reading within the lines: naturally processed peptides displayed by MHC class I molecules. Curr Opin Immunol. 1998;10:137–144. doi: 10.1016/s0952-7915(98)80241-0. [DOI] [PubMed] [Google Scholar]
- Shastri N, Schwab S, Serwold T. Producing nature’s gene-chips. The generation of peptides for display by MHC class I molecules. Annu Rev Immunol. 2002;20:463–493. doi: 10.1146/annurev.immunol.20.100301.064819. [DOI] [PubMed] [Google Scholar]
- Townsend ARM, Bastin J, Gould K, Brownlee GG. Cytotoxic T lymphocytes recognize influenza haemagglutinin that lacks a signal sequence. Nature. 1986;324:575–577. doi: 10.1038/324575a0. [DOI] [PubMed] [Google Scholar]
- Trombetta ES, Mellman I. Cell biology of antigen processing in vitro and in vivo. Annu Rev Immunol. 2005;23:975–1028. doi: 10.1146/annurev.immunol.22.012703.104538. [DOI] [PubMed] [Google Scholar]
- Yan J, Parekh VV, Mendez-Fernandez Y, Olivares-Villagomez D, Dragovic S, Hill T, Roopenian DC, Joyce S, Van Kaer L. In vivo role of ER-associated peptidase activity in tailoring peptides for presentation by MHC class Ia and class Ib molecules. J Exp Med. 2006;203:647–659. doi: 10.1084/jem.20052271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yewdell JW, Reits E, Neefjes J. Making sense of mass destruction: quantitating MHC class I antigen presentation. Nat Rev Immunol. 2003;3:952–961. doi: 10.1038/nri1250. [DOI] [PubMed] [Google Scholar]
- York IA, Chang SC, Saric T, Keys JA, Favreau JM, Goldberg AL, Rock KL. The ER aminopeptidase ERAP1 enhances or limits antigen presentation by trimming epitopes to 8-9 residues. Nat Immunol. 2002;3:1177–1184. doi: 10.1038/ni860. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.