

Abstract
This review provides detailed procedures for the crystallization of membrane proteins via the lipidic cubic phase method. Bacteriorhodopsin-specific, hands-on protocols are given for (i) the preparation of bacteriorhodopsin from purple membrane by monomerization in octylglucoside and gel filtration chromatography or by selective extraction after pre-treatment with dodecyl-tri-methylammonium bromide, (ii) the incorporation of bacteriorhodopsin into lipidic cubic phases by mixing in vials or within coupled syringes and, (iii) the crystallization of bacteriorhodopsin in the lipidic matrix by adding a solid salt or an overlaying with a solution. References for further useful procedures and materials are listed in order to provide biochemists and crystallographers with all information that is necessary to grow crystals of the membrane protein bacteriorhodopsin.
1. Introduction
It is the purpose of this review to familiarize the experimental scientist with the practicalities involved in the use of lipidic cubic phases for membrane protein crystallization purposes. The detailed protocols given here enable the reader to readily generate crystals of the membrane protein bacteriorhodopsin. After mastering the crystallization of bacteriorhodopsin, further resources may be consulted, e.g., those listed in Table 1. It lists references that describe useful procedures and lipidic cubic phase related crystallization reports which may be instrumental in designing crystallization screens, handling of non-colored proteins, or the retrieval of crystals for X-ray diffraction purposes. It is hoped that the simple protocols in this review serve as a basis for benchmark crystallization experiments that embolden the crystallographer to experiment with more difficult proteins, in particular those that have not yielded crystals employing conventional crystallization methods.
Table 1.
Useful methods and procedures have been developed to aid in various aspects of the lipidic cubic phase crystallization methodology
| Topic | Description | Reference |
|---|---|---|
| Lipidic cubic phase based crystallization | Pioneering bacteriorhodopsin and lysozyme crystallization report, glass vials are employed | [9,10] |
| Monoolein as matrix for crystallization of five different membrane proteins | [3] | |
| Review of the vial-based technique | [21] | |
| Micro crystallization | Syringe and manual rachet dispenser are employed, 200 nl volume; use of liquid instead of solid salt as crystallization inducing agent | [15] |
| Reproduction of above procedure | [22] | |
| Refinement of above procedure and further reduction of volume | [5] | |
| Observation of crystallization setups by microscopy | Observation options and guide to interpretation of crystallization results | [16] |
| Demonstration that Hoffman modulation contrast and polarization microscopy are useful for the detection of small, colorless protein crystals | [17] | |
| Crystal harvesting | Detergent mediated release | [12] |
| Enzymatic release | [13] | |
| Overview with protocols similar to the ones given here | [18] | |
| Cubic phase forming lipids | Lists of lipids and lipid mixes that support bacteriorhodopsin crystallization are given in Appendix A of the review | [11] |
| Crystallization in absence of any detergent | [14] | |
| Screening conditions | A list with compatible salts and solutions is given | [18] |
| The destabilization effect of a set of crystallization solutions is investigated | [4] | |
| Boundaries on protein size and crystallization conditions are estimated from a crystallization model | [7] | |
| Temperature-dependent phase behaviour of monoolein-detergent mixes | [24] | |
| Lyo- and thermotropic monoolein phase diagram | [1] |
The track record of the lipidic cubic phase crystallization method is summarized in Table 2. It has been particularly successful for membrane proteins with seven transmembrane α-helices (bacteriorhodopsin, halorhodopsin, sensory rhodopsin II, and sensory rhodopsin II with its transducer domain). It is therefore believed that heptahelical membrane proteins that are of non-bacterial origin, namely G-protein coupled receptors (GPCR), may be appropriate targets for this crystallization method. After its inception and description in 1996 [9], the crystallization of bacteriorhodopsin has been repeated in many laboratories (Table 2). The reproduction of well-diffracting bacteriorhodopsin crystals, the application of the method to other membrane proteins, and the continuing development of lipidic cubic phase-based crystallization technology demonstrate the value and future promise of this approach. Numerous original research reports and reviews have been published on the topic of lipidic cubic phase-based protein crystallization. For an introduction to this subject, consult Table 1.
Table 2.
Track record of the lipidic cubic phase-assisted protein crystallization method
| Protein | Resolution (Å) | PDB accession code or reference |
|---|---|---|
| Bacteriorhodopsin | 3.7 | [9] |
| Bacteriorhodopsin | 2.35 | 1AP9 |
| Bacteriorhodopsin | 2.3 | 1BRX |
| Bacteriorhodopsin | 1.55 | 1C3W |
| Bacteriorhodopsin D96M | 2.0 | 1C8S |
| Bacteriorhodopsin, K-state | 2.5 | 1DZE |
| Bacteriorhodopsin, K-state | 2.6 | 1IXF |
| Bacteriorhodopsin, L-state | 2.1 | 1EOP |
| Bacteriorhodopsin, L-state | 2.3 | 1R3P |
| Bacteriorhodopsin E204Q | 1.8 | 1F4Z |
| Bacteriorhodopsin E204Q | 1.7 | 1F50 |
| Bacteriorhodopsin | 2.3 | 1IW6 |
| Bacteriorhodopsin D85S/F219L | 2.0 | 1JV6 |
| Bacteriorhodopsin early M state | 2.0 | 1KG8 |
| Bacteriorhodopsin D85S O state | 2.25 | 1JV7 |
| Bacteriorhodopsin mock early M | 1.81 | 1KG9 |
| Bacteriorhodopsin | 1.65 | 1KGB |
| Bacteriorhodopsin, K-state | 1.43 | 1MOK |
| Bacteriorhodopsin | 1.47 | 1MOL |
| Bacteriorhodopsin M-1 | 1.43 | 1MOM |
| Bacteriorhodopsin | 1.9 | 1QHJ |
| Bacteriorhodopsin early state | 2.1 | 1QKO |
| Bacteriorhodopsin early state | 2.1 | 1QKP |
| Sensory rhodopsin II | 2.4 | 1JGJ |
| Sensory rhodopsin II K state | 2.27 | 1GU8 |
| Sensory rhodopsin II K state | 2.27 | 1GUE |
| Sensory rhodopsin II | 2.1 | 1H68 |
| Sensory rhodopsin II | 2.4 | 1JGJ |
| SRII with transducer complex | 1.93 | 1H2S |
| Halorhodopsin | 1.8 | 1E12 |
| Halorhodopsin | 3.2 | [3] |
| Photosynthetic reaction centre Rhodopseudomonas viridis | 3.7 | [3] |
| Photosynthetic reaction centre Rhodobacter Sphaeroides | 2.35 | 1OGV |
| Photosynthetic reaction centre Rhodobacter Sphaeroides | ca. 6 | [3] |
| Light harvesting complex 2, Rp. acidophila | ca. 25 | [3] |
| Nicotinic acetylcholine receptor | [19] |
2. Protocols: crystallization of bacteriorhodopsin in practice
In general, lipidic cubic phase-based crystallizations are often carried out as a two-step process. At first the protein solution is mixed with the lipid, and a lipidic cubic phase forms spontaneously. This first step is considered complete when the material is transparent, non-birefringent, and very viscous. Then the second step, crystallization, is started by the addition of a crystallant. The latter may be a solid salt mix or a solution. Depending on the precise conditions, crystals typically appear within days or weeks 1.
Bacteriorhodopsin is the major protein component in ‘purple membrane,’ a membrane fraction obtained from Halobacterium salinarum. Purple membrane preparations may be obtained from cultivated H. salinarum strain ET1001 or strain S9 [20]. Alternatively, they are available from several commercial sources, i.e., as lyophilized powders from Sigma (Product No. B0184) Table 3. A total of 10 mg of purple membrane should be sufficient for initial crystallization experiments. Purple membrane is fairly stable when exposed to daylight and room temperature. In contrast, detergent solubilized bacteriorhodopsin should be kept cold and in the dark whenever possible.
Table 3.
Materials that are used in the protocols for the preparation of bacteriorhodopsin from purple membrane and for the crystallization of bacteriorhodopsin in a monoolein lipidic cubic phase
| Purpose | Item | Order information |
|---|---|---|
| Microcrystallization | 250 μl gas-tight syringe | Model 1725, Hamilton, Reno, NV, USA |
| 10 μl microsyringe | Model 701-26s, Hamilton, Reno, NV, USA | |
| Semi-automatic ratchet dispenser | Model PB600, Hamilton, Reno, NV, USA | |
| Terasaki plate | Nunc, Rochester, NY, USA | |
| Clear transparent tape | Crystal Clear, Manco, USA | |
| Cubic phase forming lipid | Monoolein, 1-monooleoyl-rac glycerol | NuCheck, MN, USA or Sigma–Aldrich, USA |
| Source of bacteriorhodopsin | Lyophilized purple membrane from S9 Halobacterium salinarum | Product No. B0184, Sigma–Aldrich, USA |
| Detergent to solubilize bacteriorhodopsin | β-Octylglucoside, n-octyl-β-d-glucopyranoside | No. O311 Anatrace, OH, USA |
| Cationic detergent for purple membrane pretreatment | DTAB, dodecyl-trimethylammonium bromide | Aldrich Chem. Metuchen, NJ, USA |
| Filtration device | Centriprep YM-50 | Millipore, Billerica, MA, USA |
2.1. Bacteriorhodopsin preparation from purple membrane
2.1.1. Avoiding the detergent solubilization step
Bacteriorhodopsin crystals may be grown directly from purple membrane which is mixed with monoolein, without any pretreatment [14]. Though conceptually simple, the timeframe for this crystallization strategy, many months, is prohibitively long to be considered as a benchmark experiment.
2.1.2. Monomerization in β-octylglucoside and gel filtration chromatography
Purple membrane may be solubilized directly with β-octylglucoside. To obtain monomeric bacteriorhodopsin, a gel filtration chromatography step is required [6].
Resuspend 10 mg lyophilized purple membrane (Sigma–Aldrich, B0184) in 10 ml of 150 mM KCl and collect the purple membrane by centrifugation (30 min, 18,000 rpm JA-20, 4 °C).
Resuspend pellet in 6 ml of 25 mM NaH2PO4, pH 6.9, and add 2 ml of 10% (w/v) β-octylglucoside.
Sonicate in bath sonicator for 1 min in the dark at room temperature and incubate over night without stirring.
Adjust pH to 5.5 with 0.1 N HCl.
Centrifuge for 45 min at 15 °C at 55,000 rpm (Ti 70 rotor) and discard the pellet.
Concentrate with Centriprep YM-50 filtration device to less than 2 ml.
Apply concentrate on a BioGel A 0.5 m column or a comparable preparative gel filtration column (e.g., TSK G3000SW, Tosoh Bioscience, Montgomeryville, PA, USA,) with 1.2% β-octylglucoside in 25 mM NaH2PO4, pH 5.5, as the stationary phase. Discard the first and collect the second peak.
Concentrate the pooled bacteriorhodopsin fraction with a Centriprep YM-50 filtration device. Adjust to a final concentration of 9 mg/ml in 25 mM NaH2PO4, pH 5.5.
2.1.3. Selective extraction after DTAB pretreatment
Alternatively, bacteriorhodpsin may be extracted from purple membranes which have been pretreated with dodecyl-trimethylammonium bromide (DTAB), a cationic detergent that is frequently used in detergent solubilization experiments [8].
Resuspend 10 mg lyophilized purple membrane (Sigma–Aldrich, B0184) in 10 ml of 150 mM KCl and collect the purple membrane by centrifugation (30 min, 18,000 rpm JA-20, 4 °C).
Resuspend pellet in 10 ml of 10 mM DTAB, 150 mM Na/K acetate. Tip-sonicate for a total of 1 min, chill on ice between 20 s pulses.
Discard supernatant after centrifugation (1 h, 18,000 rpm JA-20, 4 °C) and resuspend purple pellet in 12 ml of 1.2% (w/v) β-octylglucoside, 25 mM NaH2PO4, pH 6.9.
Sonicate again as above and adjust pH to 5.5 with 0.1 N HCl.
Clear by centrifugation (1 h, 18,000 rpm JA-20, 4 °C) and discard pellet. The supernatant contains monomerized, β-octylglucoside solubilized bacteriorhodopsin.
Concentrate bacteriorhodopsin solution with a Centriprep YM-50 filtration device (Millipore, Billerica, MA, USA). Adjust the fnal concentration to 9 mg/ml in 25 mM NaH2PO4, pH 5.6.
The concentration of bacteriorhodopsin is usually determined spectrophotometrically. The extinction coefficient of β-octylglucoside solubilized bacteriorhodopsin at 560 nm is ca. 63,000 cm2/mmol (MWbacteriorhodopsin 26.8 kDa). An absorbance ratio A280/A560 of 1.5–2.0 is acceptable. The concentrations of β-octylglucoside and those of residual purple membrane lipids in bacteriorhodopsin preparations are substantial. They depend on the type of pretreatment and they typically vary from batch to batch. This variation may be the source of slightly different outcomes of crystallization experiments with respect to crystal growth kinetics, crystal size, and X-ray diffraction quality.
2.2. Crystallization setup: mix protein and lipid, add crystallant
In the first step of the crystallization experiment, the bacteriorhodopsin solution is mixed with the lipid to form a lipidic cubic phase (ca. 3.5 mg/ml bacteriorhodopsin, ca. 60% monoolein). This procedure may be carried out in a glass vial (Fig. 1) or in two coupled syringes [15] as shown in Fig. 2. Protocols are given below for both options. For best results the crystallization experiments should be carried out under dim light at room temperature. The crystallization materials should be kept in total darkness after preparation.
Fig. 1.

Growth of bacteriorhodopsin crystals within a monoolein-based lipidic cubic phase matrix in a glass vial. (A) The 10 μl crystallization experiment was set up in a glass vial as lined out in 2.2.1.1. At first the purple bacteriorhodopsin is distributed homogeneously in the lipidic cubic phase. The bottom of the 3 mm diameter tube is filled with solid Sørensen salt. (B) Bacteriorhodopsin microcrystals formed within 1 month. (C) Schematic close-up of purple bacteriorhodopsin crystals with sizes up to ca. 100 μm along their longest dimension.
Fig. 2.
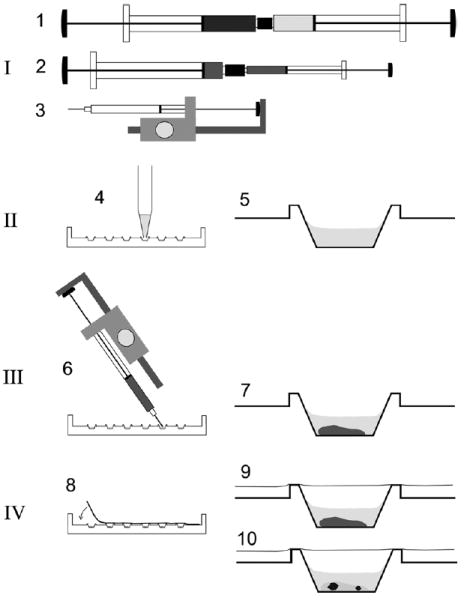
Schematic procedure for the rapid preparation of micro crystallization experiments using the cubic phase method. Crystallization setups are prepared using syringes in combination with a semi-automatic dispenser-driven micro syringe and Terasaki plates. The procedure involves four basic steps: [I] Preparation of the proper lipidic cubic phase using two 250 μl gas-tight syringes coupled together as described [2] (1). Typically, the cubic phase consists of ca. 60% monoolein, ca. 40% water and protein—i.e., bacteriorhodopsin—at a concentration of ca. 3.5 mg/ml in 20 mM Sørensen phosphate buffer, pH 5.6. For dispensing into microwells, the material is transferred into a microsyringe with 10 μl total volume either directly into the syringe barrel or via an appropriate adapter (2). This microsyringe is assembled into a semi-automatic dispenser (3) which allows dispensation in 50 steps of ca. 0.2 μl each. [II] Prior to dispensation of the lipid material, six microwells in one lane of a 72-well Terasaki plate are filled with 1 μl of the precipitant solutions (4) and dried optionally at 60 °C for 30 min. The latter resembles crystallization of bacteriorhodopsin using dry salt as the precipitation agent as described previously [9,11,23]. A single well is shown on the right (5). [III] Ca. 0.2 μl of the preformed lipidic material is added to each microwell by positioning the shortened syringe needle at an angle over the well bottom and by triggering the semi-automatic dispenser (6). The average weight of a single shot of 60% (w/v) monoolein containing cubic phase using the dispenser was determined to be 222 ± 10.3 μg. The cubic phase is stable in an excess of an overlaying liquid (7). [IV] The microwells are sealed with a clear transparent tape (8) and the plates may be stored (9) at different temperatures. Inspection of crystals (10) with a microscope can be carried out without disturbance from both sides simply by flipping the entire plate.
2.2.1. Step 1: Incorporation of bacteriorhodopsin into a lipidic cubic phase
2.2.1.1. Vials as mixing and crystallization containers
Transfer 6 mg of dry monoolein (monooleoyl-rac-glycerol [C18:1c9]) into a vial (e.g., a PCR tube or a glass tube with an inner diameter of 3 mm).
Add 4 μl of bacteriorhodopsin solution (9 mg/ml) from 2.1.2 or 2.1.3 to the lipid and seal the vial.
Mix monoolein and bacteriorhodopsin solution by repeated centrifugation in a benchtop centrifuge. Rotate by 180° about the long axis between short runs to provide mixing. Continue until a homogeneous purple, transparent, non-birefringent, and gel-like material has formed.
2.2.1.2. Coupled syringes as mixing devices
Transfer 60 mg of dry monoolein (monooleoyl-rac-glycerol [C18:1c9]) into a 250 μl Hamilton syringe (gastight, RN).
Add 40 μl of bacteriorhodopsin solution (9 mg/ml) from 1.2 or 1.3 into a second 250 μl Hamilton syringe (gastight, RN).
Join both syringes via a coupler (Fig. 2).
Mix monoolein and bacteriorhodopsin solution by repeated transfer of the material from one syringe into the other. Continue until a homogeneous purple, transparent, non-birefringent, and gel-like material has formed.
Transfer all of the material into one syringe and attach a suitable dispenser, e.g., a manual rachet dispenser with a needle (Fig. 2).
Dispense portions of 0.2 μl into a pre-filled well (see below) and cover with transparent tape.
2.2.2. Step 2: Crystal nucleation and growth
Bacteriorhodopsin nucleation and crystallization can be initiated by the addition of a mix of solid salt to the preformed lipidic cubic phase. Since weighing of very small amounts of salt is cumbersome, solutions have been formulated that can likewise trigger the crystallization process [18]. Typically such solutions are used for multi-well experiments, whereas salts are used primarily for ‘large-scale’ crystallizations in vials.
2.2.2.1. Addition of a solid salt
Prepare Sørensen salt mix by grinding 94.8 g KH2PO4 and 5.2 g Na2HPO4 · H2O.
Remove seal from vial (containing 10 mg of lipidic cubic phase, from 2.2.1.1) and place on balance.
Add 2–3 mg of Sørensen phosphate. Seal vial.
Centrifuge once in benchtop centrifuge until salt crystals combine in bottom of vial (Fig. 1A).
2.2.2.2. Pre-filling with a solution
Prepare the crystallization solution, 27% (w/v) PEG 2000 in 100 mM Sørensen phosphate buffer, pH 5.6.
Fill 1 μl of the crystallization solution into a single well and cover it with transparent tape. To this well-based setup, a portion of lipidic cubic phase is added according to Section 2.2.1.2. See Figs. 2 and 3.
Fig. 3.
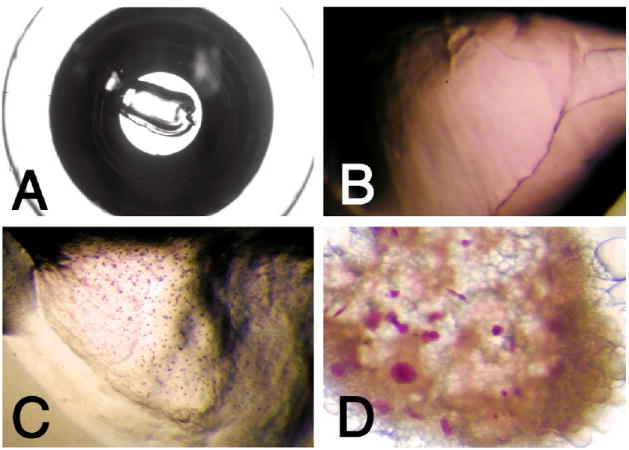
Growth of bacteriorhodopsin crystals using the syringe and microwell-based crystallization procedure. Shown are photographs of cubic phase crystallization setups in a microwell of a Terasaki plate. (A) Overview of well containing 0.2 μl cubic phase dispensed with manual syringe dispenser. Well bottom diameter is ca. 1.2 mm. (B) Close-up view of crystallization setup prior to crystallization. The purple bacteriorhodopsin is homogeneously distributed in the lipidic cubic phase. (C) Bacteriorhodopsin microcrystals formed in 0.2 μl of cubic phase when solid salt was used as a crystallization-inducing agent. (D) Purple bacteriorhodopsin crystals with sizes up to ca. 75 μm along the longest dimension were grown by adding 0.2 μl of initial monoolein cubic phase containing 3.5 mg/ml bacteriorhodopsin to 1 μl of 27% PEG 2000 in 100 mM Sørensen phosphate buffer, pH 5.6.
2.3. Observation of bacteriorhodopsin crystallization setups
Bacteriorhodopsin crystallization setups should be stored in darkness at constant temperature (e.g., room temperature). Often crystals can be detected within a few days after setup. It is simple to spot bacteriorhodopsin crystals in setups because the crystals are colored purple (Figs. 1 and 3). Although large crystals can be seen by the naked eye, it is advisable to inspect crystallization setups with a dissecting stereo microscope. When investigating crystallization setups, care must be taken to keep the temperature constant, i.e., prevent warming the setups during manipulation and by microscope light. Otherwise liquid bubbles form inside the lipidic cubic phase [16,17]. These are detrimental to observation and disrupt the crystallization process.
References
- 1.Briggs J, Chung H, Caffrey M. J Phys II France. 1997;6:723–751. [Google Scholar]
- 2.Chen AH, Hummel B, Qiu H, Caffrey M. Chem Phys Lip. 1998;95:11–21. doi: 10.1016/s0009-3084(98)00060-7. [DOI] [PubMed] [Google Scholar]
- 3.Chiu ML, Nollert P, Loewen MC, Belrhali H, Pebay-Peyroula E, Rosenbusch JP, Landau EM. Acta Cryst D. 2000;56:781. doi: 10.1107/s0907444900004716. [DOI] [PubMed] [Google Scholar]
- 4.Cherezov V, Fersi H, Caffrey M. Biophys J. 2001;81:225–242. doi: 10.1016/S0006-3495(01)75694-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cherezov V, Caffrey M. J Appl Cryst. 2003;36:1372–1377. [Google Scholar]
- 6.Dencher NA, Heyn MP. Methods Enzymol. 1982;88:5–10. [Google Scholar]
- 7.Grabe M, Neu J, Oster G, Nollert P. Biophys J. 2003;84:854–868. doi: 10.1016/S0006-3495(03)74904-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kiweon C, Reeves PJ, Khorana HG. Proc Natl Acad Sci USA. 2000;97(7):3016–3021. doi: 10.1073/pnas.97.7.3016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Landau EM, Rosenbusch JP. Proc Natl Acad Sci USA. 1996;93:14532. doi: 10.1073/pnas.93.25.14532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Landau EM, Rummel G, Cowan-Jacob SW, Rosenbusch JP. J Phys Chem B. 1997;101:1935–1937. [Google Scholar]
- 11.Loewen M, Chiu ML, Widmer C, Landau EM, Rosen-busch JP, Nollert P. G-Protein coupled receptors. In: Haga T, Berstein G, editors. Methods in Signal Transduction. CRC-Press; London: 2000. pp. 365–388. [Google Scholar]
- 12.Luecke H, Schobert B, Richter HT, Cartailler JP, Lanyi JK. J Mol Biol. 1999;291:899. doi: 10.1006/jmbi.1999.3027. [DOI] [PubMed] [Google Scholar]
- 13.Nollert P, Landau EM. Biochem Soc Trans. 1998;26(4):708–713. doi: 10.1042/bst0260709. [DOI] [PubMed] [Google Scholar]
- 14.Nollert P, Royant A, Pebay-Peyroula E, Landau EM. FEBS Lett. 1999;457:205. doi: 10.1016/s0014-5793(99)01014-5. [DOI] [PubMed] [Google Scholar]
- 15.Nollert P. J Appl Cryst. 2002;35:637–640. [Google Scholar]
- 16.Nollert P. In: Methods and Results in Crystallization of Membrane Proteins. Iwata S, editor. 2003. pp. 59–73. [Google Scholar]
- 17.Nollert P. J Appl Cryst. 2003;36:1295–1296. doi: 10.1107/S0021889803013724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nollert P, Navarro J, Landau EM. Methods Enzymol. 2002;343:183–199. doi: 10.1016/s0076-6879(02)43135-7. [DOI] [PubMed] [Google Scholar]
- 19.Paas Y, Cartaud J, Recouvreur M, Grailhe R, Dufresne V, Pebay-Peyroula E, Landau EM, Changeux JP. Proc Natl Acad Sci USA. 2003;100(20):11309–11314. doi: 10.1073/pnas.1834451100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Packer L. Methods Enzymol. 1982;88:1–27. [Google Scholar]
- 21.Pebay-Peyroula E, Neutze R, Landau EM. BBA. 2000;1460(1):119–132. doi: 10.1016/s0005-2728(00)00134-1. [DOI] [PubMed] [Google Scholar]
- 22.Rouhani S, Facciotti MT, Woodcock G, Cheung V, Cunningham C, Nguyen D, Rad B, Lin CT, Lunde CS, Glaeser RM. Biopolymers. 2002;66(5):300–316. doi: 10.1002/bip.10310. [DOI] [PubMed] [Google Scholar]
- 23.Rummel G, Hardmeyer A, Widmer C, Chiu M, Nollert P, Locher K, Pedruzzi I, Landau EM, Rosenbusch J. J Struct Biol. 1998;121:1–11. doi: 10.1006/jsbi.1997.3952. [DOI] [PubMed] [Google Scholar]
- 24.Sennoga C, Heron A, Seddon JM, Templer RH, Hankamer B. Acta Cryst D. 2003;59:239–246. doi: 10.1107/s0907444902020772. [DOI] [PubMed] [Google Scholar]