

Abstract

An investigation into Pd-catalyzed C–N cross-coupling reactions of aryl iodides is described. NaI is shown to have a significant inhibitory effect on these processes. By switching to a solvent system in which the iodide byproduct was insoluble, reactions of aryl iodides were accomplished with the same efficiencies as aryl chlorides and bromides. Using catalyst systems based on certain biarylphosphine ligands, aryl iodides were successfully reacted with an array of primary and secondary amines in high yields. Lastly, reactions of heteroarylamines and heteroaryliodides were also conducted in high yields.
In Pd-catalyzed C–C cross-coupling reactions aryl iodides have generally been better substrates than the corresponding aryl bromides or chlorides.1 However, this has not been the case for Pd-catalyzed C–N bond-forming processes,2 in which aryl iodides have traditionally been the least successful substrates of these three aryl halides. Recent advances in this field have allowed for reactions of aryl chlorides to reach the same efficiencies as aryl bromides,3 but relatively little progress has been made in increasing the proficiency of Pdcatalyzed C–N cross-coupling processes using aryl iodides.3b,4
Herein, we describe an investigation into the cause of the modest results obtained with aryl iodides as substrates. We also report that catalysts using certain biarylphosphine ligands allow for the C–N cross-coupling reactions of aryl iodides to be accomplished with the same (or better) efficiencies as with other aryl halides.

Oxidative addition complexes, using P(o-tol)3 as the ligand, exist as stable bridging halide dimers in solution.5 While the bridging bromide dimers are readily converted to [(o-tol)3P]2Pd(amine)Br upon treatment with an amine, the process with the corresponding bridging iodide dimers is endergonic.5a It was proposed that the stability of the bridging iodide dimers could be the cause of the lower observed efficiency in reactions involving aryl iodides. Preventing dimer formation would be expected to render amine binding more favorable, resulting in comparable reactivity between aryl iodides, bromides, and chlorides.
Recently we disclosed a catalyst system, comprised of ligand 1 (BrettPhos), which showed excellent reactivity for the C–N cross-coupling reactions of aryl chlorides.3a We found that the aryl bromide and aryl chloride oxidative addition complexes, 1•Pd(Ar)X, existed as monomers in solution. Based on this, we hypothesized that the use of 1 could retard the formation of the bridging dimers in the couplings of aryl iodides and improve the overall efficiency of these reactions.
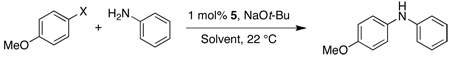
We began our studies by examining several biarylphosphine ligands for the reaction of 4-iodoanisole and aniline (Figure 1). Using a precatalyst based on ligand 1 (5), the reaction gave a 99% yield (GC) after 10 minutes at room temperature. When a precatalyst of 2 (6), which was previously our best biarylphosphine ligand for the coupling of aryl iodides, was employed, a 7% yield of product was detected. Two other precatalysts, with ligands 3 (7) and 4 (8), which have been shown to yield highly active catalysts for the Pd-catalyzed C–N cross-coupling reactions of aryl bromides and aryl chlorides, each gave <14% of the desired product. These results show that for the amination reactions of aryl iodides, the catalyst derived from 1 is highly active and displays marked differences from catalysts based on other biarylphosphine ligands.
Figure 1.

Results observed for the coupling of aniline and 4-iodoanisole in toluene using 1 mol% Pd at rt for 10 min with various ligands.
Given that Pd-catalyzed C–N bond-forming reactions are carried out in a variety of solvents, we investigated the coupling of aniline with 4-bromanisole and 4-iodoanisole in three of those most commonly employed (toluene, dioxane and DME) to probe the effect of this parameter. In toluene, the coupling of the two aryl halides with aniline proceeded at roughly the same rate (Table 1). In dioxane and DME, however, the reactions with 4-bromoanisole were 2 and 7 times faster than the reactions with 4-iodoanisole. We would not expect the bridging dimers to be more reactive in toluene than in DME or dioxane. Moreover, kinetic studies indicated that the reaction in toluene is first order in 5. Taken together, these results indicate that the bridging iodide dimers are not playing a critical role in the overall process and that there must be another cause for the observed solvent dependence and iodide effect. It is possible that bridging dimers are important when other ligands are used (vide supra).
Table 1.
Solvent effects on the coupling of aniline and 4-haloanisolea
 | |||
|---|---|---|---|
| Solvent | Halide | Reaction Time (min) | Solubility of NaI (mM) |
| Toluene | Br | 6 | 0.33 |
| Toluene | I | 6 | 0.33 |
| Dioxane | Br | 10 | 0.8 |
| Dioxane | I | 23 | 0.8 |
| DME | Br | 10 | 797 |
| DME | I | 70 | 797 |
ArX (1 mmol), aniline (1.2 mmol), NaOt-Bu (1.2 mmol), solvent (2 mL/mmol), 5 (1 mol%).
It has previously been shown that NaI can inhibit the C–N cross-coupling reactions of aryl bromides in DME.3b We postulated that the solvent dependence that we were detecting for the cross-coupling of aryl iodides could be due to the solubility of the NaI byproduct in the reaction medium. To test this hypothesis, we first determined the solubility of NaI in the three solvents used in our study.6 As expected, the greater the solubility of NaI in the solvent, the slower the reaction proceeded (Table 1). In toluene, where NaI is sparingly soluble, there is not enough of the salt in solution to inhibit the reaction. This is consistent with our observation that the rates of reaction for both the aryl iodide and aryl bromide are comparable. These solvent effects are opposite of what has been seen for C–N cross-coupling reactions using a catalyst system based on a bidentate ligand;3b this suggests that these results are only relevant for catalysts based on biarylphosphine ligands.
To further understand the mechanism of iodide inhibition, reaction calorimetry7 was used to study the effect of added NaI on the initial rates for the reaction of aniline and 4-bromoanisole in DME. As expected, an inverse order in NaI was observed, which showed saturation at ca. 10% added iodide (Figure 2). We believe that these results suggest that the iodide could be inhibiting the reaction by binding to a Pd(II) intermediate and forming a Pd ate complex (Scheme 1). This inhibition could occur at two different points on the catalytic cycle. First, the iodide could compete with the amine for binding to the Pd(II) oxidative addition complex (9). Second, the iodide could bind to the Pd(II) amido complex (10), slowing the rate of reductive elimination.8
Figure 2.

Effect of added NaI on the cross-coupling of aniline and 4-bromoanisole in DME with 1 mol% 5.
Scheme 1.

Iodide inhibition pathways.
We also wanted to further verify our postulate that these reactions were most efficient in toluene due to the lack of solubility of the NaI byproduct. Using reaction calorimetry the coupling of aniline and 4-iodoanisole in toluene was shown to be zeroth order in added NaI (Figure 3,  ). The same reaction, with added tetrahexylammonium iodide, showed an inverse order in added iodide (Figure 3,
). The same reaction, with added tetrahexylammonium iodide, showed an inverse order in added iodide (Figure 3,  ).9 This supports our hypothesis that the reactions are most effective in toluene because the NaI byproduct precipitates and is not available to inhibit the reaction.
).9 This supports our hypothesis that the reactions are most effective in toluene because the NaI byproduct precipitates and is not available to inhibit the reaction.
Figure 3.

Effect of different iodide sources on the cross-coupling of aniline and 4-iodoanisole in toluene with 1 mol% 5.
Having found a catalyst and conditions that showed high activities for the cross-coupling reactions of amines with aryl iodides, the scope of this system was next examined (Table 2). The reaction of aniline with 4-iodoanisole was successfully performed in high yield at 0.01 mol% catalyst loading in as little as 5 minutes. This rate is >10 fold faster than we previously reported for the reaction of 4-chloroanisole and aniline under identical conditions.3a It is also notable that this is the first reported C–N cross-coupling reaction using 4-iodoanisole to be carried out below 0.5 mol% catalyst loading.3b Ortho substitution on the aryl iodide and electronwithdrawing groups on the aniline were also well tolerated. The coupling of linear and branched primary aliphatic amines could also be carried out at low catalyst loadings and in high yields with <1% diarylation observed. This system also showed high chemoselectivity for the coupling of an iodide over a chloride. By switching to Cs2CO3 as the base, a number of functional groups were all well tolerated. These substrates could be transformed using 0.1–0.2 mol% catalyst; these are the first examples of C–N cross-coupling reactions to be reported with a weak base using <0.5 mol% catalyst (most of the examples reported in the literature use 1 mol% or more catalyst).3b
Table 2.
Cross-coupling of primary amines and aryl iodides
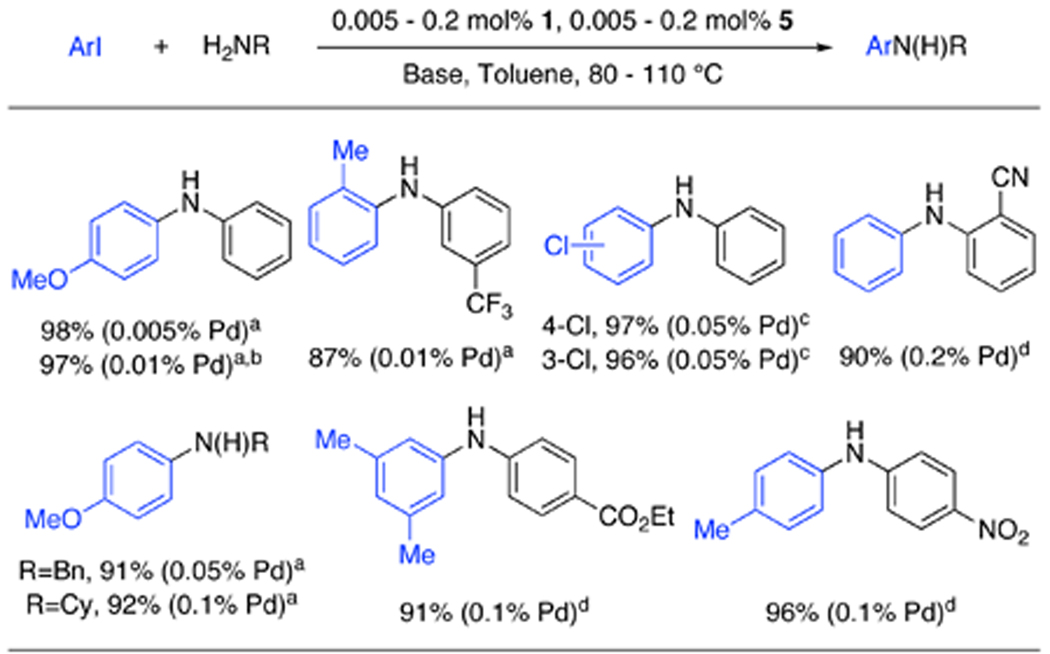 |
ArI (1.0 equiv), amine (1.4 equiv), NaOt-Bu (1.4 equiv), 1 (0.005 – 0.2 mol%), 5 (0.005 – 0.2 mol%), toluene, 80–110 °C.
5 min reaction time.
1.0 equiv of amine was used.
Cs2CO3 was used as the base.
We next set out to extend the efficiency we had seen for the coupling of aryl iodides with primary amines to reactions of secondary amines. We found that the precatalyst 8, based on RuPhos (4), showed similar reactivity for the coupling of aryl iodides and aryl bromides with secondary amines.3c,10 Further, for the coupling of 4-iodoanisole and morpholine precatalyst 8 gave a 99% yield, whereas catalyst systems based on other biarylphosphine ligands gave a maximum of only 36% of the desired product (Figure 4). Using 8, both cyclic and acyclic secondary aliphatic amines in combination with aryl iodides gave products in excellent yields with 0.025 – 0.1 mol% catalyst. N-Methylaniline and diphenylamine were also successfully coupled with aryl iodides with higher turnover numbers than any previously reported system. Functional group tolerance was also achieved by using Cs2CO3 as the base with 0.1 mol% 8. Also, as shown for the reaction of piperidine with 4-iodoanisole, these couplings proceed efficiently in dioxane as well as in toluene, although a slightly higher quantity of catalyst is required.
Figure 4.

Results observed for the coupling of morpholine and 4-iodoanisole in toluene using 1 mol% Pd at 80 °C for 3 min with various ligands.
Finally, we broadened the scope of this process to include the reactions of heteroarylamines and heteroaryliodides (Table 4). The lack of solubility of these very polar substrates in toluene necessitated a switch to t-BuOH. In this solvent aminopyrazine, 2-aminopyrimidine, and 2-aminopyridine were all coupled with aryl iodides for the first time in good to excellent yields.11 An array of heteroaryliodides, which have been poor coupling partners in the past, were also effectively transformed to product (Table 4).3b,12
Table 4.
Cross-coupling of heteroaryliodides and heteroarylaminesa
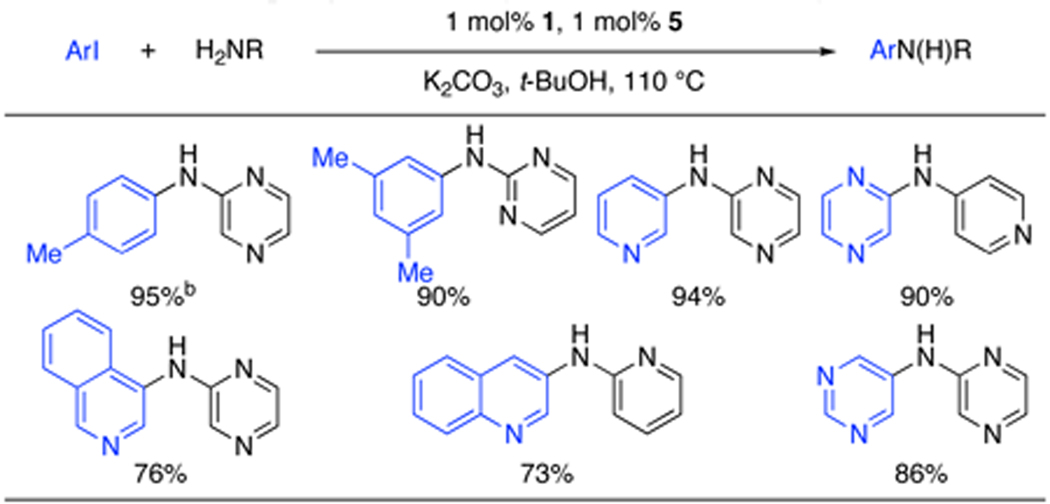 |
ArI (1.0 equiv), amine (1.4 equiv), K2CO3 (1.4 equiv), 1 (1 mol%), 5(1 mol%), t-BuOH (2 mL/mmol), 110 °C.
0.5 mol% Pd was used.
In summary, precatalysts 5 and 8 have been shown to be highly active in Pd-catalyzed amination reactions of aryl iodides. With ligand 1 (a component of 5) the formation of the bridging iodide dimers was retarded, enhancing the reactivity of the system. Inhibition by NaI was also shown to have a strong effect on these reactions. By switching to a solvent in which the iodide salt was insoluble no inhibition was observed and the reactions of aryl iodides became as efficient as other aryl halides. These catalyst systems were then applied to the coupling of aryl iodides in high yields and with low catalyst loadings with an array of both primary and secondary amines. Finally, reactions of heteroarylamines and heteroaryliodides were performed in high yields. This system is complimentary to previously reported Cu based catalysts13 and allows the synthetic chemist to choose from aryl chlorides, bromides, or iodides, depending on their availability or ease of synthesis, for use in Pd-catalyzed C–N cross-coupling processes.
Supplementary Material
Table 3.
Cross-coupling of secondary amines and aryl iodidesa
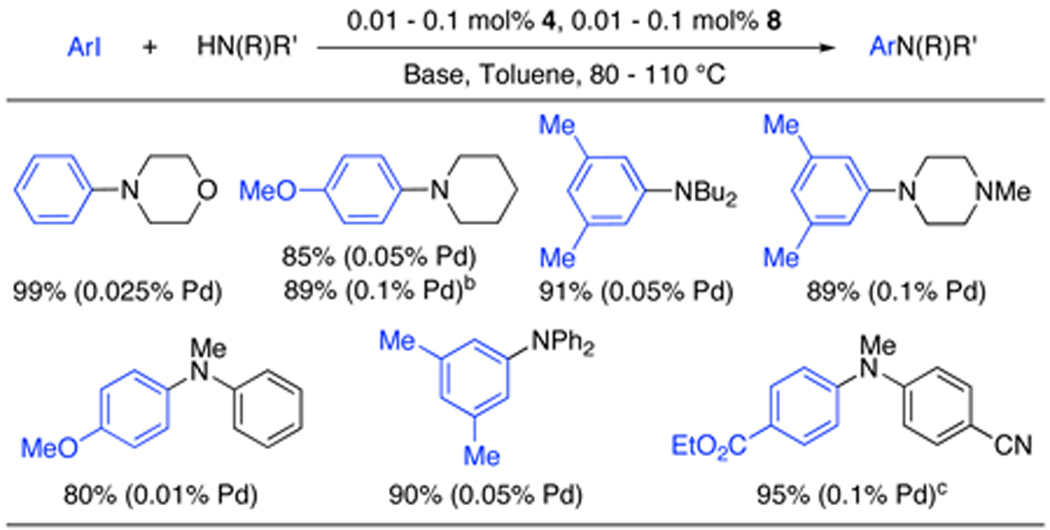 |
ArI (1.0 equiv), amine (1.4 equiv), NaOt-Bu (1.4 equiv), 4 (0.01 - 0.1 mol%), 8 (0.01 - 0.1 mol%), toluene, 80–110 °C.
Dioxane was used as the solvent.
Cs2CO3 was used as the base.
ACKNOWLEDGMENT
We thank the National Institutes of Health (NIH) for financial support of this project (Grant GM-58160). We thank Merck, BASF (Pd compounds), Chemetall (Cs2CO3), and Nippon Chemical for additional support. BPF thanks the William Asbornsen Albert Memorial Fund for a fellowship. We thank Dr. Mark R. Biscoe for helpful discussions. The Varian NMR instrument used was supported by the NSF (Grants CHE 9808061 and DBI 9729592).
Footnotes
SUPPORTING INFORMATION. Procedural, and spectral data is available free of charge via the Internet at http://pubs.acs.org.
REFERENCES
- 1.(a) Farina V. Adv. Synth. Catal. 2004;346:1553. [Google Scholar]; (b) Stanforth SP. Tetrahedron. 1998;54:263. [Google Scholar]; (c) Miyaura N, Suzuki A. Chem. Rev. 1995;95:2457. [Google Scholar]
- 2.(a) Urgaonkar S, Xu J, Verkade JG. J. Org. Chem. 2003;68:8416. doi: 10.1021/jo034994y. [DOI] [PubMed] [Google Scholar]; (b) Bolm C, Hildebrand JP. J. Org. Chem. 2000;65:169. doi: 10.1021/jo991342u. [DOI] [PubMed] [Google Scholar]; (c) Wolfe JP, Buchwald SL. J. Org. Chem. 1996;61:1133. [Google Scholar]; (d) Kosugi M, Kameyama M, Migita T. Chem. Lett. 1983:927. [Google Scholar]
- 3.(a) Fors BP, Watson DA, Biscoe MR, Buchwald SL. J. Am. Chem. Soc. 2008;130:13552. doi: 10.1021/ja8055358. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Shen Q, Ogata T, Hartwig JF. J. Am. Chem. Soc. 2008;130:6586. doi: 10.1021/ja077074w. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Biscoe MR, Fors BP, Buchwald SL. J. Am. Chem. Soc. 2008;130:6686. doi: 10.1021/ja801137k. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Marion N, Navarro O, Mei J, Stevens ED, Scott NM, Nolan SP. J. Am. Chem. Soc. 2006;128:4101. doi: 10.1021/ja057704z. [DOI] [PubMed] [Google Scholar]
- 4.(a) Urgaonkar S, Nagarajan M, Verkade JG. J. Org. Chem. 2003;68:452. doi: 10.1021/jo0205309. [DOI] [PubMed] [Google Scholar]; (b) Ali MH, Buchwald SL. J. Org. Chem. 2001;66:2560. doi: 10.1021/jo0008486. [DOI] [PubMed] [Google Scholar]; (c) Hamann BC, Hartwig JF. J. Am. Chem. Soc. 1998;120:7369. [Google Scholar]
- 5.(a) Widenhoefer RA, Buchwald SL. Organometallics. 1996;15:2755. [Google Scholar]; (b) Louie J, Hartwig JF. Tetrahedron Lett. 1995;36:3609. [Google Scholar]
- 6.Solubilities were measured volumetrically in 1 L of solvent at room temperature.
- 7.Blackmond DG. Angew. Chem. Int. Ed. 2005;44:4302. doi: 10.1002/anie.200462544. [DOI] [PubMed] [Google Scholar]
- 8.The initial results of experiments were inconclusive as to whether one or both modes of inhibition were taking place during the reaction. Additional studies to differentiate between the two will be carried out.
- 9.7.5% tetrahexylammonium iodide was the maximum amount used in the inhibition studies due to its low solubility in toluene.
- 10.Milne JE, Buchwald SL. J. Am. Chem. Soc. 2004;126:13028. doi: 10.1021/ja0474493. [DOI] [PubMed] [Google Scholar]
- 11.(a) Anderson KW, Tundel RE, Ikawa T, Altman RA, Buchwald SL. Angew. Chem. Int. Ed. 2006;45:6523. doi: 10.1002/anie.200601612. [DOI] [PubMed] [Google Scholar]; (b) Yin J, Zhao MM, Huffman MA, McNamara JM. Org. Lett. 2002;4:3481. doi: 10.1021/ol0265923. [DOI] [PubMed] [Google Scholar]; (c) Jonckers THM, Maes BUW, Lemière GLF, Dommisse R. Tetrahedron. 2001:7027. [Google Scholar]
- 12.Parrot I, Ritter G, Wermuth CG, Hibert M. Synlett. 2002;7:1123. [Google Scholar]
- 13.(a) Ma D, Cai Q. Acc. Chem. Res. 2008;41:1450. doi: 10.1021/ar8000298. [DOI] [PubMed] [Google Scholar]; (b) Ley SV, Thomas AW. Angew. Chem. Int. Ed. 2003;42:5400. doi: 10.1002/anie.200300594. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.