

Abstract
The regulation of multipotent cardiac progenitor cell (CPC) expansion and subsequent differentiation into cardiomyocytes, smooth muscle, or endothelial cells is a fundamental aspect of basic cardiovascular biology and cardiac regenerative medicine. However, the mechanisms governing these decisions remain unclear. Here, we show that Wnt/β-Catenin signaling, which promotes expansion of CPCs1–3, is negatively regulated by Notch1-mediated control of phosphorylated β-Catenin accumulation within CPCs, and that Notch1 activity in CPCs is required for their differentiation. Notch1 positively, and β-Catenin negatively, regulated expression of the cardiac transcription factors, Isl1, Myocd and Smyd1. Surprisingly, disruption of Isl1, normally expressed transiently in CPCs prior to their differentiation4, resulted in expansion of CPCs in vivo and in an embryonic stem (ES) cell system. Furthermore, Isl1 was required for CPC differentiation into cardiomyocyte and smooth muscle cells, but not endothelial cells. These findings reveal a regulatory network controlling CPC expansion and cell fate that involve unanticipated functions of β-Catenin, Notch1 and Isl1 that may be leveraged for regenerative approaches involving CPCs.
Keywords: β-Catenin, Notch1, Isl1, cardiac progenitors, Myocd
Heart malformation is the most frequent form of human birth defects and heart disease remains the number one killer of adults in the developed world, largely because of the limited regenerative capacity of the heart. Recent advances have provided insights into potential therapies based on multipotent cardiac progenitor cells (CPCs). Such CPCs can be isolated from early embryos or embryonic stem (ES) cells and cultured to differentiate into numerous cardiac cell types4–12. For instance, Nkx2.5+, Flk1+ or Isl1+ CPCs purified from embryoid bodies (EBs) can each give rise to cardiomyocyte, endothelial, and smooth muscle lineages7, 8, 10, 12.
Nkx2.5 is an ancient cardiac gene activated in CPCs of early embryos13. Nkx2.5+ cells and their progeny populate the precardiac mesoderm located dorsal to the cardiac region and the developing heart tube in vivo14. Isolated Nkx2.5+ cells spontaneously differentiate into distinct cardiac cell lineages including cardiomyocytes, smooth muscle cells and endothelial cells in vitro7, 12. These cardiac cell lineages can also be generated from cells expressing Flk1, a marker of the primitive streak in early embryogenesis10 or Isl1, a CPC marker8, 15. All of these CPCs exhibit overlapping expression patterns in precardiac mesodermal cells in vivo8 and have similar differentiation potential in vitro7, 8, 10, 12, suggesting that they comprise a similar CPC population. Although these multipotent CPCs hold great potential for cardiac repair, the mechanisms that regulate their self-renewal, expansion, and differentiation remain elusive.
We, and others, recently reported that canonical Wnt signaling is a critical regulator of Nkx2.5+ and Isl1+ CPCs and is responsible for their expansion in vivo and in vitro1–3. The inactivation of β-Catenin, the transcriptional mediator of canonical Wnt signaling, in precardiac mesoderm resulted in nearly complete loss of Isl1 cells that contribute to the right ventricle2. Conversely, stabilization of β-Catenin in the same cells led to an expansion in the number of CPCs2 in vivo, while Wnt/β-Catenin signaling promoted the renewal of CPCs isolated from ES cells2, 3. Notch signaling reciprocally affects Wnt signals in many contexts16 and is thought to inhibit cardiac differentiation17, 18, although its function within CPCs in vivo is unknown. Ultimately, these and other early signals must be integrated with a network of transcriptional regulators that influence CPCs.
To examine the CPC-autonomous role of Notch1 signaling in vivo, we deleted Notch1 in precardiac mesodermal progenitors by crossing Notch1flox mice19 with mice containing Cre recombinase in the Isl1 locus (Isl1Cre)20, resulting in Cre-mediated recombination in early CPCs by E7.75. The resulting Notch1-null embryos failed to populate the developing right ventricle segment, which is derived from Isl1+ CPCs (Fig. 1a–c, g–i). Strikingly, the affected Isl1+ CPC pool dorsal to the developing heart was expanded with an increased percentage of proliferating cells marked by a phosphohistone H3 (PH3) antibody (Fig. 1d–f, j–m). The accumulation and proliferation of CPCs behind the developing heart was similar to the effect of stabilized β-Catenin on CPCs2, although in the latter CPCs also migrated into the heart.
Figure 1.
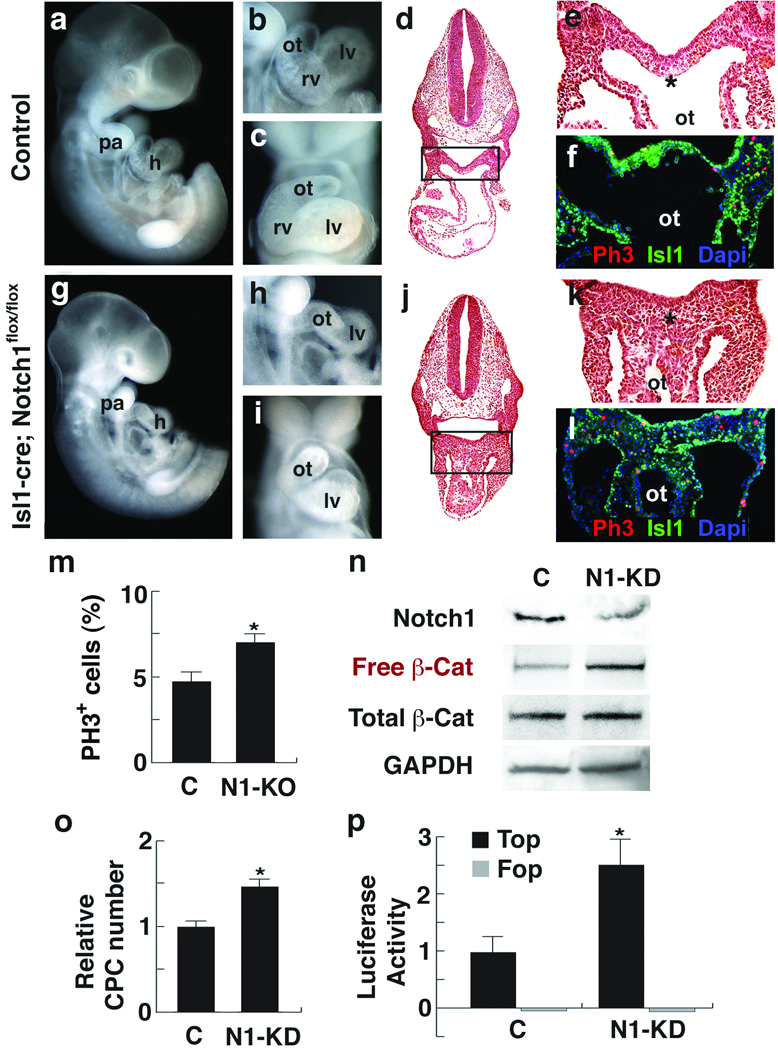
Notch1 loss-of-function causes CPC expansion and increases free β-Catenin levels. a–f, Control embryos. g–l, Isl1Cre, Notch1flox/flox embryos (N1-KO). a,g, Lateral views of ED10.5 embryos. b, c, h, i, Lateral (b,h) or frontal (c,i) view of embryos focused on cardiac regions showing absence of right ventricle (rv) in mutants. d,e, j, k, Transverse sections (H&E) of embryos (d, j) with enlargement of boxed areas (e, k) showing hyperplasia of precardiac progenitors (asterisk). f, l, Phosphohistone3 (Ph3, red) and Isl1 (green) immunostaining of transverse sections through the precardiac region. To compensate for the severe downregulation of Isl1 in Notch1 mutant embryos, Isl1 signals were amplified with the TSA system. DAPI (blue) was used to counterstain the nuclei. m, Percentage of ph3-positive cells in precardiac mesoderm region shown in e and k (mean ± s. d.; n=4; *P < 0.01). n, Western analyses of FACS-purified CPCs transfected with control siRNA (C) or Notch1 siRNA (N1-KD) using Notch1, free or total β-Catenin antibodies. Free β-Catenin antibodies detect dephosphorylated β-Catenin, the effector molecule of the Wnt/β-Catenin signaling pathway. GAPDH antibody was used as a control. o, Relative number of cells on the 2nd day after transfecting CPCs with control or Notch1 siRNA (mean ± s. d.; n=6; *P < 0.01). p, Top/Fop flash activity in CPCs transfected with indicated siRNA. Top flash is a luciferase reporter with Tcf binding sites to read Wnt/β-Catenin signaling activity. Fop flash contains mutated Tcf binding sites. Luciferase values were normalized to Renilla activity (mean ± s. d.; n=3; *P < 0.01). h, heart; pa, pharyngeal arch; ot, outflow tract; lv, left ventricle. Scale bars, 250 µm (a, g) or 100 µm (b–e, h–k).
The striking similarity of Notch1 loss-of-function and β-Catenin gain-of-function mutants in CPCs led us to hypothesize that Notch and β-Catenin signaling intersect during CPC fate or expansion decisions. No significant expression changes of genes involved in the Notch signal transduction pathway were observed in β-Catenin stabilized mice (not shown), suggesting it is unlikely that β-Catenin regulates Notch signaling in CPCs. Using an ES cell line with a bacterial artificial chromosome (BAC) containing green fluorescent protein (GFP) in the Nkx2.5 locus21, we isolated Nkx2.5-GFP+ cells by fluorescent-activated cell sorting (FACS). The Nkx2.5-GFP+ cells expressed high levels of Isl1 (Supp. fig. 1a), consistent with these cells representing CPCs. We knocked down (KD) Notch1 levels with Notch1 siRNAs in Nkx2.5-GFP+ CPCs cultured in a monolayer. Endogenous levels of Notch1 were considerably reduced by the siRNA transfection, determined by Western blot analysis (Fig. 1n). Consistent with the in vivo data, Notch1 KD resulted in an increased number of CPCs (Fig. 1o). Notch1 KD did not affect the levels of total β-Catenin in CPCs (Fig. 1n). However, the levels of dephosphorylated (free) β-Catenin, a form required to mediate Wnt/β-Catenin signaling, were considerably higher in the Notch1-KD CPCs (Fig. 1n). Consistent with this, Notch1-KD CPCs showed significantly increased levels of Topflash activity, a luciferase-based reporter system for Wnt/β-Catenin signaling (Fig. 1p). Increased levels of nuclear β-Catenin were also observed in the cardiac mesoderm of the Notch1 mutant embryo (Supp. fig. 1b). These findings suggest that Notch1 normally represses CPC expansion and negatively regulates the active form of β-Catenin.
To search for genes responsible for CPC expansion in an unbiased manner, we performed gene expression analyses of β-Catenin-stabilized CPCs in vivo. For this analysis, we generated RosaYFP; Isl1Cre; β-Catenin(ex3)loxPloxP embryos that express YFP in descendants of Isl1+ progenitors in the cardiac region with stabilized β-Catenin (Fig. 2a). YFP+ cells from embryonic (E) day 9.0 embryos, before cardiac dysfunction, were purified by FACS (Fig. 2b) and used for mRNA expression arrays.
Figure 2.
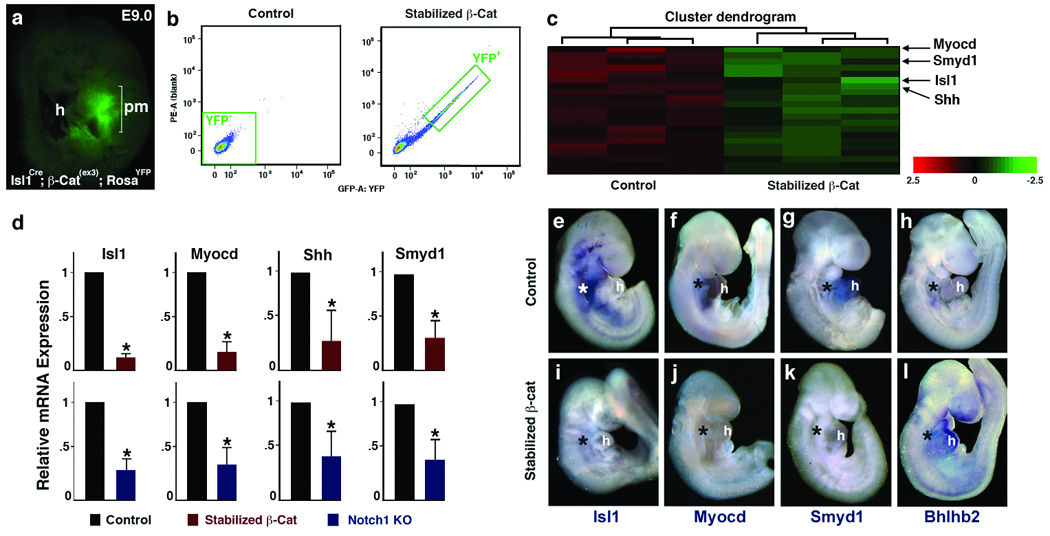
Identification of genes affected by stabilized β-Catenin in cardiac progenitors. a, Lateral view of RosaYFP; Isl1Cre; β-catenin(ex3)loxP embryo at E9.0 showing YFP+ cells in precardiac mesoderm (pm). b, Histograms of YFP+ cell populations from control (Isl1Cre, left) and stabilized β-cat (Isl1Cre; β-catenin(ex3)loxP, right) embryos. c, A heatmap of expression arrays showing significantly downregulated cardiac genes (green) in stabilized β-catenin pm cells. Color bar indicates fold change in log2 scale. d, qPCR data of downregulated genes in FACS-purified cardiac progenitors with stabilized β-Catenin (Top). These genes were similarly affected in pm of Notch1 loss-of-function embryos (Bottom). Data are mean ± s. d.; n=3; *P < 0.01. l, Whole-mount in situ hybridization of genes indicated from control (top) and stabilized β-Catenin (bottom) embryos at E 9.5. Asterisks indicate precardiac mesoderm. h, heart. Scale bars, 100 µm.
Many known targets of canonical Wnt signaling, including components of the Wnt signaling pathway, were upregulated in mutants, supporting the quality of the data set (Supp. table 1, Supp. fig. 1c). We found that the expression of genes implicated in cell proliferation and differentiation (e.g., Ndrg1, Bhlhb2, and Fgfs) was highly upregulated (4–11 fold) in mutants (Supp. table 1, Supp. fig. 1c). Unexpectedly, several genes essential for CPC development, including Isl1, Myocd, Shh and Smyd1 were significantly downregulated in the mutants and this was validated by quantitative real-time PCR (qPCR) (Fig. 2c, d). It was curious that Isl1 was downregulated upon stabilization of β-Catenin. In agreement with the array analyses, Isl1 transcripts were barely detectable in CPCs of β-Catenin-stabilized embryos by in situ hybridization (Fig. 2e, I, Supp. fig. 1d). Smyd1 and Myocd transcripts were also significantly downregulated in β-Catenin-stabilized embryos, while Bhlhb2 was upregulated specifically in the Isl1Cre domain (Fig. 2f–h, j–l, Supp. fig. 1d). Consistent with the opposing functions of Notch1 and β-Catenin described above, Isl1, Myocd, Shh and Smyd1 were significantly downregulated and Bhlhb2 was upregulated in Notch1 mutant embryos (Fig. 2d, Supp. fig. 1e).
Isl1 is a homeodomain-containing factor that is transiently expressed in CPCs prior to their migration into the heart tube, but is extinguished as further migration and differentiation proceed4. Although Isl1 is intuitively thought to promote CPC expansion based on its temporal expression, we investigated whether Isl1 downregulation mediates the expansion of CPCs observed in embryos with stabilized β-Catenin. To test this possibility, we used the Isl1Cre line described above which contains an IRES-Cre cassette inserted into the exon encoding the second LIM domain of Isl1, resulting in an Isl1-null allele20. The Isl1Cre mice were bred with RosaYFP mice to generate Isl1Cre/Cre; RosaYFP embryos. We quantified the number of YFP+ cells at E8.0 (5 somite stage), before Isl1Cre expression is initiated in neural cells, by FACS. Surprisingly, Isl1-null embryos had a significantly higher percentage of YFP+ cells than control embryos (Fig. 3a, b). The results suggest that Isl1 negatively regulates the number of CPCs in vivo. The significant increase is unlikely from higher Cre expression in Isl1-null embryos, since heterozygous Cre mice mediate recombination as efficiently as homozygous Cre mice.
Figure 3.
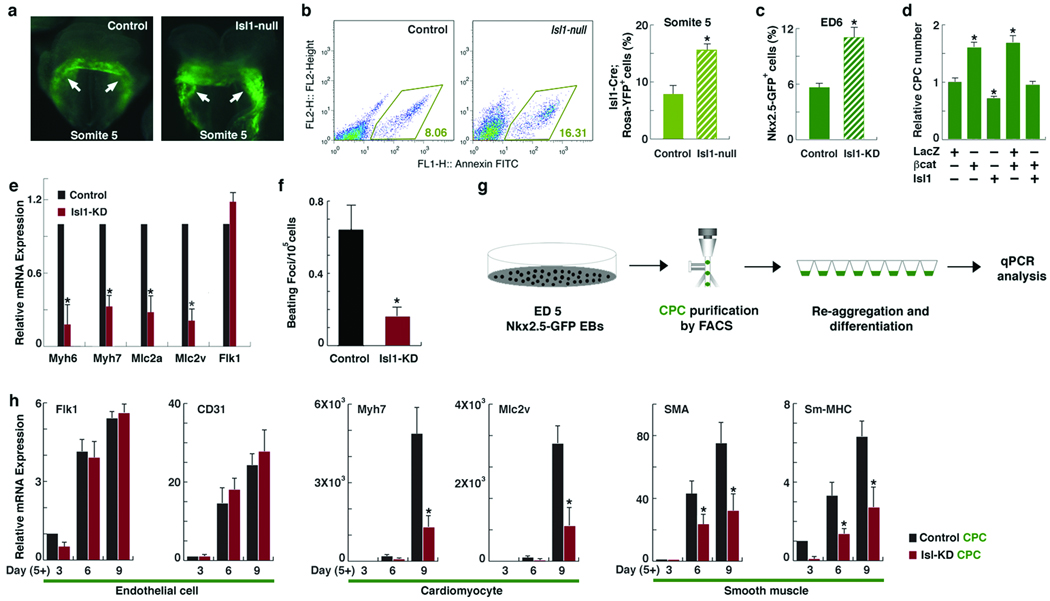
Isl1 loss-of-function results in expansion of CPCs and suppression of their myocardial and smooth muscle lineages.a, YFP expression in control (RosaYFP, Isl1Cre/+, left) and Isl1-null (RosaYFP, Isl1Cre/Cre, right) embryos at the 5-somite stage. Arrows indicate YFP+ CPCs. Scale bars, 50 µm. b, Quantification of YFP+ cells in indicated embryos at somite 5 (mean ± s. d.; n=3; *P < 0.01). c, Quantification of GFP+ cells in ED6 Nkx2.5-GFP EBs with or without Isl1 KD (mean ± s. d.; n=3; *P < 0.01). d, Relative number of cells on the 2nd day after transfecting EB-derived CPCs with lacZ, β-catenin, or Isl1 (mean ± s. d.; n=6; *P < 0.01). e, Relative mRNA expression of indicated genes in control or Isl1-KD EBs at ED 9, determined by qPCR (mean ± s. d.; n=4; *P < 0.01). f, Number of beating foci per 105 cells in control or Isl1-KD EBs at ED12. g, Schematic diagram of isolating CPCs from ES cells and their differentiation. h, Relative mRNA expression of endothelial (Flk1, CD31), cardiomyocyte (Myh7, Mlc2v) or smooth muscle (Sma, Sm-mhc) genes during CPC differentiation, determined by qPCR (mean ± s. e. m. ; n=4; *P < 0.05).
To determine if Isl1 also negatively regulates expansion of CPCs derived from pluripotent ES cells, we transiently knocked down Isl1 levels in the Nkx2.5-GFP ES cell line by introducing an Isl1-shRNA construct, which efficiently reduced Isl1 transcripts by ~75% (Supp. fig. 1f). We then quantified the number of Nkx2.5-GFP+ CPCs in EBs from embryoid day (ED) 6 as cardiac progenitors begin to emerge and differentiate from primitive mesoderm7, 8. The KD of Isl1 from ED0–3 did not change the number of Nkx2.5+ progenitors (data not shown). However, the KD of Isl1 from ED3–6, just after emergence of mesoderm, resulted in an increased cardiac progenitor population at ED6–8 (Fig. 3c, Supp. fig. 2a), consistent with our in vivo data.
These findings prompted us to test if Isl1 downregulation was required for CPC expansion induced by β-Catenin. We transfected Nkx2.5-GFP+ FACS-purified CPCs from day 5 EBs with a stabilized β-Catenin expression construct22 with or without an Isl1 expression construct. As previously reported, increased CPC expansion was evident two days after transfection with stabilized β-Catenin (Fig. 3d). However, co-transfection with Isl1 restored the number of CPCs to normal levels (Fig. 3d). This suggests that the decrease in Isl1 is necessary for Wnt/β-Catenin signaling–mediated expansion of CPCs.
Because Isl1 appeared to be involved in repressing expansion of CPCs, we investigated whether Isl1 promotes differentiation in the ES cell system. We generated a stable Isl1-KD ES cell line by introducing an Isl1 shRNA construct into Nkx2.5-GFP ES cells and clonally isolating cells with effective (~80%) Isl1-KD (Supp. fig. 1f). Similar to the transient Isl1-KD, the number of Nkx2.5-GFP+ cardiac progenitors was significantly increased at ED6 (Supp. fig. 2b). However, cells differentiated from the Isl1-KD ES cells showed severely reduced beating frequencies with compromised expression of cardiac sarcomeric genes (Myh6, Myh7, Mlc2a, Mlc2v) from ED9 (Fig. 3e, f). To determine the CPC-autonomous role of Isl1 during cardiac differentiation, we FACS-purified Nkx2.5-GFP+ CPCs from ED5 EBs and differentiated them by re-aggregating in suspension (Fig. 3g). Nkx2.5-GFP+ CPCs are multipotent and differentiate into myocardial, smooth muscle, and endothelial lineages7, 12. Normal levels of endothelial gene expression (CD31, Flk1) were observed in differentiating Isl1-KD CPCs (Fig. 3h). However, expression of cardiomyocyte and smooth muscle genes was severely downregulated (Fig. 3h). This suggests that Isl1 not only represses expansion of CPCs, but is also necessary for proper differentiation of CPCs into the myocardial and smooth muscle, but not endothelial, cell lineages.
Given that Isl1 loss-of-function suppressed cardiomyocyte differentiation, we sought to determine if Isl1 conversely plays an instructive role in myocardial lineage formation. Isl1 expression levels were upregulated from ED4–5 EBs (Supp. fig. 3a). To prematurely increase Isl1 expression levels in a temporally and physiologically relevant way, we transiently transfected an Isl1 expression construct (30 ng/105 cells) into dissociated ED2 EB cells and re-aggregated them for further differentiation (Fig. 4a). This resulted in about a twofold increase in Isl1 levels at ED6 (Fig. 4b). Myocardial differentiation was monitored by sarcomeric gene (e.g., Myh7, Mlc2v, Actc1) expression over the course of EB differentiation. Sarcomeric gene expression levels did not change during the early phase of CPC differentiation (data not shown). However, by ED8, Isl1-transfected EBs expressed higher levels of cardiac muscle genes than control EBs (Fig. 4c). To determine the effect of excess Isl1 on the number of cardiomyocytes, we utilized the Myh7-GFP ES cell line to quantify cardiomyocytes. We observed a 25% increase in Myh7+ cells in Isl1-overexpressed EBs (Fig. 4d, Supp. fig. 3b). This suggests that Isl1 can promote myocardial differentiation of CPCs in an instructive manner.
Figure 4.

Increased levels of Isl1 promote myocardial differentiation. a, Schematic diagram of differentiation of Myh7-GFP ES cells with Isl1 overexpression. b, c, Relative expression levels of Isl1 on ED6 EBs (b), and endothelial (Flk1), cardiac sarcomeric (Actc1, Mlc2v, Myh7) and smooth muscle (Sma) genes on day 8 EBs (c), determined by qPCR. d, FACS analyses on ED 9 EBs to identify % of cells entering myocardial-lineage. Data are mean ± s. e. m.; n=3; *P < 0.005.
In addition to Isl1, Myocd and Smyd1 are important genes for cardiogenesis23–27 that were downregulated in CPCs with increased β-Catenin (Fig. 2c–g, i–k). Myocd is a potent coactivator for serum response factor regulation of smooth muscle24 and cardiac gene expression27. Smyd1 is a muscle-restricted histone methyltransferase essential for cardiomyocyte differentiation in vivo23, 25. To determine whether Isl1 regulates these genes in CPCs, we used Nkx2.5-GFP+ CPCs purified from the stable Isl1-KD ES cell line. Smyd1 levels did not change, but Myocd levels were significantly reduced in the Isl1-KD CPCs (Fig. 5a). To determine if this is also the case in vivo, we performed in situ hybridization for Myocd transcripts in Isl1-null embryos. In agreement with in vitro data, Myocd levels were severely compromised in Isl1-null embryos, while Smyd1 levels did not change (Fig. 5b–i). This suggests that Isl1 is required for normal Myocd expression.
Figure 5.
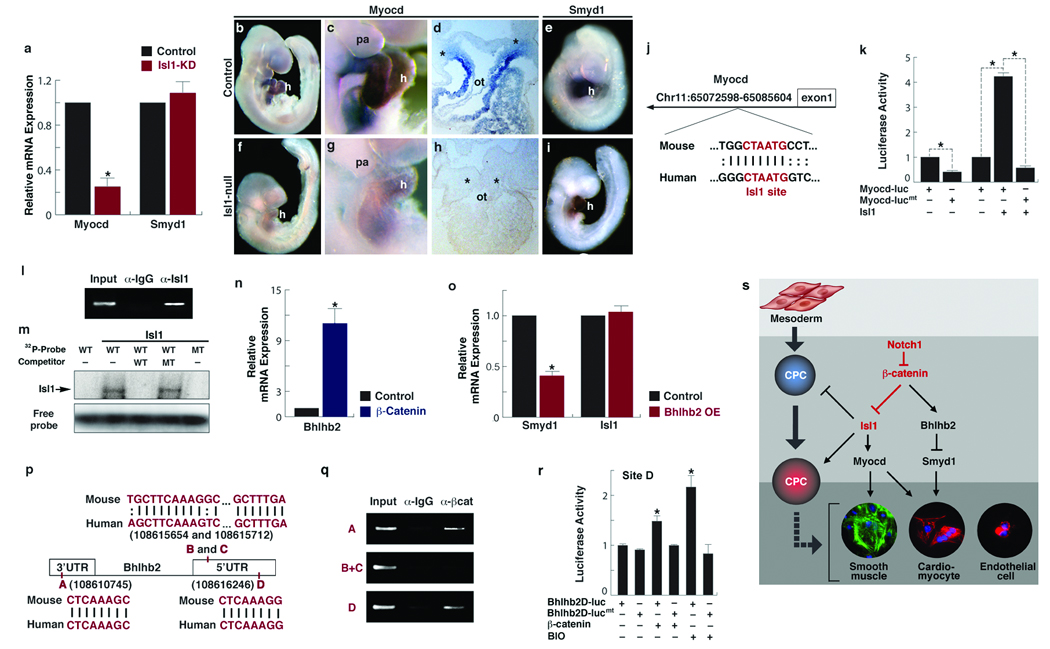
Isl1 targets Myocd and β-Catenin regulates Bhlhb2 to repress Smyd1. a, Relative expression levels of Myocd and Smyd1 in FACS-purified control and Isl1 knockdown (KD) CPCs, determined by qPCR (mean ± s. d.; n=4; *P < 0.005). b–i, Control (b–e) and Isl1-null (f–i) embryos at E 9.5 after in situ hybridization with Myocd (b–d, f–h), or Smyd1 (e, i) riboprobes. c, g, Lateral views focused on heart (h) and pharyngeal arch (pa) regions. d, h, Transverse section through the outfow tract. Asterisks indicate pre-cardiac mesoderm. Scale bars, 100 µm. j, Location of the conserved island containing Isl1 binding site (red) in the Myocd locus. k, Relative luciferase activity determined with luciferase reporters linked to the conserved island with the intact Isl1 site (Myocd-luc) or with a mutant Isl1 site (Myocd-lucmt) in the presence or absence of Isl1 (mean ± s. d.; n=3; *P < 0.005). l, Chromatin immunoprecipitation (ChIP) assay shows specific PCR amplification of the Isl1 consensus site shown in j, representing association with Isl1 protein. m, Electophoretic mobility shift assay with in-vitro synthesized Isl1 protein and radiolabeled probes (Probe) spanning the Isl1 site shown in j. Unlabeled probes were used as competitors. WT, wildtype; MT, mutant. n, Relative expression levels of Bhlhb2 in CPCs with stabilized β-Catenin, determined by qPCR (mean ±s. d.; n=3; *P < 0.005).. o, Relative expression levels of Smyd1 and Isl1 after transfecting FACS-purified CPCs with Bhlhb2 and differentiating them for 3 days (mean ± s. d.; n=3; *P < 0.005). p, The Bhlhb2 locus showing four conserved Lef/Tcf binding sites. q, ChIP assays performed with Lef/Tcf consensus sites shown in p. β-Catenin forms complexes with sites A and D as revealed by amplification of those sites. r, Relative luciferase activity determined with luciferase reporters containing the intact Lef/Tcf site D (Bhlhb2D-luc) or with a mutant Lef/Tcf site D (Bhlhb2D-lucmt) in the presence or absence of β-Catenin or BIO (2µM). Data are mean ± s. d.; n=3; *P < 0.005. s, A molecular cascade involving Notch1/ β-Catenin/ Isl1 during CPC fate determination. Notch1 functions to negatively regulate accumulation of free β-Catenin, which regulates Myocd and Smyd1 through Isl1 and Bhlhb2, respectively, to determine CPC fates. Relationships indicated may be direct or indirect.
Through bioinformatic searches, we identified an Isl1 consensus site in an evolutionarily conserved island (555 bp) located in the first intron of the Myocd locus (Fig. 5j). We observed robust transactivation of luciferase when the element was linked to luciferase reporter and introduced into ED6–8 EBs (when endogenous Isl1 is enriched and biologically functional) (Fig. 5k). However, the luciferase activity was significantly reduced when the Isl1 site was mutated (Fig. 5k). In addition, excessive Isl1 further increased luciferase activity with the Isl1 site intact but not with the site mutated (Fig. 5k). Chromatin immunoprecipitation (ChIP) with anti-Isl1 antibodies in ED8 EBs revealed that the site was associated with Isl1 protein (Fig. 5l). This association was further confirmed by electrophoretic mobility shift analyses that showed the specific binding of Isl1 to the site (Fig. 5m). Together, these data suggest that Isl1 may directly regulate Myocd expression.
Since Isl1 did not affect Smyd1 expression, we hypothesized that β-Catenin might activate a transcriptional repressor to downregulate Smyd1 expression. Among the transcriptional repressors affected by β-Catenin in our array, Bhlhb2 was the most highly upregulated. Bhlhb2 is a basic helix-loop-helix (bHLH)-containing DNA-binding repressor that is involved in many biological processes, including proliferation, differentiation and regulation of circadian rhythms28–30. qPCR confirmed that Bhlhb2 was highly upregulated in embryos with stabilized β-Catenin (Fig. 5n) consistent with the upregulation by in situ hybridization in the cardiac area and other domains of Isl1Cre activity (Fig. 2h, l). Overexpression of Bhlhb2 in Nkx2.5-GFP+ CPCs mimicked the Smyd1 repression observed with β-Catenin stabilization (Fig. 5o). Isl1 expression was not affected by Bhlhb2, providing an important control (Fig. 5o). We identified four conserved Lef/Tcf consensus sites in the 5′ and 3′ UTRs of Bhlhb2 (Fig. 5p) and tested whether any were directly bound by β-Catenin. ChIP with anti-β-Catenin antibodies in ED8 EBs revealed that two of the four sites (A and D) were indeed associated with β-Catenin (Fig. 5q). To determine which site can mediate Wnt/β-Catenin signaling, conserved elements encompassing the Lef/Tcf sites were individually inserted upstream of luciferase reporter and examined luciferase activity in ED8 EBs. We found that the construct containing site D, but not A, resulted in a significant increase in luciferase activity upon stimulation with β-Catenin or 6-bromoindirubin-3′-oxime (BIO), a Wnt/β-Catenin signaling activator (Fig. 5r). This increase was, however, not observed in cells transfected with a mutant construct (Fig. 5r). These data suggest that Bhlhb2 may be a direct target of the Wnt signal.
Through use of mouse genetics and the embryonic stem cell system, we have shown that Wnt/β-Catenin signaling functions as a central regulator of CPCs by integrating signals from the Notch pathway and regulating a cascade of downstream transcriptional events involving Isl1, Myocd and Smyd1 (Fig. 5s). We found that Notch1 activity within CPCs was required for their exit from the expansive state into the differentiated state, providing the first evidence for Notch signaling requirement within multipotent CPCs in vivo. Consistent with Notch1’s negative regulation of active β-Catenin, Notch1 loss-of-function and β-Catenin gain-of-function had similar effects on expression of the cardiac transcription factors, Isl1, Myocd, Smyd1 and Bhlhb2. Our finding that CPCs in vivo and in vitro had greater expansion upon disruption of Isl1 and that Isl1 could promote differentiation suggests that despite its very transient expression, Isl1 triggers the further development of CPCs into cardiac cells rather than promoting its renewal state. Strikingly, Isl1 downregulation induced by β-Catenin was necessary for Wnt/β-Catenin-induced expansion of CPCs. These findings reveal a regulatory network controlling CPC expansion and cell fate that involve unanticipated functions of β-Catenin, Notch1 and Isl1 that may be leveraged for regenerative approaches involving CPCs.
Methods
Mouse Genetics and CPC and ES Cell Culture
The control (RosaYFP/+; Isl1Cre/+) or mutant (RosaYFP/+; Isl1Cre/+; β-Catenin(ex3)loxPloxP/+) embryos were obtained by crossing RosaYFP/+; β-Catenin(ex3)loxPloxP/+ with Isl1Cre/+ mice20, 31. YFP+ cells from the resulting embryos were purified by FACS and used for gene expression analyses. To quantify embryonic CPCs, RosaYFP/+; Isl1Cre/+ were crossed with Isl1Cre/+ mice, and YFP+ cells from the resulting embryos were counted by FACS. To generate Isl1Cre/+; Notch1loxP/loxP, Isl1Cre/+; Notch1loxP/+ mice were crossed with Notch1loxP/loxP mice19. Genotyping was done as described2. To identify Isl1-het (Isl1Cre/+) or null (Isl1Cre/Cre) embryos, DNA was isolated from individual embryos, and qPCR was done using SYBR Green (Applied Biosystems) with control Isl1 and Cre primers shown in Supp. table 2. ES cells and purified Nkx2.5-GFP+ CPCs were propagated undifferentiated or differentiated as previously described2. For CPC differentiation, the FACS-purified GFP+ cells were re-aggregated in suspension (105 cells per well) in ultra-low-attachment 24-well plates (Corning).
Flow Cytometry and Gene Expression Analysis
A Becton Dickinson FACS Diva flow cytometer and cell sorter were used for quantifying and purifying Nkx2.5-GFP+ or Myh7-GFP+ cells. For the microarray analysis and qPCR, total RNA was amplified with the WT-Ovation Pico RNA Amplification System, fragmented and labeled with the FL-Ovation cDNA Biotin Module V2 (Nugen). The hybridization, staining and scanning of the Affymetrix GeneChips were performed in the Gladstone Genomics Core Lab. Raw data generated from at least three independent experiments were further analyzed by the group of Dr. Ru-Fang Yeh at the Center for Informatics and Molecular Biostatistics, UCSF. To quantify gene expression in Notch1 mutant embryos, total RNA was isolated from hearts and pharyngeal arches from E10.0 embryos. qPCR was performed with the ABI Prism system (7900HT, Applied Biosystems). TaqMan primers used in this study are listed in Supp. table 2. All samples were run at least in triplicate. Real-time PCR data were normalized and standardized with SDS2.2 software.
Constructs, siRNA, Transfection, EMSA and Luciferase Assays
For Isl1-KD experiments, an Isl1 shRNA construct set (RMM4534-NM_021459, Open Biosystems) was used to transiently transfect EBs and to generate stable KD ES cell lines. For Isl1 or Bhlhb2 overexpression studies, their full-length cDNAs (Open Biosystems) were amplified and cloned into the pEF-DEST51 vector (pDEST51-Isl1 or Bhlhb2) through the pENTR vector (pENTR-Isl1 or Bhlhb2) using the Gateway system (Invitrogen). pEF-lacZ (Invitrogen) was used as a control. For Notch1-KD studies, Block-iT Alexa Fluor Red (46–5318, Invitrogen) or Notch1 siRNA (M-041110-00-0005, Dharmacon) was used at concentration of 50 or 100 nM. Myocd-luc was generated by cloning their corresponding regions into the pGL3 luciferase vector (Promega). Myocd-lucmt was generated using QuikChange Site-Directed Mutagenesis Kit (Stratagene). For Bhlhb2D-luc and Bhlhb2D-lucmt generation, oligonucleotides containing the Tcf/Lef site were cloned into the pGL3 vector. All the oligonucleotide sets are listed in Supp. table 2. Stabilized β-Catenin and Top/Fop-flash luciferase constructs were kindly provided by Dr. A. Barth (Stanford University) and the laboratory of Dr. R. Moon (University of Washington), respectively. ES cells, EBs or CPCs were transfected with indicated constructs or siRNA using Lipofectamine 2000 (Invitrogen) after generating single-cell suspension with Accutase (Chemicon). EMSAs and luciferase assays were performed as described previously32, 33. For EMSAs, the pCITE-ISL134 construct containing the truncated Isl1 cDNA with the homeodomain was kindly provided by Dr. B. Black (University of California, San Francisco) and used to generate Isl1 protein. All EMSA probes are listed in Supp. table 2. For luciferase assays, Renilla was used as an internal normalization control.
In Situ Hybridization, Immunostaining and Western Analysis
Whole-mount in situ hybridization was performed as described with designated antisense probes4, 23, 26. Bhlhb2 antisense riboprobe was synthesized and purified from pENTR-Bhlhb2. To detect proliferating cells in CPCs, embryo sections were stained with anti-Phospho-histone H3 (Upstate) and anti-Isl1 (DSHB). To visualize Isl1 protein in Notch1 mutant embryos, the TSA System (PerkinElmer) was used to amplify Isl1 signals. Nuclear β-Catenin was detected with anti-PY489 antibody (DSHB). For western blotting, lysates from day 3 CPCs after transfection with indicated siRNAs were analyzed using antibodies against Notch1 (DSHB), Dephospho β-Catenin (Calbiochem), and GAPDH (Santa Cruz Biotechnology).
Chromatin Immunoprecipitation Assays
For chromatin immunoprecipitation (ChIP) assay, EBs were treated with BIO (2.5 uM) or transfected with Isl1 or β-Catenin constructs22 (100 ng/ 105 cells) from ED 5–7, and harvested at ED 8. Cross-linking of histones to DNA, chromatin extraction, immunoprecipitation and elution were performed using the ChIP Assay Kit (Upstate) with anti-IgG-HRP, Isl1 (Abcam) or β-Catenin (Santa Cruz Biotechnology). PCR primer sets spanning the indicated Lef/Tcf binding sites in the Bhlhb2 locus are shown in Supp. table 2.
Supplementary Material
a, Relative Isl1 expression levels in GFP− and GFP+ cells isolated from Day 5 Nkx2.5-GFP EBs, determined by qPCR (mean ± s. d.; n=3). b, Immunostaining of transverse sections through the pre-cardiac mesoderm and outflow tract (ot) of indicated E 9.5 mouse embryos for nuclear β-Catenin. Higher levels of β-Catenin are observed in precardiac regions (arrowheads) in Notch1 mutants. Numerous Wnts are expressed in the ectodermal cells and form a gradient pattern of active β-Catenin (arrows), providing a positive control. c, qPCR data of positively affected genes in cardiac progenitors with stabilized β-Catenin (mean ± s. d.; n=3; *P< 0.01). d, Transverse sections of corresponding embryos (Fig. 2-l), focused on precardiac mesoderm (asterisk) and outflow tract (ot) area. h, heart. e, Relative Bhlhb2 expression levels in hearts and precardiac mesoderm from E10.0 control or Isl1Cre; Notch1flox/flox embryos (Notch KO), determined by qPCR (mean ± s. d.; n=3; *P < 0.01). f, Relative Isl1 expression levels in EBs 2 days after transfection with an Isl1 siRNA constuct (transient Isl1-KD, left) and in ED6 EBs differentiated from control and stable Isl1-KD lines (stable Isl1-KD, right), determined by qPCR (mean ± s. d.; n=3; *P < 0.01), Scale bars, 100 µm.
a, Histograms showing percentages of GFP+ cells of ED6, 7, and 8 EBs after transient transfection with an Isl1 siRNA construct on ED3. b, Histograms showing percentages of GFP+ cells of ED6 EBs differentiated from control and stable Isl1-KD lines.
a, Relative Isl1 expression levels in EBs at indicated days of differentiation (ED), determined by qPCR. b, Histograms showing percentages of Myh7+ cells entering myocardial-lineage in ED9 EBs.
Acknowledgements
We thank R. Kopan (Washington Univeristy, St. Louis, MO) and M. M. Taketo (Kyoto University, Kyoto, Japan) for providing Notch1flox and β-Catenin/loxP(ex3)loxP mice, respectively. The authors thank G. Howard and S. Ordway for editorial assistance, R.F. Yeh for statistical analyses, K. Cordes for graphical assistance and Srivastava lab members for helpful discussions. C.K. was supported by a fellowship from the American Heart Association (AHA) and California Institute for Regenerative Medicine (CIRM); D.S. was an Established Investigator of the AHA and was supported by grants from NHLBI/NIH and CIRM. This work was also supported by NIH/NCRR grant (C06 RR018928) to the Gladstone Institutes.
Footnotes
Accession Number
The full microarray data performed in this study are available in NCBI Gene Expression Omnibus (GEO, accession number: GSE15232).
Competing interests statement: The authors declare no competing financial interests.
References
- 1.Cohen ED, et al. Wnt/beta-catenin signaling promotes expansion of Isl-1-positive cardiac progenitor cells through regulation of FGF signaling. J Clin Invest. 2007;117:1794–1804. doi: 10.1172/JCI31731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kwon C, et al. Canonical Wnt signaling is a positive regulator of mammalian cardiac progenitors. Proc Natl Acad Sci U S A. 2007;104:10894–10899. doi: 10.1073/pnas.0704044104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Qyang Y, et al. The renewal and differentiation of Isl1+ cardiovascular progenitors are controlled by a Wnt/beta-catenin pathway. Cell Stem Cell. 2007;1:165–179. doi: 10.1016/j.stem.2007.05.018. [DOI] [PubMed] [Google Scholar]
- 4.Cai CL, et al. Isl1 identifies a cardiac progenitor population that proliferates prior to differentiation and contributes a majority of cells to the heart. Dev Cell. 2003;5:877–889. doi: 10.1016/s1534-5807(03)00363-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yang L, et al. Human cardiovascular progenitor cells develop from a KDR+ embryonic-stem-cell-derived population. Nature. 2008;453:524–528. doi: 10.1038/nature06894. [DOI] [PubMed] [Google Scholar]
- 6.Buckingham M, Meilhac S, Zaffran S. Building the mammalian heart from two sources of myocardial cells. Nat Rev Genet. 2005;6:826–835. doi: 10.1038/nrg1710. [DOI] [PubMed] [Google Scholar]
- 7.Wu SM, et al. Developmental origin of a bipotential myocardial and smooth muscle cell precursor in the mammalian heart. Cell. 2006;127:1137–1150. doi: 10.1016/j.cell.2006.10.028. [DOI] [PubMed] [Google Scholar]
- 8.Moretti A, et al. Multipotent embryonic isl1+ progenitor cells lead to cardiac, smooth muscle, and endothelial cell diversification. Cell. 2006;127:1151–1165. doi: 10.1016/j.cell.2006.10.029. [DOI] [PubMed] [Google Scholar]
- 9.Laugwitz KL, et al. Postnatal isl1+ cardioblasts enter fully differentiated cardiomyocyte lineages. Nature. 2005;433:647–653. doi: 10.1038/nature03215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kattman SJ, Huber TL, Keller GM. Multipotent flk-1+ cardiovascular progenitor cells give rise to the cardiomyocyte, endothelial, and vascular smooth muscle lineages. Dev Cell. 2006;11:723–732. doi: 10.1016/j.devcel.2006.10.002. [DOI] [PubMed] [Google Scholar]
- 11.Srivastava D. Making or breaking the heart: from lineage determination to morphogenesis. Cell. 2006;126:1037–1048. doi: 10.1016/j.cell.2006.09.003. [DOI] [PubMed] [Google Scholar]
- 12.Christoforou N, et al. Mouse ES cell-derived cardiac precursor cells are multipotent and facilitate identification of novel cardiac genes. J Clin Invest. 2008;118:894–903. doi: 10.1172/JCI33942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lyons I, et al. Myogenic and morphogenetic defects in the heart tubes of murine embryos lacking the homeo box gene Nkx2-5. Genes Dev. 1995;9:1654–1666. doi: 10.1101/gad.9.13.1654. [DOI] [PubMed] [Google Scholar]
- 14.Stanley EG, et al. Efficient Cre-mediated deletion in cardiac progenitor cells conferred by a 3′UTR-ires-Cre allele of the homeobox gene Nkx2-5. Int J Dev Biol. 2002;46:431–439. [PubMed] [Google Scholar]
- 15.Prall OW, et al. An Nkx2-5/Bmp2/Smad1 negative feedback loop controls heart progenitor specification and proliferation. Cell. 2007;128:947–959. doi: 10.1016/j.cell.2007.01.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hayward P, Kalmar T, Arias AM. Wnt/Notch signalling and information processing during development. Development. 2008;135:411–424. doi: 10.1242/dev.000505. [DOI] [PubMed] [Google Scholar]
- 17.Rones MS, McLaughlin KA, Raffin M, Mercola M. Serrate and Notch specify cell fates in the heart field by suppressing cardiomyogenesis. Development. 2000;127:3865–3876. doi: 10.1242/dev.127.17.3865. [DOI] [PubMed] [Google Scholar]
- 18.Nemir M, Croquelois A, Pedrazzini T, Radtke F. Induction of cardiogenesis in embryonic stem cells via downregulation of Notch1 signaling. Circ Res. 2006;98:1471–1478. doi: 10.1161/01.RES.0000226497.52052.2a. [DOI] [PubMed] [Google Scholar]
- 19.Radtke F, et al. Deficient T cell fate specification in mice with an induced inactivation of Notch1. Immunity. 1999;10:547–558. doi: 10.1016/s1074-7613(00)80054-0. [DOI] [PubMed] [Google Scholar]
- 20.Srinivas S, et al. Cre reporter strains produced by targeted insertion of EYFP and ECFP into the ROSA26 locus. BMC Dev Biol. 2001;1:4. doi: 10.1186/1471-213X-1-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hsiao EC, et al. Marking embryonic stem cells with a 2A self-cleaving peptide: a NKX2-5 emerald GFP BAC reporter. PLoS ONE. 2008;3:e2532. doi: 10.1371/journal.pone.0002532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Votin V, Nelson WJ, Barth AI. Neurite outgrowth involves adenomatous polyposis coli protein and beta-catenin. J Cell Sci. 2005;118:5699–5708. doi: 10.1242/jcs.02679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gottlieb PD, et al. Bop encodes a muscle-restricted protein containing MYND and SET domains and is essential for cardiac differentiation and morphogenesis. Nat Genet. 2002;31:25–32. doi: 10.1038/ng866. [DOI] [PubMed] [Google Scholar]
- 24.Li S, Wang DZ, Wang Z, Richardson JA, Olson EN. The serum response factor coactivator myocardin is required for vascular smooth muscle development. Proc Natl Acad Sci U S A. 2003;100:9366–9370. doi: 10.1073/pnas.1233635100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tan X, Rotllant J, Li H, De Deyne P, Du SJ. SmyD1, a histone methyltransferase, is required for myofibril organization and muscle contraction in zebrafish embryos. Proc Natl Acad Sci U S A. 2006;103:2713–2718. doi: 10.1073/pnas.0509503103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ueyama T, Kasahara H, Ishiwata T, Nie Q, Izumo S. Myocardin expression is regulated by Nkx2.5, and its function is required for cardiomyogenesis. Mol Cell Biol. 2003;23:9222–9232. doi: 10.1128/MCB.23.24.9222-9232.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang D, et al. Activation of cardiac gene expression by myocardin, a transcriptional cofactor for serum response factor. Cell. 2001;105:851–862. doi: 10.1016/s0092-8674(01)00404-4. [DOI] [PubMed] [Google Scholar]
- 28.Zawel L, et al. DEC1 is a downstream target of TGF-beta with sequence-specific transcriptional repressor activities. Proc Natl Acad Sci U S A. 2002;99:2848–2853. doi: 10.1073/pnas.261714999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Shen M, et al. Basic helix-loop-helix protein DEC1 promotes chondrocyte differentiation at the early and terminal stages. J Biol Chem. 2002;277:50112–50120. doi: 10.1074/jbc.M206771200. [DOI] [PubMed] [Google Scholar]
- 30.Honma S, et al. Dec1 and Dec2 are regulators of the mammalian molecular clock. Nature. 2002;419:841–844. doi: 10.1038/nature01123. [DOI] [PubMed] [Google Scholar]
- 31.Harada N, et al. Intestinal polyposis in mice with a dominant stable mutation of the beta-catenin gene. Embo J. 1999;18:5931–5942. doi: 10.1093/emboj/18.21.5931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kwon C, Hays R, Fetting J, Orenic TV. Opposing inputs by Hedgehog and Brinker define a stripe of hairy expression in the Drosophila leg imaginal disc. Development. 2004;131:2681–2692. doi: 10.1242/dev.01127. [DOI] [PubMed] [Google Scholar]
- 33.Kwon C, Han Z, Olson EN, Srivastava D. MicroRNA1 influences cardiac differentiation in Drosophila and regulates Notch signaling. Proc Natl Acad Sci U S A. 2005;102:18986–18991. doi: 10.1073/pnas.0509535102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dodou E, Verzi MP, Anderson JP, Xu SM, Black BL. Mef2c is a direct transcriptional target of ISL1 and GATA factors in the anterior heart field during mouse embryonic development. Development. 2004;131:3931–3942. doi: 10.1242/dev.01256. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
a, Relative Isl1 expression levels in GFP− and GFP+ cells isolated from Day 5 Nkx2.5-GFP EBs, determined by qPCR (mean ± s. d.; n=3). b, Immunostaining of transverse sections through the pre-cardiac mesoderm and outflow tract (ot) of indicated E 9.5 mouse embryos for nuclear β-Catenin. Higher levels of β-Catenin are observed in precardiac regions (arrowheads) in Notch1 mutants. Numerous Wnts are expressed in the ectodermal cells and form a gradient pattern of active β-Catenin (arrows), providing a positive control. c, qPCR data of positively affected genes in cardiac progenitors with stabilized β-Catenin (mean ± s. d.; n=3; *P< 0.01). d, Transverse sections of corresponding embryos (Fig. 2-l), focused on precardiac mesoderm (asterisk) and outflow tract (ot) area. h, heart. e, Relative Bhlhb2 expression levels in hearts and precardiac mesoderm from E10.0 control or Isl1Cre; Notch1flox/flox embryos (Notch KO), determined by qPCR (mean ± s. d.; n=3; *P < 0.01). f, Relative Isl1 expression levels in EBs 2 days after transfection with an Isl1 siRNA constuct (transient Isl1-KD, left) and in ED6 EBs differentiated from control and stable Isl1-KD lines (stable Isl1-KD, right), determined by qPCR (mean ± s. d.; n=3; *P < 0.01), Scale bars, 100 µm.
a, Histograms showing percentages of GFP+ cells of ED6, 7, and 8 EBs after transient transfection with an Isl1 siRNA construct on ED3. b, Histograms showing percentages of GFP+ cells of ED6 EBs differentiated from control and stable Isl1-KD lines.
a, Relative Isl1 expression levels in EBs at indicated days of differentiation (ED), determined by qPCR. b, Histograms showing percentages of Myh7+ cells entering myocardial-lineage in ED9 EBs.