

Abstract
Besides its role in translation and ribosome maturation, human ribosomal protein S3 (hS3) is implicated in DNA damage recognition as reflected by its affinity for abasic sites and 7, 8-dihydro-8-oxoguanine (8-oxoG) residues in DNA in vitro. Here, we demonstrate that hS3 is capable of carrying out both roles by its ex vivo translocation from the cytoplasm to the nucleus as a consequence of genotoxic stress. The translocation of hS3 is dependent on ERK1/2-mediated phosphorylation of a threonine residue (T42) of hS3. Two different ectopically expressed site-directed mutants of T42 failed to respond to conditions of genotoxic stress, thus providing a link between DNA damage and ERK1/2 dependent phosphorylation of hS3. Lastly, hS3 was traced in exposed cells to its co-localization with 8-oxoG foci, raising the possibility that hS3 is a member of a cellular DNA damage response pathway that results in its interaction with sites of DNA damage.
Keywords: Base excision repair, Ribosomal protein S3, ERK, Genotoxic stress, Nuclear translocation
1. Introduction
Among the numerous DNA lesions formed under conditions of oxidative stress, 7, 8-dihydro-8-oxoguanine (8-oxoG) is notable as it is mutagenic if not repaired [1–4] where its persistence in the genome can lead to a number of pathological states. The primary means to remove 8-oxoG from DNA is via the action of N-glycosylases that participate in the base excision repair (BER) pathway. In human cells, the most potent activity for the removal of 8-oxoG is through the action of OGG1, which possesses an N-glycosylase activity for the liberation of the modified base. The BER protein also contains an intrinsic apurinic/apyrimidinic (AP) lyase, β-elimination activity that leaves an unsaturated sugar residue at the 3′-terminus of newly cleaved DNA.
The interaction of human OGG1 with 8-oxoG is not so robust as that seen of other OGG1-like activities found in Escherichia coli and Drosophila [5–7]. For example, OGG1 compares unfavorably in both its affinity for 8-oxoG in DNA [7], and its turn-over rate (Kcat) at 8-oxoG-containing substrates [8]. This has led several investigators to suggest that human OGG1 remains bound to the AP site formed as an intermediate in the excision repair process and that other BER proteins may be necessary for dislodging OGG1 to produce a more vigorous turnover rate. Indeed, APE/ref-1, the major AP endonuclease in human cells, does displace OGG1, resulting in a more robust turn-over rate of the N-glycosylase step in BER [9].
Using surface plasmon resonance (SPR), we have recently shown that human ribosomal protein S3 (hS3) positively interacts with OGG1 [10]. However, SPR analysis also showed that hS3 has an unprecedented high apparent binding affinity for 8-oxoG DNA lesions [7]. This led us to speculate that the positive interaction of hS3 with OGG1 was conditional, and would occur only under circumstances where hS3 was not bound to 8-oxoG residues in DNA. This apparent conundrum was recently solved through the generation of a site-directed mutant (hS3-K132A) that completely abrogated the DNA binding affinity of hS3 for 8-oxoG [11]. Using 5′ end labeled oligonucleotides containing a single 8-oxoG residue (8-oxoG-37mer), it was shown that incubations of OGG1 with hS3-K132A resulted in a two-fold stimulation of OGG1 N-glycosylase activity, whereas pre-incubations with 8-oxoG-37mer and wild-type hS3 completely inhibited the subsequent liberation of 8-oxoG by OGG1.
Since hS3 is associated with ribosomes, a question that is dealt here is if and when hS3 is translocated from the cytoplasm to the nucleus to bind DNA adducts such as 8-oxoG, as suggested by our previous in vitro studies. Here, we show that hS3 is translocated to the nucleus when cells are exposed to DNA damaging agents, a process that is dependent upon phosphorylation of hS3 by ERK1/2. Moreover, hS3 co-localizes with foci of nuclear 8-oxoG DNA lesions as part of the translocation process.
2. Materials and methods
2.1. Cell culture
HEK 293 cells (ATCC) were cultured in EMEM (Sigma) supplemented with 10% FBS (Sigma), penicillin (250 IU/ml), and streptomycin (250 μ g/ml). Cells were seeded in complete media at approximately 30–40% confluence, 24 h prior to each experiment. Exposure of HEK 293 cells to genotoxic agents, hydrogen peroxide (H2O2, CAS#7722-84-1), methyl methanesulfonate (MMS, CAS#66-27-3) (Fluka), and N,N′,N″-Triethylenethiophosphoramide (Thio-TEPA, CAS#52-24-4) (Sigma), was performed by adding 1000× stock solutions. Exposure of cells to hydrogen peroxide in the presence of protein kinase inhibitors was performed by pre-treating the cells with the inhibitor for 1 h prior to the addition of hydrogen peroxide. For the induction of serum starvation, cells grown to approximately 50% confluence in complete medium were changed to serum-free medium and incubated for 6 h.
2.2. Subcellular fractionation
Cytosolic and nuclear fractions were prepared as described previously [12] with minor modifications. Briefly, cell pellets were resuspended in buffer A (25 mM HEPES [pH 7.4], 50 mM KCl, 1 mM EDTA, 0.5% NP-40, 1 mM DTT, 25 mM NaF, 1 mM NaO3V, 1 mM PMSF, 10 μg/ml aprotinin, 10 μg/ml leupeptin, and 1% phosphatase inhibitor cocktail I), and incubated on ice for 30 min. Samples were then centrifuged at 500 × g for 1 min and the supernatants were collected as cytosolic fractions. The pellets were washed in buffer B (buffer A without NP-40) and centrifuged at 500 × g for 1 min. The recovered nuclear pellets were re-suspended in nuclei extraction buffer (buffer B, 450 mM KCl, and 50% glycerol), and frozen at −70 °C for 30 min. The nuclear suspensions were then thawed, clarified by centrifugation (16,000 × g for 15 min), and the supernatants were retained as the nuclear fraction.
2.3. Subfractionation of nuclei
Subfractionation of purified nuclei was performed as described previously with minor modifications [13,14]. Briefly, cytosolic and crude nuclear fractions were prepared from ~1 × 107 cells, and purified nuclei were subsequently isolated from crude nuclei. Purified nuclear pellet was resuspended in 20 mM Tris (pH 7.4), 5 mM MgCl2 containing 10 μg/ml of DNase and 8 μg/ml of RNase, and subjected to nuclease digestion by incubating at 37 °C for 15 min. The nuclease supernatant was collected by centrifugation (1000 × g for 10 min), and the pellet was resuspended in 290 mM sucrose, 10 mM Tris (pH 7.4), 0.1 mM MgCl2, 2% Triton X-100. After 10 min incubation on ice, Triton X-100 supernatant was collected by centrifugation (1000 × g for 10 min). The pellet was resuspended in 100 mM Tris (pH 7.4), 290 mM sucrose, 0.1 mM MgCl2 and 1M NaCl, and incubated on ice for 10 min. The supernatant was collected by centrifugation (10,000 × g for 10 min), and the NaCl extraction was repeated with the pellet. The final pellet containing the subnuclear matrix was solubilized in phosphate-buffered saline (PBS) containing 8 M urea.
2.4. Western blot analysis
Western blot analysis was performed as described previously [15]. Briefly, whole cell lysates and sub-cellular fractions were resolved by SDS-PAGE and transferred onto a nitrocellulose membrane by electrotransfer technique. Membranes were then blocked in 5% non-fat dairy milk, and immunoblotted with anti-hS3 (custom-made by Proteintech Group, Chicago, IL), anti-poly(ADP)ribose polymerase (PARP) (Cell Signaling Technology), or anti-nuclear pore complex protein 62 (NPC62, clone MAB414) primary antibodies. For the detection of phosphorylated ERK1/2 proteins, a rabbit polyclonal antibody (Cell Signaling Technology) that specifically detects phosphorylated forms of ERK1/2 was used. After treating the membranes with appropriate secondary antibodies, antigen bands were visualized by enhanced chemiluminescence technique using western lightning chemiluminescence reagent plus (PerkinElmer). Densitometric tracing of the protein bands was performed by using AlphaEaseFc software (Alpha Innotech Corporation).
2.5. Detection of hS3 and 8-oxoG co-localization by immunofluorescence microscopy
Cells were fixed in 10% formalin for 30 min and permeabilized in 0.1% Triton X-100 for 10 min. After washing in PBS, cells were blocked in 1% BSA for 1 h and incubated with anti-hS3 and anti-8-oxoG (clone 4E9, Trevigen) antibodies for an additional hour. Cells were then washed in PBS, and further incubated with rhodamine red-anti-rabbit (Molecular Probes) and FITC-anti-mouse (Zymed) secondary antibodies for 1 h. After washing again in PBS, cells were stained with 4′-6-diamidino-2-phenylindole (DAPI) (1 μg/ml in PBS) (Sigma), and localization of hS3, 8-oxoG, and nuclei was performed by fluorescence microscopy (Nikon Eclipse TE2000-U inverted microscope with 40× objective), using rhodamine, FITC, and UV filters, respectively. Images were captured using a CoolSNAPcf digital camera (Photometrics) and MetaMorph software (Universal Imaging Corp). Co-localization of hS3 with 8-oxoG foci was analyzed by using ImageJ software (http://rsb.info.nih.gov/ij). This analysis generates a scatter plot and co-localization coefficients, ranging from 0–1, 1 being the maximum co-localization between the 2 channels.
2.6. Analysis of cell survival
Cell survival after treatment with DNA damaging compounds was evaluated as described previously [11,16,17], by using the Cell Counting Kit-8 (CCK-8) (Dojindo Molecular Technologies Inc, Gaithersburg, MD). Briefly, cells seeded in a 96 well plate (10,000–15,000/well) were exposed to various genotoxic agents in triplicate. After 24 h, 10 μl of CCK-8 solution was added to each well and the plate was incubated at 37 °C for additional 3 h. Cell survival was then determined by reading the absorbance at 450 nm (Bio-Rad, Benchmark Plus spectrophotometer), which is a measure of the activity of cellular dehydrogenases that in turn is directly proportional to the number of living cells in the sample [17].
2.7. Overexpression and purification of human GST-S3
E. coli strain RPC501 was used for overexpressing human GST-S3 [18]. Overnight E. coli cultures bearing human GST-S3 plasmid construct were grown at 37 °C up to A595 of 0.5 OD and induced with 1 mM Isopropyl-1-thio-β-D-galactopyranoside (IPTG) for 3 h at 27 °C. Cells were harvested by centrifugation at 12,000 × g for 10 min at 4 °C and resuspended in PBS containing 1 mM DTT and protease inhibitors (10 μg/ml aprotinin and 10 μg/ml leupeptin). After 30 min incubation on ice, cells were lysed by 15 sec constant pulse sonication three times with 2 min intervals. Cell debris was removed by centrifugation at 16,000 × g for 15 min at 4 °C and the supernatant was applied to a glutathione-agarose column. Human GST-S3 protein was eluted from the column using 10 mM reduced glutathione in 50 mM Tris (pH 9.0).
2.8. In Vitro phosphorylation and dephophorylation of human GST-S3 by phospho-ERK1/2
In vitro phosphorylation of GST-S3 was performed using the phospho-ERK1/2, immunoprecipitated from HEK 293 cells following the induction of phospho-ERK1/2 by serum starvation [19,20]. Cells were grown to 50% confluence in DMEM supplemented with 10% FBS, and the medium was changed to serum-free medium. After 6 h incubation in serum deficient medium, cell lysates were prepared in modified RIPA buffer (50 mM Tris [pH 7.5], 150 mM NaCl, 1% NP-40, 1 mM EDTA, 25 mM NaF, 2.5 mM sodium pyrophosphate, 1 mM NaO3V, 1% [vol/vol] phosphatase inhibitor cocktail I, 10 μg/ml aprotinin, 10 μg/ml leupeptin and 1 mM PMSF). After determining the protein concentration by Bradford assay, protein extract (400 μg) was subjected to phospho-ERK1/2 immunoprecipitation by overnight incubation with anti-phospho ERK1/2 antibody (1:100, Cell Signaling Technology), followed by incubation with 40 μl of protein A/G plus agarose beads (Santacruz Biotechnology) for additional 3 h. The immunoprecipitates were washed four times with lysis buffer, twice with kinase buffer (20 mM Tris [pH 7.5], 10 mM MgCl2, 1 mM EGTA, and 1 mM DTT) and resuspended in 20 μl of kinase buffer containing 2 μg of purified human GST-S3, 50 μM ATP, and 2 μCi γ-[32P]ATP. After 30 min incubation at 30 °C, the kinase reaction was stopped by placing the samples on ice. The supernatant was collected by centrifugation (16,000 × g for 30 sec) and mixed with equal volume (15 μl) of 2× Laemmli sample buffer. For dephosphorylation, supernatant from the kinase reaction was incubated with 50 units of λ-protein phosphatase (New England Biolabs) in reaction buffer (50 mM Tris [pH 7.5], 2 mM MnCl2, 100 mM NaCl, 0.1 mM EGTA, 2 mM DTT, and 0.01% Brij 35) at 30 °C for 30 min. The reaction was stopped by adding equal volume (15 μl) of 2× Laemmli sample buffer. After boiling for 5 min, samples were subjected to SDS-PAGE and autoradiography.
2.9. Site-directed mutagenesis
The ERK1/2 phosphorylation site of hS3 is a threonine residue at position 42. This site was mutated to either an alanine residue, to prevent phosphorylation, or an aspartic acid residue, to mimic phosphorylation, by the site directed mutagenesis approach. The oligonucleotide primers used for mutagenesis were designed based on the amino acid sequence of an internal peptide of wild type hS3 (N′-VRVTPTR-C′). The sequences of primers are as following (the mismatched bases are underlined); for aspartic acid mutant 5′-GGTGCGAGTTGATCCAACCAGG-3′ (forward), 5′-CCTGGTTGGATCAACTCGC ACC-3′ (reverse) and for alanine mutant 5′-GGTGCGAGTTGCGCCAACCAGG-3′ (forward), 5′-CCTGGTTGGCGCAACTCGC ACC-3′ (reverse). Wild type human S3 (pcDNA 3.1-hS3) was used as a template, and mutagenesis was performed using Quickchange site-directed mutagenesis kit (Stratagene) according to the manufacturer’s instructions. After transforming XL10-Gold ultracompetent E. coli (Stratagene) with the mutant DNA, plasmid DNA from individual clones was purified using QIAprep spin miniprep kit (Qiagen), and introduction of desired mutation was confirmed by sequencing. The aspartic acid and alanine mutants of hS3 in pcDNA 3.1 vector are referred as pcDNA 3.1-hS3-T42D and pcDNA 3.1-hS3-T42A, respectively, and used to construct GFP tagged expression vectors.
2.10. Construction of GFP tagged hS3, hS3-T42D, and hS3-T42A expression vectors
Plasmids encoding GFP-hS3, GFP-hS3-T42D, and GFP-hS3-T42A were constructed by subcloning the PCR amplified hS3, hS3-T42D, and hS3-T42A coding sequences from pcDNA 3.1 vector into pEGFP-N1 vector. The forward (5′-GCCGGCGAATTCCATGGCAGTG CAA ATATC CAAG-3′) and reverse (5′-CGCCGCGGATC CGCTGCTGTGG GG ACTGGC-3′) end primers used for PCR were designed in such a way that they will introduce a 5′-EcoRI and a 3′-BamHI restriction sites into hS3, hS3-T42D, and hS3-T42A sequences. PCR amplification of hS3, hS3-T42D, and hS3-T42A coding sequences was performed using pcDNA 3.1 constructs as templates, Taq DNA polymerase and 1.5 mM MgCl2 in 30 cycles of 30 sec at 95 °C, 30 sec at 55 °C, and 1.5 min at 72 °C. PCR products were gel purified using the QIAquick gel extraction kit (Qiagen) and subjected to EcoRI-BamHI digestion in EcoRI buffer. pEGFP-N1vector was also digested using EcoRI-BamHI restriction enzymes. The digestion reactions were cleaned with QIAquick nucleotide removal kit (Qiagen) and subsequently used for ligating hS3, hS3-T42D, and hS3-T42A fragments into pEGFP-N1 vector in the presence of T4 DNA ligase. Chemically competent TOP10 E.coli (Invitrogen) was transformed using 5 μl of the ligation mixture, and bacterial colonies bearing the insert were selected on LB-agar plates containing 50 μg/ml of kanamycin. Plasmid DNAs were purified from individual clones using a QIAprep spin miniprep kit (Qiagen) and the presence of the expected insert was confirmed by sequencing.
2.11. Transient transfection
HEK 293 cells (2 × 105 cells/well) were seeded in a 12 well plate, 24 h prior to transfection experiments. Cells were transiently transfected with plasmid DNAs, GFP-hS3, GFP-hS3-T42D, or GFP-hS3-T42A, using FuGENE6 transfection reagent (Roche Applied Science) according to the manufacturer’s instructions, and incubated at 37 °C in a CO2 incubator for 24 h.
2.12. Fluorescence tracing of sub-cellular localization of GFP-hS3, GFP-hS3-T42A, and GFP-hS3-T42D
HEK 293 cells transfected with expression plasmids GFP-hS3, GFP-hS3-T42A, or GFP-hS3-T42D for 24 h, and exposed to hydrogen peroxide. After 24 h incubation, cells were washed twice with PBS and fixed in 10% formalin solution for 30 min. After washing cells in PBS, sub-cellular localization of GFP-hS3, GFP-hS3-T42A, and GFP-hS3-T42D was detected by fluorescence microscopy (FITC filter with excitation at 490 nm, emission at 525 nm) using a Nikon Eclipse TE2000-U inverted microscope with 40× objective (Nikon). Images were captured using a CoolSNAPcf digital camera (Photometrics) and MetaMorph software (Universal Imaging Corp).
3. Results
3.1. Translocation of cytoplasmic hS3 to the nucleus depends upon exposure to DNA damaging agents
While the hS3 is associated with ribosomal functions [21–23], it is also involved with BER [7,10,11,15,24,25]. We therefore tested whether agents that damage DNA could result in hS3 moving from its primary location in the cytoplasm to the nucleus where it would be poised to interact with DNA. HEK 293 cells were exposed to increased concentrations of hydrogen peroxide, MMS, and Thio-TEPA and then subjected to subcellular fractionation procedures that separate the cytosolic components from those ingredients that reside in the nucleus. Western analysis with antibody specific for hS3 was then applied to the purified cytosolic and nuclear preparations. Greater than 85% of hS3 was found in the cytoplasm of unexposed HEK 293 cells (Fig. 1A). However, exposure of HEK 293 cells to 0.0625 mM hydrogen peroxide (80% survival; see Materials and methods) resulted in 40% of hS3 being present in the nucleus (Fig. 1A). Higher concentration of hydrogen peroxide treatments showed a persistent presence of hS3 in the nucleus, and at 1 mM hydrogen peroxide, hS3 appeared to be exclusively located in the nucleus, even though cell survival was dramatically reduced at this high hydrogen peroxide concentration (10% survival). Controls with PARP, a nuclear marker [26], and NPC62, a marker for the cytosolic nuclear membrane [27], established that our subcellular fractionations resulted in an authentic separation of nuclear and cytoplasmic fractions.
Fig. 1.

Nuclear translocation of hS3 in HEK 293 cells exposed to hydrogen peroxide. HEK 293 cells were exposed to various concentrations of hydrogen peroxide for 24 h (A) or to 0.125 mM hydrogen peroxide for various periods of time (B). Cytosolic (C) and nuclear (N) fractions were prepared by sub-cellular fractionation technique, as explained under Experimental Procedures. A 10 μg aliquot of protein extract from each sample was immobilized by SDS-PAGE and electrotransfer techniques. Subcellular localization of hS3 was detected by immunoblot analysis utilizing a monoclonal anti-hS3 antibody. The same blots were stripped and re-probed with antibodies against PARP (nuclear marker) and NPC62 (nuclear envelope marker), in order to exclude the cross-contamination of the subcellular fractions during sample processing. Human S3 levels in cytosolic and nuclear fractions are shown as percent of total cellular hS3 protein, which was determined by summing the integrated density values (IDV) of cytosolic and nuclear bands from each sample.
We also examined the time course of the translocation of hS3 after exposure to 0.125 mM hydrogen peroxide (Fig. 1B). The nuclear presence of hS3 rose from an initial 10% to roughly double that after 1 h. Within 2 h, 40% of the total hS3 cross-reacting material was located in the nucleus (Fig. 1B). This value did not change with an additional hour of hydrogen peroxide exposure.
Two other DNA damaging agents were examined to see if they also elicited the translocation of hS3. Exposure of HEK 293 cells to MMS (Fig. 2A) led to a translocation of hS3 from the cytoplasm to the nucleus, as did exposure to the chemotherapeutic alkylating agent Thio-TEPA (Fig. 2B). These agents form various alkylated DNA bases, some of which spontaneously give rise to AP sites.
Fig. 2.
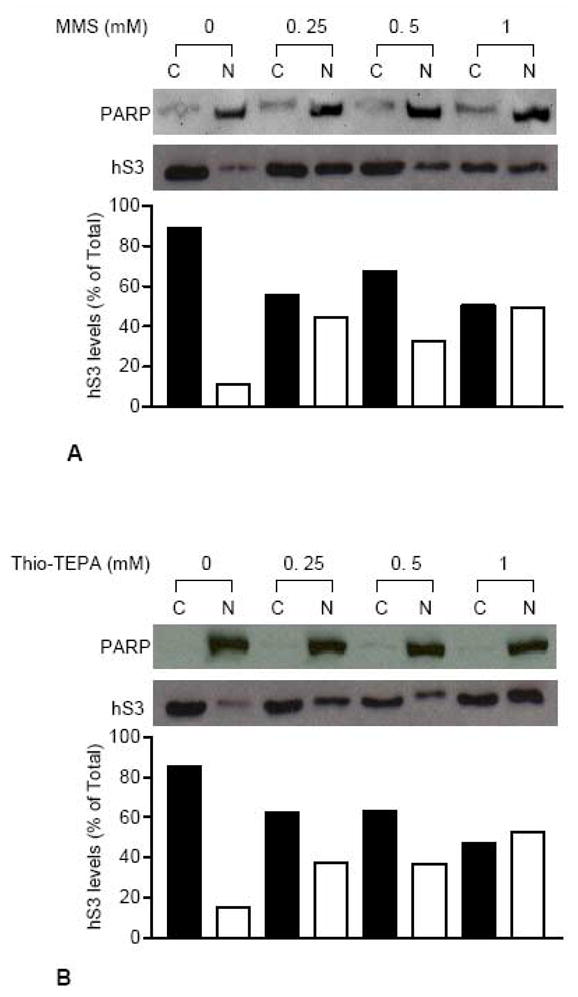
Nuclear translocation of hS3 in response to MMS and Thio-TEPA treatment. Cytosolic (C) and nuclear (N) fractions were prepared from HEK 293 cells exposed to various concentrations of MMS (A) or Thio-TEPA (B) for 24 h. After immunoblotting analysis of fractions using anti-hS3 antibody, sub-cellular levels of hS3 were evaluated from integrated density values, obtained by densitometry tracing of each band. Cytosolic and nuclear density values for each sample were added to obtain total cellular hS3 and hS3 levels in individual fractions are represented as percent of total hS3. Localization of nuclear protein PARP on both blots was traced by stripping and re-probing with anti-PARP antibody.
It should be noted that the two alkylating agents used in this study were not as effective in eliciting the translocation of hS3 as was hydrogen peroxide. Since all of the agents used in this study were fairly toxic, we examined the relative abundance of nuclear hS3 under conditions where survival of HEK 293 cells was roughly 70–80%, which was the case for cells exposed to 0.0625–0.125 mM genotoxic agents. Densitometric analysis revealed that 0.0625 mM hydrogen peroxide treatment resulted in 40–45% of total hS3 residing in the nucleus. On the other hand, exposure to 0.125 mM alkylating agents resulted in 15–20% nuclear hS3 (not shown). It should be noted that higher concentrations of alkylating agents, as seen in Fig. 2, did result in roughly 40% of hS3 residing in the nucleus.
3.2. Translocation of hS3 to the nucleus is dependent upon ERK1/2-mediated phosphorylation
Previous studies have shown that ribosomal protein function can depend upon phosphorylation and dephosphorylation. Phosphorylation of ribosomal proteins often involve diverse classes of kinases that include PKC, CKII and ERK [28–31]. We therefore examined the effect that the inhibition of these kinases would have on hS3 translocation. HEK 293 cells were exposed to specific kinase inhibitors prior to 0.125 mM hydrogen peroxide treatment, and the subcellular location of hS3 subsequently determined by SDS-PAGE and Western analysis (Fig. 3A). Pre-treatment with MEK1/2 inhibitor U0126, which disrupts ERK1/2 phosphorylation [32], clearly led to the failure of genotoxicity-dependent hS3 translocation to the nucleus (Fig. 3A, compare lanes 3 and 4 and lanes 7 and 8), in which 100% of hS3 was found in the cytoplasm in the presence of UO126. Control experiments using antibody to phosphorylated ERK1/2 failed to detect activated, phosphorylated ERK1/2 in hydrogen peroxide/U0126 treated cells, thus confirming that the loss of activated ERK1/2 leads to hS3 remaining in the cytosol (not shown). On the other hand, Staurosporine (Fig. 3A, lanes 11 and 12), a PKC inhibitor [33], and 5, 6 Dichlorobenzimidazole riboside (DRB; Fig. 3A, lanes 15 and 16), a CK II inhibitor [34], had no effect on the translocation of hS3 to the nucleus.
Fig. 3.

ERK1/2 mediated phosphorylation is involved in nuclear translocation of hS3 in response to hydrogen peroxide treatment. (A) Cells were pre-treated with vehicle alone (lanes 1–4), 25 μM UO126 (lanes 5–8), 10 nM Staurosporine (lanes 9–12) or 10 μM DRB (lanes 13–16) for 1 h, and then exposed to 0.125 mM hydrogen peroxide. At 24 h post hydrogen peroxide treatment, cells were fractionated into cytosol (C) and nuclei (N), and hS3 levels in the fractions were determined by the western blot analysis and densitometry tracing of protein bands. Human S3 levels in sub-cellular fractions are expressed as a percent of total cellular hS3, which is a sum of nuclear and cytosolic hS3 in each sample. The blots were stripped and re-probed with anti-PARP antibody in order to detect the presence of this nuclear marker. (B) Whole cell lysates were prepared from serum starved HEK 293 cells and phospho-ERK1/2 was immunoprecipitated using an anti-phospho-ERK1/2 antibody. The resulting immunoprecipitate was subsequently used to phosphorylate purified GST-hS3 protein by immunocomplex kinase reaction. Supernatant from the kinase reaction was subjected to SDS-PAGE and autoradiography. Top panel demonstrates the immunoprecipitation of phospho-ERK1/2 (detected by western blot analysis using the anti-phospho-ERK1/2 antibody). The bottom panel shows the autoradiogram demonstrating the phosphorylation of hS3. Kinase reactions digested with λ-phosphatase (lane 2, bottom panel) and reactions carried out in the absence of ERK1/2 immunocomplex (lane 3, bottom panel) were also included on the gel. (C) HEK 293 cells were transiently transfected with GFP-hS3, GFP-hS3-T42D or GFP-hS3-T42A, and then treated with vehicle alone or 0.25 mM hydrogen peroxide for 24 h. Sub-cellular localization of hS3, hS3-T42D, and GFP-hS3-T42A was traced by fluorescence microscopy using FITC filter.
We subsequently tested whether hS3 was indeed susceptible to ERK phosphorylation. To accomplish this, we conducted experiments in which HEK 293 cells were serum starved to induce ERK1/2 phosphorylation, and cell lysates subsequently immunoprecipitated (IP) with antibody specific for phospho-ERK1/2 (Fig. 3B, upper panel). The IP was in turn used to test whether it could phosphorylate purified GST-hS3. As can be seen in Fig. 3B (lower panel), GST-hS3 was susceptible to phosphorylation by IP preparations of phospho-ERK1/2 (lane 1). On the other hand, kinase reaction digested with λ-phosphatase (lane 2) failed to show a phospho-hS3 band. Kinase reaction carried out in the absence of ERK1/2 immunocomplex was also included on the gel (lane 3) to rule out the possibility of autophosphorylation of human S3 by ATP.
As an alternative means of showing the dependence of hS3 translocation on phosphorylation by ERK1/2, we traced by immunofluorescence the subcellular localization of two different site directed mutants of the threonine residue at position 42 residing within the ERK1/2 phosphorylation motif of hS3 [30]. In the first case, threonine was mutated to alanine, and hS3 linked to GFP to form GFP-hS3-T42A. This mutant, when compared to wild-type GFP-hS3, failed to translocate into the nucleus when cells were exposed to hydrogen peroxide (Fig. 3C, compare middle right panel with upper right panel). We also produced a site directed mutant of the hS3 threonine residue (GFP-hS3-T42D) to convert it to an aspartic acid residue, which has been shown in previous studies for other proteins to mimic a site that is phosphorylated [35–37]. Notably, GFP-hS3-T42D was found in the nucleus, and importantly did not depend upon cells being exposed to hydrogen peroxide (Fig. 3C, lower left panel).
3.3. The final destination of nuclear hS3 is its co-localization with foci of 8-oxoG
To establish the sub-nuclear destination of hS3, nuclei were purified [13,14] from HEK 293 cells that were either untreated or exposed to hydrogen peroxide (0.125 mM). Subfractionation of nuclei was then performed [14] with the end result being the purification of the subnuclear matrix (Fig. 4, lane M). As expected, in unexposed cells, hS3 remained in the cytosolic fraction in route to the purification of nuclei (Fig. 4, lane C). Conversely, the subnuclear matrix from exposed cells revealed an obvious and substantial presence of hS3 (Fig. 4, lane M).
Fig. 4.
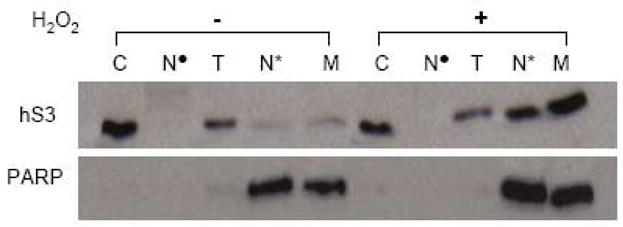
Subnuclear localization of hS3 in cells treated with hydrogen peroxide. Cytosol (C) and nuclear fractions were isolated from HEK 293 cells exposed to vehicle alone or 0.125 mM hydrogen peroxide for 24 h. Purified nuclei were further subjected to subfractionation as described under Experimental Procedures. Cytosol as well as various subnuclear fractions (Nuclease supernatant (N•), Triton X-100 extraction (T), NaCl extraction (N*), Subnuclear matrix (M) were separated by SDS-PAGE and immunoblotted against hS3 (top panel). The membrane was further stripped and re-probed with the anti-PARP antibody.
To determine the precise nuclear localization of hS3, we performed immunofluorescence microscopy with HEK 293 cells that had been exposed to hydrogen peroxide (0.25 mM), or left untreated, for 24 h. Human S3 was subsequently detected by staining the cells with a rabbit monoclonal anti-hS3 primary antibody and rhodamine red labeled secondary antibody (red fluorescence). For 8-oxoG localization, anti-8-oxoG primary antibody and FITC labeled secondary antibody (green fluorescence) were used. Samples were also stained with the nuclear fluorochrome, DAPI. A representative of images obtained with rhodamine (hS3), FITC (8-oxoG), and UV filters (nuclei) are shown in Fig. 5. Additionally, a merge of rhodamine, FITC, and UV images is shown (Fig. 5). Exposure of cells to hydrogen peroxide resulted in the co-localization of hS3 (red) and 8-oxoG (green) foci in the nuclei (blue), which is pointed by arrows in the magnified area of the merged image. The degree of co-localization between hS3 and 8-oxoG was determined by ImageJ software, which generated a scatter plot (not shown) and co-localization coefficient of 0.957 and 1 for the red (hS3) and green (8-oxoG) channels, respectively, where a coefficient of 1 being the maximum co-localization attainable.
Fig. 5.

Human S3 co-localizes with 8-oxoG residues in cells exposed to hydrogen peroxide. HEK 293 cells were exposed to 0.25 mM hydrogen peroxide for 24 h, and 8-oxoG/hS3 co-localization was evaluated by immunofluorescence microscopy. Human S3 was detected by staining the cells with a rabbit monoclonal anti-hS3 primary antibody and rhodamine red labeled secondary antibody (red fluorescence). For 8-oxoG localization, anti-8-oxoG primary antibody and FITC labeled secondary antibody (green fluorescence) were used. After staining with DAPI, samples were analyzed by fluorescence microscopy. A representative of images obtained with rhodamine (hS3), FITC (8-oxoG), and UV (nuclei) filters are shown. Additionally, a merge of rhodamine, FITC, and UV images is shown. Arrows in the magnified area of the merged image indicate foci of the co-localization of hS3 and 8-oxoG within the nuclei.
4. Discussion
For hS3 to act in BER and also be involved in protein synthesis would require its displacement from cytoplasmic ribosomes and translocation to the nucleus. Here we show that predominately cytoplasmic hS3 is indeed translocated to the nucleus, but only after exposure of human cells to DNA damaging agents. This translocation appears to be dependent on the activation of ERK1/2, which in turn phosphorylates hS3 as part of the translocation process. The phosphorylation of hS3 is in agreement with previous results that showed that a threonine residue at position 42 is phosphorylated both in vitro and in vivo [30]. Furthermore, we have constructed site-directed mutants at T42 that blocks phosphorylation (T42A) and one that mimics constitutive phosphorylation (T42D). Fusion constructs of these mutants to GFP show that the ectopic expression of T42A does not enter the nucleus, whereas T42D is present in the nucleus even in the absence of exposure to DNA damaging agents (Fig. 3C). This confirms that the translocation of hS3 into the nucleus is dependant on ERK1/2 phosphorylation.
ERK is known to respond to both oxidative stress and alkylating agents [38]. Notably, our data would suggest that ERK is perhaps more sensitive to oxidative stress than alkylating agents since HEK 293 cells exposed to hydrogen peroxide accumulated roughly twice as much nuclear S3 as did exposure to the two alkylating agents when cell survival was at 70%. We have further traced the DNA damage-induced nuclear translocation of hS3 to the subcellular matrix. Significantly, immunofluorescent microscopy showed that hS3 colocalized to foci of 8-oxoG.
We have hypothesized in the past that the persistence of hS3 at sites of 8-oxoG could have adverse consequences on cell survival. Recent results using RNA interference assays have reinforced that notion. These studies showed that a 40% knockdown of hS3 expression results in as much as a seven-fold increase in the 24-h survival-rate of HEK 293 cells exposed to hydrogen peroxide [15]. The same study showed that the ectopic expression of hS3 could accentuate the cytotoxic effect of various DNA damaging agents. Overall, these results raise the possibility that hS3 influences the repair of 8-oxoG and perhaps other DNA lesions such as AP sites. Based upon its high binding affinity for 8-oxoG and AP sites [7], one likely outcome is that hS3 creates a blockade to enzymes involved in BER.
Previous studies have shown that hS3 is involved in apoptosis as the overexpression of hS3 leads to distinct cellular morphological and biochemical changes including fragmented nuclei, and the activation of caspases 8 and 3 [39]. Another study has shown that hS3 interacts with NM23-H1 [40], which is a nucleoside diphosphate kinase as well as an endonuclease that produces single-stranded DNA damage. Notably, NM23-H1 is part of the SET complex that is activated by Granzyme A (GzmA) and translocated from the endoplasmic reticulum to the nucleus [41]. GzmA generates reactive oxygen species and activates a caspase-independent cell death pathway by cleaving SET [42] that in turn activates the endonuclease possessed by NM23-H1, which in concert with TREX1 degrades DNA, leading to cell death with morphological features similar to apoptosis [43].
In summary, the data presented here show that hS3 undergoes an ERK1/2-mediated translocation to the nucleus in cells exposed to genotoxic stress. We have also shown that hS3 co-localizes with 8-oxoG foci. Its interaction with NM23-H1 and possible association with apoptosis therefore raises the possibility that the translocation of hS3 to damaged bases may act as a DNA-sentinel for programmed cell death in cells experiencing oxidative stress.
Acknowledgments
We especially want to thank Dr. Stuart Linn for his continued interest in the multifunctional roles of hS3. We thank Dr. Weihong Pan, Blood Brain Barrier laboratory, PBRC for use of the microscopy facility. This research was supported by US Public Health Service Grant CA 109798 to WAD.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Cabrera M, Nghiem Y, Miller JH. MutM, a second mutator locus in Escherichia coli that generates G.C->T.A transversions. J Bacteriol. 1988;170:5405–5407. doi: 10.1128/jb.170.11.5405-5407.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wood ML, Dizdaroglu M, Gajewski E, Essigman JM. Mechanistic studies of ionizing radiation and oxidative mutagenesis genetic effects of a single 8-hydroxyguanine (7-hydro-8-oxoguanine) residue inserted at a unique site in a viral genome. Biochemistry. 1990;29:7024–7032. doi: 10.1021/bi00482a011. [DOI] [PubMed] [Google Scholar]
- 3.Moriya M, Ou C, Bodepudi V, Johnson F, Takeshita M, Grollman AP. Site-specific mutagenesis using a gapped duplex vector:a study of translesion synthesis past 8-oxodeoxyguanosine in E.coli. Mutat Res. 1991;254:281–288. doi: 10.1016/0921-8777(91)90067-y. [DOI] [PubMed] [Google Scholar]
- 4.Shibutani S, Takeshita M, Grollman AP. Insertion of specific bases during DNA synthesis past the oxidation-damaged base 8-oxodG. Nature. 1991;349:431–434. doi: 10.1038/349431a0. [DOI] [PubMed] [Google Scholar]
- 5.Jaruga P, Dizdaroglu M. Repair of products of oxidative DNA base damage in human cells. Nucleic Acids Res. 1996;24:1389–1394. doi: 10.1093/nar/24.8.1389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cappelli E, Degan P, Frosina G. Comparative repair of the endogenous lesions 8-oxo-7, 8-dihydroguanine (8-oxoG), uracil and abasic site by mammalian cell extracts: 8-oxoG is poorly repaired by human cell extracts. Carcinogenesis. 2000;21:1135–1141. [PubMed] [Google Scholar]
- 7.Hegde V, Wang M, Deutsch WA. Characterization of human ribosomal protein S3 binding to 7,8-dihydro-8-oxoguanine and abasic sites by surface plasmon resonance. DNA Repair (Amst) 2004;3:121–126. doi: 10.1016/j.dnarep.2003.10.004. [DOI] [PubMed] [Google Scholar]
- 8.Asagoshi K, Yamada T, Terato H, Ohyama Y, Monden Y, Arai T, Nishimura S, Aburatani H, Lindahl T, Ide H. Distinct repair activities of human 7,8-dihydro-8-oxoguanine DNA glycosylase and formamidopyrimidine DNA glycosylase for formamidopyrimidine and 7,8-dihydro-8-oxoguanine. J Biol Chem. 2000;275:4956–4964. doi: 10.1074/jbc.275.7.4956. [DOI] [PubMed] [Google Scholar]
- 9.Hill JW, Hazra TK, Izumi T, Mitra S. Stimulation of human 8-oxoguanine-DNA glycosylase by AP-endonuclease: potential coordination of the initial steps in base excision repair. Nucleic Acids Res. 2001;29:430–438. doi: 10.1093/nar/29.2.430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hegde V, Wang M, Deutsch WA. Human ribosomal protein S3 interacts with DNA base excision repair proteins hAPE/Ref-1 and hOGG1. Biochemistry. 2004;43:14211–14217. doi: 10.1021/bi049234b. [DOI] [PubMed] [Google Scholar]
- 11.Hegde V, Wang M, Mian IS, Spyres L, Deutsch WA. The high binding affinity of human ribosomal protein S3 to 7,8-dihydro-8-oxoguanine is abrogated by a single amino acid change. DNA Repair (Amst) 2006;5:810–815. doi: 10.1016/j.dnarep.2006.04.001. [DOI] [PubMed] [Google Scholar]
- 12.Olson VA, Wetter JA, Friesen PD. Baculovirus transregulator IE1 requires a dimeric nuclear localization element for nuclear import and promoter activation. J Virol. 2002;76:9505–9515. doi: 10.1128/JVI.76.18.9505-9515.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fisher PA, Berrios M, Blobel G. Isolation and characterization of a proteinaceous subnuclear fraction composed of nuclear matrix, peripheral lamina, and nuclear pore complexes from embryos of drosophila melanogaster. J Cell Biol. 1982;92:674–686. doi: 10.1083/jcb.92.3.674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wilson D, Deutsch WA, Kelley MR. Drosophila ribosomal protein S3 contains an activity that cleaves DNA at apurinic/apyrimidinic sites. J Biol Chem. 1994;269:25359–25364. [PubMed] [Google Scholar]
- 15.Hegde V, Yadavilli S, Deutsch WA. Knockdown of ribosomal protein S3 protects human cells from genotoxic stress. DNA Repair (Amst) 2007;6:194–199. doi: 10.1016/j.dnarep.2006.09.004. [DOI] [PubMed] [Google Scholar]
- 16.Okumura M, Tsuzuki H, Tomita B. A rapid detection method for paralytic shellfish poisoning toxins by cell bioassay. Toxicon. 2005;46:93–98. doi: 10.1016/j.toxicon.2005.03.018. [DOI] [PubMed] [Google Scholar]
- 17.Maeda H, Hori S, Nishitoh H, Ichijo H, Ogawa O, Kakehi Y, Kakizuka A. Tumor growth inhibition by arsenic trioxide (As2O3) in the orthotopic metastasis model of androgen-independent prostate cancer. Cancer Res. 2001;61:5432–5440. [PubMed] [Google Scholar]
- 18.Hegde V, Kelley MR, Xu Y, Mian IS, Deutsch WA. Conversion of the bifunctional 8-oxoguanine/beta-delta apurinic/apyrimidinic DNA repair activities of Drosophila ribosomal protein S3 into the human S3 monofunctional beta-elimination catalyst through a single amino acid change. J Biol Chem. 2001;276:27591–27596. doi: 10.1074/jbc.M101213200. [DOI] [PubMed] [Google Scholar]
- 19.Isono M, Cruz MC, Chen S, Hong SW, Ziyadeh FN. Extracellular signal-regulated kinase mediates stimulation of TGF-beta1 and matrix by high glucose in mesangial cells. J Am Soc Nephrol. 2000;11:2222–2230. doi: 10.1681/ASN.V11122222. [DOI] [PubMed] [Google Scholar]
- 20.Jung YD, Nakano K, Liu W, Gallick GE, Ellis LM. Extracellular signal-regulated kinase activation is required for up-regulation of vascular endothelial growth factor by serum starvation in human colon carcinoma cells. Cancer Res. 1999;59:4804–4807. [PubMed] [Google Scholar]
- 21.Bommer UA, Lutsch G, Stahl J, Bielka H. Eukaryotic initiation factors eIF-2 and eIF-3: interactions, structure, and localization in ribosomal initiation complexes. Biochimie. 1991;73:1007–1019. doi: 10.1016/0300-9084(91)90142-n. [DOI] [PubMed] [Google Scholar]
- 22.Westermann P, Heumann W, Bommer UA, Bielka H, Nygard O, Hultin T. Crosslinking of initiation factor eIF-2 to proteins of the small subunit of rat liver ribosomes. FEBS Lett. 1979;97:101–104. doi: 10.1016/0014-5793(79)80061-7. [DOI] [PubMed] [Google Scholar]
- 23.Tolan DR, Hershey JW, Traut RT. Crosslinking of eukaryotic initiation factor eIF3 to the 40S ribosomal subunit from rabbit reticulocytes. Biochimie. 1983;65:427–436. doi: 10.1016/s0300-9084(83)80062-5. [DOI] [PubMed] [Google Scholar]
- 24.Kim J, Chubatsu LS, Admon A, Stahl J, Fellows R, Linn S. Implication of mammalian ribosomal protein S3 in the processing of DNA damage. J Biol Chem. 1995;270:13620–13629. doi: 10.1074/jbc.270.23.13620. [DOI] [PubMed] [Google Scholar]
- 25.Kim SH, Lee JY, Kim J. Characterization of a wide range base-damage-endonuclease activity of mammalian rpS3. Biochem Biophys Res Commun. 2005;328:962–967. doi: 10.1016/j.bbrc.2005.01.045. [DOI] [PubMed] [Google Scholar]
- 26.Samuel T, Okada K, Hyer M, Welsh K, Zapata JM, Reed JC. cIAP1 Localizes to the nuclear compartment and modulates the cell cycle. Cancer Res. 2005;65:210–218. [PubMed] [Google Scholar]
- 27.Lopez-Soler RI, Moir RD, Spann TP, Stick R, Goldman RD. A role for nuclear lamins in nuclear envelope assembly. J Cell Biol. 2001;154:61–70. doi: 10.1083/jcb.200101025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Parker PJ, Katan M, Waterfield MD, Leader DP. The phosphorylation of eukaryotic ribosomal protein S6 by protein kinase C. Eur J Biochem. 1985;148:579–586. doi: 10.1111/j.1432-1033.1985.tb08879.x. [DOI] [PubMed] [Google Scholar]
- 29.Hasler P, Brot N, Weissbach H, Parnassa AP, Elkon KB. Ribosomal proteins P0, P1, and P2 are phosphorylated by casein kinase II at their conserved carboxyl termini. J Biol Chem. 1991;266:13815–13820. [PubMed] [Google Scholar]
- 30.Kim HD, Lee JY, Kim J. Erk phosphorylates threonine 42 residue of ribosomal protein S3. Biochem Biophys Res Commun. 2005;333:110–115. doi: 10.1016/j.bbrc.2005.05.079. [DOI] [PubMed] [Google Scholar]
- 31.Ballif BA, Roux PP, Gerber SA, MacKeigan JP, Blenis J, Gygi SP. Quantitative phosphorylation profiling of the ERK/p90 ribosomal S6 kinase-signaling cassette and its targets, the tuberous sclerosis tumor suppressors. Proc Natl Acad Sci U S A. 2005;102:667–672. doi: 10.1073/pnas.0409143102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shi Y, Hsu JH, Hu L, Gera J, Lichtenstein A. Signal pathways involved in activation of p70S6K and phosphorylation of 4E-BP1 following exposure of multiple myeloma tumor cells to interleukin-6. J Biol Chem. 2002;277:15712–15720. doi: 10.1074/jbc.M200043200. [DOI] [PubMed] [Google Scholar]
- 33.Chen CC, Wang JK, Chen WC, Lin SB. Protein kinase C eta mediates lipopolysaccharide-induced nitric-oxide synthase expression in primary astrocytes. J Biol Chem. 1998;273:19424–19430. doi: 10.1074/jbc.273.31.19424. [DOI] [PubMed] [Google Scholar]
- 34.Chantome A, Pance A, Gauthier N, Vandroux D, Chenu J, Solary E, Jeannin JF, Reveneau S. Casein kinase II-mediated phosphorylation of NF-kappaB p65 subunit enhances inducible nitric-oxide synthase gene transcription in vivo. J Biol Chem. 2004;279:23953–23960. doi: 10.1074/jbc.M313731200. [DOI] [PubMed] [Google Scholar]
- 35.Koteiche HA, McHaourab HS. Mechanism of chaperone function in small heat-shock proteins. Phosphorylation-induced activation of two-mode binding in alphaB-crystallin. J Biol Chem. 2003;278:10361–10367. doi: 10.1074/jbc.M211851200. [DOI] [PubMed] [Google Scholar]
- 36.Luciano BS, Hsu S, Channavajhala PL, Lin LL, Cuozzo JW. Phosphorylation of threonine 290 in the activation loop of Tpl2/Cot is necessary but not sufficient for kinase activity. J Biol Chem. 2004;279:52117–52123. doi: 10.1074/jbc.M403716200. [DOI] [PubMed] [Google Scholar]
- 37.Murakami M, Kataoka K, Fukuhara S, Nakagawa O, Kurihara H. Akt-dependent phosphorylation negatively regulates the transcriptional activity of dHAND by inhibiting the DNA binding activity. Eur J Biochem. 2004;271:3330–3339. doi: 10.1111/j.1432-1033.2004.04267.x. [DOI] [PubMed] [Google Scholar]
- 38.Hayakawa J, Depatie C, Ohmichi M, Mercola D. The activation of c-Jun NH2-terminal kinase (JNK) by DNA-damaging agents serves to promote drug resistance via activating transcription factor 2 (ATF2)-dependent enhanced DNA repair. J Biol Chem. 2003;278:20582–20592. doi: 10.1074/jbc.M210992200. [DOI] [PubMed] [Google Scholar]
- 39.Jang CY, Lee JY, Kim J. RpS3, a DNA repair endonuclease and ribosomal protein, is involved in apoptosis. FEBS Lett. 2004;560:81–85. doi: 10.1016/S0014-5793(04)00074-2. [DOI] [PubMed] [Google Scholar]
- 40.Kim SH, Kim J. Reduction of invasion in human fibrosarcoma cells by ribosomal protein S3 in conjunction with Nm23-H1 and ERK. Biochim Biophys Acta. 2006;1763:823–832. doi: 10.1016/j.bbamcr.2006.03.011. [DOI] [PubMed] [Google Scholar]
- 41.Fan Z, Beresford PJ, Oh DY, Zhang D, Lieberman J. Tumor suppressor NM23-H1 is a granzyme A-activated DNase during CTL-mediated apoptosis, and the nucleosome assembly protein SET is its inhibitor. Cell. 2003;112:659–672. doi: 10.1016/s0092-8674(03)00150-8. [DOI] [PubMed] [Google Scholar]
- 42.Martinvalet D, Zhu P, Lieberman J. Granzyme A induces caspase-independent mitochondrial damage, a required first step for apoptosis. Immunity. 2005;22:355–370. doi: 10.1016/j.immuni.2005.02.004. [DOI] [PubMed] [Google Scholar]
- 43.Chowdhury D, Beresford PJ, Zhu P, Zhang D, Sung JS, Demple B, Perrino FW, Lieberman J. The exonuclease TREX1 is in the SET complex and acts in concert with NM23-H1 to degrade DNA during granzyme A-mediated cell death. Mol Cell. 2006;23:133–142. doi: 10.1016/j.molcel.2006.06.005. [DOI] [PubMed] [Google Scholar]